Back to Journals » International Journal of Nanomedicine » Volume 21
Advancing Drug Discovery with AI: Machine and Deep Learning Strategies for Target Identification and Precision Nanomedicine
Authors Chakraborty A, Gholap AD ![]() , Khuspe PR, Sundaram G, Webster TJ
, Khuspe PR, Sundaram G, Webster TJ ![]() , Khalid M, Haris MS
, Khalid M, Haris MS ![]() , Faiyazuddin M
, Faiyazuddin M ![]()
Received 2 February 2026
Accepted for publication 1 May 2026
Published 9 June 2026 Volume 2026:21 600651
DOI https://doi.org/10.2147/IJN.S600651
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Eng San Thian
Ananya Chakraborty,1,* Amol D Gholap,2,* Pankaj R Khuspe,3 Gowri Sundaram,4 Thomas J Webster,5– 8 Mohammad Khalid,9– 11 Muhammad Salahuddin Haris,12 Md Faiyazuddin12,13
1Department of Biotechnology, Maulana Abul Kalam Azad University of Technology, Haringhata, Nadia, West Bengal, 741249, India; 2Department of Pharmaceutics, St. John Institute of Pharmacy and Research, Palghar, Maharashtra, 401404, India; 3Department of Pharmaceutics, Shriram Shikshan Sanstha’s College of Pharmacy, Paniv, Solapur, Maharashtra, 413113, India; 4PG & Research Department of Physics, Cauvery College for Women (Autonomous), Tiruchirappalli, Tamil Nadu, India; 5School of Health Sciences and Biomedical Engineering, Hebei University of Technology, Tianjin, People’s Republic of China; 6Program in Materials, Universidade Federal do Piauí, Teresina, Brazil; 7Division of Pre-college and Undergraduate Studies, Brown University, Providence, RI, USA; 8Department of Pharmacy, University of the Basque Country, Vitoria-Gasteiz, Spain; 9James Watt School of Engineering, University of Glasgow, Glasgow, G12 8QQ, UK; 10Sunway Centre for Electrochemical Energy and Sustainable Technology (SCEEST), Faculty of Engineering and Technology, Sunway University, Selangor, Malaysia; 11University Centre for Research and Development, Chandigarh University, Mohali, Punjab, 140413, India; 12Universiti Kuala Lumpur, Royal College of Medicine Perak (UniKL RCMP), Faculty of Pharmacy and Health Sciences, Perak, Malaysia; 13Centre for Global Health Research, Saveetha Institute of Medical and Technical Sciences, Chennai, Tamil Nadu, India
*These authors contributed equally to this work
Correspondence: Md Faiyazuddin, Email [email protected] Thomas J Webster, Email [email protected]
Abstract: The integration of machine learning (ML) and deep learning (DL) into drug discovery and target identification has catalyzed a paradigm shift in pharmaceutical research, enhancing efficiency and translational potential for nano-enabled therapeutics. ML models have demonstrated up to 85% accuracy in predicting drug–target interactions, whereas DL frameworks, such as convolutional neural networks (CNNs), graph neural networks (GNNs), and transformer architectures, can improve molecular property predictions by 40%. AI-driven drug discovery workflows have curtailed drug candidate attrition rates by up to 30% and accelerated discovery timelines by 20%– 40%, accentuating their rising industrial and clinical impact. This critical review evaluates the transformative roles of ML and DL in the drug discovery pipeline, emphasizing their capacity to accelerate development timelines and advance precision nano medicine. We analyzed predictive modelling techniques, including quantitative structure–activity relationship (QSAR) and absorption, distribution, metabolism, and excretion (ADME) predictions, which streamline the identification of viable drug candidates, including nanocarrier-enabled drug systems. Virtual screening and bioactivity prediction further refine candidate prioritization, whereas target identification and validation leverage protein–ligand interaction modelling and biological pathway analysis to ensure therapeutic specificity. Additionally, we discuss the profound impact of DL on medical image analysis, genomic data interpretation, and protein structure prediction (PSP), which collectively advance structural bioinformatics and enable optimized targeted nano medicine. By synergizing ML and DL, multi-modal data fusion, explainable artificial intelligence (XAI), and nanotechnology-driven datasets, the drug discovery process is evolving into a more efficient, predictive, and patient-centric endeavor, paving the way for ground-breaking therapies and improved clinical outcomes. A multi-box infographic titled “Machine learning and deep learning interventions in drug discovery and drug targeting”. A left section shows a human torso silhouette beside a network of computer screens around an Rx symbol, with the text “Machine learning (ML) and deep learning (DL) applications in pharmaceuticals”, plus “T TECHNOLOGY” and a chip icon. Top row, three titled boxes. “Predictive modeling & screening” lists “Quantitative Structure-Activity Relationship”, “Ligand and structure based virtual screening”, “Absorption, distribution, metabolism and excretion”, and “Bioactivity predictions”, illustrated with a QR-like block, a receptor diagram, a blood vessel and a lab scientist with glassware. “Target identification and validation” lists “Protein-ligand interactions”, “Molecular Docking”, “Molecular Dynamics Simulations”, and “Biological pathway analysis”, illustrated with a lab worker, a person at a computer, a gear-like symbol and a pathway graphic. “DL in Pharmaceuticals” lists “Medical diagnoses”, “Microscopy data for cellular analysis”, “Medical imaging for drug delivery optimization”, and “Genomic data analysis for personalized medicine”, illustrated with a joint image, a checklist, a brain with gears and DNA. Bottom row, three titled boxes. “ML and DL in Pharma Industry” lists “Protein structure prediction”, “Text and literature mining process”, “Biopharmaceutical Manufacturing”, and “Personalized Medicine”, illustrated with a protein ribbon, a grid, manufacturing equipment and a laptop with chat bubbles. “ML in biosimilar & drug delivery” lists “Predictive Toxicology”, “Drug Target Identification”, “De Novo Drug Design”, and “Biotechnological products”, illustrated with blood cells, a target-like symbol, a chemical structure and a funnel. “Challenges for ML in Pharma” lists “Ethics & legal issues”, “Regulatory compliance”, “Transparent interpretation”, and “Data availability & quality”, illustrated with a suited figure, a discussion icon, a person at a desk and a group of people. No numeric statistics are shown.An infographic mapping ML and DL uses in pharmaceuticals across modeling, targets, delivery and challenges.
