Back to Journals » International Journal of Nanomedicine » Volume 14
Alzheimer’s disease: pathogenesis, diagnostics, and therapeutics
Authors Tiwari S ![]() , Atluri V, Kaushik A, Yndart A
, Atluri V, Kaushik A, Yndart A ![]() , Nair M
, Nair M
Received 5 January 2019
Accepted for publication 2 March 2019
Published 19 July 2019 Volume 2019:14 Pages 5541—5554
DOI https://doi.org/10.2147/IJN.S200490
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Thomas Webster
Sneham Tiwari, Venkata Atluri, Ajeet Kaushik, Adriana Yndart, Madhavan Nair
Department of Immunology and Nano-Medicine, Institute of NeuroImmune Pharmacology, Herbert Wertheim College of Medicine, Florida International University, Miami, FL 33199, USA
Abstract: Currently, 47 million people live with dementia globally, and it is estimated to increase more than threefold (∼131 million) by 2050. Alzheimer’s disease (AD) is one of the major causative factors to induce progressive dementia. AD is a neurodegenerative disease, and its pathogenesis has been attributed to extracellular aggregates of amyloid β (Aβ) plaques and intracellular neurofibrillary tangles made of hyperphosphorylated τ-protein in cortical and limbic areas of the human brain. It is characterized by memory loss and progressive neurocognitive dysfunction. The anomalous processing of APP by β-secretases and γ-secretases leads to production of Aβ40 and Aβ42 monomers, which further oligomerize and aggregate into senile plaques. The disease also intensifies through infectious agents like HIV. Additionally, during disease pathogenesis, the presence of high concentrations of Aβ peptides in central nervous system initiates microglial infiltration. Upon coming into vicinity of Aβ, microglia get activated, endocytose Aβ, and contribute toward their clearance via TREM2 surface receptors, simultaneously triggering innate immunoresponse against the aggregation. In addition to a detailed report on causative factors leading to AD, the present review also discusses the current state of the art in AD therapeutics and diagnostics, including labeling and imaging techniques employed as contrast agents for better visualization and sensing of the plaques. The review also points to an urgent need for nanotechnology as an efficient therapeutic strategy to increase the bioavailability of drugs in the central nervous system.
Keywords: amyloid beta, amyloidogenesis, amyloid precursor proteins, β-secretases, γ-secretases, tau phosphorylation
Introduction
Alzheimer’s disease (AD) is a neurodegenerative and prominent protein-conformational disease (PCD)1,2 primarily caused by the aberrant processing and polymerization of normally soluble proteins.3 When misfolded, soluble neuronal proteins attain altered conformations, due to genetic mutation, external factors, or aging, and aggregate, leading to abnormal neuronal functions and loss.4 AD’s discovery as a neurodegenerative disease is attributed to Alois Alzheimer, a German neurologist who examined a 51-year-old woman named Auguste Deter, who was suffering with loss of memory, language, disorientation, and hallucinations. Her autopsy revealed plaques and tangles in the cerebral cortex,5 which convinced him that this went beyond typical dementia. His discovery was followed by further research that revealed the presence of neuritic amyloid β (Aβ) plaques in dementia patients.6 Young onset of the disease is attributed to predisposition to PS1 genetic mutation, which is a rare but potent cause.7 Other neurodegenerative diseases associated with abnormal protein conformations are Parkinson’s disease, Creutzfeldt–Jakob disease, Huntington’s disease, and Machado–Joseph disease, which are caused by abnormalities in the α-synuclein, Cellular Prion protein (PrPc), Scrapie prion protein (PrPSc), Htt, and Ataxin3 proteins, respectively. Upon understanding the causal factors and pathogenesis mechanism of the disease, it becomes of the utmost importance to address such fields as AD mechanisms, pathogenesis, and diagnosis, and finally how to design novel therapeutics against it (Figure 1).
 |
Figure 1 Overview of fields of research that need to be elucidated to understand the pathophysiology of Alzheimer’s disease and develop therapeutic strategies against it. |
Diagnostic and imaging techniques include nanoparticle (NP)-based sensitive early-phase detection of AD biomarkers like Aβ and τ in cerebrospinal fluid (CSF) samples from patients. Nanomaterials can also be used as contrast agents for imaging aggregated Aβ plaques. It is imperative to understand the role of NPs in increasing the efficacy and bioavailability of the drug across the blood–brain barrier (BBB) into the central nervous system (CNS). This review includes a detailed analysis of the pathogenic pathway leading toward full-blown AD, addresses current diagnostics and therapeutics available, and emphasizes the potential role of nanotechnology in therapeutics against disease progression.
AD pathogenesis
The field of research toward understanding AD pathogenesis and designing efficient therapies is vast. AD is a highly complex and progressive neurodegenerative disease.8 It is one of the leading cause of dementia cases globally. In the US alone, approximately 5.3 million Americans have AD, of which 5.1 million are aged 65 years or older and 200,000 have younger-onset AD.9 Reported histopathological characteristics of AD are extracellular aggregates of Aβ plaques and intracellular aggregations of neurofibrillary tangles (NFTs), composed of hyperphosphorylated microtubule-associated τ. Aβ plaques develop initially in basal, temporal, and orbitofrontal neocortex regions of the brain and in later stages progress throughout the neocortex, hippocampus, amygdala, diencephalon, and basal ganglia. In critical cases, Aβ is found throughout the mesencephalon, lower brain stem, and cerebellar cortex as well. This concentration of Aβ triggers τ-tangle formation, which is found in the locus coeruleus and transentorhinal and entorhinal areas of the brain. In the critical stage, it spreads to the hippocampus and neocortex.10 Aβ and NFTs are considered the major players in disease progression, and this review focuses on the cause, pathogenesis, and factors associated with progression of AD.
Amyloid β and AD pathogenesis
Amyloid pathogenesis starts with altered cleavage of amyloid precursor protein (APP), an integral protein on the plasma membrane, by β-secretases (BACE1) and γ-secretases to produce insoluble Aβ fibrils. Aβ then oligomerizes, diffuses into synaptic clefts, and interferes with synaptic signaling.11,12 Consequently, it polymerizes into insoluble amyloid fibrils that aggregate into plaques. This polymerization leads to activation of kinases, which leads to hyperphosphorylation of the microtubule-associated τ protein, and its polymerization into insoluble NFTs. The aggregation of plaques and tangles is followed by microglia recruitment surrounding plaques. This promotes microglial activation and local inflammatory response, and contributes to neurotoxicity.
