Back to Journals » Clinical Interventions in Aging » Volume 20
Aging and Intestinal Fibrosis: Mechanisms, Implications, and Therapeutic Strategies
Authors Shi Y, Zheng H, Zhu X ![]() , Lv J, Zhou M, Zhang S
, Lv J, Zhou M, Zhang S
Received 25 May 2025
Accepted for publication 13 November 2025
Published 26 November 2025 Volume 2025:20 Pages 2177—2194
DOI https://doi.org/10.2147/CIA.S542671
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Nandu Goswami
Yujie Shi,1,* Han Zheng,1,* Xiaxin Zhu,1 Jianyu Lv,2 Mi Zhou,1 Shuo Zhang1
1Department of Gastroenterology, The Second Affiliated Hospital of Zhejiang Chinese Medical University, Hangzhou, People’s Republic of China; 2Department of General Surgical Science, Gunma University, Graduate School of Medicine, Maebashi, Japan
*These authors contributed equally to this work
Correspondence: Shuo Zhang, The Second Affiliated Hospital of Zhejiang Chinese Medical University, No. 318 Chaowang Road, Gongshu District, Hangzhou, 310005, People’s Republic of China, Tel +86-13957192066, Email [email protected]
Abstract: With the global aging population, the impact of aging on various organ systems is becoming increasingly significant. The gastrointestinal tract, a key site of immune activity and microbial colonization, undergoes functional decline that is closely associated with a range of intestinal and systemic diseases. While aging-related fibrosis has been extensively studied in organs such as the lungs, liver, heart, and kidneys, its role in intestinal fibrosis remains underexplored. This review discusses mechanisms by which aging may promote or increase the risk of intestinal fibrosis, including immunosenescence, cellular senescence, gut microbiota dysbiosis, and dysregulated growth factor signaling. Additionally, both traditional and emerging therapeutic strategies are summarized to guide future interventions.
Keywords: immunosenescence, cellular senescence, gut microbiota, TGF-β signaling pathway
Introduction
Aging is widely defined as a complex natural process involving a gradual decline in organismal function.1 It is estimated that the proportion of the global population aged 65 years and older will increase from 10% in 2022 to 16% in 2050, and aging has become a major risk factor for multiple chronic diseases. In recent years, the molecular and cellular features of aging have been progressively summarized into multiple “hallmarks of aging”. The twelve classical hallmarks include: genomic instability, telomere attrition, epigenetic alterations, loss of proteostasis, deregulated nutrient sensing, mitochondrial dysfunction, cellular senescence, stem cell exhaustion, altered intercellular communication, impaired autophagy, chronic inflammation, and microecological dysregulation.2 In 2025, researchers further proposed potential new hallmarks such as extracellular matrix remodeling and psychosocial isolation, suggesting that aging is a multidimensional process involving biological, environmental, and psychological interactions.3 Notably, all of these hallmarks have been linked to alterations in the alternative splicing (AS) process, indicating its central role in regulating age-associated phenotypes2,4 Although these features provide a theoretical framework for understanding aging, the precise organ-specific mechanisms and interconnections remain unclear, and effectively delaying aging-related diseases remains a major challenge.5
The gastrointestinal tract is one of the most immunologically active organs in the human body and serves as a major reservoir for complex microbial communities.6 Its robust immune activity and continuous interactions with the gut microbiota play a pivotal role in maintaining systemic homeostasis. With advancing age, the gastrointestinal system undergoes significant changes, including compromised mucosal barrier function, alterations in microbial composition, dysregulated immune responses, and persistent low-grade inflammation.7 These age-related changes not only impair intestinal function but also have widespread effects on overall health.
Studies have shown that fibrosis in organs such as the lungs, liver, heart, and kidneys increases significantly in the elderly, and aging may contribute to fibrotic remodeling through shared underlying mechanisms.8 In the gastrointestinal tract, aging-related symptoms such as constipation and reduced motility may be associated with structural changes and fibrotic alterations. Moreover, intestinal fibrosis is commonly observed in patients with chronic inflammatory conditions, such as inflammatory bowel disease (IBD). Notably, the aging gut shares several biological features with IBD—including immune dysregulation, chronic inflammation, and dysbiosis—which may predispose older individuals to intestinal fibrogenesis.9,10 Thus, intestinal fibrosis could represent an important hallmark of both intestinal and overall aging, highlighting the significance of understanding its mechanisms and treatment strategies. This review explores how aging promotes intestinal fibrosis and discusses potential therapeutic approaches.
The Process of Intestinal Fibrosis
Under physiological conditions, tissue repair after intestinal injury proceeds through a tightly regulated and dynamic sequence involving four overlapping phases: hemostasis, inflammation, proliferation, and remodeling.11 Initially, clot formation prevents blood loss, while neutrophils and monocytes are recruited to eliminate pathogens and cellular debris, forming the inflammatory response. During the proliferative phase, fibroblasts are activated to secrete type I and III collagen and to support the formation of granulation tissue, while angiogenesis restores vascular supply to the injured site. In the final remodeling phase, myofibroblasts, which emerge transiently to facilitate tissue contraction and ECM remodeling, undergo apoptosis or revert to a quiescent state once re-epithelialization is complete. This resolution is crucial for preventing excessive scarring and maintaining tissue homeostasis. Throughout this process, the extracellular matrix (ECM) exists in a highly dynamic and tightly balanced state, constantly synthesized, degraded, and remodeled by fibroblasts to meet the evolving demands of tissue recovery.12 Upon completion of repair, the inflammatory response resolves, cellular activity subsides, and ECM composition returns to baseline, restoring tissue architecture and function.
However, when intestinal injury is chronic, recurrent, or unresolved—or when the wound healing response itself becomes dysregulated—this orderly process fails. Structural reconstruction is impaired, and mesenchymal cells (including fibroblasts, myofibroblasts, and smooth muscle cells) undergo transient or sustained expansion.13 These activated cells continuously produce ECM components, and when ECM synthesis exceeds its degradation, excessive matrix deposition ensues. This eventually leads to scarring, loss of function, and irreversible architectural disruption, marking the transition from normal tissue repair to pathological intestinal fibrosis. Mechanistically, this process can be divided into three major stages: initiating triggers, activation of myofibroblasts, and ECM accumulation.14
Triggers
The process of intestinal fibrosis usually begins with the initiation of intestinal inflammation, which may be triggered by intestinal damage due to infection, autoimmunity, mechanical injury, or other external factors. Intestinal cell death triggers an inflammatory response that leads to destruction of the surrounding ECM.15 However, although fibrosis begins with inflammation, once the fibrotic process has begun, inhibition of inflammation alone is not sufficient to control the process.16 In a fibrotic mouse model of Salmonella typhimurium infection eradication of pathogens causing inflammation did not prevent the fibrotic process from occurring,17 and the severity of inflammation was not associated with collagen deposition,18 suggesting that although inflammation may be a prerequisite for triggering fibrosis, inflammation is not the only driver of fibrotic progression, which also explains the limited efficacy of many anti-inflammatory therapeutic agents.19
Activation of Myofibroblasts
In response to tissue injury, quiescent fibroblasts differentiate into myofibroblasts, a transitional phenotype between fibroblasts and smooth muscle cells (SMCs). Myofibroblasts acquire contractile properties through the expression of cytoskeletal proteins such as α-smooth muscle actin (α-SMA), facilitating wound contraction and tissue closure. In parallel, they synthesize large amounts of extracellular matrix (ECM) components, particularly type I collagen, which provide mechanical support, stabilize the myofibroblast phenotype, and contribute to tissue remodeling and force transmission.20,21
However, under conditions of chronic inflammation or repetitive injury, the normal wound healing process is disrupted. Persistent exposure to proinflammatory cytokines, oxidative stress, cell death, and tissue hypoxia reshapes the local signaling microenvironment, ultimately shifting the tissue response from repair to fibrosis. Key cytokines such as transforming growth factor-β (TGF-β), interleukin-4 (IL-4), and interleukin-13 (IL-13) are upregulated in chronic inflammatory settings and act through signaling pathways including Smad, STAT6, and PI3K/Akt to activate fibroblasts and SMCs.22 These cells are thereby driven toward a myofibroblast phenotype, leading to excessive production of collagen and fibronectin and the sustained activation of fibrotic processes. Moreover, during fibrosis progression, myofibroblasts exhibit not only persistent activation and proliferation but also resistance to apoptosis. This apoptosis-resistant phenotype enables their prolonged survival within tissues, contributing to excessive ECM accumulation and scar formation, which eventually result in irreversible structural remodeling and organ dysfunction12 (Figure 1).
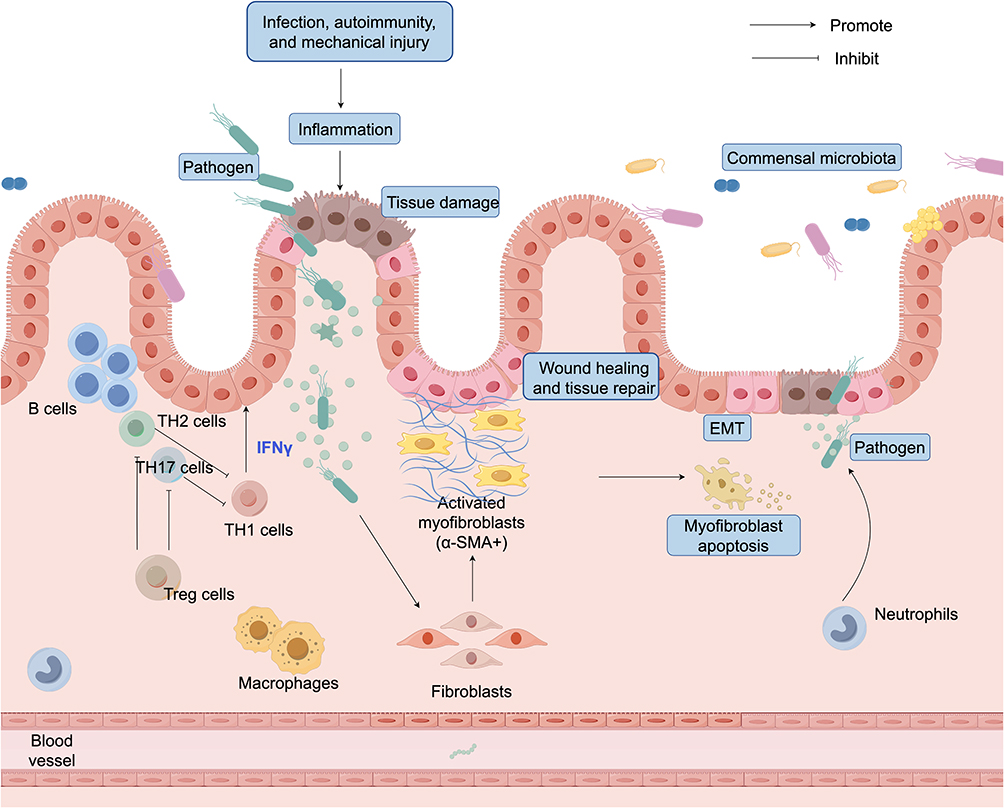 |
Figure 1 Healthy gut: normal tissue repair. External factors such as infection, autoimmunity, or mechanical injury can cause intestinal injury, leading to epithelial disruption and ECM degradation. Fibroblasts differentiate into activated myofibroblasts (α-SMA⁺), which secrete ECM proteins to promote wound healing and tissue repair. During resolution, myofibroblasts undergo apoptosis or revert to a quiescent state, thereby restoring tissue homeostasis. In this process, epithelial-mesenchymal transition (EMT)—where epithelial cells transiently acquire mesenchymal features—contributes to wound closure. Under normal physiological conditions, EMT is tightly regulated and does not result in fibrosis. T cells are also involved in coordinating this repair process. A dynamic balance among different T cell subsets helps regulate inflammation, limit tissue damage, and promote resolution. Together with microbiota homeostasis and controlled EMT, these immune interactions ensure effective repair without progression to fibrosis. |
Deposition of ECM
Continuous ECM deposition leads to progressive intestinal fibrosis, causing structural remodeling and impaired gut function. Matrix metalloproteinases (MMPs) and their tissue inhibitors (TIMPs) regulate ECM turnover. In fibrosis, an imbalance between MMPs and TIMPs facilitates excessive ECM accumulation. Additionally, transforming growth factor-beta (TGF-β) promotes ECM deposition by enhancing the contractile phenotype of mesenchymal cells, further contributing to fibrotic stiffening. Mechanistically, progressive tissue stiffening reinforces a pathological positive feedback loop mediated by the Ras homolog family member A (RhoA)/Rho-associated coiled-coil-containing protein kinase (ROCK)/myocardin-related transcription factor (MRTF) signaling pathway.23,24 This axis enhances actomyosin contractility and sustains myofibroblast activation, thereby driving fibrotic progression. Specifically, increased matrix stiffness and profibrotic stimuli such as TGF-β activate RhoA, which subsequently triggers ROCK. This promotes actin polymerization and stress fiber formation, facilitating MRTF activation and nuclear translocation, ultimately leading to enhanced transcription of profibrotic genes.25,26 Furthermore, persistent collagen accumulation results in chronic tissue hypoxia. Elevated vascular endothelial growth factor (VEGF) expression promotes pathological neoangiogenesis, which further exacerbates fibrotic remodeling and perpetuates disease progression.13
Factors in the Aging Gut That Promote Fibrosis
Immunosenescence and Fibrosis
Macrophage Senescence
Macrophages play a pivotal role in intestinal tissue repair and homeostasis maintenance, particularly during the overlapping phases of inflammation, proliferation, and remodeling. Their functional plasticity is essential for preserving the integrity of the intestinal microenvironment. Based on phenotypic characteristics, surface marker expression, and cytokine secretion profiles, macrophages are broadly categorized into classically activated M1 (pro-inflammatory) and alternatively activated M2 (anti-inflammatory) subsets.27 M1 macrophages are primarily engaged in the initiation of inflammation and are characterized by high expression levels of inducible nitric oxide synthase (iNOS/NOS2), interleukin (IL)-1β, IL-6, C-C motif chemokine ligand 5 (CCL5), C-X-C motif chemokine ligand 9 (CXCL9), and matrix metalloproteinase-9 (MMP9). These factors contribute to the disruption of epithelial tight junctions and the induction of apoptosis, thereby amplifying mucosal inflammation.28,29 Moreover, M1 macrophages actively modulate extracellular matrix (ECM) dynamics by regulating ECM turnover and MMP activity, thereby influencing tissue remodeling either directly or indirectly. In the context of inflammatory bowel disease (IBD), particularly during active disease stages, intestinal macrophages exhibit a pronounced shift toward the M1 phenotype. This polarization is accompanied by activation of multiple pro-inflammatory pathways, including those mediated by Toll-like receptors, IL-8, interferon-γ (IFN-γ), and tumor necrosis factor (TNF). Notably, upregulation of the triggering receptor expressed on myeloid cells 1 (TREM1) signaling pathway has been shown to promote fibroblast and myofibroblast proliferation, thereby exacerbating intestinal fibrosis30 (Figure 2).