Keywords: machine learning, deep learning, drug discovery, drug targeting, predictive modelling, virtual screening, genomics, precision nanomedicine
Introduction
Brief Overview of the Drug Discovery Process in Nano Medicine
Drug discovery is a protracted and complex process aimed at developing novel compounds and pharmaceuticals for the diagnosis and treatment of diseases, thereby enhancing human health, with increasing emphasis on precision nanomedicine and nanomedicine-based therapeutic strategies. The process commences with the precise identification of a biological target, such as a protein or enzyme, that contributes to the pathogenesis of the disease. These targets can be identified through genetic studies, research on disease pathology, or computational modelling and are increasingly informed by multi-omics data and nano–bio interaction studies.1 Once a genetic target is identified, researchers attempt to validate it by confirming its role in the condition and considering it for future drug treatments, including its suitability for nano-enabled targeting and delivery. In the hit generation step, molecules or compounds that engage the target and modify its action are identified using techniques such as high-throughput chemical screening, virtual screening using computer models, and rationalized drug design. Once these primary hits are discovered, the next step is to ensure that the lead compounds are stable, potent, selective, and possess essential pharmacological properties (Figure 1).2
 |
Figure 1 Overview of (A) the nano medicine and drug discovery process for lead optimization and target identification, validation, and lead optimization, (B) the critical role of advanced technologies in drug discovery, (C) types of techniques used in drug discovery, and (D) advantages of ML and DL in accelerating and enhancing efficiency, accuracy, and decision-making, with implications for precision nano medicine in the drug discovery process (Created using BioRender). |
Medicinal chemistry techniques are employed to refine these hits by making structural modifications to improve their drug-like profiles. The drug developer then initiates a New Drug Application (NDA) or Marketing Authorization Application (MAA) seeking approval from regulatory authorities, such as the U.S. These regulatory bodies evaluate the data to assess the drug’s safety, efficacy, and quality; once approved, the drug can be marketed and sold in the respective countries.3
A collaborative interdisciplinary process involving scientists from multiple fields, such as chemistry, biology, pharmacology, and nanotechnology, is critical for successful drug discovery. Furthermore, advances in science and technology continue to influence drug discovery, providing additional tools for drug refinement and supporting the clinical translation of precision nano-medicine stages.
Role of Technology in Accelerating Drug Discovery in Nano Medicine
Each year, drug discovery efforts screen millions of compounds, identifying tens of thousands of hits and leads, with approximately 1, 000–1, 500 candidates entering Phase-I clinical trials. Despite this scale, only 30–50 new drugs are approved annually, reflecting high attrition rates in development, which are mainly pronounced for complex biological targets and delivery challenges.3 The widespread utilization of technology in drug discovery is primarily attributed to its efficacy in expediting the process and enhancing its precision and scope (Figure 1). The implementation of automation, robotics, imaging, and sensing methodologies has augmented the rapidity of analyzing chemical libraries with greater breadth against biological targets. Advanced imaging and nanoscale characterization tools further allow for real-time monitoring of nanoparticle behavior, cellular uptake, and biodistribution. Consequently, a substantial number of possibilities that would have previously required years to screen can now be evaluated more quickly. Deterministic approaches, including molecular modelling, virtual screening, and structure-based drug design, employ advanced algorithms and computational resources to predict the associations between small molecules and biological targets.4 This tactic shortens the process of off-target identification and explores molecules with specific characteristics, thereby enhancing medicinal chemistry.
Genomics, proteomics, and other “omics” technologies are driving the next generation of precision nanomedicine by enabling comprehensive biological profiling and providing deeper insights into disease mechanisms, biomarker discovery, and drug target identification. Advanced methodologies, such as high-throughput sequencing and mass spectrometry, support large-scale analyses of genetic, proteomic, and metabolomic data, paving the way for novel drug targets and personalized therapeutic strategies.5 To maximize the impact of these technologies, integrated platforms have been developed to facilitate data sharing, analysis, and interpretation. These platforms leverage advanced artificial intelligence (AI) tools and foster collaboration among multidisciplinary teams and external partners, enhancing multimodal data accessibility and accelerating decision-making in drug discovery pipelines. By streamlining communication and expediting knowledge dissemination, these integrative systems significantly enhance the efficiency of translational research and the development of targeted and nano-enabled therapies.