Structure and function of APP
APP belongs to a family of associated proteins that includes mammalian amyloid precursor like proteins (APLP1 and APLP2), and Amyloid precursor protein-like (APPL) in Drosophila. It is an integral transmembrane protein with extracellular domains (Figure 2). In a diseased state, APP generates amyloidogenic fragments through differential cleavage by enzymes.7 The physiological functions of APP remain less understood. Studies with transiently transfected cell lines show that APP moderates cell survival, growth, and motility, along with neurite outgrowth and functions, which are attributed to the release of soluble ectodomains upon normal cleavage of APP.13,14 The importance of APP has been highlighted by studies where neuronal abnormalities have been reported in animals injected with APP RNAi,15 and APP-ectodomain intracerebral injections have shown improved cognitive function and synaptic density.16 APP encodes type 1 transmembrane glycoprotein, which is cleaved either via a nonamyloidogenic pathway (normal state) or via an amyloidogenic pathway (diseased state).17 APP releases various polypeptides that arise possibly due to alternative splicing, glycosylation, phosphorylation, or complex proteolysis.18,19
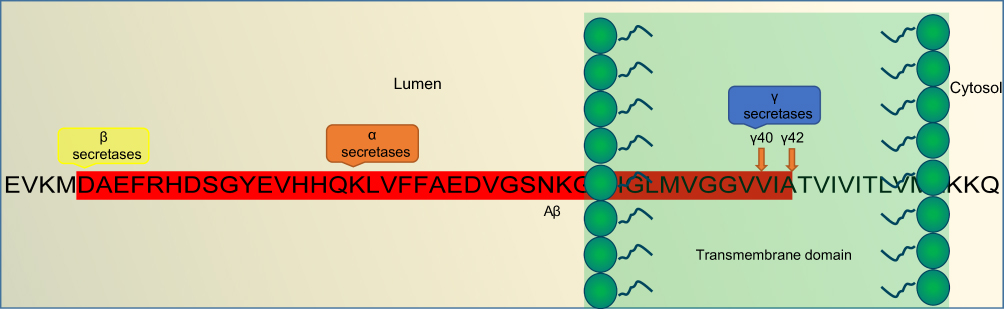 |
Figure 2 An overview of the Aβ-pathogenesis hypothesis. Note: Amino-acid sequence of the Aβ fragment and location of action of α-, β-, and γ-secretases in diseased neurons within a diseased amyloidogenic pathway.Abbreviation: Aβ, amyloid β. |
APP comprises 770 amino acids, of which Aβ includes 28 residues and an additional 14 residues from the transmembrane domain of APP. At the cleavage site, α-secretase cleaves and secretes large soluble ectodomain APPsα into the medium and the C-terminal fragment C83 is retained in the membrane, which is further cleaved by γ- secretase at residue 711, releasing soluble P3 peptide. Alternatively, in a diseased state, abnormal cleavage is done by β-secretase releasing truncated APPsβ and C-terminal fragment C99 is retained in the membrane and further cleaved by γ-secretase, releasing insoluble Aβ peptides. Cleavage of both C83 and C99 by γ-secretase releases the APP intracellular domain into the cytoplasm, which is soluble and translocates to nuclei for further gene-expression function.5
Nonamyloidogenic pathway
APP undergoes constitutive and regulated cleavage. The α-secretase enzyme cleaves APP at residues 16–17 of the Aβ domain and yield soluble and nonpathogenic precursors. In neurons, ADAM10 and ADAM17 (metalloprotease) are considered the major α-secretases. Processing by α-secretase and γ-secretase generates the small hydrophobic fragment p3, which is soluble and has a role in normal synaptic signaling, but its exact functions are still to be elucidated. It has been reported that cell-surface APP may get endocytosed as well, resulting in endosomal production of Aβ, which leads to extracellular release and aggregation of Aβ. The α-secretase processing releases the large soluble ectodomain APPsα, which acts a neuroprotective factor and also has a role in cell–substrate adhesion. The presence of APPsα associates with normal synaptic signaling and adequate synaptic plasticity, learning, memory, emotional behavior, and neuronal survival. Further, sequential processing releases the APP intracellular domain, which translocates into nuclei and facilitates nuclear signaling and gene-expression and -regulation pathways.20
Amyloidogenic pathway
APP is cleaved differently in the diseased state. Aβ is released from APP through sequential cleavages by BACE-1, a membrane-spanning aspartyl protease with its active site situated in lumen, and γ-secretase, an intramembrane aspartyl protease that is made up of four proteins: presenilin, nicastrin, anterior pharynx-defective 1 (Aph1), and Psen2 complexed together.21 This complex contributes to the activity of γ-secretase, which produces insoluble and neurotoxic Aβ fragments. β-secretase cleavage is the first and rate-limiting step, making a cut at the N-terminus of Aβ. It removes the majority of the extracellular portion of the protein, leaving the C-terminal of APP,22 which is further cleaved at the C-terminus of Aβ, resulting in formation of the Aβ oligomers that further polymerize, forming aggregated plaques (Figure 3).
 |
Figure 3 Alternative splicing of APP in amyloidogenic and nonamyloidogenic pathways. Note: Cleavage of APP by α- and γ-secretases in normal state and alternative cleavage by β- and γ- secretases in diseased state. Abbreviations: C83, 83-amino-acid carboxyterminal; C99, 99-amino-acid membrane-bound fraction; AICD, APP intracellular domain. |
There are two main types of Aβ polymers that have direct a role in plaque formation and induced neurotoxicity: Aβ40 and Aβ42. Aβ40 is abundant and less neurotoxic than Aβ42, which is less abundant, highly insoluble, severely neurotoxic, and more aggregation-prone and acts as a toxic building fraction of Aβ assembly. Aβ40/Aβ42 aggregation results in blocked ion channels, altered calcium homeostasis, increased mitochondrial oxidative stress, and diminished energy metabolism and glucose regulation, which contributes to deterioration of neuronal health and finally to neuronal cell death.
Hyperphosphorylation of τ and AD
AD is also characterized by the presence of NFTs. These tangles are the result of hyperphosphorylation of the microtubule-associated τ protein.23 NFTs are fragments of paired and helically wound protein filaments in the cell cytoplasm of neurons and also in their processes. The τ protein has a microtubule-binding domain and coassembles with tubulin to form matured and stable microtubules.24,25 It has the capability of stabilizing microtubules and forming interconnecting bridges between contiguous microtubules to form a proper stable network of microtubules and hold them together. When the τ protein comes into contact with the kinases released, due to the abundance of Aβ in the environment, it gets hyperphosphorylated. Its hyperphosphorylation leads to its being oligomerized. The tubule gets unstable, due to dissociation of tubule subunits, which fall apart and then convert into big chunks of τ filaments, which further aggregate into NFTs. These NFTs are straight, fibrillary, and highly insoluble patches in the neuronal cytoplasm and processes, leading to abnormal loss of communication between neurons and signal processing and finally apoptosis in neurons (Figure 4).26 It has been reported that soluble Aβ controls cleavage and phosphorylation of τ for NFT generation.7
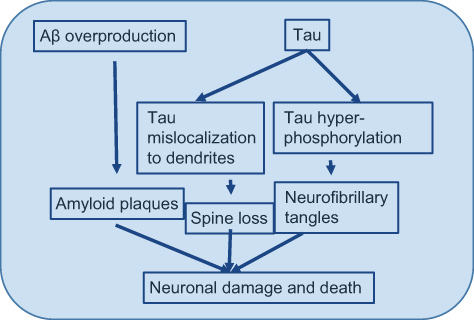 |
Figure 4 Hyperphosphorylationof τ. Note: Mechanism by which τ hyperphosphorylation leads to instability of the microtubule and finally microtubule subunits fall apart leading to formation of insoluble and big neurofibrillary tangles.Abbreviation: Aβ, amyloid β. |
Further, phosphorylation of τ is regulated by several kinases, including Glycogen Synthase kinase 3 (GSK3β) and cyclin-dependent kinase 5 (CDK5) activated by extracellular Aβ. Even though GSK3β and CDK5 are primarily responsible kinases for τ hyperphosphorylation, other kinases like Protein Kinase C, Protein Kinase A, ERK2, a serine/threonine kinase, caspase 3, and caspase 9 have prominent roles too, which may be activated by Aβ.27
GSK3β and CDK5 in AD
GSK3β regulates the cleavage of APP carboxyterminal fragments. Lithium and kenpaullone (two GSK3 inhibitors) prevent GSK3 expression and contribute to inhibition of Aβ production.28 As such, GSK3 inhibitors might indirectly interfere with the generation of both Aβ plaques and tangles in AD. GSK3β activity in mitochondria has been associated with increased oxidative stress.29 As such, GSK3β plays a significant role in AD pathogenesis, contributing to Aβ production and Aβ-mediated neuronal death by increasing τ hyperphosphorylation. Additionally, it has been reported that τ phosphorylation gets affected by Aβ–CDK5 interaction. This interaction leads to cleavage of adjacent proteins, releasing cleaved peptides with lower solubility and longer half-lives, which may also phosphorylate distant proteins. Substantial research focusing on identifying and classifying kinases accountable for pathogenic τ hyperphosphorylation points toward the primary pathogenic kinases GSK3β and CDK5, in addition to mitogen-activated protein kinase (MAPK), ERK1 and -2, MAP Kinase (MEK), microtubule affinity-regulating kinase (MARK), c-Jun NH(2)-terminal kinases (JNKs), p38, and PKA, among others.30,31 Abnormal processing of APP leads to secretion of Aβ, which affects GSK3 kinases, leading phosphorylation of the τ protein. This leads to aggregation of τ filaments that are insoluble and finally formation of huge masses of NFTs in neurons.32
