Back to Journals » Journal of Experimental Pharmacology » Volume 10
Airway remodeling disease: primary human structural cells and phenotypic and pathway assays to identify targets with potential to prevent or reverse remodeling
Authors Rosethorne EM, Charlton SJ ![]()
Received 1 September 2018
Accepted for publication 30 October 2018
Published 29 November 2018 Volume 2018:10 Pages 75—85
DOI https://doi.org/10.2147/JEP.S159124
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Bal Lokeshwar
Elizabeth M Rosethorne,1–3 Steven J Charlton1–3
1School of Life Sciences, Queen’s Medical Centre, University of Nottingham, Nottingham NG7 2UH, UK; 2Centre of Membrane and Protein and Receptors (COMPARE), University of Birmingham and University of Nottingham, Midlands, UK; 3Excellerate Bioscience Ltd, MediCity, Nottingham NG7 2UH, UK
Abstract: Airway remodeling is a characteristic of many chronic respiratory diseases and occurs when there are significant changes to the architecture of the small and large airways leading to progressive loss of lung function. Some common features include airway smooth muscle and goblet cell hyperplasia, basement membrane thickening and subepithelial fibrosis. To explore the mechanisms driving airway remodeling and identify novel targets to treat this aspect of respiratory disease, appropriate models must be used that will accurately predict the pathology of disease. Phenotypic assays can be used in primary human lung cells to measure changes in cell behavior that are associated with particular disease pathology. This is becoming increasingly popular when targeting chronic pathologies such as airway remodeling, where phenotypic assays are likely to model disease in vitro more accurately than traditional second messenger assays. Here we review the use of primary human lung structural cells in a range of disease-relevant chronic phenotypic assays, and how they may be used in target identification/validation and drug discovery.
Keywords: respiratory, fibroblast, smooth muscle, differentiation, hyperplasia
Introduction
Respiratory diseases are among the leading causes of death in the world, with an estimated 4 million people dying prematurely of chronic respiratory disease each year. They remain a significant health burden and account for 10% of all disability-adjusted life-years, which is second only to the cardiovascular diseases.1 A pathological feature of many chronic respiratory diseases is the alteration of healthy lung architecture that leads to progressive loss of lung function, reduction in normal gas exchange and a decrease in the ability of patients to breathe effectively. This loss of pulmonary function is driven by structural changes that occur in the lung in a process known as airway remodeling. In severe disease, airway remodeling can result in fixed or irreversible airflow obstruction that is refractory to commonly used bronchodilators such as β2-adrenoceptor agonists and muscarinic antagonists in asthma and can contribute to clinical presentation and outcome.2
A number of respiratory diseases are associated with a degree of airway remodeling, which can be broadly categorized into two different pathologies. Those that involve the conducting airways including severe asthma3 and COPD, and those which involve the acinar airways or lung parenchyma such as idiopathic pulmonary fibrosis (IPF), acute lung injury, acute respiratory distress syndrome and lymphangioleiomyomatosis. For simplicity, we use the term airway remodeling to encompass any structural changes associated with chronic respiratory disease.
Although the causes and types of airway remodeling that occur in these diseases are different, they share commonalities, with uncontrolled migration, proliferation and differentiation of resident cells playing a key role. These include airway smooth muscle (ASM) hypertrophy and hyperplasia, subepithelial fibrosis, myofibroblast accumulation, mucus gland and goblet cell hyperplasia and epithelial disruption.4 In addition to the airways, remodeling may also be detected in the vasculature. This is a key pathological feature of pulmonary arterial hypertension,5 where increased stiffness and muscuralization of proximal pulmonary arteries occurs due to hyperproliferation and transdifferentiation of pulmonary artery smooth muscle cells. However, it is now known that vascular remodeling may also occur in asthma and COPD where a degree of neovascularization is observed leading to enhanced tissue perfusion, which may affect normal gas exchange and airway dynamics.6–8
It was originally thought that the repeat cycles of chronic inflammation and repair in the lung was the key driver for the initiation of airway remodeling; however, this paradigm has recently been challenged. Airway remodeling often occurs subsequent to, or even contemporaneously with inflammation of the airways, and recent work with pediatric asthma has demonstrated that airway remodeling is not simply a result of a chronic inflammatory response.9–13 In fact, it has been found that infants exhibit the same degree of basement membrane thickening, a hallmark of remodeling, as adults with chronic disease. This demonstrates that airway remodeling is not related to the duration of disease and therefore airway inflammation, but rather develops concurrently with the inflammatory response.9,10 In addition, while there is increasing evidence that inflammation is important in driving pathology, it is often the associated remodeling that drives disease severity. In severe asthma, the thickness of subepithelial fibrosis observed was found to be correlated with disease severity, but not the length of asthma history.14,15 Similarly, ASM hypertrophy, bronchial wall thickening and fibroblast accumulation have been linked to the persistent airflow obstruction observed in severe asthma.16–19 Similar to the conclusions from severe asthma, the prevailing view is now that IPF pathology is driven by epithelial damage and an aberrant wound healing response from fibroblasts. There is no convincing evidence to suggest that IPF is a result of a chronic inflammatory response, and it is likely that the inflammation observed in IPF occurs as a result of fibrosis. In support of this, clinical measurements of inflammation fail to correlate with stage or outcome, and potent anti-inflammatory therapy does not improve outcome.20 This may help explain, in part, why therapies such as corticosteroids that target inflammation are not effective in certain patients with chronic respiratory disease.21,22 For this reason, we will not focus on inflammation in this review, as novel therapeutics that will prevent progression or reverse airway remodeling are a more attractive target class.
Airway remodeling
To develop novel therapeutics that specifically target the airway remodeling observed in chronic respiratory diseases, we need to understand not only the molecular mechanisms of remodeling but also identify the key cell types involved as well. Resident cells, such as epithelial cells, smooth muscle cells and fibroblasts, are generally considered to be responsible for most pathological features of remodeling, with different cell types playing key roles in specific diseases. For example, ASM cell hyperplasia is predominantly responsible for remodeling of conducting airways leading to airway obstruction in COPD23,24 and asthma,16,18,25 whereas pulmonary artery smooth muscle cell hyperplasia results in vascular remodeling in pulmonary arterial hypertension.26 In contrast, it is the epithelial cells and fibroblasts that drive the remodeling of acinar airways and the excessive wound healing response, or fibrosis that is observed in IPF.27–29 The damaged epithelium is also a source of growth factors (GFs) such as TGFβ that promote epithelial cell migration into the site of the wound and subsequent fibroblast activation.30 The epithelial cells line the exterior of the airways and as such act as a barrier to the external environment, making them particularly susceptible to damage by a range of factors including mechanical stress, pathogens, allergens or environmental irritants.31 This is a unique feature of epithelial cells that can be mimicked in culture by growing polarized epithelial cells at air liquid interface..
Although it is thought to be the resident ASM cells and fibroblasts that drive airway remodeling, there is some evidence that circulating fibroblast precursor cells, or fibrocytes, are recruited to areas of ongoing tissue damage or inflammation where they then differentiate into fibroblasts or myofibroblasts. These cells express a distinct set of cell surface markers, including the hematopoietic stem cell antigen CD34, as well as mesenchymal markers such as collagen and fibronectin.32,33 As the circulating fibrocytes are not yet resident cells, and they are rare and difficult to identify, they fall out of the scope of this review and therefore will not be discussed further.