Introduction to Machine Learning and Deep Learning
The integration of machine learning (ML) and deep learning (DL) technologies is transforming the nanomedicine pharmaceutical industry by enabling more comprehensive analyses of biomedical data, predicting drug–target interactions, and accelerating the development of new drugs.6 ML, a specialized branch of artificial intelligence (AI), involves training computers to learn autonomously from structured datasets without explicit programming.7 In drug discovery, ML-driven data mining facilitates the identification of biological, chemical, and clinical patterns, thereby allowing the development of predictive models for small molecules and nanomedicines. DL, a subset of ML, uses artificial neural networks (ANNs) with multiple layers to extract complex hierarchical features from data.8 This capability makes DL particularly effective for recognizing intricate patterns within large datasets (high-dimensional omics data, medical imaging data, and nanoparticle characteristics). Together, ML and DL serve as transformative tools that redefine drug research methodologies, enhance decision-making through big data insights, and expedite the identification of novel therapeutic candidates.9 Drug discovery is a multifaceted process aimed at developing novel compounds and pharmaceutical agents to diagnose, treat, and prevent diseases, while improving overall human health. This review systematically examines the drug discovery pipeline, encompassing target identification, drug development, and regulatory approval, and critically evaluates the current methodologies and their limitations, emphasizing AI and nano medicine-driven approaches.10
The key objectives of this review are as follows: First, it provides a comparative analysis of the essential drug discovery stages, including target validation, hit-to-lead identification, lead optimization, and clinical trials. Second, it highlights the interdisciplinary nature of drug discovery, spanning AI and nanotechnology. Third, it examines emerging technological advancements that are poised to reshape the field towards precision nanomedicine. The structure of this review is as follows: The “Introduction” section presents a comprehensive overview of the drug discovery process, emphasizing the revolutionary roles of ML and DL. The “Methodology” section details the data sources, study selection criteria, and statistical approaches employed in this review. The subsequent section, “Applications of ML in Drug Discovery”, explores predictive modelling techniques, compound screening methods, and target identification strategies that enhance the efficiency of drug development. The “DL Applications in Drug Discovery and Drug Targeting” section delves into specialized applications, such as medical image analysis, genomic data interpretation, protein structure prediction, and nano-enabled drug delivery optimization.
A key highlight of this review is the “Case Studies and Reports”, which illustrates the real-world applications of ML and DL in drug discovery. This includes four major case studies: (1) the use of DL to predict protein-ligand binding affinities, accelerating drug design by accurately forecasting binding strength; (2) ML-driven drug repurposing, identifying existing drugs for new therapeutic uses, such as COVID-19 treatment; (3) target identification through biological network analysis, leveraging ML to uncover novel drug targets in complex diseases; and (4) recent innovations in ML for drug discovery, showcasing state-of-the-art approaches, including graph neural networks for drug design and nanodrug delivery. These case studies highlight the profound impact of ML/DL on pharmaceutical research, providing data-driven insights and tangible success.
The subsequent section discusses “Ethical and Regulatory Considerations”, addressing challenges related to data privacy, model transparency, and regulatory compliance in AI-driven drug discovery and nano-enabled therapeutics. The review then explores “Challenges and Future Directions”, emphasizing the need for enhanced data accessibility, improved interpretability of ML/DL models, and stronger collaboration between ML experts and domain specialists. The manuscript concludes with “Limitations and Constraints of ML-enabled Research”, followed by a “Conclusion” that emphasizes the significant potential of ML and DL in transforming pharmaceutical sciences to advance precision nano medicine.
The integration of artificial intelligence, particularly machine learning and deep learning, into nanomedicine-enabled drug discovery is highly dependent on the availability, quality, and standardization of data. In contrast to small-molecule drug discovery, nanomedicine has distinct data-related issues owing to the inherent physicochemical complexity and heterogeneity of nanoscale systems. Such issues have a major impact on the reliability, interpretability, and translational applicability of AI-driven models.11
One of the greatest weaknesses of nanomedicine data science is heterogeneity in nanoparticle formulations, such as particle size and morphology, surface charge, composition, functionalization, and batch-to-batch reproducibility.12 These parameters play a critical role in biological interactions, including cellular uptake, biodistribution, immune recognition, and toxicity. Nevertheless, these characteristics are not consistently reported in the literature or assessed using non-standardized protocols, resulting in disjointed data that are challenging to incorporate into AI models. For example, two nanoparticles with nearly identical chemical compositions but slightly different sizes or surface chemistries can have significantly different pharmacokinetics and toxicities. Such variability adds noise and bias to the ML/DL training datasets, thereby reducing predictive accuracy and generalizability.13
To overcome these problems, curated databases such as NanoTox14 have become useful tools for standardizing nanotoxicology data. These systems incorporate experimentally tested data related to the physicochemical properties of nanoparticles, exposure parameters, and biological measurements, thus aiding in the establishment of robust predictive tools for nanotoxicity and biosafety evaluation. Nevertheless, despite these databases, there are issues related to the insufficient amount of data, incomplete metadata tagging, and the absence of interoperability among datasets. In addition, existing datasets tend to be biased toward a particular type of nanoparticle; therefore, polymeric, lipid-based, and hybrid nanocarrier systems that are frequently used in drug delivery are missing.15,16
Another key challenge is the integration of multi-modal data, such as omics (genomics, proteomics, and metabolomics), imaging, pharmacokinetics, and nanomaterial characterization data. Although ML/DL models can process high-dimensional data, the lack of harmonized frameworks for incorporating diverse data types discourages the creation of unified predictive models. Other recent techniques, such as multimodal learning or knowledge graphs, are under development; however, their use in nanomedicine is still in its infancy. In addition, the black-box character of most DL models raises interpretability issues, especially when making predictions about complex nano–bio interactions that must be explained mechanistically.17,18