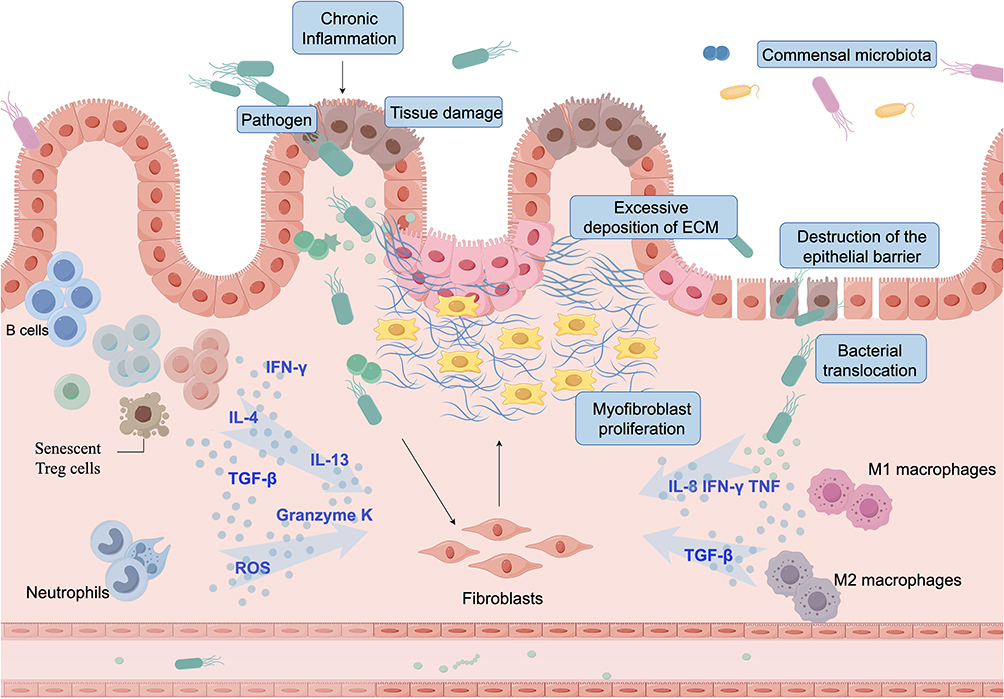 |
Figure 2 Aging Gut: Fibrosis. Chronic inflammation caused by pathogens, immune imbalance, or persistent injury drives abnormal proliferation and activation of fibroblasts, leading to excessive extracellular matrix (ECM) deposition and fibrosis. Senescent T cells release profibrotic cytokines (TGF-β, IL-4, IL-13), while neutrophils produce reactive oxygen species (ROS), enhancing oxidative stress and tissue damage. Together, they promote myofibroblast activation and ECM accumulation. Macrophages also play dual roles: M1 macrophages secrete IFN-γ, TNF, and IL-8 to amplify inflammation, whereas M2 macrophages, though typically linked to repair, may under chronic conditions release TGF-β and sustain fibrosis. Epithelial barrier disruption and bacterial translocation further aggravate mucosal injury, ultimately shifting the gut from normal repair toward irreversible fibrosis. |
Aging further exacerbates this process. Several studies have demonstrated that aging does not significantly alter the total number of macrophages in tissues such as the macula, skeletal muscle, and intestinal muscularis; however, it induces a phenotypic shift from anti-inflammatory M2 macrophages toward a pro-inflammatory M1 phenotype, thereby contributing to heightened tissue inflammation and fibrosis.31,32 Senescent macrophages exhibit elevated expression of senescence-associated β-galactosidase (SA-β-Gal), cyclin-dependent kinase inhibitor 2A (p16Ink4a), and a typical senescence-associated secretory phenotype (SASP). These alterations are believed to enhance fibrosis by promoting the secretion of pro-inflammatory mediators and disrupting intercellular communication.33,34 Notably, impaired crosstalk between senescent macrophages and myofibroblasts may contribute to insufficient tissue repair and further drive fibrotic progression.35 These findings suggest that age-related changes in macrophage phenotype and function are critical factors in the pathogenesis of fibrosis in aging tissues.
Although M2 macrophages are traditionally considered to have anti-inflammatory and tissue repair functions, increasing evidence suggests that they may play a dual role in fibrosis. M2 macrophages, through the secretion of cytokines like TGF-β and PDGF, not only promote tissue regeneration but also activate fibroblasts and induce the differentiation of myofibroblasts, thereby driving fibrosis progression.36 Further studies found that M2 type can differentiate into functionally distinct subtypes, among which M2a and M2c are closely related to fibrosis. For instance, M2a macrophages have been found to promote collagen deposition in kidney fibrosis through the “macrophage-to-myofibroblast transition” (MMT) mechanism.37 Similar phenomena have been observed in fibrosis in the lungs, heart, and skin, with the infiltration of M2 macrophages correlating with the severity of the disease.38
In intestinal fibrosis, studies have shown that in the TNBS-induced rat colitis model, the stenotic regions contain a high density of CD163⁺, CD206⁺, and arginase-positive M2 macrophages. These cells promote excessive proliferation of intestinal smooth muscle cells by secreting TGF-β1 and enhance their survival under hypoxic conditions.39 Similar findings have been observed in Crohn’s disease (CD) patients. Moreover, compared to newly diagnosed patients, these fibrosis-promoting M2 macrophages are more abundant in chronic CD patients, indicating that they not only persist in inflamed tissues but also accumulate further as the inflammation becomes chronic.40,41 However, the role of M2 macrophages in intestinal fibrosis is not always consistent. For instance, in a DSS-induced colitis model, although M2 macrophages contribute to fibrosis progression, exosomes derived from the exosomes derived from M2b macrophages have been shown to reduce the severity of colitis and promote mucosal healing.42,43 Despite these observations, the precise mechanisms underlying the role of M2 macrophages in intestinal fibrosis remain unclear, and further studies are needed to delineate how different subtypes and the microenvironment modulate the fibrosis process.44
T-Lymphocyte Senescence
T lymphocytes are central leukocytes of the immune system and play a pivotal role in adaptive immune responses, encompassing various T helper (Th) cell subsets such as Th1, Th2, and Th17, as well as regulatory T cells (Tregs), and coordinating functions with B cells. Th1 cells produce interferon-gamma (IFN-γ), which exerts a dual role in inflammation regulation and tissue repair. Recent studies have revealed that, although IFN-γ is a key pro-inflammatory cytokine, it can exert broad anti-fibrotic effects in chronic fibrotic environments by downregulating TGF-β and its downstream connective tissue growth factor (CTGF), upregulating indoleamine 2,3-dioxygenase 1 (IDO-1), and improving the immune microenvironment, with evidence across the kidney, lung, liver, and intestine.45–48 Th1-deficient murine models exhibit exacerbated fibrosis following abdominal irradiation, which is closely associated with markedly reduced expression of the hallmark Th1 cytokine IFN-γ, further confirming its critical role in intestinal anti-fibrotic responses.49 In contrast to Th1 cells, Th2 and Th17 cells are generally considered pro-fibrotic. Th2 cells regulate Th1 cell-derived IFN-γ expression and promote the secretion of cytokines such as IL-4, IL-5, and IL-13, driving type 2 immune responses and contributing to fibrosis in multiple organs, including the lungs and heart.50–52 Th17 cells additionally promote fibroblast activation, enhance TGF-β1 expression, and drive myofibroblast differentiation through the secretion of inflammatory mediators such as IL-17, thereby exacerbating tissue remodeling and extracellular matrix (ECM) deposition under chronic inflammatory conditions.50 Tregs, as negative regulators of immune responses, typically mitigate inflammation and tissue damage through upregulation of Foxp3 and IL-10 and suppression of Th17-mediated IL-17 secretion, exerting anti-fibrotic effects.53 However, recent evidence indicates that under TGF-β stimulation, Tregs can secrete Amphiregulin (AREG), promoting fibroblast proliferation and matrix deposition, suggesting a potential pro-fibrotic role in specific inflammatory or tissue repair contexts.54
With aging, the diversity and functionality of the T cell repertoire gradually decline, impairing the clearance of damaged cells and maintenance of mucosal homeostasis.55 Tregs are particularly prone to senescence compared with effector T cells, with a KLRG1⁺ Treg subset accumulating significantly in elderly humans and mice, exhibiting mitochondrial dysfunction, DNA damage accumulation, and impaired suppressive capacity, thereby representing key mediators of “inflammaging” and fibrosis progression.56 Moreover, senescent T cells persistently secrete IFN-γ and TNF, activating senescence-associated signaling pathways and promoting Th17 and certain Th1-like cytokine expression, exacerbating tissue injury in chronic inflammatory environments.55 Enhanced Th1-like pro-inflammatory cytokines have been confirmed in the intestines of aged rhesus macaques, and pathogenic gut microbiota can further drive fibrosis by augmenting Th1/Th17 responses.49,57 Furthermore, a subset of GZMK⁺ CD8⁺ T cells (Taa cells) accumulates with age, amplifying fibroblast senescence-associated secretory phenotype (SASP) responses—such as increased IL-6, CCL2, and CXCL1 secretion—via persistent Granzyme K production, thereby exacerbating tissue inflammation and injury and acting as a crucial driver of age-related disease progression.58 Collectively, T cells exhibit marked subset- and context-dependent roles in fibrosis, and the loss of immune homeostasis and sustained inflammatory activation during aging represents a critical mechanism driving intestinal fibrogenesis.
