Back to Journals » Therapeutics and Clinical Risk Management » Volume 14
Amantadine extended-release capsules for levodopa-induced dyskinesia in patients with Parkinson's disease
Authors Sharma VD, Lyons KE, Pahwa R
Received 25 January 2018
Accepted for publication 16 March 2018
Published 12 April 2018 Volume 2018:14 Pages 665—673
DOI https://doi.org/10.2147/TCRM.S144481
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Garry Walsh
Vibhash D Sharma, Kelly E Lyons, Rajesh Pahwa
Department of Neurology, University of Kansas Medical Center, Kansas City, KS, USA
Abstract: Levodopa-induced dyskinesia (LID) is a common motor complication in patients with Parkinson’s disease on chronic levodopa therapy. The management of LID is important as dyskinesia can be disabling and impair quality of life. Currently, there are limited treatment options for the medical management of LID. Amantadine extended-release capsules (Gocovri™) is the first medication that received US Food and Drug Administration approval for the treatment of LID. The following is a review of the pharmacodynamics, efficacy and safety profile, and current state of treatment of amantadine for LID.
Keywords: Parkinson’s disease, levodopa-induced dyskinesia, amantadine, amantadine extended-release
Introduction
Parkinson’s disease (PD) is a neurodegenerative disorder characterized by a loss of dopaminergic neurons in the substantia nigra pars compacta. Deficiency of dopamine within the striatum leads to subsequent development of motor symptoms, including bradykinesia, tremor, rigidity, and gait difficulty.1 The current management of PD relies on symptomatic control with medications acting on the dopaminergic system by either increasing dopamine concentrations in the brain or stimulating dopamine receptors.2 Levodopa is considered the most effective gold standard therapy for controlling motor symptoms in PD; however, its chronic use is associated with the development of motor complications, including motor fluctuations and dyskinesia.3
Levodopa-induced dyskinesia (LID) can have varied clinical presentations and are typically characterized by choreic or dystonic movements involving the limbs, orofacial–buccal musculature, and the trunk. LID can present as “peak-dose dyskinesia,” “diphasic dyskinesia (onset and end of dose dyskinesia),” and “OFF-period dystonia.”3 Approximately 40% of patients have a likelihood of developing LID after 4–6 years of treatment with levodopa and >90% of patients can develop dyskinesia after treatment for 10 years.4 Dyskinesia can become disabling to patients over time, impairing quality of life, affecting activities of daily living, and increasing caregiver burden.4–7 Even though dyskinesia is a well-recognized complication of chronic levodopa therapy, there are limited medical treatment options for LID and the management of dyskinesia is often challenging.
Pathophysiology of LID
Induction of LID involves complex pathophysiological changes, which are not well understood. The degree of nigro-striatal degeneration and exposure to levodopa are important factors in the origin of LID.3,8 Loss of dopaminergic neurons in the substantia nigra causes dysfunction of dopamine storage and release pre-synaptically, and alterations in the striatal output pathways post-synaptically.9 Normally dopamine has an excitatory response through D1 receptors on direct pathways and an inhibitory response through D2 receptors on the indirect pathway. Motor movements are facilitated by activation of direct pathways and reduced by activation of indirect pathways. In PD, with the loss of dopamine, there is underactivity of direct pathways and overactivity of indirect pathways, which, in turn, leads to a decrease in thalamocortical glutamatergic output and results in hypokinetic movements.10 It is thought that with chronic levodopa therapy, there is overstimulation of D1 and D2 receptors, which leads to overactivity of direct pathways and inhibition of