One of the key principles supporting nanomedicine is the nanobiosurface, particularly the development of the protein corona.19 Once materials are exposed to biological fluids, they immediately adsorb biomolecules, creating a dynamic corona, which determines the biological identity of the nanoparticle. This corona causes considerable modification of nanoparticle behavior, affecting cellular recognition, immune clearance, targeting capability, and therapeutic efficacy. Notably, the protein corona composition is highly context-specific and depends on the properties of the nanoparticle surfaces, biological context, and disease conditions.20,21
In the context of AI, modeling the protein corona is challenging in the field of AI because of its dynamic and stochastic nature. Mass spectrometry and proteomics are examples of experimental characterization methods that produce massive datasets of corona composition; however, these datasets are commonly condition-dependent and unstandardized.22 Consequently, protein corona data must be carefully incorporated into predictive ML models, considering experimental variability and context. More recent developments in DL and network-based modelling have demonstrated promise in predicting protein adsorption patterns and corona evolution; however, these methods require large high-quality datasets, which are currently scarce. Protein corona dynamics is a topic of study that is still open to be incorporated into AI-based ADME and toxicity modeling; however, its incorporation is essential for developing accurate and clinically relevant nanomedicine models.23,24
In addition to scientific and technical issues, the economic cost of nanomedicine development is another obstacle to translation. Nanomedicine requires further characterization steps, such as physicochemical profiling, stability analysis, scalability, and regulatory compliance related to nanomaterials, compared to traditional drug development.25 These conditions add significant expenses and timeframes to development. In particular, detailed in vitro and in vivo research is required to assess the toxicity, biodistribution, and safety of nanoparticles over the long term, especially in light of the possibility of nanoparticle accumulation in organs such as the liver and spleen.26
Manufacturing complexity is another source of economic problems. Advanced equipment and strict quality control practices are required to scale and reproducibly produce nanocarriers, particularly those that require complicated architectures, such as multilayered or functionalized nanoparticles. The variability of batches is a serious problem, and additional validation studies may be required, which is expensive. The economic burden is also unrealized through regulatory uncertainties, as the rules governing nanomedicine are still developing and could differ in different regions, necessitating massive documentation and compliance. AI-based solutions have the potential to alleviate such economic limitations by optimizing formulation design, predicting early stage toxicity, and eliminating the use of expensive experimental screening. For example, ML models can be used to determine the best nanoparticle parameters, which increase bioavailability, decrease off-target effects, and reduce trial-and-error experimentation. Similarly, high-risk candidates can be identified at an early stage using predictive toxicology models, leading to fewer late-stage failures. However, the success of such methods depends on the quality and standardization of the datasets and the strength of the validation frameworks.27,28
Although AI can enable nanomedicine to achieve various breakthroughs, its effectiveness is closely associated with addressing major issues related to data heterogeneity, the complexity of the nano–bio interface, and financial viability. To resolve these concerns, data standardization, the creation of multifaceted nanomedicine databases, data multi-modeling, and the development of regulatory frameworks that enable innovation and safety are necessary. These developments will play a pivotal role in bringing AI-enabled nanomedicine out of the laboratory and into the clinical setting.29,30
This review provides a critical evaluation of the limitations and challenges associated with modern drug discovery. Additionally, it highlights emerging tools and technologies, such as virtual screening and AI-driven drug design, which could serve as catalysts for future research. By offering insights into contrasting technological approaches, this review seeks to assist scholars in optimizing methodologies for target validation, drug-like molecule optimization, nanoformulation design, and the expedited development of safe and effective therapeutics.
Methodology
The Databases, Keywords, and Selection Criteria
The databases used were PubMed, Scopus, Web of Science, Food Science & Technology Abstracts (FSTA), and Google Scholar. These databases were selected to ensure inclusive coverage of pharmaceutical, biomedical, computational, and nanomedicine-related literature. The search utilized the following keywords: “Machine learning and deep learning for drug discovery,” “Machine learning and deep learning for drug targeting,” “Predictive modeling,” “Lead optimization,” “Machine learning and deep learning for target identification,” “Machine learning and deep learning for personalized therapeutics,” “Machine learning-driven nanomedicine,” and “Drug repurposing.” The selection criteria for including or excluding studies are outlined in Table 1, which provides a structured framework for assessing research focused on the application of ML and DL in drug discovery and targeting, with particular emphasis on precision medicine, nano medicine, and translational relevance.
 |
Table 1 The Criteria for Including or Excluding Studies |
The Process of Study Selection and Data Extraction
This systematic review process began with a comprehensive literature search conducted via all selected databases using predefined keywords relevant to ML and DL applications in drug discovery. The retrieved records were subsequently screened for duplicates, which were removed to ensure data integrity. The titles and abstracts of the remaining entries were evaluated against the pre-established inclusion and exclusion criteria to identify potentially eligible studies for inclusion. Full-text articles were retrieved for all records meeting the initial screening criteria, followed by a detailed assessment to confirm their suitability for inclusion in this review. Data extraction was performed independently and focused on study objectives, AI/ML methodologies, datasets used, therapeutic area, relevance to precision or nanomedicine, and reported outcomes. A comprehensive statistical analysis of published manuscripts, including research articles, reviews, patents, and copyright documents, focusing on ML/DL in drug discovery and targeting between 2014 and 2024 is presented in Figure 2.