In addition to the different cell types implicated in airway remodeling, there are a number of diverse pathological processes that these cells can undergo, with some common features appearing in many of these respiratory diseases. Some of the more common features include hyperplasia and/or hypertrophy, differentiation, protein secretion and apoptosis.
Current therapeutic research is now focusing on the development of novel approaches to treat chronic respiratory diseases that aim to target this remodeling in order to prevent further disease progression. Ultimately the goal is to prevent further development of disease by halting the progression of airway remodeling, and potentially even to reverse aspects of existing remodeling. To this aim, research is focusing on understanding the exact molecular mechanisms responsible for driving remodeling, in an attempt to discover novel targets that will impact on disease progression. A key part of this research is developing alternative screening assays for use in both target ID/validation and selection of appropriate candidate drug molecules.
Assaying airway remodeling
Intracellular signaling assays are often used in recombinant cell systems to measure activation or inactivation of specific enzymes or receptors, due to their robustness, ease of use and amenability to high-throughput screening. More recently, efforts have been made to perform these assays in relevant human primary cells in order to increase their translatability to the clinical situation; however, they still represent an acute response that may not be predicative of chronic cellular events. Phenotypic assays can, therefore, be used to measure changes in cell behavior that are associated with particular disease pathology. This is becoming increasingly important when targeting chronic pathologies such as airway remodeling, where phenotypic assays are likely to model disease in vitro more accurately than traditional second messenger assays. This has recently been demonstrated for IPF, where the inhibition of human lung fibroblast proliferation and differentiation was not predicted by the magnitude of the acute cAMP response in the same cells.34
Many, if not all, of these processes are now amenable to plate-based assays, so this review will focus on techniques that can be performed in either 96-well or 384-well format, and novel techniques that offer more in-depth characterization of cell phenotypes.
Proliferation
One way to model the remodeling of conducting airways is to measure the hyperproliferative response of the ASM that leads to thickening of the airways in asthma or COPD. This ASM hypertrophy/hyperplasia can be monitored using proliferation and imaging assays. There a number of proliferation assays available, ranging from simple cell counts to more complex assay kits. Nuclear DNA can be stained with dyes such as Hoechst and Draq5, which can then be used in combination with high-content imaging (HCI) to give a measure of the increase in cell numbers, and hence proliferation of cells over time. As a surrogate marker of proliferation, artificially introduced nucleosides such as bromodeoxyuridine (BrdU) or tritiated thymidine can be used to monitor DNA replication. They are incorporated into newly synthesized DNA of replicating cells during the S phase of the cell cycle in the place of endogenous thymidine. Incorporated BrdU can then be monitored using selective antibodies35 and tritiated thymidine incorporation by autoradiography. BrdU incorporation assays (Figure 1) are more in vogue compared with adenine incorporation, due to both their sensitivity and the lack of requirement for radioactivity. These assays are also amenable to automation, allowing the screening of small- and medium-sized compound libraries against human primary cells. These techniques have been successfully used in a range of different cells, including airway and vascular smooth muscle cells, fibroblasts and epithelial cells.34,36–38
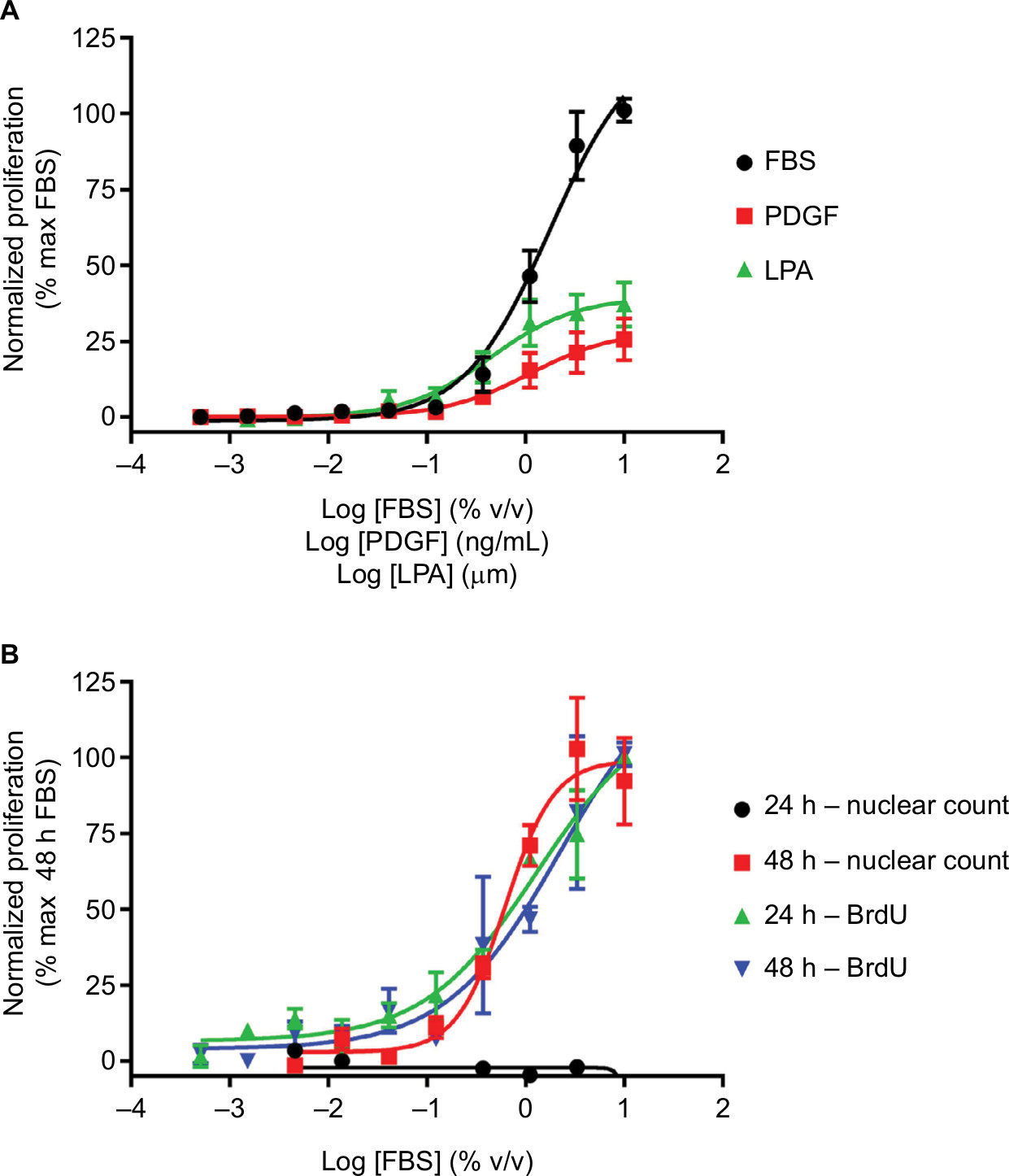 | Figure 1 Fibroblast proliferation. Notes: (A) Concentration-dependent increases in human lung fibroblast proliferation following treatment with FBS, PDGF or LPA for 24 hours, assayed using BrdU incorporation. (B) Concentration-dependent increases in proliferation following treatment with FBS for 24 or 48 hours, assayed using BrdU incorporation or nuclear count. Data were normalized to maximal proliferation observed with 10% serum, and expressed as mean ± SEM for >3 independent experiments. Abbreviations: BrdU, bromodeoxyuridine; LPA, lysophosphatidic acid; PDGF, platelet-derived growth factor. |
There are a number of stimuli available to promote proliferation, perhaps the most robust being FBS, which is commonly used during cell maintenance to drive cell growth. While FBS will promote robust proliferation of most cell types, it has no causative link to human airway remodeling disease, therefore more disease relevant stimuli should be used. These may include GFs such as platelet-derived growth factor (PDGF)39 or G protein-coupled receptor agonists such as lysophosphatidic acid (LPA)40 (Figure 1A). It is important to choose the correct stimulus, as some GFs that have been linked to airway remodeling, such as TGFβ, do not directly promote proliferation. In addition, the duration of stimulation required to drive proliferation must be determined for the assay and cell type being studied. As the BrdU incorporation assays are more sensitive, they may require only 24 hours stimulation, whereas the less sensitive models may require up to 72 hours stimulation before a robust response is observed (Figure 1B).34
Differentiation
To explore novel targets for remodeling that occurs within the parenchyma of the lungs, such as in IPF, we can model certain aspects of cellular fibrosis. Subepithelial fibrosis consists of an increase in either the recruitment or the activation of fibroblasts to the site of inflammation, followed by their differentiation to myofibroblasts forming fibroblast foci. These myofibroblasts have a more contractile phenotype and deposit a number of extracellular matrix (ECM) proteins into the reticular basement membrane. This results in the thickening and stiffening of the basement membrane, creating a rigid environment where bronchodilation or vascular dilation is impaired.