indirect pathways. These changes lead to increased glutamatergic thalamocortical output and result in excess motor movements or LID (Figure 1). However, these changes, based on the classic basal ganglia model, are not sufficient to fully explain the induction of dyskinesia.9 Dyskinesia, in part, also involve changes in glutamatergic receptors (including N-methyl-D-aspartate [NMDA]) within the striatum, which lead to maladaptive plasticity across the glutamatergic corticostriatal synapse.11 It is believed that multiple changes also occur in the glutamatergic neurotransmitter systems as PD progresses. There is increased glutamate release leading to more glutamate in the post-synaptic receptors and upregulation in the number and activity of glutamatergic receptors.11 These changes lead to increased glutamatergic signaling and neurotransmission enhancing excitatory signaling in both direct and indirect pathways.11 Higher levels of exogenous dopamine in the form of levodopa, bias glutamatergic signaling toward the direct pathway leading to excessive movements or dyskinesia.12 Hence, blocking or reducing glutamatergic signaling may help reduce motor complications. The role of glutamatergic receptors in dyskinesia is supported by the fact that NMDA receptor antagonists, such as dextromethorphan and amantadine reduce LID.9,13
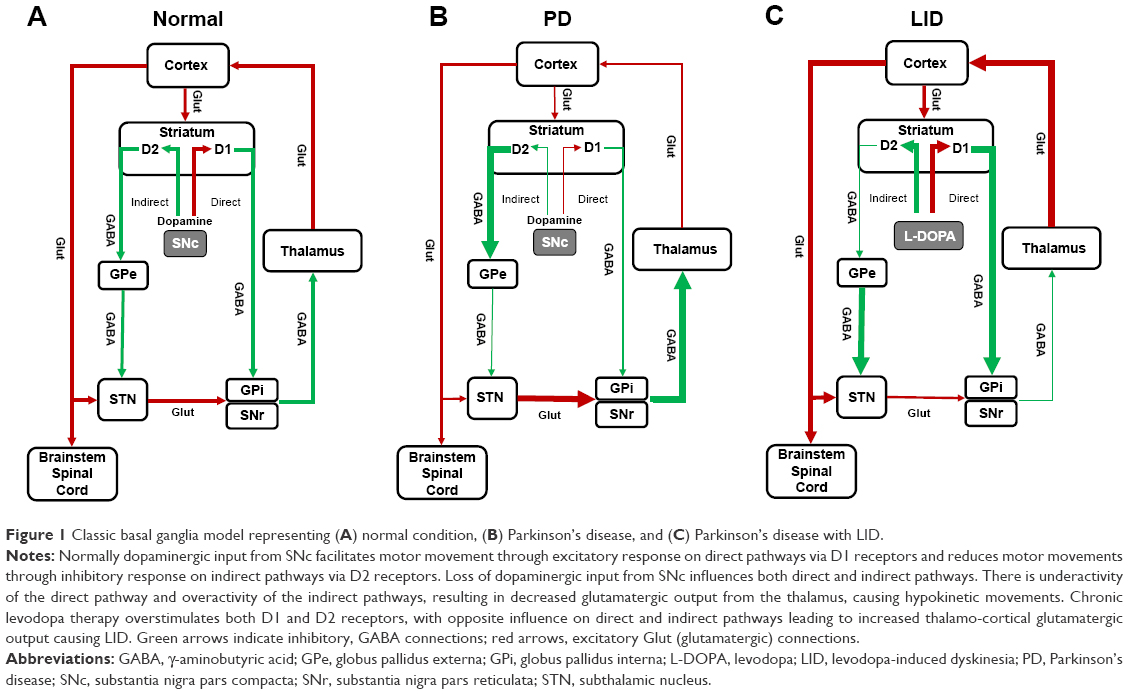 | Figure 1 Classic basal ganglia model representing (A) normal condition, (B) Parkinson’s disease, and (C) Parkinson’s disease with LID. |
Pharmacological treatment of dyskinesia
Treatment of LID should be considered when symptoms become disabling and impair quality of life. A detailed evaluation of patients with regard to the type of dyskinesia and dopaminergic medications is required for the management of LID.
OFF-period dystonia, which commonly occurs at night or early morning, can be improved by adding a long-acting levodopa formulation at bedtime. For OFF-period dystonia during the day, the addition of a dopamine agonist, monoamine oxidase type B inhibitor (MAO-B) or a catechol-O-methyltransferase (COMT) inhibitor can be considered.3,14 Peak-dose dyskinesia frequently require adjustment of dopaminergic medications. Management of peak-dose dyskinesia involves adjustment of levodopa to a smaller and/or more frequent dose and/or reduction or discontinuation of dopamine-enhancing medications, such as MAO-B or COMT inhibitors. Patients who are on longer acting levodopa (controlled release) can have erratic absorption of levodopa, which is implicated in LID, thus switching them to an immediate release (IR) form of levodopa throughout the day can improve LID.3,14 Diphasic dyskinesia can be challenging to treat. In patients with diphasic dyskinesia, reducing levodopa dose and increasing or adding a dopamine agonist can improve dyskinesia. Reducing or discontinuing MAO-B or COMT inhibitors can also be considered.14 Evaluation for deep brain stimulation (DBS) therapy or carbidopa–levodopa enteral suspension, which provide a steady concentration of levodopa,15 can be considered in selected cases. Anti-dyskinetic medications, such as amantadine or clozapine can be added to improve LID. The clinical benefits of amantadine in dyskinesia are discussed later. Clozapine has been shown to improve dyskinesia, however, its use is limited by potential adverse events, including agranulocytosis, seizures, and myocarditis. Mandatory monitoring of complete blood count is needed due to concern for agranulocytosis.14
Amantadine
Amantadine is an oral NMDA receptor antagonist originally approved as a prophylactic agent against influenza. Later, it was serendipitously discovered to have an anti-parkinsonian effect with improvement in bradykinesia, rigidity, and tremor.16 Amantadine has been shown to have anti-dyskinetic effects. In 2017, amantadine extended-release (ER) was the first medication that received US Food and Drug Administration approval for the treatment of LID.