Genetic mutations: presenilin 1 mutation and AD
APP is not the only gene associated with AD. Presenilin gene (PSEN1 and PSEN2), which are part of the γ-secretase family, also mutate.33 Moreover, AD patients may be predisposed to PS1 mutation leading to familial AD at a young age.34 The γ-secretase complex is made up of four proteins: Psen1, Psen2, Aph1, and nicastrin. Psen, an aspartyl protease, attributes to the catalytic core of the complex. Psen2 facilitates the maturation of PSEN, whereas Aph1 stabilizes the complex.35 Nicastrin acts as a receptor for γ-secretase substrates. There are 179 PSEN1 and 14 PSEN2 gene mutations that participate in early-onset autosomal-dominant AD. These mutations favor production of more toxic forms of amyloid, eg, Aβ42 as opposed to Aβ40, which contributes in disease progression.36
Epigenetics and AD
Epigenetics deals with the study of interactions between genes, expression of genotypes, and various molecular pathways that modify genotype expression into respective phenotypes.37 Epigenetics exploring neurological diseases, neuroepigenetics, has developed fairly well and been widely studied in CNS-associated diseases comprising learning, motor, behavior, and cognition pathologies and disorders.38,39 Epigenetics is important to understand the depth of effect of environment or paternal genes, nutritional habits, trauma, stress or learning disabilities, exposure to chemicals or drug addiction on DNA and resultant structural disturbances, mutations, or changes.40,41 The involvement of epigenetics has recently been explored in one of the most complex aging-related neurological diseases — AD.42 The onset of AD and its progress involves a complex interplay of various factors like aging, genetic mutations, metabolic and nutritional disorders, effect of and exposure to environmental variables, and most importantly the involvement of social factors.43 There is a fair chance that factors in addition to aging, eg, hypertension, diabetes, obesity, and inflammatory disorders, may have an effect on AD and be inducing epigenetic changes as well or might induce AD-like pathogenesis at a young age. Associations between DNA-methylation patterns in the brain and aging are possible44 and have been reported in various regions of the brain.45 Since DNA epigenetic mechanisms have a role in memory formation and its maintenance, just as decrease in DNA methylation deteriorates neuronal plasticity, leading to memory loss, it is speculated that understanding of epigenetic mechanisms is important to understand aging and associated complexities in AD patients.46 In addition to DNA methylation, histone modifications may also play an important role. Studies have explored histone acetylation in APP–PSEN1 double-mutant transgenic mice, where impairment in associative learning was connected to H4K14 histone-acetylation reduction.47 Additionally Histone deacetylase (HDAC) inhibitors also have an effect on Aβ production and aggregation in AD mice. Studies involving their inhibitors, such as trichostatin A, valproic acid, and vorinostat, are promising. Therefore, it becomes of the utmost importance to understand epigenetic mechanisms involved in aging, in order to target AD-associated mechanisms and complexities.48
Microglial infiltration during plaque formation leading to neurodegeneration
In addition to extracellular Aβ plaques and NFTs due to τ hyperphosphorylation, microglial infiltration in response to these aggregates exacerbates AD pathogenesis. In addition to plaques and tangles, a diversity of morphological variants of Aβ deposits is found in the AD brain. Extracellular and intracellular Aβ and tangles cause extreme toxicity, resulting in synaptic damage and increased reactive oxidative stress, which then leads to microglial infiltration around the plaque areas. Microglia are resident phagocytes in the CNS and play a vital role in the maintenance of neuronal plasticity and synapse remodeling.49 Microglia get activated by protein accumulation, which acts as a pathological trigger, migrate, and initiate innate immunresponses (Figure 5).50 Aβ plaques activate Toll-like receptors on microglia, leading to microglial activation and secretion of proinflammatory cytokines and chemokines.50
 |
Figure 5 Mechanism of neuronal damage and Alzheimer's disease (AD) progression. Note: Extracellular and intracellular amyloid β and tangles cause extreme toxicity, resulting in synaptic damage and increased reactive oxidative stress that then leads to microglial infiltration around the plaque areas. |
In AD, microglia can bind to Aβ via cell-surface receptors, including SCARA1, CD36, CD14, α6β1 integrin, CD47, and Toll-like receptors.51,52 Following receptor binding, microglia endocytose Aβ oligomers and NFT fibrils, which are eliminated by endolysosomal degradation. Microglial proteases like neprilysin and insulin-degrading enzyme play major roles in the degradation.53 However, in severe cases of AD, microglial clearance of Aβ is inefficient, due to increased localized cytokine concentrations, which downregulate the expression of Aβ-phagocytosis receptors and decrease Aβ clearance.54 One of the factors behind compromised AD clearance by microglia is Triggering receptor expressed on myeloid cells 2 (TREM2) mutation. TREM2 mutations are associated with increased AD severity. TREM2 is a cell-surface receptor of the Ig superfamily highly expressed on microglia and involved in mediating phagocytic clearance of neuronal debris. It also binds anionic carbohydrates, bacterial products, and phospholipids and transmits intracellular signals through the associated transmembrane adaptor DAP1255 and further phosphorylation of downstream mediators.56
During AD, a rare mutation of TREM2 (R47H) has been reported that plays a potent role in aggravating the risk of developing AD.57 This mutation leads to inability of the receptors to clear Aβ from the CNS, contributing to Aβ accumulation and further intensification of pathogenesis in AD patients.
Aβ and HIV1-associated neurological disorders
Currently, disease-associated neurological disorders are the biggest area of concern. In this era of antiretroviral therapy (ART), with the increase number of aged HIV patients, the incidence of dementia or other neurocognitive functions is increasing in aged patients when compared to younger patients.58 In AD, there are neurological dysfunctions due to abnormal accumulation of extracellular Aβ produced by alternate cleavage of APP. This Aβ deposition is also reported to occur in the cortices of HIV patients when compared to age-matched non-HIV controls.59–62 The increased AD-like indications, with increased Aβ levels, during HIV infection are not well understood. It is hypothesized that Aβ deposition may be a common factor aggravating in HIV1 infection, thus contributing toward HIV1-associated neurocognitive disorders. If Aβ is the common factor between AD and HIV1-disease scenarios, it becomes imperative to address targeting of the Aβ pathway and end products with a single efficacious drug molecule. With the increase in aging in HIV patients, due to the introduction of ART, a significantly higher occurrence of dementia/neurocognitive dysfunctions has been observed in aged HIV1-infected individuals than younger patients, and HIV1-associated dementia risk in these patients is three times that of younger people.58 The prevalence of HIV1-associated neurocognitive disorders is increasing, as continuing ART medication causes subtle neurodegeneration, especially in hippocampal neurons. Additionally, increased Aβ deposition is characteristic of HIV1-infected brains, and it has been hypothesized that brain vascular dysfunction contributes to this phenomenon, with a critical role suggested for the BBB in brain Aβ homeostasis.