Fibroblast differentiation can be monitored using assays that detect increases in α-smooth muscle actin (αSMA), a key marker of fibroblast to myofibroblast transdifferentiation (FMT).41 This can be performed using In-Cell Western or HCI with fluorescently labelled secondary antibodies (Figure 2). The advantage of HCI over traditional Western blotting techniques is that the assays are performed on intact cells, allowing determination of both the expression of αSMA as well as its filamentous reorganization. The advances in HCI platforms allowing simultaneous detection of different fluorescent wavelengths makes this assay amenable to multiplexing, allowing detection of multiple phenotypic readouts in a single well. This multiplexing can give more detailed information about the complex processes that are driving airway remodeling.
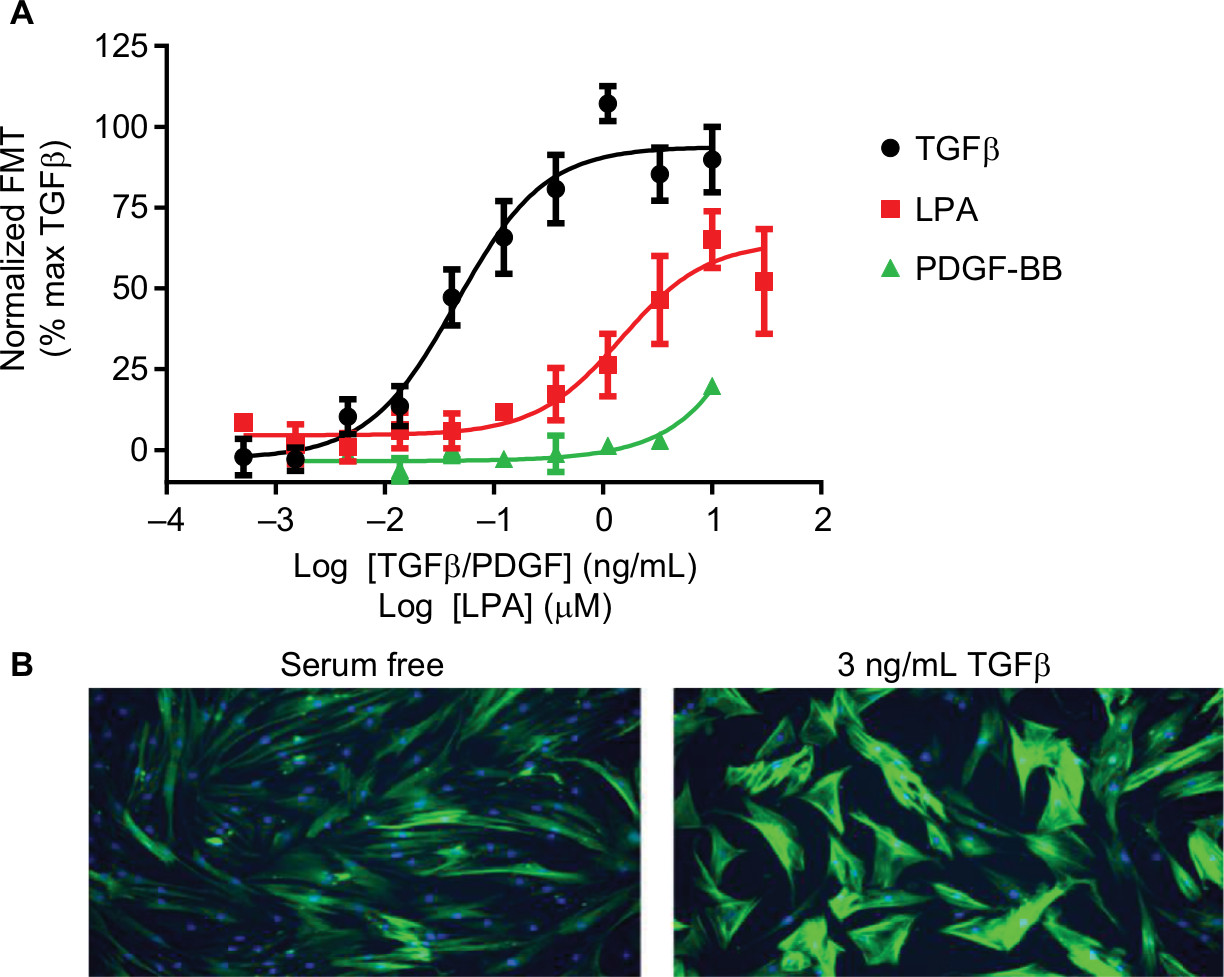 | Figure 2 FMT. Notes: (A) Concentration-dependent increases in αSMA expression indicative of FMT were determined in human lung fibroblasts following treatment with TGFβ, PDGF or LPA for 48 hours. For each individual experiment, data were normalized to the maximal levels of FMT observed with TGFβ, and are expressed as mean ± SEM for four independent experiments. (B) Representative images of αSMA expression using HCI after serum free or 3 ng/mL TGFβ treatment. Mouse anti-αSMA (1:1000) and FITC-conjugated Affini-pure goat anti-rabbit IgG (1:1000) were used to measure αSMA expression (green) and nuclei staining using 1 µM Hoechst333 (blue). Images were acquired on a widefield ImageXpress Micro microscope. Abbreviations: αSMA, α-smooth muscle actin; FMT, Fibroblast to myofibroblast transdifferentiation; HCI, high-content imaging; LPA, lysophosphatidic acid; PDGF, platelet-derived growth factor; TGFβ, transforming growth factor β. |
Similar to proliferation, the correct stimulus must be selected in order to drive the differentiation of fibroblasts, rather than promoting their proliferation. The most well-described stimulus of FMT is TGFβ, whose role in fibrosis has been well characterized,42 although LPA has also been shown to drive FMT43 (Figure 2). In contrast to the proliferation assay, PDGF plays no role in the differentiation of fibroblasts, highlighting the need to choose the correct stimulus for each individual assay, as different GFs will stimulate different pro-remodeling phenotypes.