Pharmacodynamics
Amantadine acts on both dopaminergic and non-dopaminergic systems. It acts pre-synaptically to enhance dopamine release17 and inhibit dopamine reuptake.18 Amantadine also acts on glutamatergic receptors and exerts an NMDA receptor antagonist effect,19 which can contribute in dopaminergic transmission at higher concentrations.20 It has been suggested that the anti-dyskinetic effects of amantadine are secondary to inhibition of NMDA receptors.9,21
Pharmacokinetics
Amantadine (1-adamantanamine) is an aliphatic primary amine, which is commonly formulated as a hydrochloride (HCL) salt for clinical use. The available strength of amantadine HCL is 100 mg tablets, which is equivalent to 81 mg of actual amantadine without salt. Median time to reach maximum concentration for amantadine HCL is around 2–4 hours and the half-life is around 17 hours. The maximum plasma concentration achieved after a single dose of amantadine HCL 100 mg is around 220 ng/mL.22 The plasma concentration of amantadine HCL IR at doses of 100 mg twice a day achieves peak plasma concentrations of <1,000 ng/mL. In human and animal studies, it has been reported that the EC50, which is the concentration of the drug that induces a response halfway between baseline and maximum response, for amantadine is 1,400 ng/mL. This would suggest that a level of amantadine 1,400 ng/mL would reduce dyskinesia by 50%.23
Amantadine ER (Gocovri™, Adamas Pharmaceuticals, Inc., Emeryville, CA, USA) is an extended-release capsule that contains beads with the active ingredient, amantadine HCL. Amantadine ER dissolves upon entry to the gastrointestinal system and releases slow-release coated beads. Pores or channels from the coating enable the drug to leave the bead in a slow fashion. The capsule can be opened and the beads can be used without the capsule as long as all the beads in the capsule are used.24 Amantadine ER capsules are available in strengths of 68.5 and 137 mg (equivalent to 85 or 170 mg amantadine HCL, respectively). It is generally initiated with 137 mg daily at bedtime and increased to the recommended dose of 274 mg daily after 1 week. The median time to reach maximum concentration is around 12 hours, and the drug achieves steady state concentration within 4 days of dose initiation. Since ~20% of the drug is released in the first 4 hours and the rest is released slowly over the next 6–8 hours, it should be dosed at bedtime. This allows the drug to reach sustained concentration during the morning and throughout the daytime, and the concentration decreases toward evening and throughout the night. Once-daily dosing of 274 mg at bedtime achieves peak plasma concentrations of ~1,500 ng/mL and sustains levels throughout the day. The drug is primarily excreted unchanged in the urine. It is contraindicated for patients with end-stage renal disease and dosing should be reduced in patients with mild-to-severe renal impairment.24,25
Amantadine in dyskinesia
The effect of amantadine HCL IR formulation on LID has been examined in multiple small studies. In a 6-week, placebo-controlled, double-blind, crossover study, Verhagen Metman et al found that PD patients (n=18) receiving amantadine IR (average dose of 350 mg/day) had around a 60% reduction in dyskinesia compared with placebo. Dyskinesia was evaluated using the modified Abnormal Involuntary Movement Scale during a steady-state levodopa infusion.26 There was significant improvement in patient-reported dyskinesia assessed by Unified Parkinson’s Disease Rating Scale (UPDRS) part IV, items 32 (duration of dyskinesia) and 33 (severity of dyskinesia). The UPDRS IV, item 39 (proportion of the day in “off” state, scale 0–4 with 0=none, 4=76%–100% of waking day) was also reduced in the amantadine group with a mean score of 1 with amantadine and 1.5 with placebo (p<0.01). The authors conducted a follow-up study and found that 13 of 17 patients originally treated in the short-term study had a 56% reduction in dyskinesia scores at 1 year.27
A similar study by Snow et al, assessed the effect of amantadine IR 200 mg per day on LID in a double-blind, crossover, placebo-controlled trial in PD patients (n=24). The authors utilized a dyskinesia rating scale (0–4 range, for each limb, trunk, head, neck, and face) and found around a 24% reduction in total dyskinesia scores at week 3 during a levodopa challenge. There was significant improvement in the UPDRS III motor score in the “OFF” state in the amantadine group compared with placebo (p=0.04).28 Although these studies provided evidence for the anti-dyskinetic effect of amantadine, the studies were not of sufficient size or duration.
The duration of the anti-dyskinetic effect of amantadine was analyzed in a few studies. In a placebo-controlled trial by Thomas et al, amantadine IR (300 mg) was found to reduce dyskinesia by about 45% after 15 days of treatment; however, the anti-dyskinetic effect wore off after around 8 months of treatment in the majority of patients.29 Two washout studies were conducted to assess the long-term anti-dyskinetic effect of amantadine. Wolf et al studied 32 PD patients on amantadine IR for a mean duration of 4.8 years. Patients were randomized to continue amantadine or switched to placebo and followed for an additional 3 weeks. In patients receiving placebo, there was a significant worsening of LID from baseline as assessed by UPDRS IV items 32 and 33 (p=0.02) compared with no significant change in patients staying on amantadine.30 In the AMANDYSK trial,31 57 PD patients with LID treated with amantadine for an average of 3.4 years were enrolled in a 3-month double-blind, placebo-controlled, washout design. There was significant worsening of dyskinesia scores in patients switched to placebo compared (+1.7 points) with those maintained on amantadine HCL (+0.2), suggesting a long-term anti-dyskinetic effect.
Amantadine ER (Gocovri™)
Amantadine ER has demonstrated safety and efficacy in reducing LID in 3 randomized, double-blinded, placebo-controlled clinical trials (Table 1).32–34 In the following section, we will review the therapeutic efficacy and safety profile of oral amantadine ER capsules for LID.