State of the art: AD therapeutics
AD involves protein misfolding, which distorts cellular systems and neuronal death. Protein misfolding results in either loss or toxic gain of function of a protein. This might occur due to abnormal protein aggregation, upon which the protein no longer performs its normal role and fails to be cleared by the cellular environment, leading to deleterious biological responses. There are constant AD studies on inhibiting the production of misfolding proteins and their aggregation and spread to limit the toxicity caused by abnormal proteins.63 The majority of AD-therapeutic approaches are focused on reducing levels of toxic forms of Aβ and τ, the broad scope of neurodegenerative processes underlying both early- and late-stage AD. Several drugs have been analyzed and have reached Phase I, II, and III clinical trials. Table 1 summarizes the drugs specific to amyloid that are being studied and which target sufficiently fundamental and proximate degenerative mechanisms.64,65
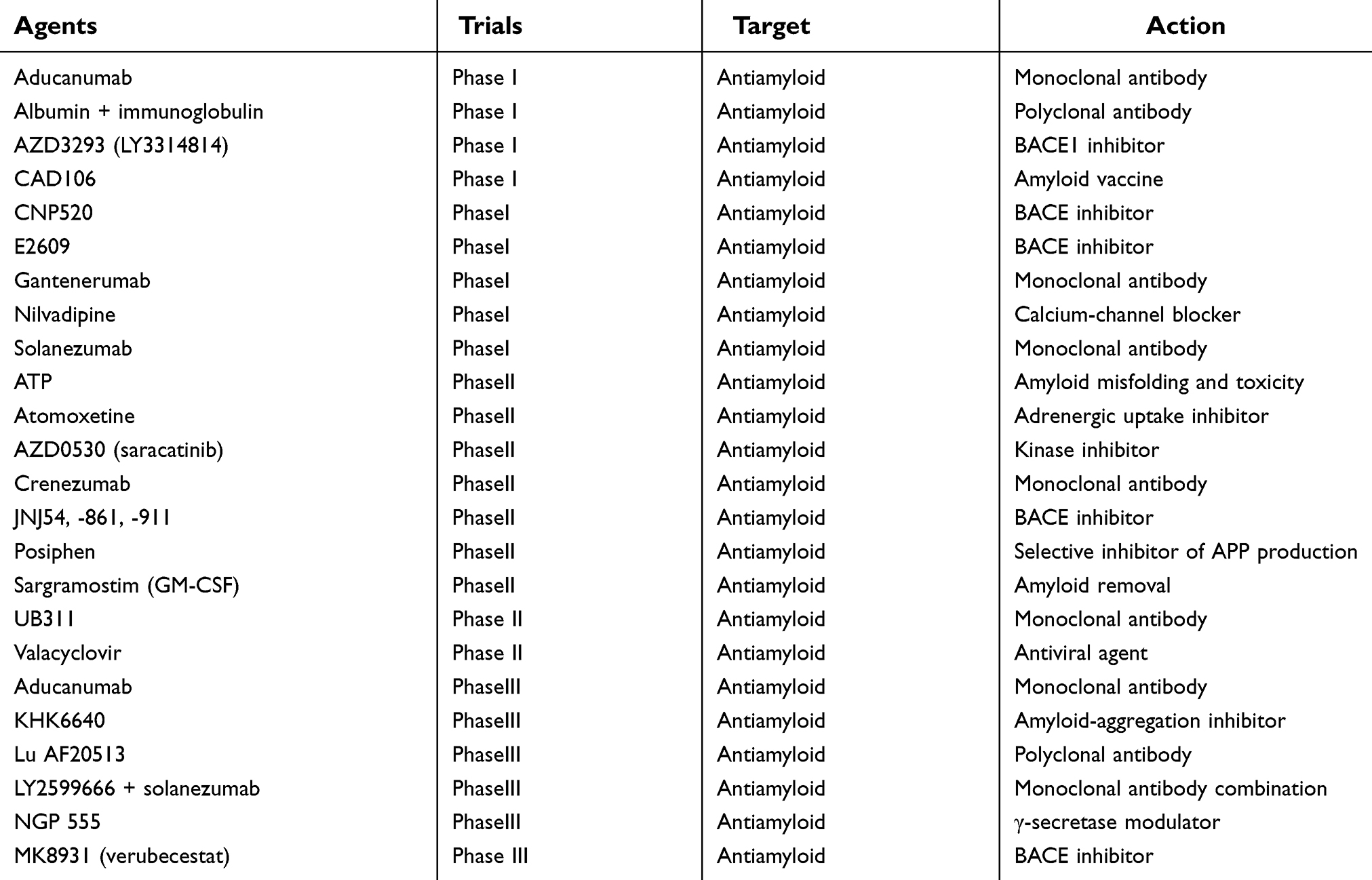 |
Table 1 Drugs specific to amyloid that target fundamental and proximate degenerative mechanisms |
However, all these current therapeutic (eg, rivastigmine, galantamine, and donepezil) targets appear secondary, and none is currently thought to be causally involved in the development of AD. Therapy failure frequently occurs due to the unfavorable pharmacokinetics and pharmacodynamics of drugs. Pharmacotherapy failure is the result of inadequate physical chemistry of drugs (such as hydrophobicity), unfavorable absorption by biological membranes, unfavorable pharmacokinetic parameters (such as intense and plasma metabolism), instability of drugs (oxidation, hydrolysis, or photolysis), and toxicity to tissue (hepatotoxicity, neurotoxicity, or kidney toxicity).
Several treatment strategies have been proposed and attempted for the removal of Aβ. Several drugs are employed for Aβ degradation, but the majority of drugs that showed promising results in in-vivo studies were not able to clear human clinical trials and failed, creating an urgent need to develop new strategies. Many of the available drugs lose their efficacy while crossing the BBB and are minimally bioavailable in the brain. This requires a new area of study that expands into efficacious neuroprotective strategies specific to the CNS. NPs are intriguing candidates for this purpose, because of their potential for multifunctionalization, enabling them to mimic the physiological mechanisms of transport across the BBB. This barrier is an important physical fence made of cells protecting the brain from potential hazardous substances in the bloodstream; however, it also prevents the passage of 98% of available neuropharmaceuticals and diagnostics.
Diagnostics for AD: labeling and imaging
Current AD diagnosis is primarily based on neuropsychological testing. A clinical diagnosis of AD requires neuroimaging and monitoring accepted biomarkers, eg, concentrations of Aβ peptides (Aβ1–42:Aβ1–40 ratio) as well as total and hyperphosphorylated τ (Thr181 and Thr231) proteins in the CSF. Amyloid oligomers and plaque accumulation can also be imaged with 18F-florbetapir (or alternatively 11C Pittsburgh compound B) positron-emission tomography (PET) but nonlinear association between Aβ content in CSF and PET scans remains of concern. However, CSF sampling is relatively invasive and is not always well tolerated or feasible in a number of elderly patients. Noninvasive imaging methods, such as fludeoxyglucose PET, which gives insights into brain metabolism, are of great clinical utility. Indeed, altered cerebral metabolism (hyper- and hypometabolism) has been associated with different stages of AD. Magnetic resonance imaging (MRI) at increasing field strength and resolution is another helpful, noninvasive approach for identification of functional abnormalities. MRI is utilized for detection and identification of amyloid plaques utilizing iron oxide NPs as contrast agents or tagged with fluorescent probes to make detection efficient.66 These iron oxide NPs are reported to bind to N terminal of Aβ , aiding their imaging. Additionally, nonfluorescent or fluorescent rhodamine tagged γFe2O3 NPs have been reported to label Aβ fibrils selectively and remove them from solubilized Aβ, by employing external magnetic field.67,68 In addition to iron NPs, there have been reports of polystyrene-block-poly (n-butyl cyanoacrylate) NPs encapsulating thioflavin T to target Aβ.69 Gold NPs have been used in MRI as contrasting agents to study structural stages in Aβ self-assembly70 and fluorescent semiconductor nanocrystals (quantum dots) for labeling.71
For sensing soluble forms of Aβ from CSF, an ultrasensitive NP-based biobarcode system that specifically detects soluble oligomers with the aid of oligonucleotide (DNA barcode)-modified AuNPs and magnetic microparticles functionalized with monoclonal/polyclonal antibodies have been used,72 as well as electrochemical sensing utilizing click chemistry, which involves AuNPs and assembled monolayers thereon to interact with Aβ peptide,73 and ultrasensitive electrical detection for Aβ1–42 using scanning tunneling microscopy.74 These recently achieved technological and conceptual achievements have considerably improved AD diagnosis. Once AD is diagnosed, the therapeutic choice concerns the treatments that are only disease-modifying and offer relatively limited benefit.
Need for nanotechnology as a therapeutic strategy across the BBB
There are promising drugs against Aβ toxicity,75 but in order to explore their maximum effect on CNS cells, there is a need of nanocarriers to be employed. Availability of drugs in the CNS is the major issue faced in the field of therapeutics against AD. The main reason is the presence of a fully functional semipermeable BBB, which poses as an obstacle for transmigration of neurotherapeutic molecules (like drugs, peptides, vectors, and molecules) across it, into the CNS. The BBB and its selective transport of molecules into the brain oppose efficacious delivery of therapeutic agents. In addition, the BBB also negatively affects drug efficacy and tolerance, because large doses of drugs are needed to reach levels above the minimum effective concentration in the brain. Nanotechnology inclusive of nanoparticulate systems offer an opportunity to overcome such problems and can be used as Trojan-horse systems for transporting active molecules across the BBB (Figure 6), thus reducing toxicity and improving therapeutic efficacy.76,77
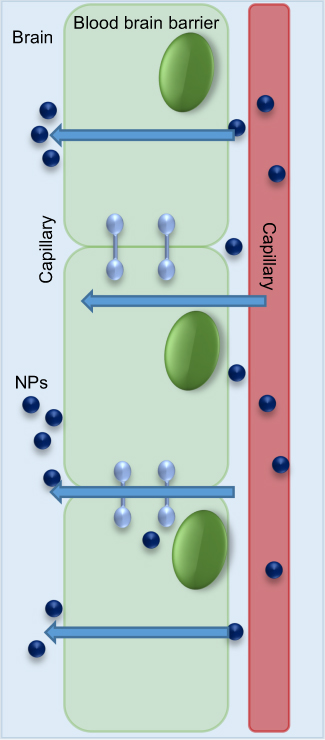 |
Figure 6 Semipermeable blood–brain barrier and transmigration route of the nanoparticles (NPs). |
The use of drugs in nanoplatforms or nanodevices results in enhancement of their pharmacokinetics and pharmacodynamics, as well as reduces the toxicity. An essential aspect in nanomedicine development is the delivery of drugs and controlled release of drugs into disease sites. Therefore, the effectiveness of a treatment can be increased by incorporating nanotechnology-based drug-delivery systems. These new platforms aim to improve bioavailability across the BBB, pharmacokinetics, and pharmacodynamics of drugs while reducing their side effects.