In addition to fibroblasts, epithelial cells may also undergo differentiation to a more migratory, mesenchymal phenotype in response to TGFβ treatment, known as epithelial–mesenchymal transdifferentiation (EMT). Similar to FMT, this can be monitored using specific antibodies to markers of EMT. The loss of E-cadherin, through either intracellular degradation or inhibition of its expression, and upregulation of vimentin can be used as a marker of EMT.44
Cell migration
Another aspect of fibroblast or epithelial cell behavior that can be monitored is the migration to either a chemoattractant or wounding of a cell monolayer. Chemoattractants may include GFs, chemokines and lipids, such as PDGF,45 CC chemokine ligand 21 acting via the CCR7 receptor46 or LPA.47,48 The chemotactic assay is based on the original Boyden Assay49 and uses plastic inserts that contain a cell-permeable membrane, such as the Transwell® Permeable Supports, that are placed in multiwell plates. Cells are placed on top of the insert and migration through the membrane to a stimulus monitored, mimicking the activities of fibroblasts on the invasion of the basement membrane. The number of migratory cells can be assessed using cell detachment and counting, or cellular staining in situ and microscopic analysis. Fibrosis itself is believed to be caused by dysregulated wound healing;20 therefore, monitoring the migration of fibroblasts to a wound is also of interest. There are a number of technologies now available to explore the migratory response of fibroblasts into a wound. The scratch wound assay can be used to monitor wound healing and is performed by mechanical disruption of a confluent monolayer of cells, often by scratching using a pipette tip or pin tool50 or removal of a physical plug around which the monolayer has formed (Figure 3) (Oris™ cell migration plates, Platypus Technologies, LLC).51 This leads to disruption of cell–cell contacts at the site of the wound, and increased secretion of GFs that drive both migration into the wound site and proliferation of cells in order to heal the wound. The time course of this movement can be monitored by fixing cells after different incubation times and imaging.52
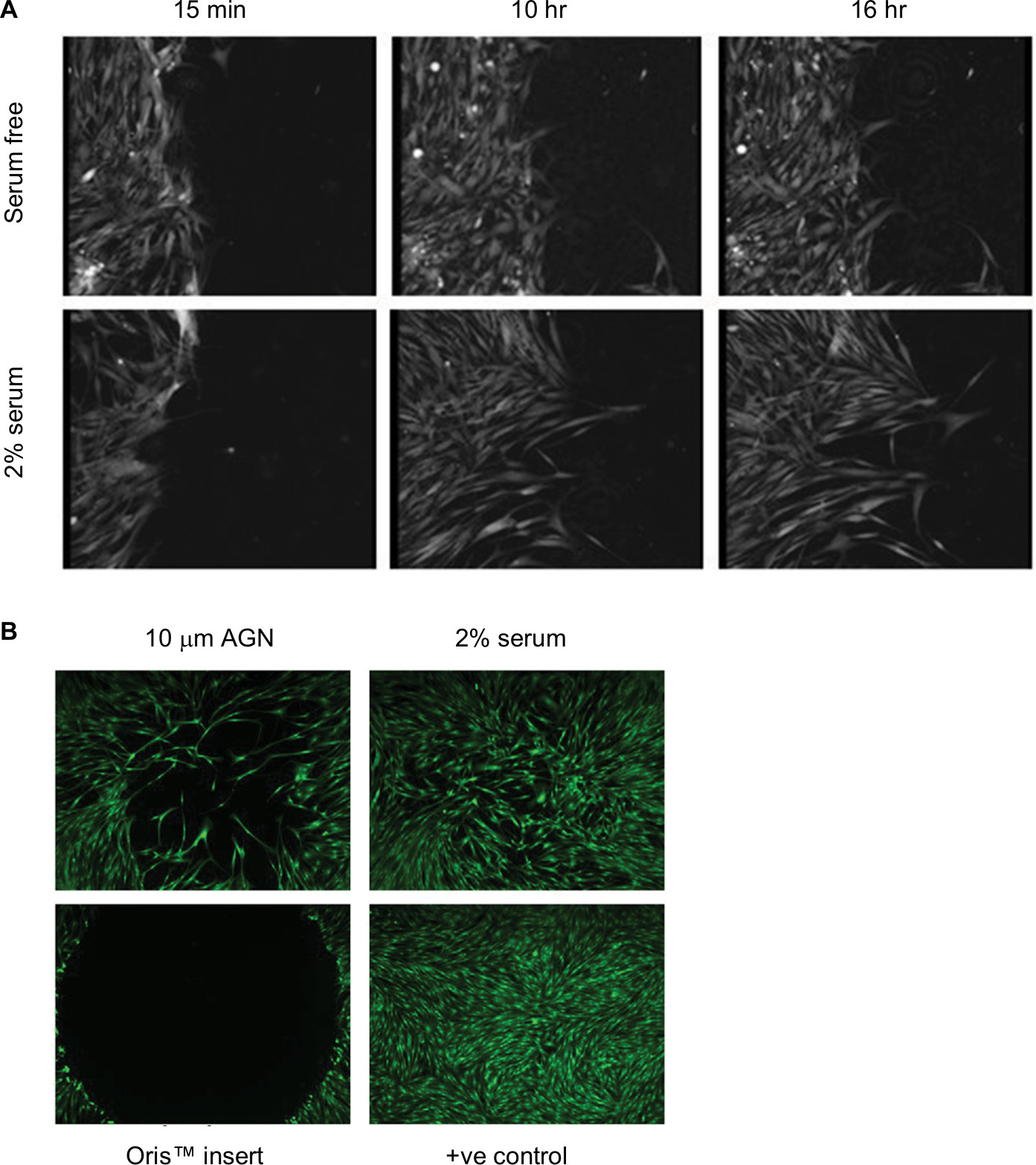 | Figure 3 Fibroblast migration. Notes: (A) Time course of migration after scratch wounding of a confluent monolayer of HLF, in response to serum-free conditions or treatment with 2% serum, measured using ptychography on the Phase Focus Livecyte. Images were acquired every 15 minutes, using 20× magnification. (B) HLF were grown to confluence in Oris™ plates, plug removed and migration measured after 48 hours treatment with either serum-free medium or 2% serum. 100 nM Cell Mask™ was used to stain cytoplasm. Images were acquired on a widefield Image Xpress Micro microscope. Abbreviation: HLF, human lung fibroblast. |
Some common detection methods include the use of fluorescent cellular stains, such as the nuclear stain Hoechst 33342, phalloidin to detect actin filaments, or cytoplasmic stains such as CellMask™, coupled with either manual microscopy or, more commonly, automated HCI platforms. Many of these biomarkers require a fixation step in order to denature proteins and preserve biological structure of the cells, meaning that they can only be performed in end-point mode at a specific time point. This may mean a more lengthy development phase to ensure that the correct stimulus and time point are chosen. However, these assays offer a significant advantage as once fixed, automated HCI platforms can be used to stack and read multiple plates at a time allowing for medium-throughput screening. An alternative method is to perform experiments in live cells, and use non-invasive, label-free imaging techniques coupled with time-lapse photography to allow tracking of phenotypic and kinetic behavior of individual cells, as well as cell populations. These can be monitored using transmitted light microscopes, phase-contrast imaging (including ptychography) or impedance assaysPlatforms include the IncuCyte® (Intellicyt53 which measures phase contrast) or the Livecyte (Phasfocus54 which uses ptychography). During ptychography light is scattered as it passes through the specimen and then captured as a diffraction pattern on a CCD camera. These diffraction patterns can then be processed using a ptychographic algorithm to provide information on the three-dimensional (3D) structure of the sample such as cell morphology.55 Similarly, impedance assays monitor changes in cell morphology, but in this case, cells are plated out onto electrodes, and the degree of electrical current transmitted through or between the cells is monitored.56 Impedance systems include ECIS Zθ™ (Applied Biosystems), xCELLigence (ACEA Biosciences) and EPIC® (Corning). Although these assays are lower throughput, they can offer advantages over the fixed time point assay in terms of the depth of information that can be gathered. For ptychography, this can also include both cell speed and directionality of movement, as well as tracking of individual cells. The choice of assay must, therefore, be a balance between the required throughput of the assay vs the detail of the biological question being asked. For example, automated wound-healing assays such as the scratch or Oris™ migration assays are more amenable to compound screening, whereas the lower throughput label-free assays can be used to generate information-rich assays that may be more suited to target ID/validation.