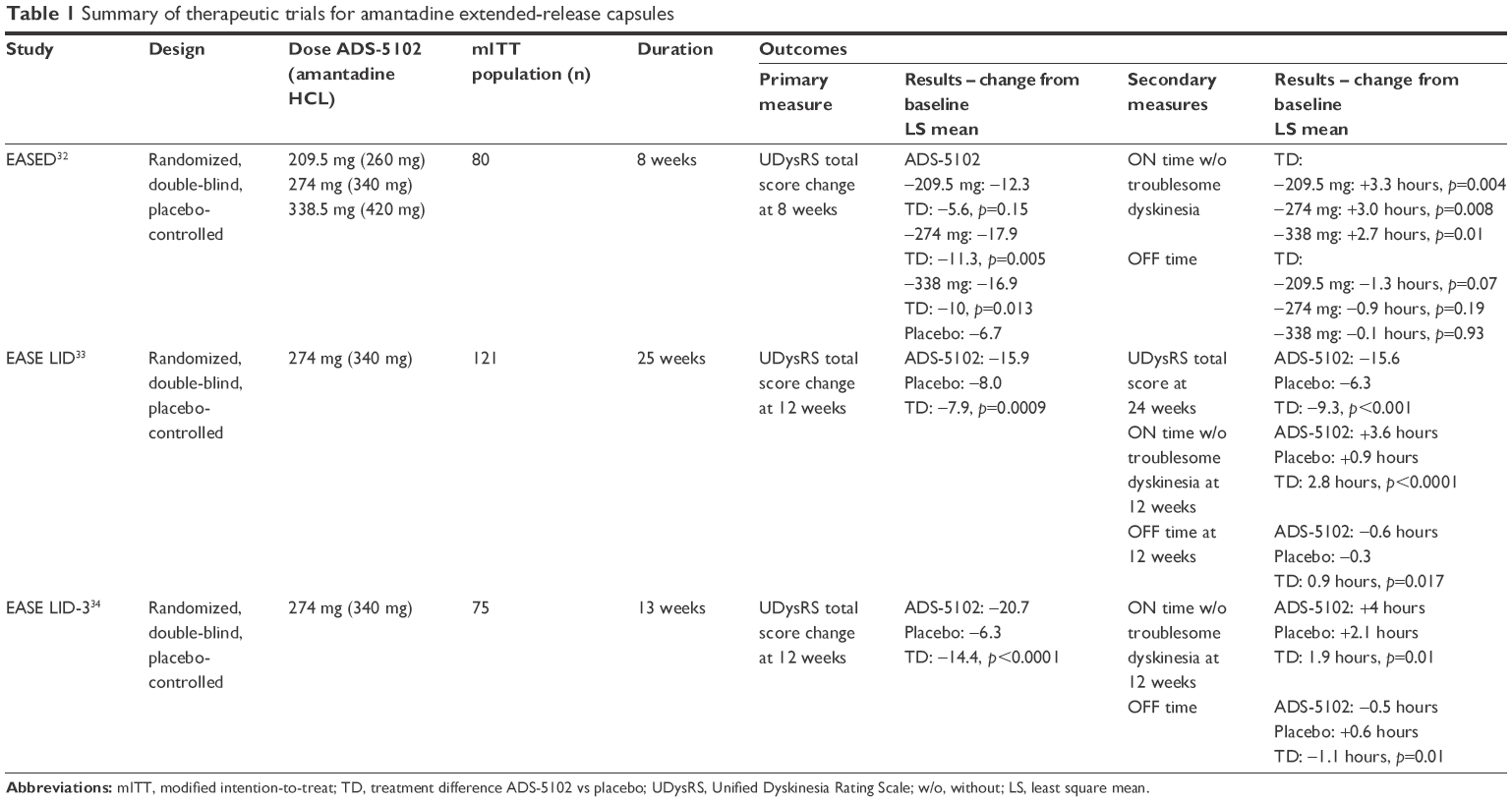 | Table 1 Summary of therapeutic trials for amantadine extended-release capsules |
Therapeutic efficacy
EASED study
EASED was the first dose-finding study to assess tolerability and efficacy with 3 different doses of amantadine ER capsules (ADS-5102). The study involved 83 PD patients between 30 and 85 years of age, who had troublesome LID. Based on the inclusion criteria, patients enrolled into this study had a score of at least 2 on part IV, item 4.2 (functional impact of dyskinesia, range 0–4) of the Movement Disorder Society (MDS)-UPDRS, and at least 2 half-hour periods of ON time with troublesome dyskinesia during the day time (9 AM–4 PM) on 2 consecutive days. All patients were on levodopa at least 3 times per day and PD medications were unchanged during the study period. Patients with diphasic, myoclonic, dystonic, or akathetic dyskinesia without peak-dose dyskinesia were excluded.32
Patients were randomized in a 1:1:1:1 ratio to placebo or 1 of 3 doses of ADS-5102 capsule containing 260, 340, and 420 mg of amantadine HCL. The primary outcome measure was the change in the Unified Dyskinesia Rating Scale (UDysRS) at week 8 compared with baseline.32 The UDysRS has 4 parts: 1) historical disability (patient perceptions) of ON-dyskinesia impact (maximum 44 points); 2) historical disability (patient perceptions) of OFF-dystonia impact (maximum 16 points); 3) objective impairment (dyskinesia severity, anatomical distribution over 7 body regions, and type (choreic or dystonic) based on 4 activities observed or video-recorded (28 points); 4) objective disability based on part III activities (maximum 16 points).35
The primary efficacy analysis compared the mean change in UDysRS total scores in the amantadine group with placebo at 8 weeks. The mean percentage improvement in UDysRS total score at 8 weeks was 43% for the amantadine group receiving 340 mg. When compared with placebo, this represented a reduction of 27% (p=0.005). Improvement was also seen in patients who received a dose of 420 mg (25% reduction compared with placebo, p=0.013), however, no significant improvement was seen in patients receiving the 260 mg dose. A significant increase in the duration of ON time without troublesome dyskinesia (assessed by 24 hour PD diaries) was noted in all the patients receiving amantadine ER capsules vs placebo, 3.3 hours for 260 mg (p=0.004), 3 hours for 340 mg (p=0.008), and 2.7 hours for 420 mg dose (p=0.018). The mean duration of OFF time reduced in patients receiving 260 mg (−1.3 hours) and 340 mg dose (−0.9 hours) compared with placebo, however, it did not reach statistical significance. In the 340 mg group, 75% of patients reported moderate-to-marked improvement in their clinical status as measured by the Clinical Global Impression of Change (CGI-C) scale.32
EASE LID study
In the EASE LID Phase III, randomized, double-blind, placebo-controlled clinical trial, the efficacy and safety of amantadine ER capsules given at the dose of 274 mg at bedtime was evaluated. This dose is equivalent to 340 mg of amantadine HCL, which was shown to be safe and efficacious in the previously discussed EASED study.32 A total of 126 patients were randomized to either placebo or amantadine ER capsules. The inclusion and exclusion criteria were similar to the EASED study.32 The total duration of treatment was 25 weeks, in which the amantadine group received 137 mg for 1 week followed by 274 mg for the duration of the study (week 2–24). The primary outcome was the change in UDysRS total score at 12 weeks compared with baseline.33