In brief, recent nanotechnology advancements propose effective diagnostic and therapeutic options. Targeted drug delivery with the aid of NPs 100 nm in size can effectively increase drug bioavailability across the BBB into the CNS with minimal or no side effects. Furthermore, these nanomaterials are designed to be biocompatible, hence reducing toxicity, plus with the advancement in their magnetic and optical properties, they may be efficient alternative agents for an early diagnosis.78 The delivery of saxagliptin via dipeptidyl peptidase 4 enzyme–inhibitor molecules is now being explored for its activity in the therapy of AD, with the aid of a chitosan–L-valine conjugate used to prepare NPs encapsulating saxagliptin. These NPs are stable and crossed the BBB efficiently.79 Furthermore, one of the most efficient nanocarriers is magnetoelectric NPs (MENPs), which have been studied well for their potency in delivering drugs across the BBB noninvasively and on-demand release of drugs to target areas without adverse effects. The on-demand release feature is really important, as it ensures delivery of exact amounts of drugs, which is efficacious physiologically without causing toxicity.80–83 Their applications in drug delivery have been well reported in the field of neuroAIDS and AD.83–86
Research interest in nanotherapeutics, ie, utilizing nanocarriers to carry drugs across the BBB, is growing continuously and positively, as these NPs aid efficient drug-delivery systems. The advantages of NPs over plain drugs or microdrug systems are many, including bigger surface area (higher drug loading) and a diverse range of biomaterials, organic (natural or synthetic polymers), and inorganic (metals) compounds for NP production. The interaction between the drug moiety and NPs is diverse. It can be covalent binding, the presence of an ionic surface charge (ionic binding), direct adsorption, or surface binding, and entrapment of the drug. NP surfaces can be modified as well to aid drug binding, such as with PEGylation, which is the process of covalent/noncovalent amalgamation of polyethylene glycol (PEG) to the surface.87–91 Additionally, they increase target specificity via ligand binding. NPs can be modified and imbued with unique physicochemical properties, ie, the addition of metal or electrical attributes, like MENPs, which facilitates drug transport across the BBB, on demand with the introduction of externally applied electric or magnetic fields, increasing the drug delivery severalfold. NPs can have their surface charges altered to interact with the BBB (negatively charged), hence introducing ionic interaction or pull toward the BBB. This charge alteration increases the drug-loading capacity of NPs and aids in on-demand release of the drugs.
MENPs are one of the most effective NP types for noninvasive and image-guided personalized therapy against CNS diseases. They have a unique magnetoelectric actuation effect, which allows longitudinal noninvasive monitoring utilizing MRI,92,93 contributing to image-guided therapy. In addition, liposomal NPs are also potent candidates in drug delivery, as they can be easily surface-modified, facilitating loading of both the hydrophilic and hydrophobic drugs, and aid sustained release across the BBB. They can also be tagged with fluorescent lipids, which can help in image-guided therapy by being able to be observed under microscopy. Plasmonic carbon nano–tube–based systems against CNS diseases have been well studied.
Challenges for clinical translation
With the advent of NPs, various types, such as gold NPs, metal NPs, silver NPs, silica, hydrogels, liposomes, and magnetic NPs, are being employed in drug-delivery studies at a rapid rate. NPs are being explored for CNS drug delivery at the clinical level. The US Food and Drug Administration (FDA) and National Institutes of Health are supporting the concept of personalized nano-medicine, which may usher in a revolution in drug delivery across the BBB, contributing to better health care and more opportunities to combat CNS diseases.94 The success of preclinical studies on CNS nanomedicine95–98 may act as a base to examine these strategies at a clinical level to test biocompatibility, toxicity, efficacy, availability at the human-patient level. Clinical translation of these NPs against CNS diseases at the patient level depends on a lot of factors, eg, patient diversity, genetic and environmental effects, combination of multiple diseases, toxicity, efficacy, and bioavailability in the brain. Based on the patient-disease profile, these NPs can be designed and modified to provide personalized nanomedicine, which can be more beneficial to the individual. This requires proper understanding of the disease mechanism, and even predictive methods utilizing bioinformatics can be utilized to understand disease progression and then design the therapeutic accordingly. With respect to CNS therapy, several studies have highlighted the importance of nanotechnology application for disease diagnosis, drug delivery, and theranostic application. Though, the majority of current research is at the preclinical level, the success of these preclinical and in vivo studies provides promising potential to be translated to clinical levels. Safety, efficacy, and regulatory issues are the major challenges for the progression of personalized nanomedicine to treat CNS diseases clinically. Novel methods like ultrasound-mediated BBB disruption by opening the BBB noninvasively applying external stimulation like focused ultrasound or electromagnetic fields can be promising, but these methods may result in side effects like neurobehavioral distortions or induced infection from entry of unwanted molecules during forced opening of the BBB.99 Therefore, controlled parameters of these stimulations are very critical at clinical levels, as not only can they modulate the intrinsic properties of the introduced NPs by heating them or modifying their surfaces they can also disrupt the homeostasis of the CNS by disturbing BBB permeability, causing inward flow of unwanted circulating molecules into the CNS, leading to neurotoxicity, dysfunction, immunohyperactivation, inflammation, release of reactive oxygen species, synaptic damage, and oxidative stress, contributing to fatal neuronal injury.96,97 Therefore, even though nanotechnology-based research is promising, it has a long way to go to be translated from bench to bedside therapy. There is an urgent need to addressing the issues of toxicity, bioavailability, pharmacokinetics, clearance, and metabolism of NPs for successful clinical trials. There challenges, highlighted by the FDA, focus on biodistribution of NPs, modes of administration, ability of NPs to carry multiple drugs, efficacious transmigration across the BBB, risk assessments, toxicity, standards, safety, procedures, and validation.100 The quest to address the biocompatibility issues, surface functionalization, endosomal entrapment, enzymatic degradation, and off-targeting issues is ongoing through the introduction of surface functionalization, preservation strategies to minimize side effects of external stimulation, and maintaining the availability of drugs in the CNS for longer periods. Progression toward personalized nanomedicine is challenging, but it is critical for successful future clinical trials to make nanotherapeutics available at the patient level.
Summary and future perspectives
AD is a neurodegenerative disease affecting people worldwide. Clinically, it is characterized by the presence of extracellular amyloid plaques and intracellular NFTs, resulting in neuronal dysfunction. Amyloid aggregation happens due to differential cleavage of APP sequentially by β-secretase and γ-secretase, leading to release of extracellular Aβ 40/Aβ 42. AD is also characterized by the presence of NFTs. These tangles are the result of hyperphosphorylation of the microtubule-associated protein τ. GSK3 and CDK5 are the kinases primarily responsible for phosphorylation of τ. In addition to plaque and tangle aggregation, microglial aggregation at the site also plays a vital role in triggering innate immunoresponses against aggregation. A rare mutation paralyzes the regular functioning of microglial surface receptors, contributing to AD intensification. Understanding all these factors and then designing therapeutics specific to targeting them is the need of the hour.
AD is one of the most common neurodegenerative diseases today, but unfortunately101 there is no cure available currently. Several treatments are being employed to combat the cognitive and behavioral deficits associated with AD. Development of a targeted efficacious therapeutic approach against AD is still in its developmental stage, and thus the need of the hour is to look at cellular factors closely associated with disease pathogenesis and target these for improvement of quality of life for AD patients. Cellular factors discussed in this paper, like Aβ, APP, secretases, CDK5, and GSK3β, could be key targets for a therapeutic approach. It is of the utmost importance to understand the limitations of drug bioavailability in the CNS due to the tightly controlled permeability of the BBB. Drugs that target Aβ synthesis or suppress formation of NFTs can stop or reverse AD. Nanomedicine offers an attractive approach to delivering drugs across the BBB.85,86,102,103 Nanotechnology pertains to nanosized drug molecules and their efficient delivery and controlled release in the brain by external magnetic fields, which could be a promising factor in therapeutics for AD. The need of the hour is to unravel the mechanisms of the genesis of AD, its early detection using state-of-the-art biosening devises, specific targeting of the molecules associated with the disease's manifestation, and efficient delivery of optimum drugs to the brain using novel nanotechnology approaches. Further, studies of comorbidities of AD with other diseases or viral infections are also very important to understand and exploit therapeutic approaches.