Mediator release
Another important aspect of airway remodeling is the secretion of GFs and cytokines that drive the progression of remodeling, as well as components of ECM that cause thickening of the basement membrane and airway stiffening. With the development of selective antibodies against these proteins, it is now possible to monitor their secretion from human primary cells in a higher throughput manner using plate-based ELISA.
Similar to HCI, there are now a number of platforms available that allow multiplexed detection of the presence of a range of GFs and cytokines, by immobilizing multiple selective antibodies within a single well. These include the Meso Scale Discovery R-Plex, the Quansys multiplex system and Luminex® technology bead-based immunoassays for flow cytometers.57–59 The major advantage of these technologies is that because samples can be simultaneously monitored for a range of different remodeling mediators, information gathering is not limited when sample sizes are small, such as with those obtained from patient populations.
ECM rearrangement
As discussed previously, myofibroblasts form foci in the fibrotic lung where they secrete components of the ECM such as collagens, proteoglycans and fibronectin. The secretion of these proteins can be monitored in conditioned medium from cultured cells using ELISA (Figure 4). The secreted collagen is laid down in highly ordered, cross-linked collagen fibers that are resistant to degradation and form permanent scarring of the lung tissue. This occurs through enzymatic covalent cross-linking, catalyzed by the lysyl oxidase and transglutaminase families of enzymes,60 to produce collagen cross-links including hydroxylysinonorleucine, dihydroxylysinonorleucine and hydroxypyridinium. The ratio of different cross-links determines severity and duration of fibrotic responses, with an elevation of dihydroxylysinonorleucin: hydroxylysinonorleucine ratio associated with acute fibrosis, and increased hydroxypyridinium indicative of chronic lung fibrosis. The importance of this has been demonstrated in bleomycin models of fibrosis, where increased cross-linking of collagen, rather than total collagen levels, was associated with disease severity.61,62 Ultimately, novel therapies for airway remodeling should not only inhibit the secretion of soluble collagen but also affect the cross-linking and reorganization of collagen into insoluble fibers.
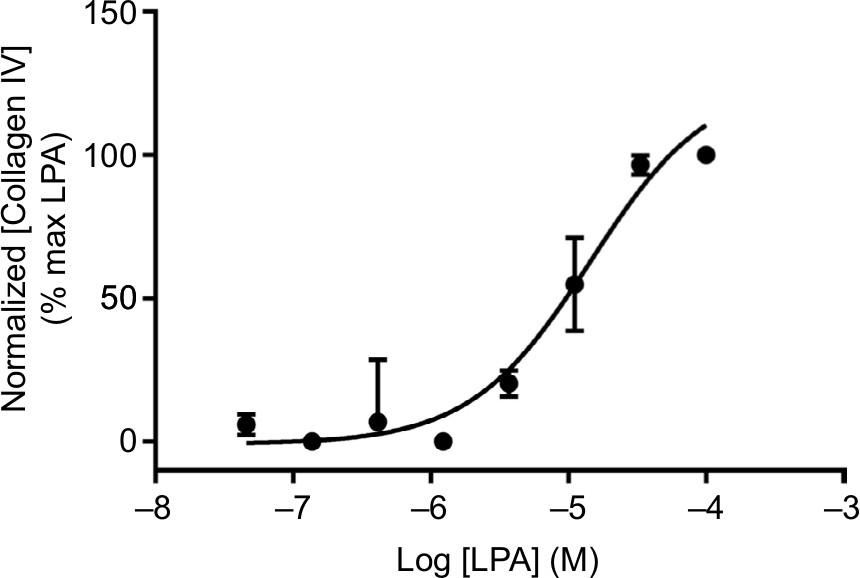 | Figure 4 Collagen secretion. Notes: Concentration-dependent increase in collagen IV secretion following treatment with LPA for 48 hours, assayed using ELISA (mouse monoclonal, clone COL-94). Data were normalized to maximal collagen IV secretion observed with LPA, and expressed as mean ± SEM for three independent experiments. Abbreviation: LPA, lysophosphatidic acid. |
Matrix architecture and stiffness has marked functional effects on fibroblasts but these parameters are not revealed by standard pathology or gene expression studies. To investigate the ability of novel therapeutics to inhibit collagen cross-linking or increase the turnover of ECM in the remodeled airway, a sensitive technique is required to monitor not just the levels of secreted soluble collagen, but the degree of cross-linked, insoluble collagen in the ECM. One such technique is the nonlinear optical method of second harmonic and multiphoton excitation fluorescence. Second harmonic generation (SHG) is a well-characterized method of label-free microscopy that utilizes the ability of a material to produce second harmonic light. In this process, the excitation photons interact with highly ordered, but noncentrosymmetric molecules in the sample, to generate a new photon with twice the energy (frequency doubled) of the initial photons. An SHG microscope analyses the variations between excitation (or incident light, around 900 nm) and the resultant emitted, frequency-doubled SHG signal (around 450 nm). SHG relies on the specific molecular orientation of the sample and is therefore highly specific for the detection of fibrillary, rather than soluble collagen, in both cell and tissue structures. For this reason, SHG can be used to monitor collagen cross-linking and 3D organization of collagen matrices both in cells and tissue structures.63,64
The lung is also subjected to significant mechanical stress during the cyclical volume changes that occur during breathing. This is especially important in chronic respiratory disease where mechanical forces within the diseased airway may further contribute to structural changes in the airway.65
Increased complexity; moving beyond single-cell populations
Single-cell multiplexing
Multiplexing approaches allow us to maximize the research from a single population of primary cells from patients with chronic respiratory diseases that have a limited lifetime during which they maintain disease phenotype in culture. In addition, as many of these assays are amenable to miniaturization, being compatible with both 96- and 384-well format, they also allow the monitoring of multiple pathways and mediators involved in remodeling. This gives us the possibility to explore novel strategies to inhibit airway remodeling in more disease-relevant cells. We are no longer restricted to targets that directly inhibit the pathways of pro-fibrotic mediators, but have scope to explore the potential of novel targets that will inhibit airway remodeling by opposing the activities of multiple pathways. For example, activating cAMP-dependent signaling pathways has been shown to be able to inhibit a diverse range of both GFs pathways and phenotypic responses.66–69
Although the use of human primary cells is useful for characterization of compounds in simple models of disease, the use of co-cultures or 3D matrices of cells/tissues can further increase the depth of information available, and better represent the complex nature of human disease.