Neutrophil Senescence
Neutrophils play both a destructive and protective role in wound healing. When the intestinal epithelial barrier is compromised, neutrophils are the first immune cells to respond quickly, arriving at the scene of injury to remove tissue debris and destroy invading microbes.59 However, with age, increased levels of pro-inflammatory cytokines such as TNF-α, IL-1β, and IL-6 prolong the life span of neutrophils. Neutrophils that cannot be cleared in a timely manner exacerbate the inflammatory response in tissues by secreting large amounts of matrix metalloproteinase-9 (MMP-9) to disrupt epithelial cell-to-cell adhesion and releasing microRNAs such as miR-23a and miR-155 to promote DNA double-strand breaks and impede colonic healing.60 In addition, senescent neutrophils exhibit impaired microbial killing, and their chemotactic response is particularly reduced following priming with granulocyte-macrophage colony-stimulating factor (GM-CSF), which normally enhances neutrophil activation and effector functions.61 These age-related changes not only diminish their role in wound healing, but also lead to prolonged inflammation and fibroblast overactivation, which promotes intestinal fibrosis.
Potential Link Between Cellular Senescence, Oxidative Stress, Autophagy, and Fibrosis
In recent years, cellular senescence has emerged as a critical factor contributing to tissue homeostasis imbalance. Oxidative stress, as an important marker of cellular senescence, is particularly prominent in aged intestinal tissues.62 With advancing age, the decline in antioxidant defense mechanisms leads to excessive accumulation of reactive oxygen species (ROS), which in turn induces oxidative stress and causes damage to nucleic acids, proteins, and lipids.63 ROS activate key inflammatory signaling pathways, such as NF-κB and AP-1, thereby enhancing the expression of pro-inflammatory cytokines like TNF-α, disrupting the intestinal epithelial barrier, and triggering chronic inflammation and tissue injury.64 Especially in patients with IBD and ulcerative colitis, NF-κB activity was significantly enhanced and correlated with elevated peroxidase levels, suggesting that oxidative stress plays an important role in intestinal inflammation.65–67 Furthermore, ROS have been shown to induce the expression of NADPH oxidase in macrophages, amplifying the inflammatory response. Animal studies have demonstrated that inhibition of ROS production alleviates colitis symptoms, suggesting that ROS are key mediators in the development of intestinal fibrosis.68,69 These findings underscore the pivotal role of oxidative stress in aging-related intestinal pathology and provide a rationale for the development of antioxidant-based therapeutic strategies.
Autophagy, a key mechanism for cellular degradation and metabolic recycling, plays a crucial role in maintaining the intestinal epithelial barrier and suppressing inflammatory responses. In aging individuals, sustained activation of TGF-β signaling has been shown to inhibit autophagy pathways, particularly through upregulation of mTORC1 signaling, which interferes with the expression and function of essential autophagy-related genes such as ATG5, ATG7, and ATG16L1, ultimately leading to reduced autophagic activity.70 Studies have demonstrated that impaired autophagy results in persistent secretion of pro-inflammatory cytokines such as IL-1β, enhanced epithelial cell apoptosis, and compromised barrier integrity.71,72 These changes promote activation of myofibroblasts and excessive extracellular matrix (ECM) deposition, thereby exacerbating fibrosis. In patients with enteric strictures associated with celiac disease, increased frequencies of ATG16L1 mutations have been reported, suggesting a potential direct link between autophagy deficiency and intestinal fibrosis in humans.73 Animal studies further support this association, as autophagy activators such as rapamycin have been shown to restore fibroblast autophagic activity by inhibiting mTORC1 signaling, significantly attenuating colonic fibrosis.74 These findings highlight the pathological relevance of autophagy dysfunction in aging-associated intestinal fibrosis and support its therapeutic potential.
Gut Microbial Changes Accompanying Aging
The human gastrointestinal tract harbors approximately 100 trillion microorganisms, which play a crucial role in host immunity, metabolism, and nutrient absorption.75 The intestinal microbiota is mainly composed of Firmicutes, Bacteroidetes, Actinobacteria, and Verrucomicrobia, maintaining a dynamic equilibrium that preserves intestinal homeostasis in healthy individuals.76 However, this balance can be disrupted by pathogenic factors, leading to dysbiosis that adversely affects health, making the maintenance of a balanced gut microbiota essential for overall health and disease prevention.77 The role of the gut microbiome in the pathogenesis of intestinal fibrosis is now being progressively revealed, and evidence has been found in the aging gut.
We have summarized the role of gut microbes in intestinal fibrosis into the following three points: 1) increase in pro-inflammatory microbiota 2) alteration of gut microbial metabolites, and 3) impaired intestinal barrier function. An increased abundance of pro-inflammatory bacterial taxa from the Enterobacteriaceae family, such as adherent-invasive Escherichia coli (AIEC) and Salmonella Typhimurium, has been observed in both patients with inflammatory bowel disease (IBD) and mouse models of intestinal fibrosis.78 These changes were accompanied by elevated levels of type I/III collagen expression and significantly enhanced expression of pro-fibrotic mediators such as TGF-β1, connective tissue growth factor (CTGF) and insulin-like growth factor I (IGF-I).79 These pro-inflammatory microbiotas trigger an inflammatory response through activation of Toll-like receptor 4 (TLR4), particularly enhancing the response of Th1 and Th17 cells, leading to excessive deposition of ECM, which in turn causes severe and persistent intestinal fibrosis.9 IL-6 and IL-1β are critical cytokines involved in the progression of inflammatory diseases, and their levels in plasma and intestinal mucosal tissues are significantly elevated in patients with intestinal fibrosis compared to healthy individuals or patients without intestinal mucosal lesions. IL-6 activates the JAK/STAT3 signaling pathway via both classical signaling (membrane-bound IL-6R/gp130) and trans-signaling (soluble IL-6R/gp130), thereby promoting the differentiation of Th17 cells and the release of pro-inflammatory factors such as IL-17 and TNF-α, exacerbating intestinal barrier disruption.80 IL-1β intensifies the inflammatory response through the NF-κB pathway, induces epithelial cell apoptosis and degradation of tight junction proteins, further impairing barrier function, and stimulates TGF-β secretion, which promotes myofibroblast activation and excessive extracellular matrix (ECM) accumulation. Additionally, studies have confirmed that blockade of IL-1β receptor signaling can reduce the risk of developing inflammatory bowel disease (IBD) and intestinal fibrosis.81
Changes in gut microbiota composition also impact microbial metabolite production. In IBD patients, the abundance of Firmicutes—particularly Faecalibacterium prausnitzii—and Bacteroidetes is significantly reduced. These bacterial groups constitute over 90% of the gut microbiota and are major producers of short-chain fatty acids (SCFAs), including acetate, propionate, and butyrate.82 SCFAs, the most abundant microbial-derived metabolites in the intestinal lumen, play a critical role in maintaining gut homeostasis. They regulate gene expression by activating G protein-coupled receptors (GPCRs) and inhibiting histone deacetylases (HDACs), thereby suppressing intestinal inflammation, preventing pathogen invasion, and preserving barrier integrity.83 Among SCFAs, butyrate is particularly important, as it enhances mucosal barrier function by promoting Treg differentiation and modulating Th cell responses in both in vitro and in vivo studies. This regulatory effect helps control immune activation and limits excessive inflammatory responses in the intestinal mucosa.84
The intestinal epithelial barrier, maintained by intestinal epithelial cells (IECs), acts as a primary defense mechanism by separating gut microbiota from host immune cells and regulating intestinal permeability. A loss of microbial diversity weakens this barrier, impairing its ability to prevent pathogen translocation and triggering chronic inflammation and immune activation.85 Persistent inflammation is a major driver of intestinal fibrosis, and prolonged exposure to inflammatory mediators further accelerates fibrotic remodeling. Additionally, mucosal immune dysfunction contributes to an overgrowth of potentially pathogenic Gram-negative bacteria and alters microbial metabolic pathways. Bacterial lipopolysaccharide (LPS) as a key endotoxin in the cell wall of Gram-negative bacteria, has been shown to activate intestinal fibroblasts via the NF-kB signaling pathway, disrupt epithelial integrity, and enhance collagen contraction, all of which contribute to the progression of intestinal fibrosis.86
The aging gut microbiota exhibits similar patterns of microbial alterations as those observed in disease-associated dysbiosis.87,88 Key characteristics of these changes include a decline in bacterial diversity and a reduced abundance of core commensal bacteria, such as Bacteroidetes, Bacillus thuringiensis, and Bifidobacterium. Concurrently, there is a significant increase in potentially pathogenic bacteria, including Escherichia, Salmonella, Yersinia, Desulfovibrio, Helicobacter pylori, Clostridium, and Ruminococcaceae, all of which are notably less abundant in healthy young individuals.89–91 When these opportunistic bacteria proliferate uncontrollably, they can trigger chronic inflammation and contribute to the onset of age-related diseases. Even in the absence of overt infection, aging is associated with elevated levels of pro-inflammatory cytokines, particularly IL-6 and TNF-α, in the serum, indicating a persistent state of low-grade systemic inflammation.92 Decreased levels of SCFA, especially age-related reduction in butyrate production, are a contributing factor to increased intestinal permeability and intestinal inflammation. Decreased mucin secretion and decreased expression of the tight junction proteins zonulin and occludin were observed in the intestinal epithelium of aged animals, suggesting that the aging intestinal mucosal barrier function is reduced, allowing microorganisms to translocate to deeper intestinal layers.93 Huang et al improved intestinal barrier function in elderly individuals by enhancing beneficial gut microbiota abundance, including Bacteroidaceae, Prevotellaceae, Coprobacillaceae, and Faecalibacterium. These beneficial bacteria produce SCFAs, such as acetate, propionate, and butyrate, thus reducing pro-inflammatory cytokines including IL-1β, IL-6, and TNF-α, enhancing antioxidant capacity, and maintaining intestinal homeostasis through metabolic pathways such as butyrate-derived GABA.94 In addition, high levels of LPS are released, further activating pro-inflammatory signaling pathways that perpetuate inflammation. This progressive disruption of the intestinal barrier is a key factor in the pathogenesis of chronic inflammation and fibrosis.95 Conway et al found that increased intestinal barrier permeability in elderly individuals is significantly associated with thymic involution and T cell aging, suggesting that changes in the intestinal microenvironment also accelerate tissue fibrosis through immune pathways.96 In conclusion, Aging-induced gut microbiota dysbiosis, metabolic alterations, and barrier dysfunction create a pro-inflammatory environment that increases intestinal permeability and promotes chronic inflammation. These changes cumulatively contribute to the development of intestinal fibrosis, establishing a mechanistic link between microbial aging and fibrotic progression (Figure 3).