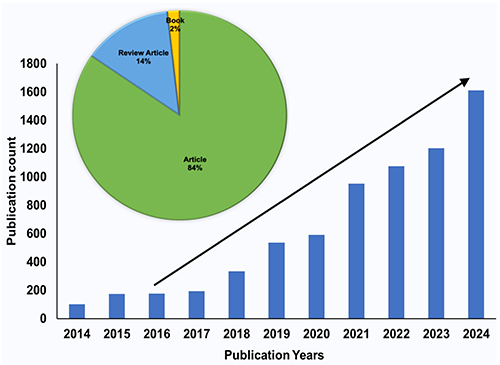 |
Figure 2 Statistical overview of published manuscripts (research, review, patent, and copyright) on ML and DL applications in drug discovery and drug targeting important for nano medicine from 2014–2024. |
Statistical Overview and Representation of Data
This review synthesizes data from diverse scientific sources, including original research articles, reviews, conference proceedings, book chapters, and early access publications retrieved from the Web of Science database with the International Journal of Nanotechnology standards. A targeted search was conducted using keywords such as machine learning, deep learning, data mining, predictive modelling, target identification, molecular docking, personalized therapeutics, nanomedicine, nanodrug delivery, and drug repurposing, focusing on titles, abstracts, and keywords in the context of drug discovery and delivery (2014–2024). The analysis identified a striking surge in ML/DL applications within this domain: publications increased from approximately 100 in 2014 to 1610 in 2024, reflecting a 16-fold increase. This swift growth parallels the growing adoption of AI-driven strategies in precision medicine and nanomedicine research. This trend aligns with advancements in computational infrastructure (eg, cloud platforms and high-performance computing) that support the training of complex ML models and the processing of large-scale datasets. The expansion of accessible chemical, biological, clinical, and nanomaterial datasets has provided critical resources for the development and refinement of AI/ML algorithms.
Recent breakthroughs in techniques, such as deep learning, reinforcement learning, and generative adversarial networks, have further broadened their applications across various research fields. Innovations in natural language processing (NLP), reinforcement learning, and generative models have enabled the robust handling of intricate biological data, improving the prediction of drug–target interactions, lead optimization, absorption, distribution, metabolism, excretion, and toxicity (ADMET) properties. Enhanced data quality from multi-omics (genomics, proteomics, and transcriptomics) and electronic health records (EHRs) has driven cost-effective and efficient drug research. Together, these developments highlight the transformative role of AI/ML in accelerating therapeutic discovery and advancing precision nanomedicine and targeted delivery of therapeutics.
Applications of ML in Drug Discovery and Drug Targeting for Nano Medicine
Predictive modelling in drug discovery for nanomedicine involves the use of computational techniques to predict the biological activity, toxicity, and pharmacokinetic properties of potential drug candidates. This approach helps streamline the drug discovery process for nanomedicine by the early identification of promising compounds, thereby reducing the need for extensive experimental testing (Figures 3 and 4).
 |
Figure 3 Key applications of computational approaches in nano medicine and drug discovery. (A) Role of predictive modelling in QSAR and ADME. (B) Importance of virtual screening for identification of potential biological activity. (C) Applications of protein-ligand interactions for prediction of binding to target proteins. (D) Biological pathway analysis. (Created using BioRender). |
Predictive Modelling
QSAR (Quantitative Structure-Activity Relationship)
QSAR is a computational methodology that establishes quantitative relationships between the structural properties of chemical compounds and their biological activities. This approach employs statistical and ML techniques to develop predictive models by analyzing datasets of structurally diverse molecules with experimentally determined activity profiles. The modelling process generally comprises four key stages: (i) data collection: gathering a dataset of chemical structures and their biological activities; (ii) descriptor calculation: converting chemical structures into numerical values called descriptors, which represent various molecular properties; (iii) model building: using statistical or ML algorithms to build a model that correlates descriptors with biological activity; and (iv) validation: Evaluating the model’s performance using a separate validation set to ensure its predictive accuracy. In drug discovery, QSAR models streamline the identification of bioactive molecules by enabling the rapid in silico screening of virtual compound libraries. These models also guide lead optimization by predicting structural modifications that improve potency, selectivity, or pharmacokinetic properties, thereby reducing experimental costs and accelerating development timelines.11
ADME (Absorption, Distribution, Metabolism, Excretion) Prediction
ADME prediction is crucial for drug development because it assesses the pharmacokinetic properties of a compound and determines its suitability for use as a drug. Predicting ADME properties helps identify compounds with favorable absorption, distribution, metabolism, and excretion profiles, thereby reducing the risk of failure in the later stages of development. Absorption predicts the extent to which a compound is absorbed into the body, considering factors such as solubility and permeability. However, distribution estimates how a compound is distributed throughout the body, including its ability to cross biological membranes and reach the target tissues. Metabolism predicts how a compound is metabolized by enzymes in the body, including potential metabolites and their effects, whereas excretion estimates how a compound is eliminated from the body, primarily through urine or feces.2 ADME prediction models use various computational techniques, including machine learning, to predict the properties of compounds based on their chemical structures. These models help prioritize compounds with desirable pharmacokinetic profiles, thereby reducing the need for extensive in vivo testing.31
A key research gap in materials and nanomedicine modeling is the trade-off between accuracy and interpretability in predictive frameworks. Conventional physics-based models offer interpretability but lack scalability and predictive power, whereas deep learning models provide high accuracy but suffer from poor transparency and computational inefficiency. Ren et al, have addressed these limitations by harmonizing physical modeling with deep learning, integrating domain knowledge into data-driven architectures to improve interpretability and efficiency. The hybrid approach reduced the computational cost while maintaining predictive accuracy. The outcomes demonstrated robust property prediction, enhanced model explainability, and improved generalization, highlighting the potential of this approach for accelerating rational design in nanomedicine and materials science.32
Compound Screening
In compound screening, large libraries of compounds are evaluated to identify those with potential biological activity against the target of interest. This process is essential in drug discovery for identifying hit compounds that can be further optimized into lead compounds and eventually drug candidates.