Abbreviations
AD, Alzheimer’s disease; Aβ, Amyloid β; BBB, blood–brain barrier; CNS, central nervous system; CSF, cerebrospinal fluid; NFTs, neurofibrillary tangles; PCD, protein-conformational disease.
Acknowledgments
The authors acknowledge financial support from NIH grant R01DA034547 and the Florida Department of Health’s Ed and Ethel Moore Alzheimer’s Disease Research Program (grant # 8AZ04). We would also like to acknowledge the Dissertation Year Fellowship 2018 awarded to ST (graduate student) by the University Graduate School, Florida International University, Miami, FL, USA.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Adav SS, Sze SK. Insight of brain degenerative protein modifications in the pathology of neurodegeneration and dementia by proteomic profiling. Mol Brain. 2016;9(1):92. doi:10.1186/s13041-016-0272-9
2. Leandro P, Gomes CM. Protein misfolding in conformational disorders: rescue of folding defects and chemical chaperoning. Mini Rev Med Chem. 2008;8(9):901–911.
3. Tran L, Ha-Duong T. Exploring the Alzheimer amyloid-β peptide conformational ensemble: A review of molecular dynamics approaches. Peptides. 2015;69:86–91. doi:10.1016/j.peptides.2015.04.009
4. Horwich A. Protein aggregation in disease: a role for folding intermediates forming specific multimeric interactions. J Clin Invest. 2002;110(9):1221–1232. doi:10.1172/JCI16781
5. Selkoe DJ. Cell biology of protein misfolding: the examples of Alzheimer‘s and Parkinson‘s diseases. Nat Cell Biol. 2004;6(11):1054. doi:10.1038/ncb1104-1054
6. Blessed G, Tomlinson BE, Roth M. The association between quantitative measures of dementia and of senile change in the cerebral grey matter of elderly subjects. Br J Psychiatry. 1968;114(512):797–811.
7. O‘Brien RJ, Wong PC. Amyloid precursor protein processing and Alzheimer‘s disease. Annu Rev Neurosci. 2011;34:185–204. doi:10.1146/annurev-neuro-061010-113613
8. Henry W, Querfurth H, LaFerla F. Mechanisms of disease Alzheimer’s disease. New Engl J Med. 2010;362:329–344. doi:10.1056/NEJMra0909142
9. Alzheimer’s A. 2015 Alzheimer‘s disease facts and figures. Alzheimer‘S Dementia. 2015;11(3):332. doi:10.1016/j.jalz.2015.02.003
10. Goedert M. Alzheimer’s and Parkinson’s diseases: the prion concept in relation to assembled Aβ, tau, and α-synuclein. Science. 2015;349(6248):1255555. doi:10.1126/science.1255555
11. Chen JX, Yan SS. Role of mitochondrial amyloid-β in Alzheimer‘s disease. J Alzheimer‘S Dis. 2010;20(s2):S569–S578. doi:10.3233/JAD-2010-100357
12. Crews L, Masliah E. Molecular mechanisms of neurodegeneration in Alzheimer‘s disease. Hum Mol Genet. 2010;19(R1):R12–R20. doi:10.1093/hmg/ddq160
13. Oh ES, Savonenko AV, King JF, et al. Amyloid precursor protein increases cortical neuron size in transgenic mice. Neurobiol Aging. 2009;30(8):1238–1244. doi:10.1016/j.neurobiolaging.2007.12.024
14. Thinakaran G, Koo EH. Amyloid precursor protein trafficking, processing, and function. J Biol Chem. 2008;283(44):29615–29619. doi:10.1074/jbc.R800019200
15. Young-Pearse TL, Bai J, Chang R, Zheng JB, LoTurco JJ, Selkoe DJ. A critical function for β-amyloid precursor protein in neuronal migration revealed by in utero RNA interference. J Neurosci. 2007;27(52):14459–14469. doi:10.1523/JNEUROSCI.4701-07.2007
16. Meziane H, Dodart J-C, Mathis C, et al. Memory-enhancing effects of secreted forms of the β-amyloid precursor protein in normal and amnestic mice. Proc Natl Acad Sci. 1998;95(21):12683–12688. doi:10.1073/pnas.95.21.12683
17. Selkoe DJ. Cell biology of the amyloid beta-protein precursor and the mechanism of Alzheimer‘s disease. Annu Rev Cell Biol. 1994;10(1):373–403. doi:10.1146/annurev.cb.10.110194.002105
18. Hefter D, Kaiser M, Weyer SW, et al. Amyloid precursor protein protects neuronal network function after hypoxia via control of voltage-gated calcium channels. J Neurosci. 2016;36(32):8356–8371. doi:10.1523/JNEUROSCI.4130-15.2016
19. Shoji M, Golde TE, Ghiso J, et al. Production of the Alzheimer amyloid beta protein by normal proteolytic processing. Science. 1992;258(5079):126–129.
20. Kimberly WT, Zheng JB, Guenette S, Selkoe DJ. The intracellular domain of the ß-amyloid precursor protein is stabilized by Fe65 and translocates to the nucleus in a notch-like manner. J Biol Chem. 2001. doi:10.1074/jbc.C100447200
21. Bergmans BA, De Strooper B. γ-secretases: from cell biology to therapeutic strategies. Lancet Neurol. 2010;9(2):215–226. doi:10.1016/S1474-4422(09)70332-1
22. Tu S, Okamoto S-I, Lipton SA, Xu H. Oligomeric Aβ-induced synaptic dysfunction in Alzheimer’s disease. Mol Neurodegener. 2014;9(1):48. doi:10.1186/1750-1326-9-48
23. Eftekharzadeh B, Daigle JG, Kapinos LE, et al. Tau protein disrupts nucleocytoplasmic transport in Alzheimer’s disease. Neuron. 2018;99(5):925–940.e927. doi:10.1016/j.neuron.2018.07.039
24. Claeysen S, Cochet M, Donneger R, Dumuis A, Bockaert J, Giannoni P. Alzheimer culprits: cellular crossroads and interplay. Cell Signal. 2012;24(9):1831–1840. doi:10.1016/j.cellsig.2012.05.008
25. Marcus JN, Schachter J. Targeting post-translational modifications on tau as a therapeutic strategy for Alzheimer‘s disease. J Neurogenet. 2011;25(4):127–133. doi:10.3109/01677063.2011.626471
26. Lee VM, Goedert M, Trojanowski JQ. Neurodegenerative tauopathies. Annu Rev Neurosci. 2001;24(1):1121–1159. doi:10.1146/annurev.neuro.24.1.1121
27. De Strooper B, Woodgett J. Alzheimer‘s disease: mental plaque removal. Nature. 2003;423(6938):392. doi:10.1038/423392a
28. Phiel CJ, Wilson CA, Lee VM-Y, Klein PS. GSK-3α regulates production of Alzheimer‘s disease amyloid-β peptides. Nature. 2003;423(6938):435. doi:10.1038/nature01640
29. Hernández F, Avila J. The role of glycogen synthase kinase 3 in the early stages of Alzheimers’ disease. FEBS Lett. 2008;582(28):3848–3854. doi:10.1016/j.febslet.2008.10.026
30. Cho J-H, Johnson GV. Glycogen synthase kinase 3β induces caspase-cleaved tau aggregation in situ. J Biol Chem. 2004;279(52):54716–54723. doi:10.1074/jbc.M403364200
31. Kuruva CS, Reddy PH. Amyloid beta modulators and neuroprotection in Alzheimer‘s disease: a critical appraisal. Drug Discov Today. 2017;22(2):223–233. doi:10.1016/j.drudis.2016.10.010
32. Bossy-Wetzel E, Schwarzenbacher R, Lipton SA. Molecular pathways to neurodegeneration. Nat Med. 2004;10(7):S2. doi:10.1038/nm1067
33. Nizzari M, Thellung S, Corsaro A, et al. Neurodegeneration in Alzheimer disease: role of amyloid precursor protein and presenilin 1 intracellular signaling. J Toxicol. 2012;
34. Dries DR, Yu G. Assembly, maturation, and trafficking of the γ-secretase complex in Alzheimer‘s disease. Curr Alzheimer Res. 2008;5(2):132–146.