Multicellular matrices
Individual cells grown on plastic multiwell plates will behave differently to cells, or co-cultures of cells, that are grown on more relevant matrices such as collagen or fibronectin.70,71 Reconstituted basement membrane can be used, such as Matrigel (Corning Life Sciences) or Cultrex® basement membrane extract (Trevigen, Inc). These are composed of laminin, collagen IV, entactin and heparin sulfate proteoglycan, and are available in GFs-rich, or reduced GFs forms. The use of these types of matrix proteins has demonstrated that adherent cells such as fibroblasts will respond differently, demonstrating increased proliferative capacity and reduced protein and collagen biosynthesis than when grown directly on plastic.72 These reconstituted matrices are useful in exploring cell behavior, but are sourced from Engelbreth-Holm-Swarm (EHS) mouse sarcoma cells meaning the constituent proteins are murine, and their relative levels may change between batches. An alternative approach is to use specific ECM components to explore their effect on cell phenotype, or ECM laid down by cells derived from patient samples.71
The most commonly used systems to explore airway remodeling are epithelial cell and fibroblast co-cultures to explore the role of epithelial damage in fibroblast activation73,74 or co-cultures between fibroblasts and inflammatory cells to investigate the role of inflammation in driving airway remodeling. The major advantage of these systems is that they allow us to study crosstalk between different cell types that occurs naturally during pathology of disease, rather than trying to artificially recapitulate the behaviors by treating a single cell type with individual GFs or cytokines.
Three-dimensional matrices
The use of complex 3D culture models has increased our knowledge of the processes involved in airway remodeling, but they still do not completely capture the complex environment that exists in the human lung. Techniques such as the use of precision-cut lung slices (PCLS) can allow us to explore either the development of airway remodeling or the functional consequences on airway responsiveness in a largely intact tissue architecture.75 A number of techniques have been used ranging from the use of chronic bronchoconstriction to promote ASM hyperplasia76 to treatment with GFs or toxins to model chronic fibrosis.77,78 Alternatively, PCLS can be taken from animals that have been exposed to challenges that will promote the development of airway remodeling.79 In combination with histology or fluorescence microscopy, the architecture and cellular make up of these tissues can then be probed. Nuclei can be stained in order to give an indication of hyperplasia in the development of fibrosis, and structural proteins such as actin filaments can be visualized using stains such as fluorescently labeled phalloidin to allow us to probe the architecture of the cells in the lung slice. Currently, PCLS have been maintained in culture for up to 10 days giving scope to model some of the more chronic biomarkers of airway remodeling that are observed in respiratory diseases. Typical measurements of ASM hyperplasia are increases in airway wall thickness through αSMA or smooth muscle myosin expression. Changes to the surrounding parenchyma (fibrosis) may also be monitored, including basement membrane thickness, or collagen and TGFβ secretion. In addition, goblet cell hyperplasia can be examined using muc5A staining.
In combination, the use of these models with novel treatment agents may therefore give us increased confidence in the future clinical efficacy of selected compounds, as they monitor compound activity against human targets, in disease appropriate cells/tissues, using pathologically relevant mediators and phenotypic read outs.
Summary
In summary, when exploring the mechanisms behind airway remodeling and investigating novel treatments for this poorly treated area, we would recommend a two-tiered approach: initial higher-throughput screening in single, relevant cell types, followed by more relevant multi-cell and matrix imaging. The use of ELISA-based techniques to monitor secreted proteins and processes such as proliferation can be useful in the initial selection of lead candidates due to their sensitivity and compatibility with high-throughput screening. These phenotypic assays are compatible with human primary cells and allow us to maximize the depth of information we can obtain from cells from patients. In combination with techniques such as siRNA or CRISPR/Cas9 to perform genetic manipulation of target genes, they are becoming increasingly useful in target ID and validation. This is an important consideration in this age of the 3Rs (replacement, reduction and refinement of animal usage) as we no longer have to rely on animal models that may not accurately mimic the human disease.
However, in order to accurately represent the complexity of human disease, multicellular or tissue models in conjunction with imaging techniques can provide a wealth of information not only on the pathogenesis of human disease, but the ability of compounds to block these processes. These allow us to monitor not just single process at a single time point, but the kinetics of those responses and the interplay between different cellular markers through multiplexing.
Although none of these systems will truly replicate the complex biology of the lung and the mechanical stress it endures during breathing and gas exchange, we believe that if designed properly these models can be used to enhance our understanding of airway remodeling and the prediction of the behavior of novel treatments in chronic respiratory disease.
The use of ex vivo isolated perfused lung or “organ on a chip”80 assays will allow us to model the mechanical aspect of the breathing lung, which may give us the most accurate model of chronic respiratory disease. These approaches may be useful to identify novel pathways and targets for new treatment options, but will be of limited use in compound screening due to the low throughput nature of these approaches. Airway remodeling remains an exciting area of research, and the advent of new and more information-rich assay technologies is allowing us to deepen our understanding of the molecular mechanisms behind disease pathology. Ultimately, this will allow us to explore novel targets that have the potential for increased efficacy in the treatment of chronic airway remodeling.
Disclosure
The authors report no conflicts of interest in this work.
References
FIRS. Forum of international respiratory societies. The Global Impact of Respiratory Disease. 2nd ed. Sheffield: European Respiratory Society; 2017. | ||
Chung KF, Godard P, Adelroth E, et al. Difficult/therapy-resistant asthma: the need for an integrated approach to define clinical phenotypes, evaluate risk factors, understand pathophysiology and find novel therapies. ERS task force on difficult/therapy-resistant asthma. European respiratory society. Eur Respir J. 1999;13(5):1198–1208. | ||
Al-Muhsen S, Johnson JR, Hamid Q. Remodeling in asthma. J Allergy Clin Immunol. 2011;128(3):451–462. | ||