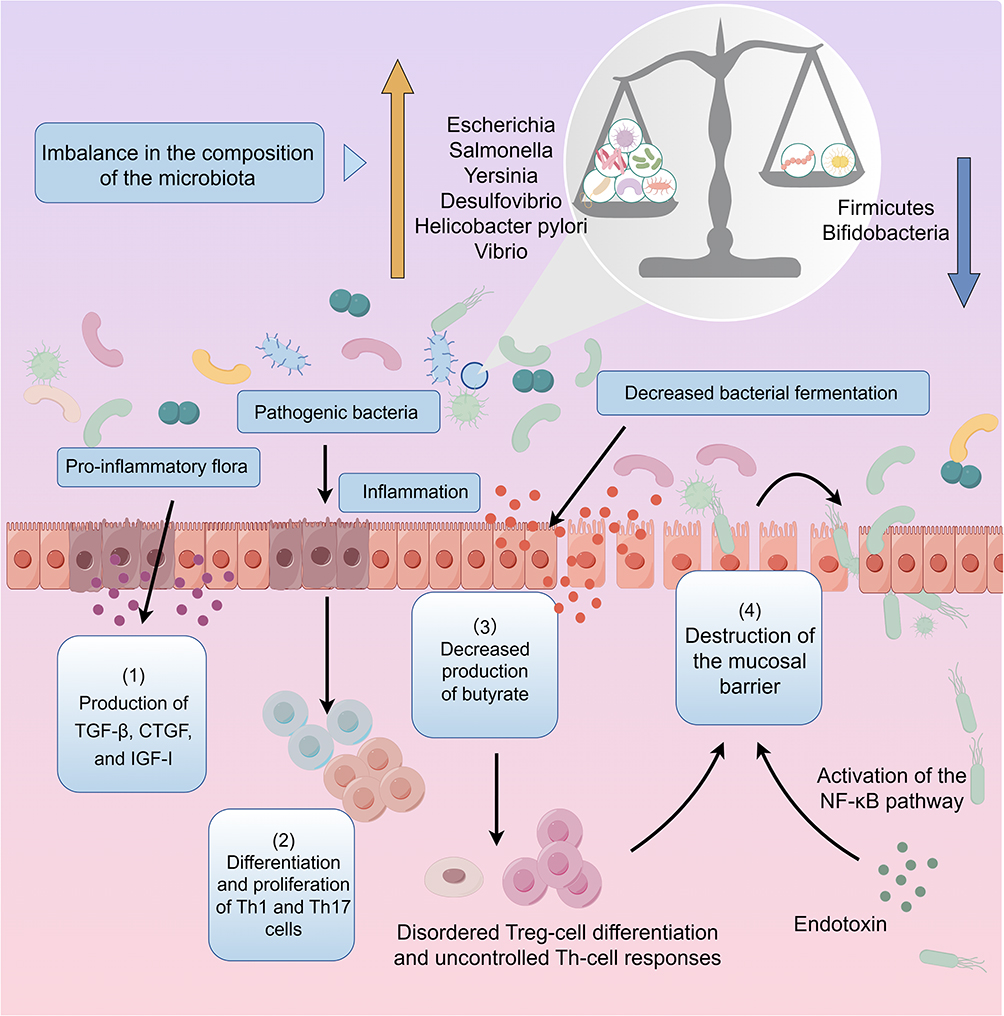 |
Figure 3 Changes in microbial species and abundance in the aging gut. In the aging gut, a reduced abundance of probiotics (blue arrow) and an expansion of harmful bacteria (yellow arrow) promote intestinal fibrosis through several mechanisms: (1) enhanced expression of various growth factors, including TGF-β, CTGF, and IGF-I, by pro-inflammatory microbiota; (2) overproliferation of TH1 and TH17 cells induced by pathogenic bacteria, leading to excessive ECM deposition; (3) decreased production of intestinal metabolites such as short-chain fatty acids (SCFAs), which impairs the regulatory functions of Treg and Th cells and results in mucosal immune dysregulation and persistent inflammation; and (4) disruption of the mucosal barrier, allowing bacterial translocation and endotoxin leakage, which further damage the intestinal mucosa and activate fibroblasts, promoting fibrosis progression. |
Growth Factors Produced by Senescence
Transforming growth factor-beta (TGF-β) is a key driver of fibrosis and is primarily produced by macrophages. It has three subtypes: TGF-β1, TGF-β2, and TGF-β3, each playing distinct roles in tissue remodeling. Among them, TGF-β/Smad signaling is considered one of the most critical pathways in organ fibrosis, as it regulates myofibroblast proliferation, fibroblast-to-myofibroblast transition, and epithelial-mesenchymal transition (EMT). Additionally, TGF-β can activate alternative signaling pathways, including the extracellular regulated protein kinase (ERK), phosphatidylinositol 3-kinase/protein kinase B (PI3K/AKT), and WNT signaling pathways,79 influencing cell proliferation, migration, inflammation, and ECM deposition.
The underlying mechanisms are as follows: First, the balance between matrix metalloproteinases (MMPs) and tissue inhibitors of metalloproteinases (TIMPs) is crucial for ECM homeostasis. MMP activity is tightly controlled by TIMPs.97 TGF-β1 and TGF-β2 promote fibrosis by stimulating collagen synthesis and upregulating TIMPs, thereby reducing collagen degradation. In contrast, exogenous TGF-β3 has been shown to reduce collagen deposition, exhibiting antifibrotic properties.98 Second, the peroxisome proliferator-activated receptor-γ (PPAR-γ) functions as an important antifibrosis pathway by regulating connective tissue fiber formation and the activation, differentiation, and survival of mesenchymal cells.99 PPAR-γ inhibits pro-inflammatory cytokines such as IL-4, IL-5, and IL-6, while also suppressing profibrotic mediators including platelet-derived growth factor (PDGF), IL-1, and TGF-β.100 Although PPAR-γ has demonstrated antifibrotic and anti-inflammatory effects in the liver, lung, heart, kidney, and skin, its role in intestinal fibrosis remains poorly understood. Notably, the TGF-β system negatively regulates PPAR-γ, thereby reinforcing the fibrotic process.101 Third, the WNT/β-catenin signaling pathway is a key regulator of cell proliferation, metabolism, apoptosis, and EMT.102 During fibrosis, WNT signaling promotes excessive ECM deposition by inhibiting PPAR-γ activity and stimulating fibroblast-driven ECM synthesis. Activation of WNT signaling has been observed in multiple fibrotic conditions, including those affecting the liver, skin, lung, kidney, and heart.103–105 Specifically, Wnt-1-induced signaling protein 1 (WISP-1) has been shown to stimulate type II alveolar epithelial cell (ATII) proliferation in the lungs and activate hepatic stellate cells, both of which drive ECM accumulation. These findings highlight the WNT/β-catenin pathway as a major contributor to tissue fibrosis and suggest its potential role in aging-related fibrosis. Additionally, TGF-β signaling enhances WNT/β-catenin activation, further amplifying fibrosis progression.101 Fourth, TGF-β promotes myofibroblast survival and resistance to apoptosis by activating FAK-PI3K-AKT and p38 MAPK pathways. This allows myofibroblasts to persist and continuously deposit ECM, thereby exacerbating fibrosis. Age-related increases in TGF-β1 levels have been demonstrated in mouse and human serum, and inhibition of overactive TGF-β1 attenuates the development of multiorgan fibrosis.106 However, data on the specific roles of TGF-β1, TGF-β2, and TGF-β3 in aging intestinal tissues remain limited. Further studies are needed to elucidate their unique roles in age-related intestinal fibrosis.
Current Medications for the Treatment of Intestinal Fibrosis
Conventional Therapy
At present, the treatment of intestinal fibrosis mainly includes drugs and surgical treatment. Commonly used drugs in the clinic primarily include anti-inflammatory drugs, such as 5-aminosalicylic acid (5-ASA) and glucocorticoids. The mechanism involves reducing the synthesis of prostaglandins; inhibiting the production of proinflammatory cytokines, oxygen free radicals, and lipoxygenase; blocking neutrophil chemotaxis and mast cell activation; and damaging the activation of the nuclear factor NF-κB in immune cells.107 The immunosuppressive agents azathioprine,108 methotrexate,109 cyclosporine,110,111 and tacrolimus112,113 block the formation and repair of nucleic acids, reduce the activation of the innate immune system, induce the apoptosis of cells in the adaptive immune system, and reduce the attack of immune cells on intestinal tissue, thereby alleviating the symptoms of fibrosis. Antifibrotic agents, such as pirfenidone (PFD) and nintedanib, exert synergistic inhibitory effects on fibrosis through multi-target mechanisms. Their core action involves blocking the canonical TGF-β/Smad pathway, inhibiting Smad phosphorylation and nuclear translocation, and downregulating key profibrotic genes such as COL1A1 and α-SMA, thereby reducing ECM synthesis.114,115 Meanwhile, these agents suppress myofibroblast activation and proliferation and induce apoptosis, decreasing the primary source of ECM.116 In addition, PFD has been shown to regulate the Wnt/GSK-3β/β-catenin and TGF-β1/Smad2/3 signaling pathways, inhibiting epithelial-mesenchymal transition (EMT) and fibroblast-mediated fibrogenesis.117,118 Furthermore, PFD can target and inversely modulate collagen triple helix repeat containing-1 (CTHRC1), thereby further suppressing TGF-β1-induced fibroblast activation.119 Since the pathogenesis of intestinal fibrosis is complex and varied and the condition varies greatly among different patients, it is often difficult to achieve ideal results with single-drug therapy, and the long-term use of drugs may lead to problems such as drug resistance and side effects, which may further affect therapeutic efficacy. Therefore, it is necessary to explore new drug types and therapeutic methods for treating intestinal fibrosis in the future to improve therapeutic efficacy and patients’ quality of life.