Virtual Screening
Virtual screening is a computational technique used to screen large compound libraries to identify compounds with potential biological activities. This method is primarily divided into two approaches:
Ligand-based virtual screening (LBVS): LBVS uses known active compounds to identify novel compounds with similar chemical structures and properties. Techniques, such as QSAR and pharmacophore modelling, are often used in ligand-based screening.
Structure-based virtual screening (SBVS) influences the 3D structure of a target protein to identify compounds that can efficiently bind to it. Molecular docking is a common technique in structure-based screening in which compounds are virtually “docked” into the active site of a protein to predict their binding affinity.
Virtual screening is a cost-effective and time-efficient method for identifying potential hit compounds. This reduces the need for extensive experimental testing and allows researchers to focus on promising candidates in future studies.
A major research gap in natural product discovery is the lack of efficient dereplication workflows to rapidly identify known compounds and avoid redundant isolation. Conventional analytical approaches that rely on manual spectral interpretation and database matching are limited by low throughput, poor integration of multi-omics data, and time-intensive processing. Sheng et al, have addressed these limitations by developing IMN4NPD, an integrated molecular networking workflow, combining MS/MS data analysis, database annotation, and network-based clustering. The platform enabled the rapid identification of known metabolites and structural relationships. The outcomes demonstrated improved dereplication efficiency, reduced redundancy, and accelerated discovery of novel natural products, supporting advanced metabolomics-driven drug discovery.33
Bioactivity Prediction
Bioactivity prediction involves the use of computational models to predict the biological activities of compounds based on their chemical structures. This approach helps prioritize compounds with the highest likelihood of exhibiting the desired biological effects. ML and DL techniques are increasingly being used to predict bioactivity. These models are trained on large datasets of compounds with known biological activities and learn to predict the activities of novel compounds based on their chemical properties. Bioactivity prediction models can identify potential off-target effects and toxicities, thereby aiding the design of safer and more effective drugs.34 These models are valuable in the early stages of drug discovery, enabling the rapid identification of promising compounds for further testing.31
Target Identification and Validation
Target identification and validation are critical steps in drug discovery, involving the identification of biological targets (eg, proteins) associated with a disease and validation of their role in the disease process.
Protein–ligand interactions: Understanding protein–ligand interactions is essential for identifying and designing compounds that can modulate the activity of target proteins. Computational techniques, such as molecular docking and molecular dynamics simulations, have been used to predict ligand (potential drug molecule) binding to target proteins.35
Molecular Docking (MD): MD predicts the preferred orientation of a ligand when it binds to the active site of a protein. Docking algorithms that score different binding pose to identify the most favorable interactions, providing insights into binding affinity and specificity.
Molecular dynamics simulations (MDS) reveal the dynamic behavior of protein-ligand complexes over time, provide detailed information about the stability and conformational changes of the complex, and help to understand the binding mechanisms for optimized ligand design.
Protein–ligand interaction studies are crucial for drug design because they guide the optimization of lead compounds to enhance their binding affinity and selectivity.
Biological Pathway Analysis
Biochemical pathway analysis involves studying complex networks of biochemical interactions within cells to understand the involvement of target proteins in disease processes.15 This analysis helps identify potential drug targets and understand the downstream effects of their modulation. Pathway analysis tools integrate data from various sources, including genomics, proteomics, and metabolomics, to construct comprehensive cellular pathway models.36 These models are used to predict the impact of targeting specific proteins on disease pathways, thereby aiding in target validation. By identifying key nodes and interactions within pathways, researchers can prioritize targets that are likely to have the most significant therapeutic effects.37 This approach also helps identify potential side effects and off-target interactions, thereby guiding the design of safer drugs.
DL Applications in Drug Discovery and Drug Targeting for Nano Medicine
The use of AI in drug development for nanomedicine has recently attracted considerable attention because it reduces the time and cost of discovering new drugs. As DL technology develops and the volume of drug-related data increases, DL-based approaches are being used more frequently throughout the drug development process for nanomedicine.38 Table 2 shows the transformative role of DL in drug discovery and targeting, illustrating how advanced algorithms can enhance the identification of potential drug candidates, optimize molecular structures, and predict biological interactions for nanomedicine.