Redington AE. Fibrosis and airway remodelling. Clin Exp Allergy. 2000;30(Suppl 1):42–45. | ||
Shimoda LA, Laurie SS. Vascular remodeling in pulmonary hypertension. J Mol Med. 2013;91(3):297–309. | ||
Wu D, Lai T, Yuan Y, et al. Elevated expression of placental growth factor is associated with airway-wall vascular remodelling and thickening in smokers with asthma. Sci Rep. 2017;7:43017. | ||
Harkness LM, Ashton AW, Burgess JK. Asthma is not only an airway disease, but also a vascular disease. Pharmacol Ther. 2015;148:17–33. | ||
Chetta A, Olivieri D. Role of inhaled steroids in vascular airway remodelling in asthma and COPD. Int J Endocrinol. 2012;2012:6–11. | ||
Payne DN, Rogers AV, Adelroth E, et al. Early thickening of the reticular basement membrane in children with difficult asthma. Am J Respir Crit Care Med. 2003;167(1):78–82. | ||
Saglani S, Mathie SA, Gregory LG, Bell MJ, Bush A, Lloyd CM. Pathophysiological features of asthma develop in parallel in house dust mite-exposed neonatal mice. Am J Respir Cell Mol Biol. 2009;41(3):281–289. | ||
Saglani S. Childhood severe asthma: new insights on remodelling and biomarkers. Paediatr Respir Rev. 2017;24:11–13. | ||
Bonato M, Bazzan E, Snijders D, et al. Clinical and pathologic factors predicting future asthma in wheezing children. A longitudinal study. Am J Respir Cell Mol Biol. 2018;59(4):458–466. | ||
Castro-Rodriguez JA, Saglani S, Rodriguez-Martinez CE, Oyarzun MA, Fleming L, Bush A. The relationship between inflammation and remodeling in childhood asthma: a systematic review. Pediatr Pulmonol. 2018;53(6):824–835. | ||
Chetta A, Foresi A, Del Donno M, Bertorelli G, Pesci A, Olivieri D. Airways remodeling is a distinctive feature of asthma and is related to severity of disease. Chest. 1997;111(4):852–857. | ||
Minshall EM, Leung DY, Martin RJ, et al. Eosinophil-associated TGF-beta1 mRNA expression and airways fibrosis in bronchial asthma. Am J Respir Cell Mol Biol. 1997;17(3):326–333. | ||
Benayoun L, Druilhe A, Dombret MC, Aubier M, Pretolani M. Airway structural alterations selectively associated with severe asthma. Am J Respir Crit Care Med. 2003;167(10):1360–1368. | ||
Pepe C, Foley S, Shannon J, et al. Differences in airway remodeling between subjects with severe and moderate asthma. J Allergy Clin Immunol. 2005;116(3):544–549. | ||
James AL, Bai TR, Mauad T, et al. Airway smooth muscle thickness in asthma is related to severity but not duration of asthma. Eur Respir J. 2009;34(5):1040–1045. | ||
Bumbacea D, Campbell D, Nguyen L, et al. Parameters associated with persistent airflow obstruction in chronic severe asthma. Eur Respir J. 2004;24(1):122–128. | ||
Selman M, King TE, Pardo A. Idiopathic pulmonary fibrosis: prevailing and evolving hypotheses about its pathogenesis and implications for therapy. Ann Intern Med. 2001;134(2):136–151. | ||
Collard HR, Ryu JH, Douglas WW, et al. Combined corticosteroid and cyclophosphamide therapy does not alter survival in idiopathic pulmonary fibrosis. Chest. 2004;125(6):2169–2174. | ||
Vestbo J, Sørensen T, Lange P, Brix A, Torre P, Viskum K. Long-term effect of inhaled budesonide in mild and moderate chronic obstructive pulmonary disease: a randomised controlled trial. Lancet. 1999;353(9167):1819–1823. | ||
Yan F, Gao H, Zhao H, Bhatia M, Zeng Y. Roles of airway smooth muscle dysfunction in chronic obstructive pulmonary disease. J Transl Med. 2018;16(1):262. | ||
Hogg JC, Chu F, Utokaparch S, et al. The nature of small-airway obstruction in chronic obstructive pulmonary disease. N Engl J Med. 2004;350(26):2645–2653. | ||
Johnson PR, Roth M, Tamm M, et al. Airway smooth muscle cell proliferation is increased in asthma. Am J Respir Crit Care Med. 2001;164(3):474–477. | ||
Morrell NW, Yang X, Upton PD, et al. Altered growth responses of pulmonary artery smooth muscle cells from patients with primary pulmonary hypertension to transforming growth factor-beta(1) and bone morphogenetic proteins. Circulation. 2001;104(7):790–795. | ||
Scotton CJ, Chambers RC. Molecular targets in pulmonary fibrosis: the myofibroblast in focus. Chest. 2007;132(4):1311–1321. | ||
Kuhn C, Boldt J, King TE, Crouch E, Vartio T, Mcdonald JA. An immunohistochemical study of architectural remodeling and connective tissue synthesis in pulmonary fibrosis. Am Rev Respir Dis. 1989;140(6):1693–1703. | ||
King TE, Schwarz MI, Brown K, et al. Idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2001;164(6):1025–1032. | ||
Thompson HG, Mih JD, Krasieva TB, Tromberg BJ, George SC. Epithelial-derived TGF-beta2 modulates basal and wound-healing subepithelial matrix homeostasis. Am J Physiol Lung Cell Mol Physiol. 2006;291(6):L1277–L1285. | ||
Davies DE. The role of the epithelium in airway remodeling in asthma. Proc Am Thorac Soc. 2009;6(8):678–682. | ||
Andersson-Sjöland A, de Alba CG, Nihlberg K, et al. Fibrocytes are a potential source of lung fibroblasts in idiopathic pulmonary fibrosis. Int J Biochem Cell Biol. 2008;40(10):2129–2140. | ||
Bucala R, Spiegel LA, Chesney J, Hogan M, Cerami A. Circulating fibrocytes define a new leukocyte subpopulation that mediates tissue repair. Mol Med. 1994;1(1):71–81. | ||
Roberts MJ, Broome RE, Kent TC, Charlton SJ, Rosethorne EM. The inhibition of human lung fibroblast proliferation and differentiation by Gs-coupled receptors is not predicted by the magnitude of cAMP response. Respir Res. 2018;19(1):56. | ||
Gratzner HG. Monoclonal antibody to 5-bromo- and 5-iododeoxyuridine: A new reagent for detection of DNA replication. Science. 1982;218(4571):474–475. | ||
Huang S, Wettlaufer SH, Hogaboam C, Aronoff DM, Peters-Golden M, Prostaglandin E. Prostaglandin E(2) inhibits collagen expression and proliferation in patient-derived normal lung fibroblasts via E prostanoid 2 receptor and cAMP signaling. Am J Physiol Lung Cell Mol Physiol. 2007;292(2):L405–L413. | ||
Sakai N, Chun J, Duffield JS, Wada T, Luster AD, Tager AM. LPA1-induced cytoskeleton reorganization drives fibrosis through CTGF-dependent fibroblast proliferation. FASEB J. 2013;27(5):1830–1846. | ||
Liu D, Huang Y, Bu D, et al. Sulfur dioxide inhibits vascular smooth muscle cell proliferation via suppressing the Erk/MAP kinase pathway mediated by cAMP/PKA signaling. Cell Death Dis. 2014;5:e1251. | ||
Hetzel M, Bachem M, Anders D, Trischler G, Faehling M. Different effects of growth factors on proliferation and matrix production of normal and fibrotic human lung fibroblasts. Lung. 2005;183(4):225–237. | ||
van Corven EJ, Groenink A, Jalink K, Eichholtz T, Moolenaar WH. Lysophosphatidate-induced cell proliferation: identification and dissection of signaling pathways mediated by G proteins. Cell. 1989;59(1):45–54. | ||
Hinz B, Celetta G, Tomasek JJ, Gabbiani G, Chaponnier C, Matsudaira PT. Alpha-smooth muscle actin expression upregulates fibroblast contractile activity. Mol Biol Cell. 2001;12(9):2730–2741. | ||
Gu L, Zhu YJ, Yang X, Guo ZJ, Xu WB, Tian XL. Effect of TGF-beta/Smad signaling pathway on lung myofibroblast differentiation. Acta Pharmacol Sin. 2007;28(3):382–391. | ||
Tang N, Zhao Y, Feng R, et al. Lysophosphatidic acid accelerates lung fibrosis by inducing differentiation of mesenchymal stem cells into myofibroblasts. J Cell Mol Med. 2014;18(1):156–169. | ||