Potential Anti-Fibrosis Treatments
Antioxidant and Anti-Inflammatory Agents
The application of antioxidants in the treatment of intestinal fibrosis represents a frontier area of research. The antioxidative effects of statins (eg, rosuvastatin), angiotensin-converting enzyme inhibitors (ACEIs), melatonin (MEL), and other agents have been demonstrated in animal models of colitis. For instance, in a dextran sulfate sodium (DSS)-induced colitis model, rosuvastatin markedly attenuated oxidative stress, inflammation, and apoptosis, suggesting its antioxidant, anti-inflammatory, and anti-apoptotic properties.120 Similarly, ACEIs121 and MEL122 have also exhibited antioxidative effects. In addition, epidemiological and experimental data have shown that a diet rich in polyphenols can alleviate disease progression.123 As natural polyphenols, the effects of resveratrol (RSV) and curcumin on ROS production may involve the direct scavenging of free radicals or the modulation of NADPH oxidase activation.124 In a rat model of Crohn’s disease, Rahal et al reported that resveratrol can reduce the levels of inflammatory cytokines and TGF-β1 and showed the potential to inhibit tissue fibrosis by activating SIRT-1 and downregulating NF-kB activation.125,126 Meng et al reported that curcumin can inhibit ROS production through the TLR4-MAPK/NF-kB pathway, thereby reducing LPS-induced vascular smooth muscle cell inflammation.127 Epstein et al used curcumin to treat the colon tissue cells of IBD patients. They reported that the activation of the p38 MAPK pathway was inhibited, the expression of IL-1β and matrix metalloproteinase 3 (MMP-3) decreased, and the expression of IL-10 increased, with anti-inflammatory properties.128 In addition, berberine, a medicinal plant, is commonly used to treat gastrointestinal diseases. Berberine effectively reduced the expression of NADPH oxidase, a key enzyme in cellular ROS production; lowered free radical levels; increased the activities of antioxidant enzymes such as superoxide dismutase (SOD), catalase (CAT), and glutathione peroxidase (GPx); restored cellular redox balance; and thereby reduced lipid peroxidation and DNA damage.129 In a Phase 1 clinical trial of berberine in the treatment of patients with ulcerative colitis, berberine was shown to potentiate the anti-inflammatory effects of mesalazine in colonic tissues.130
Mesenchymal Stem Cell Therapy
Current studies have highlighted the multifaceted therapeutic potential of mesenchymal stem cells (MSCs) in intestinal fibrosis. MSCs may exert anti-fibrotic effects via multiple mechanisms, including modulation of immunity, restoration of the intestinal barrier, regulation of gut microbiota, and suppression of fibroblast activation. In IBD models, bone marrow-derived MSCs reduce collagen deposition and epithelial-mesenchymal transition (EMT), downregulate pro-fibrotic cytokines IL-1β, IL-6, and IL-13, upregulate anti-fibrotic IL-10, and suppress pro-inflammatory gene expression in macrophages and ERK phosphorylation in neutrophils, thereby alleviating colitis and mitigating fibrosis.79,131 MSCs also protect and repair the intestinal barrier, with umbilical cord-derived MSCs (UC-MSCs) enhancing tight junction proteins such as occludin, downregulating LC3A/B, and upregulating VEGF-A and VEGFR-1, strengthening barrier integrity and reducing colitis severity and mortality.132,133 Additionally, MSCs may exert indirect anti-fibrotic effects by modulating gut microbiota and metabolism; in TNBS-induced colitis mice, UC-MSCs restore microbiota α-diversity, reverse dysbiosis (eg, increasing Bacteroidetes/Firmicutes ratio and decreasing Proteobacteria), and regulate sulfur metabolism, amino acid, and bile acid synthesis.134 Importantly, MSCs directly suppress fibroblast activation and fibrogenesis, largely mediated by their secretome. Conditioned medium (CM) derived from umbilical cord-and placenta-derived MSCs (UC/PL-MSCs) significantly inhibits TGF-β1-induced activation of human primary intestinal myofibroblasts (HIMFs) by reducing collagen synthesis, downregulating α-SMA, and blocking the Rho/MRTF/SRF pathway.135 MSCs may also secrete hepatocyte growth factor (HGF) and tumor necrosis factor-stimulated gene 6 (TSG-6) to modulate extracellular matrix (ECM) remodeling and downregulate pro-fibrotic gene expression,136 a mechanism also reported in liver and lung fibrosis.137,138
Microbial Therapy
Fecal Microbial Transplantation (FMT)
FMT uses a variety of techniques and models to collect feces from healthy donors and transfer them to the patient’s gastrointestinal tract (GI) through colonoscopy, a nasogastric tube, or an enema, refilling the intestinal tract with healthy microbiota to restore the intestinal flora balance. FMT has been shown to be very effective in the treatment of recurrent C. difficile infection, with an effective rate of more than 80%.139,140 A meta-analysis revealed that the clinical remission rate of FMT for the treatment of ulcerative colitis was 36%, that of Crohn’s disease was 50.5%, and that of colitis was 21.5%.141 This range is similar to that of many currently approved biological therapies widely used for ulcerative colitis.142 Owing to the chronic changes in the microbiome of IBD patients, repeated FMT may be needed. In a UC treatment study, 32% of participants who received FMT treatment achieved clinical remission at 8 weeks, and patients who received more FMT infusions were more likely to achieve remission.143 Although FMT has not been specifically used for patients with fibrosis, limited evidence shows the potential of FMT in the treatment of IBD patients. At present, the research scope of FMT, including the mode of administration, dosage, donor selection, and safety, is not wide enough.
Probiotic Therapy
Compared with FMT, probiotic therapy regulates the intestinal flora by adding “healthy” probiotics to the flora.144 The mechanism by which probiotics maintain intestinal stability mainly includes improving intestinal barrier function, regulating glucose and lipid metabolism, and altering intestinal thickness.145 Studies have shown that germ-free mice can maintain a more intact intestinal barrier, mitigate thymic atrophy, and reduce the impact of aging on the immune system. These findings suggest that modulating the gut microbiota may be a potential strategy to improve immunosenescence and alleviate intestinal fibrosis.96 Scott et al developed yeast-based engineered probiotics that can sense proinflammatory molecules and self-regulate the proportion of engineered yeast probiotics, which has been shown to inhibit intestinal inflammation in IBD mouse models and reduce intestinal fibrosis and ecological disorders.146 Kühbacher et al reported that VSL#3, a probiotic preparation mixed with lactic acid bacteria, Bifidobacterium, and Streptococcus salivarius, can increase the total number of intestinal bacterial cells and the richness and diversity of the bacterial microbiota (especially the anaerobic microbiota) and inhibit the diversity of fungi.147 This probiotic preparation has shown some benefit in treating remission in patients with mild to moderate UC by increasing the number of mucosal regulatory T lymphocytes and decreasing the expression of the proinflammatory cytokine IL-1b mRNA. In addition, E. faecalis prausnitzii has anti-inflammatory properties due to its ability to produce butyrate, and its metabolites have been widely noted to prevent colitis in mice by blocking the activation of the nuclear factor κ-chain enhancer of activated B cells (NF-kB) and the production of IL-8. In patients with celiac disease, the abundance of this anti-inflammatory thick-walled bacterium is low and is associated with an increased risk of postoperative ileal disease recurrence.148 Low levels of Faecalibacterium prausnitzii in feces are thought to predict CD recurrence in patients in remission. Probiotics, prebiotics, and synthetic bacteria have been used to supplement anti-inflammatory bacteria and their substrates.149 At present, probiotics have shown potential in the treatment of fibrosis in the kidney, lung and liver. However, considering that the microbiomes of individuals are very different, a single classification standard and probiotic formula is unlikely to be applicable to everyone, and further exploration is needed (Table 1).
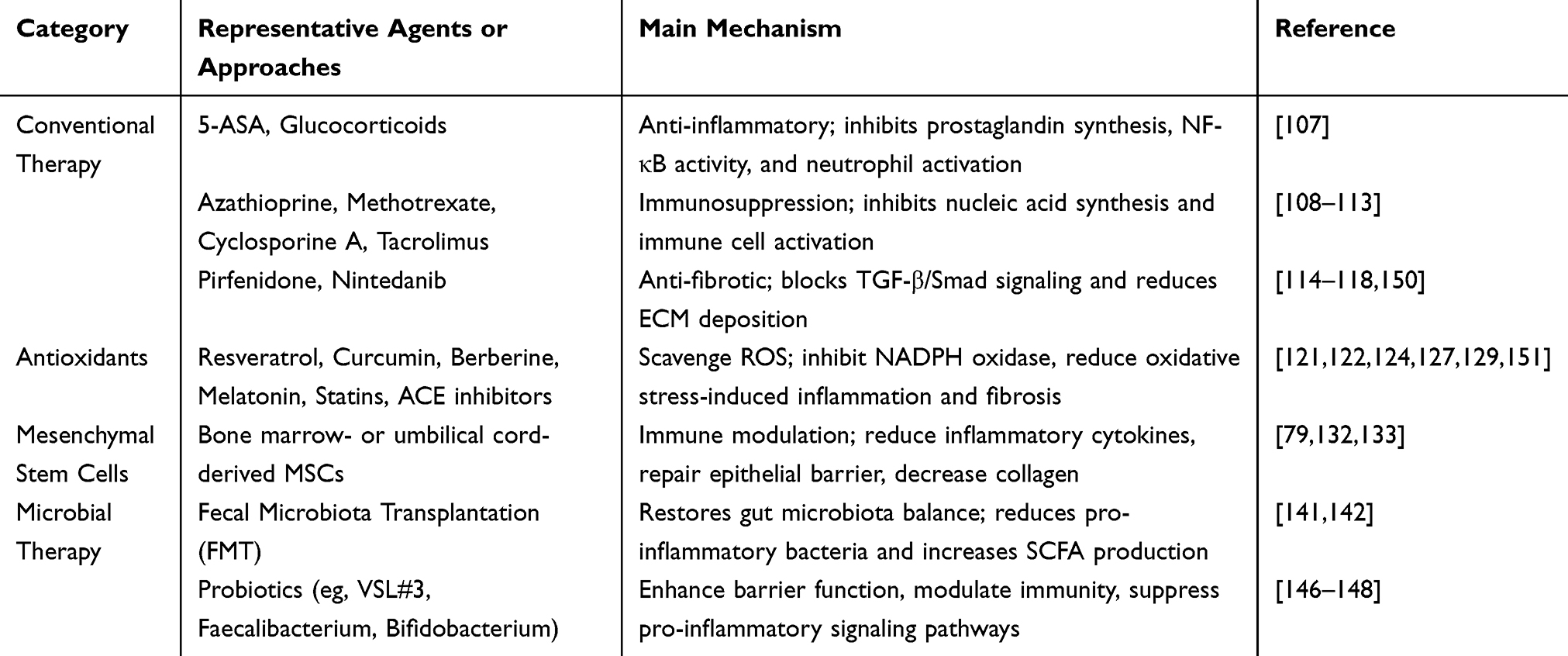 |
Table 1 Therapeutic Strategies and Mechanisms in the Management of Intestinal Fibrosis |
Discussion
As the global population ages, the impact of aging on various health conditions, including intestinal fibrosis, is receiving increasing attention. While direct evidence linking aging to intestinal fibrosis remains limited, the biological characteristics of the aging gut suggest that aging may elevate the risk of developing intestinal fibrosis. The apoptosis of immune cells, impaired intestinal barrier function, microenvironmental alterations, oxidative stress, chronic inflammation, and insufficient autophagy observed during aging may all contribute to this heightened risk. Intestinal fibrosis, characterized by abnormal proliferation of fibrous tissue, is influenced by several factors, with TGF-β being a critical mediator. TGF-β promotes fibrosis by modulating the balance of MMPs and TIMPs, as well as through pathways such as PPARγ and WNT/β-catenin. Conventional treatments for fibrosis are often limited by side effects and drug resistance, whereas emerging therapies, including antioxidant treatments, mesenchymal stem cell therapy, and microbiota-based interventions, offer promising alternative strategies. A comprehensive understanding of the molecular mechanisms linking aging and intestinal fibrosis is essential for developing effective preventive and therapeutic approaches in the future.
Consent for Publication
All participants have provided their consent for publication.
Acknowledgments
Thanks to all authors for their contributions to the manuscript.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
This work was supported by the Fourth Batch of Zhejiang Province Ten Thousand People Program Scientific and Technological Innovation Leading Talents (2020R52024), the Research Fund of the National Health Commission (WKJ-ZJ-2435), and the Chinese Medicine Clinical Research Program (2024ZL103). Funding was also provided by the Zhejiang Province Chinese Medicine Science and Technology Program for Young Talents (2021ZQ086).
Disclosure
The authors declare that there are no conflicts of interest, financial or otherwise, that could influence the objectivity of this manuscript.