The UDysRS total score in the patients receiving medication improved significantly compared with placebo, with a difference of 7.9 points at week 12, which improved to 9.3 at week 24. The mean duration of ON time without troublesome dyskinesia in the amantadine ER arm compared with placebo improved significantly, with a treatment difference of 2.8 hours at 12 weeks and 2.2 hours at 24 weeks (p<0.001). There was a reduction in the mean duration of ON time with troublesome dyskinesia, with a treatment difference of 1.6 hours at 12 weeks and 1.5 hours at 24 weeks. There was also a significant reduction in OFF time in the amantadine ER group with a treatment difference of 0.9 hours at 12 weeks and 0.8 hours at 24 weeks (p<0.05). There were no significant differences in MDS-UPDRS combined or individual scores (part I, II, and III) between the amantadine and placebo groups. The CGIC results showed that 81% of patients at 12 weeks and 68.3% at 24 weeks in the amantadine group reported improvement in PD symptoms and dyskinesia. The majority of patients tolerated amantadine ER without serious complications.33
EASE LID-3 study
EASE LID-3 was the second pivotal trial to evaluate the safety and efficacy of amantadine ER capsules. The trial design, inclusion and exclusion criteria were similar to the EASE LID study.33 The study was conducted for 13 weeks and 77 PD patients were randomized to either placebo or amantadine ER. In the amantadine group, patients received 137 mg of amantadine ER for 1 week followed by 274 mg from weeks 2 to12. The primary outcome measure was the change in UDysRS total score at week 12 compared with baseline.34
The UDysRS total score in the patients receiving amantadine improved significantly by 20.7 points at 12 weeks compared with baseline. When compared with placebo, a difference of 14.4 points (p<0.0001) was found. The mean duration of ON time without troublesome dyskinesia improved significantly by 1.9 hours (treatment difference, p=0.01) and ON time with troublesome dyskinesia reduced by 1.3 hours in the amantadine group compared with placebo. The mean duration of OFF time was reduced by 0.5 hours in the amantadine group, which corresponds to a difference of 1.1 hours when compared with placebo (p=0.019). When comparing MDS-UPDRS scores between the 2 groups at 12 weeks, a significant treatment effect was found for the amantadine group in MDS-UPDRS part II (p=0.01) and combined part I, II, and III scores (p=0.04). The combined part I, II, and III mean MDS-UPDRS scores reduced by 8.4 points in the amantadine group. Approximately 51% in the amantadine group reported moderate-to-marked improvement in overall PD symptoms at 12 weeks.34
EASE LID-2 study
The EASE LID-2 is a long-term open-label study of the patients who participated in the controlled studies. In addition, it included patients who had undergone DBS treatment for PD. The study population (n=223) included patients who received amantadine ER (n=61) or placebo (n=79) in EASE LID and EASE LID-3 and prior eligible patients who were excluded from trials due to DBS therapy (n=61). Patients from previous trials (EASED, EASE LID, and EASE LID-3) with a time gap to enrollment in the current study were also included (n=22). Patients were treated with amantadine ER for up to 101 weeks. The primary outcome measure was adverse events (AEs) and efficacy was assessed using the MDS-UPDRS, part IV (motor complications) and its subparts (range 0–4 for each question).
The mean MDS-UPDRS, part IV scores were compared from baseline to up to week 64. In patients who originally received placebo (n=78), there was a reduction of 3.4 points at week 8 and 2.3 points at week 64 with amantadine ER. In the DBS group, there was a reduction of 4 points at week 8 and 4.5 points at week 64. Patients who were on amantadine previously had low baseline scores, which were maintained through week 64. In the overall group, the MDS-UPDRS, part IV, item 4.1 (time spent with dyskinesia) reduced by 0.6 at week 8 and 0.7 at week 64, and MDS-UPDRS part IV, item 4.2 (functional impact of dyskinesia) improved by 0.8 at week 8 and 0.9 at week 64. At 64 weeks, MDS-UPDRS part IV, item 4.3 (time spent in OFF state), improved by 0.3 points in the patients who were previously on placebo and by 0.5 points in patients who had prior DBS.