 |
Table 2 An Overview of DL Applications in Drug Discovery and Targeting |
Image Analysis for Medical Diagnoses
Early approaches to medical image processing relied on conventional techniques, such as edge tracing, region expansion, and thresholding. With the exponential growth in medical imaging data, ML algorithms have gradually become integral to image assessments. However, these early ML methods depended on manually engineered features, making algorithm development time-consuming and labor-intensive. The limitations of traditional ML have prompted the adoption of ANNs, the development of which has been driven by increased data availability and enhanced computational capabilities.57 The integration of CNNs and other DL architectures has revolutionized the automated interpretation of medical images. For instance, pretrained CNN models, such as Google’s Inception v3, originally trained on the large-scale ImageNet dataset, have demonstrated remarkable efficacy in diagnosing melanoma subtypes, including acrolentiginous, amelanotic, and lentigo melanomas, through dermoscopic image analysis, as evidenced by recent studies.44,58–61
Microscopy Data for Cellular Analysis
In high-throughput imaging-based screening, morphological changes in cells can be accurately interpreted using DL-based classifiers. These models are used to extract feature vectors from images, which may subsequently be clustered to identify new cell phenotypes after training on classification tasks. The limited Boltzmann machine model, neural network with a convolution model, and sparse model are the most popular DL models. These algorithms share similarities in classification and recognition, despite minor differences in feature extraction.62 The digital images used to test, train, and assess the effectiveness of the computer vision algorithms comprise an image dataset. Using image datasets, algorithms can learn to recognize information in images and perform relevant cognitive operations. Advanced DL models, particularly U-Net and convolutional neural networks with deep max-pooling layers, have been used to analyze fluorescence microscopy images to examine cellular structures and behaviors.41 Google Inception v3-based architectures have been used for pancreatic cancer tumor/normal classification, aiding the identification of cancer cell types in breast and colon cancer histology images.41,42
Medical Imaging for Drug Delivery Optimization
DL has revolutionized medical image analysis, achieving remarkable results in tasks such as image registration, classification, feature extraction, and noise reduction.34,37 Among DL architectures, CNNs are particularly impactful because of their ability to process spatial hierarchies in image data. CNNs employ convolutional layers to automatically detect local features via filter operations, followed by pooling layers for dimensionality reduction and fully connected layers for high-level decision making.63 These networks mimic hierarchical processing in the human visual cortex, progressively abstracting input data through nonlinear transformations across multiple layers.64 In drug delivery optimization, DL-driven medical imaging techniques enhance the precision of treatment planning and efficacy assessment. For instance, CNNs, RL, and SVMs have been applied to analyze MRI data, improving targeted chemotherapy delivery by optimizing doxorubicin administration, refining tumor localization, and predicting therapeutic outcomes. Open-source frameworks (such as TensorFlow and PyTorch) facilitate the implementation of CNNs and RL for real-time medical imaging data (MRI) analysis, enabling adaptive drug delivery strategies37,39,43–45,52 Libraries such as Keras and Scikit-learn simplify tumor classification and treatment protocol forecasting through user-friendly interfaces for model development.44 Advanced computational tools further expand these capabilities: COMSOL Multiphysics and BioImageXD support multiparametric simulations and 3D image analysis to predict optimal drug targeting,37,43 while platforms like NVIDIA Clara and MATLAB’s ML Toolbox offer AI-driven solutions for real-time image processing and clinical decision support.65,66
Sequence Analysis
Genomic Data Analysis for Personalized Nano Medicine
Recent advancements in deep learning (DL) and machine learning (ML) have transformed the integration of genomic data into clinical practice by enabling precise patient stratification through the differentiation of normal and pathogenic gene expression patterns. These AI-driven methodologies support real-time diagnostic evaluations and therapeutic decisions, particularly when genetic variants influence the pharmacological effects of drugs. For example, genomic profiling can inform personalized adjustments to drug dosing or selection, thereby enhancing treatment efficacy.67 DL and ML frameworks have been applied across three primary domains in genomic analysis: variant detection, functional annotation, and predictive modelling. These approaches rely on high-quality reference genomes and large-scale sequencing datasets. However, while DL models achieve high predictive accuracy with extensive training data, their “black-box” nature often obscures the interpretability of input-output relationships, hindering their clinical adoption.68 Additionally, many AI tools prioritize statistical correlations over causal inferences, necessitating experimental validation to establish biological mechanisms.69 To address these limitations, bioinformatics pipelines (eg. GATK and STAR) have been developed to streamline genomic data processing, enabling the detection of mutations and molecular biomarkers that are used in predictive models. For instance, DL/ML frameworks are increasingly employed to analyze tumor genomic profiles, predict patient responses to immunotherapies such as nivolumab, and identify biomarkers associated with treatment efficacy.45,46
A critical research gap in drug repurposing is the limited ability to systematically predict drug–disease associations from heterogeneous biomedical data. Conventional methods often relied on isolated datasets or similarity-based approaches, which failed to capture complex biological network relationships and showed limited predictive accuracy. Zhou et al, have addressed these limitations by developing NEDD, a network embedding-based computational framework, which integrated multi-source data into a unified network representation and learned low-dimensional feature embeddings. The model effectively captured hidden associations between drugs and diseases. The outcomes demonstrated improved prediction accuracy and the identification of novel therapeutic links, supporting efficient drug repositioning and accelerated biomedical discovery.70
Protein Structure Prediction (PSP)
PSP is a key area of research in structural bioinformatics. Since the 1960s, various levels of complexity in protein structure information have been predicted using statistical methods, increasingly complex ML techniques, and most recently, DL techniques. To predict protein structures using DL, a multiple sequence alignment (MSA) comprising homologues of the target protein is typically built first. Large feature sets produced from MSAs are combined using the most effective methods, and a significant amount of computational work is required to derive input features. The Alphabet subsidiary DeepMind created an AI program, AlphaFold, which predicts protein structures.71 Although this prediction is precise concerning some alterations, it is resilient to others; however, it is difficult to forecast protein structures from amino acid sequences. In contrast, slight changes in the protein sequence can completely alter its structure and function. DL-based tools (AlphaFold, DeepMind’s Evoformer, DeepFold, Modeller, RoseTTAFold, I-TASSER, TrRosetta, and QUARK) predict 3D protein structures from amino acid sequences, particularly focusing on human proteins, as demonstrated in the CASP13 and CASP14 datasets.47,48 Researchers have predicted the structure of nearly every known protein worldwide using AlphaFold, a groundbreaking AI network that covers the structures of more than 200 million proteins from approximately one million species.72