References
1. López-Otín C, Blasco MA, Partridge L, Serrano M, Kroemer G. Hallmarks of aging: an expanding universe. Cell. 2023;186(2):243–278. doi:10.1016/j.cell.2022.11.001
2. López-Otín C, Blasco MA, Partridge L, Serrano M, Kroemer G. The hallmarks of aging. Cell. 2013;153(6):1194–1217. doi:10.1016/j.cell.2013.05.039
3. Kroemer G, Maier AB, Cuervo AM, et al. From geroscience to precision geromedicine: understanding and managing aging. Cell. 2025;188(8):2043–2062. doi:10.1016/j.cell.2025.03.011
4. Han M, Han P, Wang Z, et al. Alternative splicing in aging and aging-related diseases: from pathogenesis to therapy. Pharmacol Ther. 2025;272:108887. doi:10.1016/j.pharmthera.2025.108887
5. Partridge L, Fuentealba M, Kennedy BK. The quest to slow ageing through drug discovery. Nat Rev Drug Discov. 2020;19(8):513–532. doi:10.1038/s41573-020-0067-7
6. Ling Z, Liu X, Cheng Y, Yan X, Wu S. Gut microbiota and aging. Crit. Rev. Food Sci. Nutr. 2022;62(13):3509–3534. doi:10.1080/10408398.2020.1867054
7. Nguyen TT, Baumann P, Tüscher O, Schick S, Endres K. The aging enteric nervous system. Int J Mol Sci. 2023;24(11):9471. doi:10.3390/ijms24119471
8. Distler JHW, Györfi AH, Ramanujam M, Whitfield ML, Königshoff M, Lafyatis R. Shared and distinct mechanisms of fibrosis. Nat Rev Rheumatol. 2019;15(12):705–730. doi:10.1038/s41584-019-0322-7
9. Costa C, Sampaio-Maia B, Araujo R, et al. Gut microbiome and organ fibrosis. Nutrients. 2022;14(2):352. doi:10.3390/nu14020352
10. Choi EL, Taheri N, Chandra A, Hayashi Y. Cellular senescence, inflammation, and cancer in the gastrointestinal tract. Int J Mol Sci. 2023;24(12). doi:10.3390/ijms24129810
11. Ahmad OF, Akbar A. Microbiome, antibiotics and irritable bowel syndrome. Br Med Bull. 2016;120(1):91–99. doi:10.1093/bmb/ldw038
12. Hinz B, Lagares D. Evasion of apoptosis by myofibroblasts: a hallmark of fibrotic diseases. Nat Rev Rheumatol. 2020;16(1):11–31. doi:10.1038/s41584-019-0324-5
13. Rieder F, Fiocchi C, Rogler G. Mechanisms, management, and treatment of fibrosis in patients with inflammatory bowel diseases. Gastroenterology. 2017;152(2):340–350e346. doi:10.1053/j.gastro.2016.09.047
14. Bamias G, Pizarro TT, Cominelli F. Immunological regulation of intestinal fibrosis in inflammatory bowel disease. Inflamm Bowel Dis. 2022;28(3):337–349. doi:10.1093/ibd/izab251
15. Xin S, Liu X, He C, et al. Inflammation accelerating intestinal fibrosis: from mechanism to clinic. Eur J Med Res. 2024;29(1):335. doi:10.1186/s40001-024-01932-2
16. Yang W, Yu T, Cong Y. Stromal cell regulation of intestinal inflammatory fibrosis. CMGH. 2024;17(5):703–711. doi:10.1016/j.jcmgh.2024.01.007
17. Johnson LA, Luke A, Sauder K, Moons DS, Horowitz JC, Higgins PD. Intestinal fibrosis is reduced by early elimination of inflammation in a mouse model of IBD: impact of a “Top-Down” approach to intestinal fibrosis in mice. Inflamm Bowel Dis. 2012;18(3):460–471. doi:10.1002/ibd.21812
18. Hünerwadel A, Fagagnini S, Rogler G, et al. Severity of local inflammation does not impact development of fibrosis in mouse models of intestinal fibrosis. Sci Rep. 2018;8(1):15182. doi:10.1038/s41598-018-33452-5
19. Baumgart DC, Le Berre C. Newer biologic and small-molecule therapies for inflammatory bowel disease. New Engl J Med. 2021;385(14):1302–1315. doi:10.1056/NEJMra1907607
20. Younesi FS, Miller AE, Barker TH, Rossi FMV, Hinz B. Fibroblast and myofibroblast activation in normal tissue repair and fibrosis. Nat Rev Mol Cell Biol. 2024;25(8):617–638.
21. Schuster R, Younesi F, Ezzo M, Hinz B. The role of myofibroblasts in physiological and pathological tissue repair. Cold Spring Harbor Perspect. Biol. 2023;15(1):a041231. doi:10.1101/cshperspect.a041231
22. Gabbiani G. The myofibroblast in wound healing and fibrocontractive diseases. J Pathol. 2003;200(4):500–503. doi:10.1002/path.1427
23. Liu F, Mih JD, Shea BS, et al. Feedback amplification of fibrosis through matrix stiffening and COX-2 suppression. J Cell Biol. 2010;190(4):693–706. doi:10.1083/jcb.201004082
24. Johnson LA, Rodansky ES, Haak AJ, Larsen SD, Neubig RR, Higgins PD. Novel Rho/MRTF/SRF inhibitors block matrix-stiffness and TGF-beta-induced fibrogenesis in human colonic myofibroblasts. Inflamm Bowel Dis. 2014;20(1):154–165. doi:10.1097/01.MIB.0000437615.98881.31
25. Niu C, Hu Y, Xu K, Pan X, Wang L, Yu G. The role of the cytoskeleton in fibrotic diseases. Front Cell Dev Biol. 2024;12:1490315. doi:10.3389/fcell.2024.1490315
26. Riches DW, Backos DS, Redente EF. ROCK and Rho: promising therapeutic targets to ameliorate pulmonary fibrosis. Am J Pathol. 2015;185(4):909–912. doi:10.1016/j.ajpath.2015.01.005
27. Mosser DM, Edwards JP. Exploring the full spectrum of macrophage activation. Nat Rev Immunol. 2008;8(12):958–969. doi:10.1038/nri2448
28. Manole E, Niculite C, Lambrescu IM, et al. Macrophages and stem cells-two to tango for tissue repair? Biomolecules. 2021;11(5):697. doi:10.3390/biom11050697
29. Zhang Y, Li X, Luo Z, et al. ECM1 is an essential factor for the determination of M1 macrophage polarization in IBD in response to LPS stimulation. Proc Natl Acad Sci U S A. 2020;117(6):3083–3092. doi:10.1073/pnas.1912774117
30. Dharmasiri S, Garrido-Martin EM, Harris RJ, et al. Human intestinal macrophages are involved in the pathology of both ulcerative colitis and Crohn disease. Inflamm Bowel Dis. 2021;27(10):1641–1652. doi:10.1093/ibd/izab029
31. Zeng W, Li F, Jin S, Ho PC, Liu PS, Xie X. Functional polarization of tumor-associated macrophages dictated by metabolic reprogramming. J Exp Clin Cancer Res. 2023;42(1):245. doi:10.1186/s13046-023-02832-9
32. Sharma R. Perspectives on the dynamic implications of cellular senescence and immunosenescence on macrophage aging biology. Biogerontology. 2021;22(6):571–587. doi:10.1007/s10522-021-09936-9
33. Wang L, Hong W, Zhu H, et al. Macrophage senescence in health and diseases. Acta Pharmaceutica Sinica B. 2024;14(4):1508–1524. doi:10.1016/j.apsb.2024.01.008
34. Xiao J, Li HS, Satyanarayanan SK, et al. Advancements in targeting macrophage senescence for age-associated conditions. Aging and Disease. 2024;16(4):2201–2236. doi:10.14336/AD.2024.0720
35. Pakshir P, Hinz B. The big five in fibrosis: macrophages, myofibroblasts, matrix, mechanics, and miscommunication. Matrix Biol. 2018;68–69:81–93. doi:10.1016/j.matbio.2018.01.019
36. Sapudom J, Karaman S, Mohamed WKE, Garcia-Sabate A, Quartey BC, Teo JCM. 3D in vitro M2 macrophage model to mimic modulation of tissue repair. NPJ Regen Med. 2021;6(1):83. doi:10.1038/s41536-021-00193-5
37. Luo L, Wang S, Hu Y, et al. Precisely regulating M2 subtype macrophages for renal fibrosis resolution. ACS nano. 2023;17(22):22508–22526. doi:10.1021/acsnano.3c05998
38. Yang H, Cheng H, Dai R, Shang L, Zhang X, Wen H. Macrophage polarization in tissue fibrosis. PeerJ. 2023;11:e16092. doi:10.7717/peerj.16092
39. Lourenssen SR, Blennerhassett MG. M2 macrophages and phenotypic modulation of intestinal smooth muscle cells characterize inflammatory stricture formation in rats. Am J Pathol. 2020;190(9):1843–1858. doi:10.1016/j.ajpath.2020.05.015
40. Lis-López L, Bauset C, Seco-Cervera M, Cosín-Roger J. Is the macrophage phenotype determinant for fibrosis development? Biomedicines. 2021;9(12):1747. doi:10.3390/biomedicines9121747
41. Cosín-Roger J, Ortiz-Masiá D, Calatayud S, et al. M2 macrophages activate WNT signaling pathway in epithelial cells: relevance in ulcerative colitis. PLoS One. 2013;8(10):e78128. doi:10.1371/journal.pone.0078128
42. Kono Y, Miyoshi S, Fujita T. Dextran sodium sulfate alters cytokine production in macrophages in vitro. Die Pharmazie. 2016;71(11):619–624. doi:10.1691/ph.2016.6688
43. Yang R, Liao Y, Wang L, et al. Exosomes derived from M2b macrophages attenuate DSS-induced colitis. Front Immunol. 2019;10:2346. doi:10.3389/fimmu.2019.02346
44. Amamou A, O’Mahony C, Leboutte M, Savoye G, Ghosh S, Marion-Letellier R. Gut microbiota, macrophages and diet: an intriguing new triangle in intestinal fibrosis. Microorganisms. 2022;10(3):490. doi:10.3390/microorganisms10030490
45. Higashi K, Inagaki Y, Fujimori K, Nakao A, Kaneko H, Nakatsuka I. Interferon-gamma interferes with transforming growth factor-beta signaling through direct interaction of YB-1 with Smad3. J Biol Chem. 2003;278(44):43470–43479. doi:10.1074/jbc.M302339200
46. Ye Y, Zhang X, Su D, et al. Therapeutic efficacy of human adipose mesenchymal stem cells in Crohn’s colon fibrosis is improved by IFN-γ and kynurenic acid priming through indoleamine 2,3-dioxygenase-1 signaling. Stem Cell Res Ther. 2022;13(1):465. doi:10.1186/s13287-022-03157-8
47. Yao Y, Zhang J, Tan DQ, et al. Interferon-γ improves renal interstitial fibrosis and decreases intrarenal vascular resistance of hydronephrosis in an animal model. Urology. 2011;77(3):761.e768–713. doi:10.1016/j.urology.2010.10.015
48. Jia M, Liu Y, Liu J, et al. Xuanfei Baidu decoction ameliorates bleomycin-elicited idiopathic pulmonary fibrosis in mice by regulating the lung-gut crosstalk via IFNγ/STAT1/STAT3 axis. Phytomedicine. 2024;135:155997. doi:10.1016/j.phymed.2024.155997
49. Linard C, Billiard F, Benderitter M. Intestinal irradiation and fibrosis in a Th1-deficient environment. Int J Radiat Oncol Biol Phys. 2012;84(1):266–273. doi:10.1016/j.ijrobp.2011.11.027
50. Zhang M, Zhang S. T cells in fibrosis and fibrotic diseases. Front Immunol. 2020;11:1142. doi:10.3389/fimmu.2020.01142
51. Gieseck RL, Wilson MS, Wynn TA. Type 2 immunity in tissue repair and fibrosis. Nat Rev Immunol. 2018;18(1):62–76. doi:10.1038/nri.2017.90
52. Hartl D, Griese M, Kappler M, et al. Pulmonary T(H)2 response in Pseudomonas aeruginosa-infected patients with cystic fibrosis. J Allergy Clin Immunol. 2006;117(1):204–211. doi:10.1016/j.jaci.2005.09.023
53. Xu J, Wang J, He Y, Chen R, Meng Q. L.acidophilus participates in intestinal inflammation induced by PM(2.5) through affecting the Treg/Th17 balance. Environmental Pollution. 2024;341:122977. doi:10.1016/j.envpol.2023.122977