Tolerability/safety of amantadine ER capsules
Overall, amantadine ER is relatively safe and well tolerated by patients with PD and dyskinesia (Table 2).
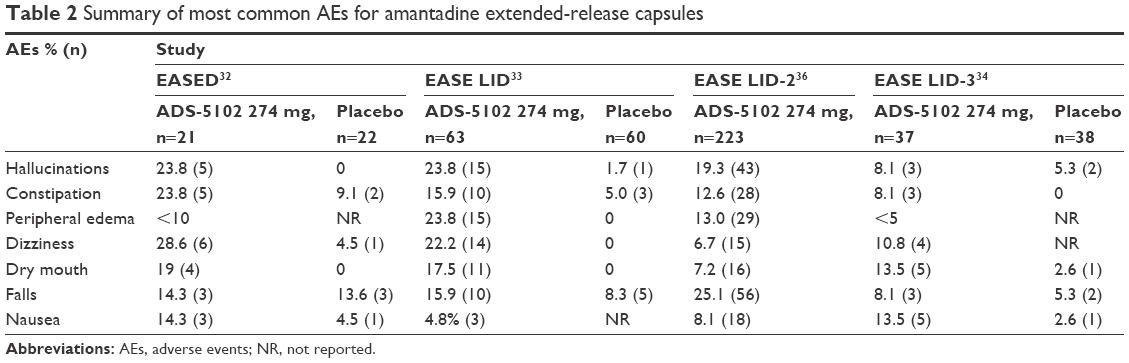 | Table 2 Summary of most common AEs for amantadine extended-release capsules |
In the EASED study, for the amantadine ER group (340 mg, n=21) the common AEs included constipation, dizziness, hallucinations, and dry mouth. In this group, 1 patient developed livedo reticularis.32 In EASE LID, the most common AEs in the amantadine ER group were visual hallucinations, peripheral edema, dizziness, dry mouth, constipation, and falls. In 15 patients who reported visual hallucinations, 5 discontinued the study, 3 had dose reduction, and 7 continued treatment. A total of 13 patients (20.6%) in the amantadine group discontinued the study drug due to AEs. Visual hallucinations were the most common reason for study discontinuation, followed by peripheral edema and dry mouth. In 3 patients, the dose was reduced to 137 mg. In the amantadine ER group, 5 patients had auditory hallucinations; of these, 2 had both visual and auditory hallucinations. There were no reports of impulse control disorder or serious AEs associated with the medication.33
In the EASE LID-3 study, the most common AEs in the amantadine ER group were dry mouth, nausea, decreased appetite, insomnia, and orthostatic hypotension. In this study, hallucinations were less common than in previous studies and were reported by 3 patients. Of the patients receiving amantadine ER, 7 patients discontinued the study and visual hallucinations were reported as the most common reason for discontinuation.33
In the EASE LID-2 study, evaluating the long-term safety and tolerability of amantadine ER 274 mg capsules, the most common AEs reported were falls, hallucinations, peripheral edema, and constipation. Of the 45 patients who experienced hallucinations, visual hallucinations occurred in the majority of patients (n=43) and/or auditory hallucinations in 5 patients were reported. Other pertinent AEs reported include livedo reticularis in 18 patients and impulse control disorder in 4 patients. In 32 patients (14.3%), AEs were the primary reason for discontinuing the study; the most common included falls (2.2%), visual hallucinations (2.2%), and peripheral edema (0.9%).36
Discussion
The introduction of levodopa therapy transformed the management of PD symptoms.37 However, the management can become challenging with disease progression, particularly with the development of motor fluctuations and dyskinesia in patients on chronic levodopa therapy. Amantadine is currently considered the most effective treatment for LID. Multiple other medications studied for LID, including dextromethorphan, memantine, quetiapine, and riluzole, are not recommended14,38,39 and different investigational drugs to reduce dyskinesia have failed in human trials either due to AEs, poor benefit and/or worsening of PD symptoms.40–42
Amantadine ER is approved in the USA “for the treatment of dyskinesia in patients with PD receiving levodopa-based therapy, with or without concomitant dopaminergic medications.”25 Amantadine ER is given at bedtime and the recommended daily dose is 274 mg.33,34 It achieves peak concentration in plasma during the daytime when patients experience bothersome dyskinesia; concentrations are lower at night, reducing the potential for sleep-related side effects.33,34 The common side effects of amantadine ER include constipation, dry mouth, hallucinations, peripheral edema, and nausea. Hallucinations can occur in around 20% of patients and may require adjustment of other dopaminergic agents. Other less common side effects include livedo reticularis and impulse control disorder.32–34,36 As the drug is renally eliminated, the dose should be adjusted in patients with impaired renal function. The maximum recommended dose in moderate and severe renal impairment is 137 mg and 68.5 mg per day, respectively. For patients with end-stage renal disease, amantadine ER is contraindicated. It is recommended to slowly discontinue the drug to avoid any withdrawal adverse reactions.25
Compared with amantadine HCL 100 mg, which is typically given 2 or 3 times per day, amantadine ER has an advantage with once-daily dosing. Amantadine ER achieves a plasma concentration of around 1,500 ng/mL, this concentration is above the concentration required to reduce dyskinesia by 50% (EC50~1,400 ng/mL) based on validated PD dyskinesia models. Amantadine HCL 100 mg is unable to achieve these concentration levels when given 2 times per day and barely reaches these concentrations when given 3 times daily.24 The clinical utility of amantadine IR is limited at higher doses due to increased adverse effects.26,29 Although, there are no comparison studies between amantadine IR and ER formulations, a sub-group analysis of the ongoing open-label EASE LID-3 study showed patients who were switched from amantadine IR to ER (n=32) had further improvement in dyskinesia and OFF time. There was an improvement of 3.4 points in MDS-UPDRS part IV scores at 8 weeks and the improvement persisted up to 64 weeks.43 No difference was found in the safety profile of patients who were switched from amantadine IR to ER. Based on these analyses, the authors suggested that patients on amantadine IR can be switched to amantadine ER without interruption.43 Also, in previous trials, amantadine ER was shown to reduce the duration of OFF time and motor scores.33,34 These results can guide clinicians who are considering switching patients to ER formulations; however, long-term experience is needed.