35. Edbauer D, Winkler E, Regula JT, Pesold B, Steiner H, Haass C. Reconstitution of γ-secretase activity. Nat Cell Biol. 2003;5(5):486. doi:10.1038/ncb960
36. Shen J, Kelleher RJ. The presenilin hypothesis of Alzheimer‘s disease: evidence for a loss-of-function pathogenic mechanism. Proc Natl Acad Sci. 2007;104(2):403–409. doi:10.1073/pnas.0608332104
37. Waddington CH. The epigenotype. Int J Epidemiol. 2011;41(1):10–13. doi:10.1093/ije/dyr184
38. Lardenoije R, Iatrou A, Kenis G, et al. The epigenetics of aging and neurodegeneration. Prog Neurobiol. 2015;131:21–64. doi:10.1016/j.pneurobio.2015.05.002
39. Sweatt JD. The emerging field of neuroepigenetics. Neuron. 2013;80(3):624–632. doi:10.1016/j.neuron.2013.10.023
40. Tsankova N, Renthal W, Kumar A, Nestler EJ. Epigenetic regulation in psychiatric disorders. Nat Rev Neurosci. 2007;8(5):355. doi:10.1038/nrn2132
41. Day JJ, Sweatt JD. DNA methylation and memory formation. Nat Neurosci. 2010;13(11):1319. doi:10.1038/nn.2511
42. Landgrave-Gómez J, Mercado-Gómez O, Guevara-Guzmán R. Epigenetic mechanisms in neurological and neurodegenerative diseases. Front Cell Neurosci. 2015;9:58.
43. Sezgin Z, Dincer Y. Alzheimer‘s disease and epigenetic diet. Neurochem Int. 2014;78:105–116. doi:10.1016/j.neuint.2014.09.012
44. Balazs R. Epigenetic mechanisms in Alzheimer‘s disease. Degener Neurol Neuromuscul Dis. 2014;4:85–102. doi:10.2147/DNND.S37341
45. Hernandez DG, Nalls MA, Gibbs JR, et al. Distinct DNA methylation changes highly correlated with chronological age in the human brain. Hum Mol Genet. 2011;20(6):1164–1172. doi:10.1093/hmg/ddq561
46. Mastroeni D, Grover A, Delvaux E, Whiteside C, Coleman PD, Rogers J. Epigenetic mechanisms in Alzheimer‘s disease. Neurobiol Aging. 2011;32(7):1161–1180. doi:10.1016/j.neurobiolaging.2010.08.017
47. Francis YI, Fà M, Ashraf H, et al. Dysregulation of histone acetylation in the APP/PS1 mouse model of Alzheimer‘s disease. J Alzheimer‘S Dis. 2009;18(1):131–139. doi:10.3233/JAD-2009-1134
48. Delgado-Morales R, Agís-Balboa RC, Esteller M, Berdasco M. Epigenetic mechanisms during ageing and neurogenesis as novel therapeutic avenues in human brain disorders. Clin Epigenetics. 2017;9(1):67. doi:10.1186/s13148-017-0365-z
49. Ji K, Akgul G, Wollmuth LP, Tsirka SE, Dunaevsky A. Microglia actively regulate the number of functional synapses. PLoS One. 2013;8(2):e56293. doi:10.1371/journal.pone.0056293
50. Heneka MT, Carson MJ, El Khoury J, et al. Neuroinflammation in Alzheimer‘s disease. Lancet Neurol. 2015;14(4):388–405. doi:10.1016/S1474-4422(15)70016-5
51. Bamberger ME, Harris ME, McDonald DR, Husemann J, Landreth GE. A cell surface receptor complex for fibrillar β-amyloid mediates microglial activation. J Neurosci. 2003;23(7):2665–2674.
52. Liu Y, Walter S, Stagi M, et al. LPS receptor (CD14): a receptor for phagocytosis of Alzheimer‘s amyloid peptide. Brain. 2005;128(8):1778–1789. doi:10.1093/brain/awh531
53. Malito E, Hulse RE, Tang W-J. Amyloid β-degrading cryptidases: insulin degrading enzyme, presequence peptidase, and neprilysin. Cel Mol Life Sci. 2008;65(16):2574–2585. doi:10.1007/s00018-008-8112-4
54. Hickman SE, Allison EK, El Khoury J. Microglial dysfunction and defective β-amyloid clearance pathways in aging Alzheimer‘s disease mice. J Neurosci. 2008;28(33):8354–8360. doi:10.1523/JNEUROSCI.0616-08.2008
55. Neumann H, Daly MJ. Variant TREM2 as risk factor for Alzheimer's disease. N Engl J Med. 2013;368(2):182–184. doi:10.1056/NEJMe1213157
56. Wang Y, Cella M, Mallinson K, et al. TREM2 lipid sensing sustains the microglial response in an Alzheimer’s disease model. Cell. 2015;160(6):1061–1071. doi:10.1016/j.cell.2015.01.049
57. Hsieh CL, Koike M, Spusta SC, et al. A role for TREM2 ligands in the phagocytosis of apoptotic neuronal cells by microglia. J Neurochem. 2009;109(4):1144–1156. doi:10.1111/j.1471-4159.2009.06042.x
58. Valcour VG, Shikuma CM, Watters MR, Sacktor NC. Cognitive impairment in older HIV-1-seropositive individuals: prevalence and potential mechanisms. Aids. 2004;18(Suppl 1):S79. doi:10.1097/00002030-200401001-00012
59. Becker JT, Lopez OL, Dew MA, Aizenstein HJ. Prevalence of cognitive disorders differs as a function of age in HIV virus infection. Aids. 2004;18:11–18. doi:10.1097/00002030-200401001-00003
60. Esiri MM, Biddolph SC, Morris CS. Prevalence of Alzheimer plaques in AIDS. J Neurol, Neurosurg Psychiatry. 1998;65(1):29–33. doi:10.1136/jnnp.65.1.29
61. Green DA, Masliah E, Vinters HV, Beizai P, Moore DJ, Achim CL. Brain deposition of beta-amyloid is a common pathologic feature in HIV positive patients. Aids. 2005;19(4):407–411.
62. Pulliam L. HIV regulation of amyloid beta production. J Neuroimmune Pharmacol. 2009;4(2):213–217. doi:10.1007/s11481-009-9151-9
63. Scannevin RH. Therapeutic strategies for targeting neurodegenerative protein misfolding disorders. Curr Opin Chem Biol. 2018;44:66–74. doi:10.1016/j.cbpa.2018.05.018
64. Cummings J, Lee G, Mortsdorf T, Ritter A, Zhong K. Alzheimer‘s disease drug development pipeline: 2017. Alzheimer‘S Dementia. 2017;3(3):367–384.
65. Simmons D, Yang T, Massa S, Longo F. Neuroprotective strategies for Alzheimer’s disease prevention and therapy. In: Wolfe MS, editor. Developing Therapeutics for Alzheimer‘s Disease. Amsterdam: Elsevier; 2016:437–458.