54. Wang L, Wang S, Lin J, et al. Treg and intestinal myofibroblasts-derived Amphiregulin induced by TGF-β mediates intestinal fibrosis in Crohn’s disease. J Transl Med. 2025;23(1):452. doi:10.1186/s12967-025-06413-6
55. Mittelbrunn M, Kroemer G. Hallmarks of T cell aging. Nat Immunol. 2021;22(6):687–698. doi:10.1038/s41590-021-00927-z
56. Soto-Heredero G, Gabandé-Rodríguez E, Carrasco E, et al. KLRG1 identifies regulatory T cells with mitochondrial alterations that accumulate with aging. Nature Aging. 2025;5(5):799–815. doi:10.1038/s43587-025-00855-9
57. She YX, Yu QY, Tang XX. Role of interleukins in the pathogenesis of pulmonary fibrosis. Cell Death Discov. 2021;7(1):52. doi:10.1038/s41420-021-00437-9
58. Liu H, Wu NC, Yuan M, et al. Cross-neutralization of a SARS-CoV-2 antibody to a functionally conserved site is mediated by avidity. Immunity. 2020;53(6):1272–1280.e1275. doi:10.1016/j.immuni.2020.10.023
59. Parkos CA. Neutrophil-epithelial interactions: a double-edged sword. Am J Pathol. 2016;186(6):1404–1416. doi:10.1016/j.ajpath.2016.02.001
60. Xue X, Falcon DM. The role of immune cells and cytokines in intestinal wound healing. Int J Mol Sci. 2019;20(23):6097. doi:10.3390/ijms20236097
61. Gomez CR, Boehmer ED, Kovacs EJ. The aging innate immune system. Curr Opin Immunol. 2005;17(5):457–462. doi:10.1016/j.coi.2005.07.013
62. Korolchuk VI, Miwa S, Carroll B, von Zglinicki T. Mitochondria in cell senescence: is mitophagy the weakest link? EBioMedicine. 2017;21:7–13. doi:10.1016/j.ebiom.2017.03.020
63. Guo Y, Guan T, Shafiq K, et al. Mitochondrial dysfunction in aging. Ageing Res Rev. 2023;88:101955. doi:10.1016/j.arr.2023.101955
64. Pasparakis M. IKK/NF-kappaB signaling in intestinal epithelial cells controls immune homeostasis in the gut. Mucosal Immunol. 2008;1 Suppl 1:S54–57. doi:10.1038/mi.2008.53
65. Nguyen VQ, Eden K, Morrison HA, et al. Noncanonical NF-κB signaling upregulation in inflammatory bowel disease patients is associated with loss of response to Anti-TNF agents. Front Pharmacol. 2021;12:655887. doi:10.3389/fphar.2021.655887
66. Amirshahrokhi K. Febuxostat attenuates ulcerative colitis by the inhibition of NF-κB, proinflammatory cytokines, and oxidative stress in mice. Int Immunopharmacol. 2019;76:105884. doi:10.1016/j.intimp.2019.105884
67. Sakthivel KM, Guruvayoorappan C. Amentoflavone inhibits iNOS, COX-2 expression and modulates cytokine profile, NF-κB signal transduction pathways in rats with ulcerative colitis. Int Immunopharmacol. 2013;17(3):907–916. doi:10.1016/j.intimp.2013.09.022
68. Banskota S, Wang H, Kwon YH, et al. Inhibition of NADPH oxidase (NOX) 2 mitigates colitis in mice with impaired macrophage AMPK function. Biomedicines. 2023;11(5):1443. doi:10.3390/biomedicines11051443
69. Piechota-Polanczyk A, Fichna J. Review article: the role of oxidative stress in pathogenesis and treatment of inflammatory bowel diseases. Naunyn-Schmiedeberg’s Arch Pharmacol. 2014;387(7):605–620. doi:10.1007/s00210-014-0985-1
70. Macias-Ceja DC, Barrachina MD, Ortiz-Masià D. Autophagy in intestinal fibrosis: relevance in inflammatory bowel disease. Front Pharmacol. 2023;14:1170436. doi:10.3389/fphar.2023.1170436
71. Cosin-Roger J, Canet F, Macias-Ceja DC, et al. Autophagy stimulation as a potential strategy against intestinal fibrosis. Cells. 2019;8(9):1078. doi:10.3390/cells8091078
72. Steen EH, Wang X, Balaji S, Butte MJ, Bollyky PL, Keswani SG. The role of the anti-inflammatory cytokine interleukin-10 in tissue fibrosis. Adv Wound Care. 2020;9(4):184–198. doi:10.1089/wound.2019.1032
73. Gazouli M, Pachoula I, Panayotou I, et al. NOD2/CARD15, ATG16L1 and IL23R gene polymorphisms and childhood-onset of Crohn’s disease. World J Gastroenterol. 2010;16(14):1753–1758. doi:10.3748/wjg.v16.i14.1753
74. Panwar V, Singh A, Bhatt M, et al. Multifaceted role of mTOR (mammalian target of rapamycin) signaling pathway in human health and disease. Signal Transduction Targeted Ther. 2023;8(1):375. doi:10.1038/s41392-023-01608-z
75. Valdes AM, Walter J, Segal E, Spector TD. Role of the gut microbiota in nutrition and health. BMJ (Clinical Research Ed). 2018;361:k2179. doi:10.1136/bmj.k2179
76. Fujisaka S, Watanabe Y, Tobe K. The gut microbiome: a core regulator of metabolism. J Endocrinol. 2023;256(3). doi:10.1530/JOE-22-0111
77. Pires L, González-Paramás AM, Heleno SA, Calhelha RC. The role of gut microbiota in the etiopathogenesis of multiple chronic diseases. Antibiotics. 2024;13(5):392.
78. Haneishi Y, Furuya Y, Hasegawa M, Picarelli A, Rossi M, Miyamoto J. Inflammatory bowel diseases and gut microbiota. Int J Mol Sci. 2023;24(4):3817. doi:10.3390/ijms24043817
79. Wang Y, Huang B, Jin T, Ocansey DKW, Jiang J, Mao F. Intestinal fibrosis in inflammatory bowel disease and the prospects of mesenchymal stem cell therapy. Front Immunol. 2022;13:835005. doi:10.3389/fimmu.2022.835005
80. Parisinos CA, Serghiou S, Katsoulis M, et al. Variation in interleukin 6 receptor gene associates with risk of Crohn’s disease and ulcerative colitis. Gastroenterology. 2018;155(2):303–306.e302. doi:10.1053/j.gastro.2018.05.022
81. Aggeletopoulou I, Kalafateli M, Tsounis EP, Triantos C. Exploring the role of IL-1β in inflammatory bowel disease pathogenesis. Front Med. 2024;11:1307394. doi:10.3389/fmed.2024.1307394
82. Ahmad MS, Krishnan S, Ramakrishna BS, Mathan M, Pulimood AB, Murthy SN. Butyrate and glucose metabolism by colonocytes in experimental colitis in mice. Gut. 2000;46(4):493–499. doi:10.1136/gut.46.4.493
83. Yoo JY, Groer M, Dutra SVO, Sarkar A, McSkimming DI. Gut microbiota and immune system interactions. Microorganisms. 2020;8(10):1587. doi:10.3390/microorganisms8101587
84. Furusawa Y, Obata Y, Fukuda S, et al. Commensal microbe-derived butyrate induces the differentiation of colonic regulatory T cells. Nature. 2013;504(7480):446–450. doi:10.1038/nature12721
85. Sokol H, Pigneur B, Watterlot L, et al. Faecalibacterium prausnitzii is an anti-inflammatory commensal bacterium identified by gut microbiota analysis of Crohn disease patients. Proc Natl Acad Sci U S A. 2008;105(43):16731–16736. doi:10.1073/pnas.0804812105
86. Burke JP, Cunningham MF, Watson RW, Docherty NG, Coffey JC, O’Connell PR. Bacterial lipopolysaccharide promotes profibrotic activation of intestinal fibroblasts. Br J Surg. 2010;97(7):1126–1134. doi:10.1002/bjs.7045
87. Hirano A, Umeno J, Okamoto Y, et al. Comparison of the microbial community structure between inflamed and non-inflamed sites in patients with ulcerative colitis. J Gastroenterol Hepatol. 2018;33(9):1590–1597. doi:10.1111/jgh.14129
88. Thursby E, Juge N. Introduction to the human gut microbiota. Biochem J. 2017;474(11):1823–1836. doi:10.1042/BCJ20160510
89. Nishino K, Nishida A, Inoue R, et al. Analysis of endoscopic brush samples identified mucosa-associated dysbiosis in inflammatory bowel disease. J Gastroenterol. 2018;53(1):95–106. doi:10.1007/s00535-017-1384-4
90. Machiels K, Joossens M, Sabino J, et al. A decrease of the butyrate-producing species Roseburia hominis and Faecalibacterium prausnitzii defines dysbiosis in patients with ulcerative colitis. Gut. 2014;63(8):1275–1283. doi:10.1136/gutjnl-2013-304833
91. Aldars-García L, Chaparro M, Gisbert JP. Systematic review: the gut microbiome and its potential clinical application in inflammatory bowel disease. Microorganisms. 2021;9(5):977. doi:10.3390/microorganisms9050977
92. Rea IM, Gibson DS, McGilligan V, McNerlan SE, Alexander HD, Ross OA. Age and age-related diseases: role of inflammation triggers and cytokines. Front Immunol. 2018;9:586. doi:10.3389/fimmu.2018.00586
93. Araújo JR, Marques C, Rodrigues C, Calhau C, Faria A. The metabolic and endocrine impact of diet-derived gut microbiota metabolites on ageing and longevity. Ageing Res Rev. 2024;100:102451. doi:10.1016/j.arr.2024.102451
94. Huang R, Zhang J, Sun M, et al. Oat beta-glucan enhances gut barrier function and maintains intestinal homeostasis in naturally aging mice. Int J Biol Macromol. 2025;305(Pt 1):141129. doi:10.1016/j.ijbiomac.2025.141129
95. Shintouo CM, Mets T, Beckwee D, et al. Is inflammageing influenced by the microbiota in the aged gut? A systematic review. Exp Gerontol. 2020;141:111079. doi:10.1016/j.exger.2020.111079
96. Conway J, De Jong EN, White AJ, et al. Age-related loss of intestinal barrier integrity plays an integral role in thymic involution and T cell ageing. Aging Cell. 2024;24(3):e14401. doi:10.1111/acel.14401
97. Biel C, Faber KN, Bank RA, Olinga P. Matrix metalloproteinases in intestinal fibrosis. J Crohns Colitis. 2024;18(3):462–478. doi:10.1093/ecco-jcc/jjad178
98. Campbell BH, Agarwal C, Wang JH. TGF-beta1, TGF-beta3, and PGE(2) regulate contraction of human patellar tendon fibroblasts. Biomech. Model. Mechanobiol. 2004;2(4):239–245. doi:10.1007/s10237-004-0041-z
99. Koo JB, Nam MO, Jung Y, et al. Anti-fibrogenic effect of PPAR-γ agonists in human intestinal myofibroblasts. BMC Gastroenterol. 2017;17(1):73. doi:10.1186/s12876-017-0627-4
100. Vetuschi A, Pompili S, Gaudio E, Latella G, Sferra R. PPAR-γ with its anti-inflammatory and anti-fibrotic action could be an effective therapeutic target in IBD. Eur. Rev. Med. Pharmacol. Sci. 2018;22(24):8839–8848. doi:10.26355/eurrev_201812_16652
101. Vallée A, Lecarpentier Y. TGF-β in fibrosis by acting as a conductor for contractile properties of myofibroblasts. Cell Biosci. 2019;9(1):98. doi:10.1186/s13578-019-0362-3
102. Heuberger J, Birchmeier W. Interplay of cadherin-mediated cell adhesion and canonical Wnt signaling. Cold Spring Harbor Perspect. Biol. 2010;2(2):a002915. doi:10.1101/cshperspect.a002915
103. Wang D, Dai C, Li Y, Liu Y. Canonical Wnt/β-catenin signaling mediates transforming growth factor-β1-driven podocyte injury and proteinuria. Kidney Int. 2011;80(11):1159–1169. doi:10.1038/ki.2011.255
104. Jiang F, Parsons CJ, Stefanovic B. Gene expression profile of quiescent and activated rat hepatic stellate cells implicates Wnt signaling pathway in activation. J Hepatol. 2006;45(3):401–409. doi:10.1016/j.jhep.2006.03.016