Disclosure
Dr Pahwa is a consultant for Adamas Pharmaceuticals and has received research support through his institution from Adamas. Dr Sharma and Dr Lyons have no conflicts of interest to disclose in this work.
References
Kalia LV, Lang AE. Parkinson’s disease. Lancet. 2015;386(9996):896–912. | ||
Connolly BS, Lang AE. Pharmacological treatment of Parkinson disease: a review. JAMA. 2014;311(16):1670–1683. | ||
Guridi J, Gonzalez-Redondo R, Obeso JA. Clinical features, pathophysiology, and treatment of levodopa-induced dyskinesias in Parkinson’s disease. Parkinson Dis. 2012;2012:943159. | ||
Ahlskog JE, Muenter MD. Frequency of levodopa-related dyskinesias and motor fluctuations as estimated from the cumulative literature. Mov Disord. 2001;16(3):448–458. | ||
Hechtner MC, Vogt T, Zollner Y, et al. Quality of life in Parkinson’s disease patients with motor fluctuations and dyskinesias in five European countries. Parkinsonism Relat Disord. 2014;20(9):969–974. | ||
Perez-Lloret S, Negre-Pages L, Damier P, et al. L-DOPA-induced dyskinesias, motor fluctuations and health-related quality of life: the COPARK survey. Eur J Neurol. 2017;24(12):1532–1538. | ||
Khlebtovsky A, Rigbi A, Melamed E, et al. Patient and caregiver perceptions of the social impact of advanced Parkinson’s disease and dyskinesias. J Neural Transm (Vienna). 2012;119(11):1367–1371. | ||
Papathanou M, Rose S, McCreary A, Jenner P. Induction and expression of abnormal involuntary movements is related to the duration of dopaminergic stimulation in 6-OHDA-lesioned rats. Eur J Neurosci. 2011;33(12):2247–2254. | ||
Iravani MM, Jenner P. Mechanisms underlying the onset and expression of levodopa-induced dyskinesia and their pharmacological manipulation. J Neural Transm (Vienna). 2011;118(12):1661–1690. | ||
DeLong MR, Wichmann T. Basal ganglia circuits as targets for neuromodulation in Parkinson disease. JAMA Neurol. 2015;72(11):1354–1360. | ||
Finlay C, Duty S. Therapeutic potential of targeting glutamate receptors in Parkinson’s disease. J Neural Transm (Vienna). 2014;121(8):861–880. | ||
Bravo SA, Rangel-Barajas C, Garduño BF. Pathophysiology of L-dopa induced dyskinesia–changes in D1/D3 receptors and their signaling pathway. In: Rana AQ, editor. A Synopsis of Parkinson’s Disease. Rijeka: InTech; 2014:Ch. 05. | ||
Elahi B, Phielipp N, Chen R. N-Methyl-D-Aspartate antagonists in levodopa induced dyskinesia: a meta-analysis. Can J Neurol Sci. 2012;39(4):465–472. | ||
Vijayakumar D, Jankovic J. Drug-induced dyskinesia, part 1: treatment of levodopa-induced dyskinesia. Drugs. 2016;76(7):759–777. | ||
Antonini A, Fung VS, Boyd JT, et al. Effect of levodopa-carbidopa intestinal gel on dyskinesia in advanced Parkinson’s disease patients. Mov Disord. 2016;31(4):530–537. | ||
Hubsher G, Haider M, Okun MS. Amantadine: the journey from fighting flu to treating Parkinson disease. Neurology. 2012;78(14):1096–1099. | ||
Von Vigtlander PF, Moore KE. Dopamine: release from the brain in vivo by amantadine. Science. 1971;174(4007):408–410. | ||
Heikkila RE, Cohen G. Evaluation of amantadine as a releasing agent or uptake blocker for H3-dopamine in rat brain slices. Eur J Pharmacol. 1972;20(2):156–160. | ||
Danysz W, Parsons CG, Kornhuber J, Schmidt WJ, Quack G. Aminoadamantanes as NMDA receptor antagonists and antiparkinsonian agents–preclinical studies. Neurosci Biobehav Rev. 1997;21(4):455–468. | ||
Mizoguchi K, Yokoo H, Yoshida M, Tanaka T, Tanaka M. Amantadine increases the extracellular dopamine levels in the striatum by re-uptake inhibition and by N-methyl-D-aspartate antagonism. Brain Res. 1994;662(1–2):255–258. | ||
Blanchet PJ, Konitsiotis S, Chase TN. Amantadine reduces levodopa-induced dyskinesias in parkinsonian monkeys. Mov Disord. 1998;13(5):798–802. | ||
Symmetrel (amantadine hydrochloride) [prescribing information]. Chadds Ford, PA: Endo Pharmaceuticals Inc; 2009. | ||