Caulín C, Scholl FG, Frontelo P, Gamallo C, Quintanilla M. Chronic exposure of cultured transformed mouse epidermal cells to transforming growth factor-beta 1 induces an epithelial-mesenchymal transdifferentiation and a spindle tumoral phenotype. Cell Growth Differ. 1995;6(8):1027–1035. | ||
Suganuma H, Sato A, Tamura R, Chida K. Enhanced migration of fibroblasts derived from lungs with fibrotic lesions. Thorax. 1995;50(9):984–989. | ||
Pierce EM, Carpenter K, Jakubzick C, et al. Idiopathic pulmonary fibrosis fibroblasts migrate and proliferate to CC chemokine ligand 21. Eur Respir J. 2007;29(6):1082–1093. | ||
Scott I. Lysophosphatidic acid is an important mediator of fibroblast recruitment in IPF. Thorax. 2008;63(7):654. | ||
Tager AM, Lacamera P, Shea BS, et al. The lysophosphatidic acid receptor LPA1 links pulmonary fibrosis to lung injury by mediating fibroblast recruitment and vascular leak. Nat Med. 2008;14(1):45–54. | ||
Boyden S. The chemotactic effect of mixtures of antibody and antigen on polymorphonuclear leucocytes. J Exp Med. 1962;115(3):453–466. | ||
Schreier T, Degen E, Baschong W. Fibroblast migration and proliferation during in vitro wound healing. A quantitative comparison between various growth factors and a low molecular weight blood dialysate used in the clinic to normalize impaired wound healing. Res Exp Med. 1993;193(4):195–205. | ||
Glenn HL, Messner J, Meldrum DR. A simple non-perturbing cell migration assay insensitive to proliferation effects. Sci Rep. 2016;6:31694. | ||
Marei H, Carpy A, Woroniuk A, et al. Differential Rac1 signalling by guanine nucleotide exchange factors implicates FLII in regulating Rac1-driven cell migration. Nat Commun. 2016;7:10664. | ||
Costa P, Gautrot JE, Connelly JT. Directing cell migration using micropatterned and dynamically adhesive polymer brushes. Acta Biomater. 2014;10(6):2415–2422. | ||
Kasprowicz R, Suman R, O’Toole P. Characterising live cell behaviour: Traditional label-free and quantitative phase imaging approaches. Int J Biochem Cell Biol. 2017;84:89–95. | ||
Marrison J, Räty L, Marriott P, O’Toole P. Ptychography – a label free, high-contrast imaging technique for live cells using quantitative phase information. Sci Rep. 2013;3(1):2369. | ||
Giaever I, Keese CR. Monitoring fibroblast behavior in tissue culture with an applied electric field. Proc Natl Acad Sci U S A. 1984;81(12):3761–3764. | ||
Ding M, Cavallin A, Hermansson NO, Berntsson P, Jinton L, Rodrigo Blomqvist S. Comparing flow cytometry QBeads plexScreen assays with other immunoassays for determining multiple analytes. SLAS Discov. 2018;23(7):676–686. | ||
Salvante KG, Brindle E, Mcconnell D, O’Connor K, Nepomnaschy PA. Validation of a new multiplex assay against individual immunoassays for the quantification of reproductive, stress, and energetic metabolism biomarkers in urine specimens. Am J Hum Biol. 2012;24(1):81–86. | ||
Dunbar SA. Applications of Luminex xMAP technology for rapid, high-throughput multiplexed nucleic acid detection. Clin Chim Acta. 2006;363(1–2):71–82. | ||
Lucero HA, Kagan HM. Lysyl oxidase: an oxidative enzyme and effector of cell function. Cell Mol Life Sci. 2006;63(19–20):2304–2316. | ||
Reiser KM, Tryka AF, Lindenschmidt RC, Last JA, Witschi HR. Changes in collagen cross-linking in bleomycin-induced pulmonary fibrosis. J Biochem Toxicol. 1986;1(1):83–91. | ||
Lin YC, Sung YK, Jiang X, Peters-Golden M, Nicolls MR. Simultaneously targeting myofibroblast contractility and extracellular matrix cross-linking as a therapeutic concept in airway fibrosis. Am J Transplant. 2017;17(5):1229–1241. | ||
Strupler M, Pena AM, Hernest M, et al. Second harmonic imaging and scoring of collagen in fibrotic tissues. Opt Express. 2007;15(7):4054–4065. | ||
Chen X, Nadiarynkh O, Plotnikov S, Campagnola PJ. Second harmonic generation microscopy for quantitative analysis of collagen fibrillar structure. Nat Protoc. 2012;7(4):654–669. | ||
Manuyakorn W. Airway remodelling in asthma: role for mechanical forces. Asia Pac Allergy. 2014;4(1):19–24. | ||
Stork PJ, Schmitt JM. Crosstalk between cAMP and MAP kinase signaling in the regulation of cell proliferation. Trends Cell Biol. 2002;12(6):258–266. | ||
Nikam VS, Wecker G, Schermuly R, et al. Treprostinil inhibits the adhesion and differentiation of fibrocytes via the cyclic adenosine monophosphate-dependent and Ras-proximate protein-dependent inactivation of extracellular regulated kinase. Am J Respir Cell Mol Biol. 2011;45(4):692–703. | ||
Kolodsick JE, Peters-Golden M, Larios J, Toews GB, Thannickal VJ, Moore BB. Prostaglandin E2 inhibits fibroblast to myofibroblast transition via E. prostanoid receptor 2 signaling and cyclic adenosine monophosphate elevation. Am J Respir Cell Mol Biol. 2003;29(5):537–544. | ||
Liu X, Ostrom RS, Insel PA. cAMP-elevating agents and adenylyl cyclase overexpression promote an antifibrotic phenotype in pulmonary fibroblasts. Am J Physiol Cell Physiol. 2004;286(5):C1089–C1099. | ||
Blaauboer ME, Boeijen FR, Emson CL, et al. Extracellular matrix proteins: a positive feedback loop in lung fibrosis? Matrix Biol. 2014;34:170–178. | ||
Philp CJ, Siebeke I, Clements D, et al. Extracellular matrix cross-linking enhances fibroblast growth and protects against matrix proteolysis in lung fibrosis. Am J Respir Cell Mol Biol. 2018;58(5):594–603. | ||
Emonard H, Grimaud JA, Nusgens B, Lapière CM, Foidart JM. Reconstituted basement-membrane matrix modulates fibroblast activities in vitro. J Cell Physiol. 1987;133(1):95–102. | ||
Zhang S, Smartt H, Holgate ST, Roche WR. Growth factors secreted by bronchial epithelial cells control myofibroblast proliferation: an in vitro co-culture model of airway remodeling in asthma. Lab Invest. 1999;79(4):395–405. | ||
Ishikawa S, Ishimori K, Ito S. A 3D epithelial-mesenchymal co-culture model of human bronchial tissue recapitulates multiple features of airway tissue remodeling by TGF-β1 treatment. Respir Res. 2017;18(1):195. | ||
Ressmeyer AR, Larsson AK, Vollmer E, Dahlèn SE, Uhlig S, Martin C. Characterisation of guinea pig precision-cut lung slices: comparison with human tissues. Eur Respir J. 2006;28(3):603–611. | ||
Oenema TA, Maarsingh H, Smit M, Groothuis GM, Meurs H, Gosens R. Bronchoconstriction induces TGF-β release and airway remodelling in guinea pig lung slices. PLoS One. 2013;8(6):e65580. | ||
Kasper M, Seidel D, Knels L, et al. Early signs of lung fibrosis after in vitro treatment of rat lung slices with CdCl2 and TGF-beta1. Histochem Cell Biol. 2004;121(2):131–140. | ||
Lin CJ, Yang PC, Hsu MT, et al. Induction of pulmonary fibrosis in organ-cultured rat lung by cadmium chloride and transforming growth factor-beta1. Toxicology. 1998;127(1–3):157–166. | ||
Kim HJ, Kim Y, Park SJ, et al. Airway smooth muscle sensitivity to methacholine in precision-cut lung slices (PCLS) from ovalbumin-induced asthmatic mice. Korean J Physiol Pharmacol. 2015;19(1):65–71. | ||
Huh D, Matthews BD, Mammoto A, Montoya-Zavala M, Hsin HY, Ingber DE. Reconstituting organ-level lung functions on a chip. Science. 2010;328(5986):1662–1668. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.