105. Königshoff M, Kramer M, Balsara N, et al. WNT1-inducible signaling protein-1 mediates pulmonary fibrosis in mice and is upregulated in humans with idiopathic pulmonary fibrosis. J Clin Invest. 2009;119(4):772–787. doi:10.1172/JCI33950
106. Carlson ME, Conboy MJ, Hsu M, et al. Relative roles of TGF-beta1 and Wnt in the systemic regulation and aging of satellite cell responses. Aging Cell. 2009;8(6):676–689. doi:10.1111/j.1474-9726.2009.00517.x
107. Neurath MF. Current and emerging therapeutic targets for IBD. Nat Rev Gastroenterol Hepatol. 2017;14(5):269–278. doi:10.1038/nrgastro.2016.208
108. Oancea I, Movva R, Das I, et al. Colonic microbiota can promote rapid local improvement of murine colitis by thioguanine independently of T lymphocytes and host metabolism. Gut. 2017;66(1):59–69. doi:10.1136/gutjnl-2015-310874
109. AlAmeel T, Al Sulais E, Raine T. Methotrexate in inflammatory bowel disease: a primer for gastroenterologists. Saudi J Gastroenterol. 2022;28(4):250–260. doi:10.4103/sjg.sjg_496_21
110. Laharie D, Bourreille A, Branche J, et al. Ciclosporin versus infliximab in patients with severe ulcerative colitis refractory to intravenous steroids: a parallel, open-label randomised controlled trial. Lancet (London, England). 2012;380(9857):1909–1915. doi:10.1016/S0140-6736(12)61084-8
111. Lu H, Lin J, Xu C, et al. Cyclosporine modulates neutrophil functions via the SIRT6-HIF-1α-glycolysis axis to alleviate severe ulcerative colitis. Clin Transl Med. 2021;11(2):e334. doi:10.1002/ctm2.334
112. Zuo L, Kuo WT, Cao F, et al. Tacrolimus-binding protein FKBP8 directs myosin light chain kinase-dependent barrier regulation and is a potential therapeutic target in Crohn’s disease. Gut. 2023;72(5):870–881. doi:10.1136/gutjnl-2021-326534
113. Wu B, Tong J, Ran Z. Tacrolimus therapy in steroid-refractory ulcerative colitis: a review. Inflamm Bowel Dis. 2020;26(1):24–32. doi:10.1093/ibd/izz068
114. Li G, Ren J, Hu Q, et al. Oral pirfenidone protects against fibrosis by inhibiting fibroblast proliferation and TGF-β signaling in a murine colitis model. Biochem. Pharmacol. 2016;117:57–67. doi:10.1016/j.bcp.2016.08.002
115. Wollin L, Maillet I, Quesniaux V, Holweg A, Ryffel B. Antifibrotic and anti-inflammatory activity of the tyrosine kinase inhibitor nintedanib in experimental models of lung fibrosis. J Pharmacol Exp Ther. 2014;349(2):209–220. doi:10.1124/jpet.113.208223
116. Cui Y, Zhang M, Leng C, et al. Pirfenidone inhibits cell proliferation and collagen i production of primary human intestinal fibroblasts. Cells. 2020;9(3):775. doi:10.3390/cells9030775
117. Lv Q, Wang J, Xu C, Huang X, Ruan Z, Dai Y. Pirfenidone alleviates pulmonary fibrosis in vitro and in vivo through regulating Wnt/GSK-3β/β-catenin and TGF-β1/Smad2/3 signaling pathways. Mol Med. 2020;26(1):49. doi:10.1186/s10020-020-00173-3
118. Dai Z, Jiang Y, Guo H, Lu Y, Chen W, Liang T. Pirfenidone ameliorates hypertrophic scar through inhibiting proliferation and migration of fibroblasts by regulating the Wnt/GSK-3β/β-catenin signaling pathway. J Burn Care Res. 2025;46(4):854–861. doi:10.1093/jbcr/iraf040
119. Jin J, Togo S, Kadoya K, et al. Pirfenidone attenuates lung fibrotic fibroblast responses to transforming growth factor-β1. Respir Res. 2019;20(1):119. doi:10.1186/s12931-019-1093-z
120. Shin SK, Cho JH, Kim EJ, et al. Anti-inflammatory and anti-apoptotic effects of rosuvastatin by regulation of oxidative stress in a dextran sulfate sodium-induced colitis model. World J Gastroenterol. 2017;23(25):4559–4568. doi:10.3748/wjg.v23.i25.4559
121. Arab HH, Al-Shorbagy MY, Abdallah DM, Nassar NN. Telmisartan attenuates colon inflammation, oxidative perturbations and apoptosis in a rat model of experimental inflammatory bowel disease. PLoS One. 2014;9(5):e97193. doi:10.1371/journal.pone.0097193
122. Sánchez A, Calpena AC, Clares B. Evaluating the oxidative stress in inflammation: role of melatonin. Int J Mol Sci. 2015;16(8):16981–17004. doi:10.3390/ijms160816981
123. Dziąbowska-Grabias K, Sztanke M, Zając P, et al. Antioxidant therapy in inflammatory bowel diseases. Antioxidants. 2021;10(3). doi:10.3390/antiox10030412
124. Sahoo DK, Heilmann RM, Paital B, et al. Oxidative stress, hormones, and effects of natural antioxidants on intestinal inflammation in inflammatory bowel disease. Front Endocrinol. 2023;14:1217165. doi:10.3389/fendo.2023.1217165
125. Hofseth LJ, Singh UP, Singh NP, Nagarkatti M, Nagarkatti PS. Taming the beast within: resveratrol suppresses colitis and prevents colon cancer. Aging. 2010;2(4):183–184. doi:10.18632/aging.100143
126. Rahal K, Schmiedlin-Ren P, Adler J, et al. Resveratrol has antiinflammatory and antifibrotic effects in the peptidoglycan-polysaccharide rat model of Crohn’s disease. Inflamm Bowel Dis. 2012;18(4):613–623. doi:10.1002/ibd.21843
127. Meng Z, Yan C, Deng Q, Gao DF, Niu XL. Curcumin inhibits LPS-induced inflammation in rat vascular smooth muscle cells in vitro via ROS-relative TLR4-MAPK/NF-κB pathways. Acta Pharmacol. Sin. 2013;34(7):901–911. doi:10.1038/aps.2013.24
128. Epstein J, Sanderson IR, Macdonald TT. Curcumin as a therapeutic agent: the evidence from in vitro, animal and human studies. Br J Nutr. 2010;103(11):1545–1557. doi:10.1017/S0007114509993667
129. Shakeri F, Kiani S, Rahimi G, Boskabady MH. Anti-inflammatory, antioxidant, and immunomodulatory effects of Berberis vulgaris and its constituent berberine, experimental and clinical, a review. Phytother Res. 2024;38(4):1882–1902. doi:10.1002/ptr.8077
130. Xu L, Zhang Y, Xue X, et al. A Phase I trial of berberine in chinese with ulcerative colitis. Cancer Prevention Res. 2020;13(1):117–126. doi:10.1158/1940-6207.CAPR-19-0258
131. Lian L, Huang Q, Zhang L, et al. Anti-fibrogenic potential of mesenchymal stromal cells in treating fibrosis in Crohn’s disease. Dig Dis Sci. 2018;63(7):1821–1834. doi:10.1007/s10620-018-5082-8
132. Pan XH, Li QQ, Zhu XQ, et al. Mechanism and therapeutic effect of umbilical cord mesenchymal stem cells in inflammatory bowel disease. Sci Rep. 2019;9(1):17646. doi:10.1038/s41598-019-54194-y
133. Rockel JS, Rabani R, Viswanathan S. Anti-fibrotic mechanisms of exogenously-expanded mesenchymal stromal cells for fibrotic diseases. Semin Cell Dev Biol. 2020;101:87–103. doi:10.1016/j.semcdb.2019.10.014
134. Yang F, Ni B, Liu Q, et al. Human umbilical cord-derived mesenchymal stem cells ameliorate experimental colitis by normalizing the gut microbiota. Stem Cell Res Ther. 2022;13(1):475. doi:10.1186/s13287-022-03118-1
135. Choi YJ, Kim WR, Kim DH, Kim JH, Yoo JH. Human umbilical cord/placenta mesenchymal stem cell conditioned medium attenuates intestinal fibrosis in vivo and in vitro. Stem Cell Res Ther. 2024;15(1):69. doi:10.1186/s13287-024-03678-4
136. Usunier B, Brossard C, L’Homme B, et al. HGF and TSG-6 released by mesenchymal stem cells attenuate colon radiation-induced fibrosis. Int J Mol Sci. 2021;22(4):1790. doi:10.3390/ijms22041790
137. Moon SH, Lee CM, Park SH, Jin Nam M. Effects of hepatocyte growth factor gene-transfected mesenchymal stem cells on dimethylnitrosamine-induced liver fibrosis in rats. Growth Factors. 2019;37(3–4):105–119. doi:10.1080/08977194.2019.1652399
138. Dong LH, Jiang YY, Liu YJ, et al. The anti-fibrotic effects of mesenchymal stem cells on irradiated lungs via stimulating endogenous secretion of HGF and PGE2. Sci Rep. 2015;5(1):8713. doi:10.1038/srep08713
139. Walter J, Shanahan F. Fecal microbiota-based treatment for recurrent Clostridioides difficile infection. Cell. 2023;186(6):1087. doi:10.1016/j.cell.2023.02.034
140. Woodhouse CA, Patel VC, Singanayagam A, Shawcross DL. Review article: the gut microbiome as a therapeutic target in the pathogenesis and treatment of chronic liver disease. Aliment Pharmacol Ther. 2018;47(2):192–202. doi:10.1111/apt.14397
141. Paramsothy S, Paramsothy R, Rubin DT, et al. Faecal microbiota transplantation for inflammatory bowel disease: a systematic review and meta-analysis. J Crohns Colitis. 2017;11(10):1180–1199. doi:10.1093/ecco-jcc/jjx063
142. Cammarota G, Ianiro G. FMT for ulcerative colitis: closer to the turning point. Nat Rev Gastroenterol Hepatol. 2019;16(5):266–268. doi:10.1038/s41575-019-0131-0
143. Claytor JD, El-Nachef N. Fecal microbial transplant for inflammatory bowel disease. Curr Opin Clin Nutr Metab Care. 2020;23(5):355–360. doi:10.1097/MCO.0000000000000676
144. Schmidt TSB, Raes J, Bork P. The human gut microbiome: from association to modulation. Cell. 2018;172(6):1198–1215. doi:10.1016/j.cell.2018.02.044
145. Wieërs G, Belkhir L, Enaud R, et al. How probiotics affect the microbiota. Front Cell Infect Microbiol. 2019;9:454. doi:10.3389/fcimb.2019.00454
146. Scott BM, Gutiérrez-Vázquez C, Sanmarco LM, et al. Self-tunable engineered yeast probiotics for the treatment of inflammatory bowel disease. Nat Med. 2021;27(7):1212–1222. doi:10.1038/s41591-021-01390-x
147. Kühbacher T, Ott SJ, Helwig U, et al. Bacterial and fungal microbiota in relation to probiotic therapy (VSL#3) in pouchitis. Gut. 2006;55(6):833–841. doi:10.1136/gut.2005.078303
148. Quévrain E, Maubert MA, Michon C, et al. Identification of an anti-inflammatory protein from Faecalibacterium prausnitzii, a commensal bacterium deficient in Crohn’s disease. Gut. 2016;65(3):415–425. doi:10.1136/gutjnl-2014-307649
149. Glassner KL, Abraham BP, Quigley EMM. The microbiome and inflammatory bowel disease. J Allergy Clin Immunol. 2020;145(1):16–27. doi:10.1016/j.jaci.2019.11.003
150. Lin SN, Mao R, Qian C, et al. Development of antifibrotic therapy for stricturing Crohn’s disease: lessons from randomized trials in other fibrotic diseases. Physiol Rev. 2022;102(2):605–652. doi:10.1152/physrev.00005.2021
151. Ananthakrishnan AN, Cagan A, Cai T, et al. Statin use is associated with reduced risk of colorectal cancer in patients with inflammatory bowel diseases. Clin Gastroenterol Hepatol. 2016;14(7):973–979. doi:10.1016/j.cgh.2016.02.017
 © 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.