Brigham EF, Johnston TH, Brown C, et al. Pharmacokinetic/pharmacodynamic analysis of amantadine for levodopa-induced dyskinesia: correlation of therapeutic plasma concentrations from multiple species with humans. Program No. 573.05. 2017 Neuroscience Meeting Planner. Washington, DC: Society for Neuroscience; 2017. | ||
Gocovrihcp.com. Available from: https://www.gocovrihcp.com/dosing-and-pharmacokinetics/. Accessed December 29, 2017. | ||
Gocovri (amantadine) extended release capsules [package inert]. Emeryville, CA: Adamas Pharmaceuticals, Inc.; 2017. | ||
Verhagen Metman L, Del Dotto P, van den Munckhof P, Fang J, Mouradian MM, Chase TN. Amantadine as treatment for dyskinesias and motor fluctuations in Parkinson’s disease. Neurology. 1998;50(5):1323–1326. | ||
Metman LV, Del Dotto P, LePoole K, Konitsiotis S, Fang J, Chase TN. Amantadine for levodopa-induced dyskinesias: a 1-year follow-up study. Arch Neurol. 1999;56(11):1383–1386. | ||
Snow BJ, Macdonald L, McAuley D, Wallis W. The effect of amantadine on levodopa-induced dyskinesias in Parkinson’s disease: a double-blind, placebo-controlled study. Clin Neuropharmacol. 2000;23(2):82–85. | ||
Thomas A, Iacono D, Luciano AL, Armellino K, Di Iorio A, Onofrj M. Duration of amantadine benefit on dyskinesia of severe Parkinson’s disease. J Neurol Neurosurg Psychiatry. 2004;75(1):141–143. | ||
Wolf E, Seppi K, Katzenschlager R, et al. Long-term antidyskinetic efficacy of amantadine in Parkinson’s disease. Mov Disord. 2010;25(10):1357–1363. | ||
Ory-Magne F, Corvol JC, Azulay JP, et al. Withdrawing amantadine in dyskinetic patients with Parkinson disease: the AMANDYSK trial. Neurology. 2014;82(4):300–307. | ||
Pahwa R, Tanner CM, Hauser RA, et al. Amantadine extended release for levodopa-induced dyskinesia in Parkinson’s disease (EASED Study). Mov Disord. 2015;30(6):788–795. | ||
Pahwa R, Tanner CM, Hauser RA, et al. ADS-5102 (amantadine) extended-release capsules for levodopa-induced dyskinesia in parkinson disease (EASE LID Study): a randomized clinical trial. JAMA Neurol. 2017;74(8):941–949. | ||
Oertel W, Eggert K, Pahwa R, et al. Randomized, placebo-controlled trial of ADS-5102 (amantadine) extended-release capsules for levodopa-induced dyskinesia in Parkinson’s disease (EASE LID 3). Mov Disord. 2017;32(12):1701–1709. | ||
Goetz CG, Nutt JG, Stebbins GT. The Unified Dyskinesia Rating Scale: presentation and clinimetric profile. Mov Disord. 2008;23(16):2398–2403. | ||
Hauser RA, Pahwa R, Tanner CM, et al. ADS-5102 (Amantadine) Extended-Release Capsules for Levodopa-Induced Dyskinesia in Parkinson’s Disease (EASE LID 2 Study): Interim Results of an Open-Label Safety Study. J Parkinsons Dis. 2017;7(3):511–522. | ||
LeWitt PA, Fahn S. Levodopa therapy for Parkinson disease: a look backward and forward. Neurology. 2016;86(14 Suppl 1):S3–S12. | ||
Pahwa R, Factor SA, Lyons KE, et al. Practice Parameter: treatment of Parkinson disease with motor fluctuations and dyskinesia (an evidence-based review): report of the Quality Standards Subcommittee of the American Academy of Neurology. Neurology. 2006;66(7):983–995. | ||
Fox SH, Katzenschlager R, Lim SY, et al. The movement disorder society evidence-based medicine review update: treatments for the motor symptoms of Parkinson’s disease. Mov Disord. 2011;26(Suppl 3):S2–S41. | ||
Goetz CG, Damier P, Hicking C, et al. Sarizotan as a treatment for dyskinesias in Parkinson’s disease: a double-blind placebo-controlled trial. Mov Disord. 2007;22(2):179–186. | ||
Lewitt PA, Hauser RA, Lu M, et al. Randomized clinical trial of fipamezole for dyskinesia in Parkinson disease (FJORD study). Neurology. 2012;79(2):163–169. | ||
Kumar R, Hauser RA, Mostillo J, et al. Mavoglurant (AFQ056) in combination with increased levodopa dosages in Parkinson’s disease patients. Int J Neurosci. 2016;126(1):20–24. | ||
Isaacson S, Pahwa R, Tanner C, et al. ADS-5102 provided reduction in motor complications in Parkinson’s disease patients with levodopa-induced dyskinesia switched from amantadine IR: subgroup analysis from open-label study (ease LID 2). J Neurol Sci. 2017;381:355. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.