66. Wadghiri YZ, Sigurdsson EM, Sadowski M, et al. Detection of Alzheimer‘s amyloid in transgenic mice using magnetic resonance microimaging. Magn Reson Med. 2003;50(2):293–302. doi:10.1002/mrm.10529
67. Skaat H, Margel S. Synthesis of fluorescent-maghemite nanoparticles as multimodal imaging agents for amyloid-β fibrils detection and removal by a magnetic field. Biochem Biophys Res Commun. 2009;386(4):645–649. doi:10.1016/j.bbrc.2009.06.110
68. Skaat H, Sorci M, Belfort G, Margel S. Effect of maghemite nanoparticles on insulin amyloid fibril formation: selective labeling, kinetics, and fibril removal by a magnetic field. J Biomed Mater Res Part A. 2009;91(2):342–351. doi:10.1002/jbm.a.32232
69. Siegemund T, Paulke B-R, Schmiedel H, et al. Thioflavins released from nanoparticles target fibrillar amyloid β in the hippocampus of APP/PS1 transgenic mice. Int J Dev Neurosci. 2006;24(2–3):195–201. doi:10.1016/j.ijdevneu.2005.11.012
70. Choi J-S, Choi HJ, Jung DC, Lee J-H, Cheon J. Nanoparticle assisted magnetic resonance imaging of the early reversible stages of amyloid β self-assembly. Chem Commun. 2008;19:2197–2199. doi:10.1039/b803294g
71. Dubertret B, Skourides P, Norris DJ, Noireaux V, Brivanlou AH, Libchaber A. In vivo imaging of quantum dots encapsulated in phospholipid micelles. Science. 2002;298(5599):1759–1762. doi:10.1126/science.1077194
72. Georganopoulou DG, Chang L, Nam J-M, et al. Nanoparticle-based detection in cerebral spinal fluid of a soluble pathogenic biomarker for Alzheimer‘s disease. Proc Natl Acad Sci. 2005;102(7):2273–2276. doi:10.1073/pnas.0409336102
73. Kolb HC, Finn M, Sharpless KB. Click chemistry: diverse chemical function from a few good reactions. Angew Chem Int Ed. 2001;40(11):2004–2021.
74. Kang D-Y, Lee J-H, Oh B-K, Choi J-W. Ultra-sensitive immunosensor for β-amyloid (1–42) using scanning tunneling microscopy-based electrical detection. Biosens Bioelectron. 2009;24(5):1431–1436. doi:10.1016/j.bios.2008.08.018
75. Tiwari S, Atluri VSR, Yndart Arias A, et al. Withaferin a suppresses beta amyloid in APP expressing cells: studies for tat and cocaine associated neurological dysfunctions. Front Aging Neurosci. 2018;10:291. doi:10.3389/fnagi.2018.00291
76. Hsiao IL, Hsieh YK, Chuang CY, Wang CF, Huang YJ. Effects of silver nanoparticles on the interactions of neuron‐and glia‐like cells: toxicity, uptake mechanisms, and lysosomal tracking. Environ Toxicol. 2017;32(6):1742–1753. doi:10.1002/tox.22397
77. Hsiao I-L, Hsieh Y-K, Wang C-F, Chen I-C, Huang Y-J. Trojan-horse mechanism in the cellular uptake of silver nanoparticles verified by direct intra-and extracellular silver speciation analysis. Environ Sci Technol. 2015;49(6):3813–3821. doi:10.1021/es504705p
78. Leszek J, Md Ashraf G, Tse WH, et al. Nanotechnology for Alzheimer disease. Curr Alzheimer Res. 2017;14(11):1182–1189. doi:10.2174/1567205014666170203125008
79. Fernandes J, Ghate MV, Mallik SB, Lewis SA. Amino acid conjugated chitosan nanoparticles for the brain targeting of a model dipeptidyl peptidase-4 inhibitor. Int J Pharm. 2018. doi:10.1016/j.ijpharm.2018.06.031
80. Lammers T, Rizzo LY, Storm G, Kiessling F. Personalized nanomedicine. Clin Cancer Res. 2012;18(18):4889–4894. doi:10.1158/1078-0432.CCR-12-1414
81. Tietjen GT, Saltzman WM. Nanomedicine gets personal. Sci Transl Med. 2015;7(314):314fs347. doi:10.1126/scitranslmed.aad3106
82. Yu D, Khan OF, Suvà ML, et al. Multiplexed RNAi therapy against brain tumor-initiating cells via lipopolymeric nanoparticle infusion delays glioblastoma progression. Proc Natl Acad Sci. 2017;114(30):
83. Jayant RD, Atluri VS, Tiwari S, et al. Novel nanoformulation to mitigate co-effects of drugs of abuse and HIV-1 infection: towards the treatment of NeuroAIDS. J Neurovirol. 2017;23(4):603–614. doi:10.1007/s13365-017-0538-8
84. Nair M, Jayant RD, Kaushik A, Sagar V. Getting in to the brain: potential of nanotechnology to manage neuroAIDS. Adv Drug Deliv Rev. 2016;103:202–217.
85. Kaushik A, Jayant RD, Nair M. Advancements in nano-enabled therapeutics for neuroHIV management. Int J Nanomedicine. 2016;11:4317. doi:10.2147/IJN.S109943
86. Nair M, Guduru R, Liang P, Hong J, Sagar V, Khizroev S. Externally controlled on-demand release of anti-HIV drug using magneto-electric nanoparticles as carriers. Nat Commun. 2013;4:1707. doi:10.1038/ncomms2717
87. Veronese FM, Mero A. The impact of PEGylation on biological therapies. BioDrugs. 2008;22(5):315–329. doi:10.2165/00063030-200822050-00004
88. Kang JS, DeLuca PP, Lee KC. Emerging pegylated drugs. Expert Opin Emerg Drugs. 2009;14(2):363–380. doi:10.1517/14728210902907847
89. Kanwal U, Irfan Bukhari N, Ovais M, Abass N, Hussain K, Raza A. Advances in nano-delivery systems for doxorubicin: an updated insight. J Drug Target. 2018;26(4):296–310. doi:10.1080/1061186X.2017.1380655
90. Dadashzadeh S, Vali A, Rezaie M. The effect of PEG coating on in vitro cytotoxicity and in vivo disposition of topotecan loaded liposomes in rats. Int J Pharm. 2008;353(1–2):251–259. doi:10.1016/j.ijpharm.2007.11.030
91. Ochi MM, Amoabediny G, Rezayat SM, Akbarzadeh A, Ebrahimi B. In vitro co-delivery evaluation of novel pegylated nano-liposomal herbal drugs of silibinin and glycyrrhizic acid (nano-phytosome) to hepatocellular carcinoma cells. Cell J (Yakhteh). 2016;18(2):135.
92. Stimphil E, Nagesetti A, Guduru R, et al. Physics considerations in targeted anticancer drug delivery by magnetoelectric nanoparticles. Appl Phys Rev. 2017;4(2):021101. doi:10.1063/1.4978642
93. Kaushik A, Jayant RD, Nikkhah-Moshaie R, et al. Magnetically guided central nervous system delivery and toxicity evaluation of magneto-electric nanocarriers. Sci Rep. 2016;6:25309. doi:10.1038/srep25309
94. Hamburg MA, Collins FS. The path to personalized medicine. N Engl J Med. 2010;363(4):301–304. doi:10.1056/NEJMp1006304
95. Theek B, Rizzo LY, Ehling J, Kiessling F, Lammers T. The theranostic path to personalized nanomedicine. Clin Transl Imaging. 2014;2(1):67–76. doi:10.1007/s40336-014-0051-5
96. Jain KK, Jain KK. The Handbook of Nanomedicine. Vol. 404. Heidelberg: Springer; 2008.
97. Fornaguera C, García-Celma M. Personalized nanomedicine: a revolution at the nanoscale. J pers med. 2017;7(4):12. doi:10.3390/jpm7040012
98. Mura S, Couvreur P. Nanotheranostics for personalized medicine. Adv Drug Deliv Rev. 2012;64(13):1394–1416. doi:10.1016/j.addr.2012.06.006
99. Yang F-Y, Lin Y-S, Kang K-H, Chao T-K. Reversible blood–brain barrier disruption by repeated transcranial focused ultrasound allows enhanced extravasation. J Control Release. 2011;150(1):111–116. doi:10.1016/j.jconrel.2010.10.038
100. Sanhai WR, Sakamoto JH, Canady R, Ferrari M. Seven challenges for nanomedicine. Nat Nanotechnol. 2008;3(5):242. doi:10.1038/nnano.2008.114
101. Association As. 2014 Alzheimer‘s disease facts and figures. Alzheimer‘S Dementia. 2014;10(2):e47–e92. doi:10.1016/j.jalz.2014.02.001
102. Kaushik A, Jayant RD, Tiwari S, Vashist A, Nair M. Nano-biosensors to detect beta-amyloid for Alzheimer‘s disease management. Biosens Bioelectron. 2016;80:273–287. doi:10.1016/j.bios.2016.01.065
103. Sagar V, Pilakka‐Kanthikeel S, Pottathil R, Saxena SK, Nair M. Towards nanomedicines for neuroAIDS. Rev Med Virol. 2014;24(2):103–124. doi:10.1002/rmv.1778
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.