AlphaFold, developed by DeepMind, has demonstrated unparalleled accuracy in structure prediction, particularly in the CASP13 and CASP14 challenges, using the Evoformer module and evolutionary data.48 Despite recent improvements in experimental methods that have considerably enhanced our ability to experimentally discover protein structures, the discrepancy between the total number of protein sequences and documented protein structures continues to grow. One technique to close this gap is the use of computational PSP. As demonstrated by the success of AlphaFold2 in the recent critical assessment of PSP (CASP14), DL-based techniques have led to significant advancements in the field of PSP. In this study, we discuss the significant achievements and developments made possible by DL-based approaches, as observed in CASP studies in the PSP. The ability to anticipate spatial constraints from sequences and/or multiple sequence alignments (MSAs) using DL approaches has greatly increased the precision of ab initio structure prediction. RoseTTAFold uses a multi-track neural network approach to handle complex protein folds,64 whereas TrRosetta integrates residue-residue distance predictions to refine models.63 The significance of the physical energy function in the era of DL has been questioned owing to the rapid advancement in the accuracy of restraint prediction. Other tools, such as I-TASSER and Modeller, rely on threading and homology modelling for proteins with known templates,73 whereas Foldit uses crowdsourcing to explore novel protein folds.47,48
Advanced Text and Literature Mining Methods
Text and literature mining tools (natural language processing, machine learning, and statistical analysis) extract meaningful insights and patterns from large datasets, making them vital for research in diverse fields of science and technology. Popular tools such as NLTK, SpaCy, Gensim, Weka, RapidMiner, and KNIME provide intuitive interfaces for tasks such as the classification and clustering of textual data and predictive modelling.49,50 A systematic review conducted by Ghosal et al explained that flow-based generative models could be used to improve the identification of potential drugs more effectively by analyzing chemical and biological data across different platforms.74 This case highlights the potential of computational techniques for drug identification, particularly for emerging health issues. Python (with pandas and scikit-learn) and “R” are widely used for custom data mining workflows,75 whereas big data platforms such as Hadoop and Apache Spark facilitate scalable analysis.49 Tableau offers powerful data visualization capabilities, whereas SAS and IBM SPSS Modeller are used for advanced analytics and predictive modelling.75,76
Drug repurposing offers the quickest route from the bench to the bedside by identifying new applications for previously approved medications. Real-world information on sizable cohorts of drug users is available from sources such as insurance claims and electronic health records (EHRs). Using a retrospective study of real-world data, we proposed an effective and easily customizable framework for generating and testing several candidates for drug repurposing.49,50 Although the expense and timing of drug design are typically prohibitive, the benefits of using the de novo technique for drug discovery are enormous, as described above. In contrast, medications with well-understood pharmacokinetics and mechanisms of action might be seen as having previous knowledge of a certain domain.77 After the potential side effects of a known medicine are discovered, the drug can be utilized more effectively and safely without having to start from scratch. Creating “old drugs for a new use” would take significantly less time and money in this scenario. Governments and pharmaceutical corporations are paying increasing attention to drug repurposing because of its excellent ability to reduce time and cost. Therefore, this approach is a viable and promising strategy.78 Emerging computational techniques can aid drug repurposing by identifying connections between various biological entities, including genes, gene components, diseases, and drugs. Massive amounts of data can be used in studies grounded in reality to reflect the real-world processes of diagnosis, therapy, and patient health.78
Owing to its extraordinary capabilities, deep learning (DL) can be used to signal and mine large amounts of real-world data. However, traditional statistical methods have difficulty processing large volumes of data. CNNs that create traditional Chinese medicine (TCM) prescriptions based on a patient’s facial picture have been proposed as a DL strategy for mining the association between a patient’s face and prescriptions.79 Investigating the polypharmacological effects of TCM formulations will help elucidate their mode of action and open new avenues for the treatment of diseases. DL methodologies can also extract more illuminating information from molecules and map molecular architectures to possible areas. Flow-based approaches can change the distribution of features; therefore, they have received significant attention. Chen et al80 established a concept of using CNN for developing TCM prescriptions from facial expressions of patients and showed how DL can correlate clinical data with a therapeutic response. Studying the polypharmacological effects of TCM formulations will aid in understanding the mechanisms of TCM and in the discovery of novel treatments for diseases. DL methodologies can also retrieve more informative information from molecules and map molecular structures to potential therapeutic areas.49,50 Tools such as Drug Repurposing in AD (DRIAD), ReDRug, Cancer DL Environment (CANDLE), PharmMapper, DrugBank, and Integrative Functional Drug Annotation (iFDA) data mining and network analysis can be used to identify drugs repurposed for different diseases.49,50 For instance, a recent clinical trial used fluoxetine, an antidepressant drug, to treat COVID-19 based on computational predictions of its antiviral potential against SARS-CoV-2.80
A significant research gap exists in the effective translation of bioactive compounds from traditional Chinese medicine (TCM) into clinically viable therapeutics because of poor solubility, low bioavailability, and inconsistent pharmacokinetics. Conventional delivery approaches often fail to stabilize natural products or achieve targeted delivery, thereby limiting their therapeutic efficacy. Zeng et al addressed these limitations by integrating nanomedicine-based delivery systems with TCM-derived compounds to enhance their solubility, stability, and controlled release. Nanocarriers, such as liposomes, nanoparticles, and micelles, improve tissue targeting and pharmacokinetic profiles. The outcomes demonstrated enhanced bioavailability, increased therapeutic efficacy, and reduced toxicity, highlighting new opportunities for modernizing TCM using advanced nanotechnology-driven drug delivery strategies.81
Miscellaneous Uses of ML and DL in Pharmaceuticals and Nano Medicine
Table 2 provides an overview of the roles of ML and DL in advancing pharmaceutical processes, with special attention to their demanding applications in drug discovery, optimizing manufacturing workflows, ensuring consistent QC, and designing targeted drug delivery systems. The key aspects of drug development are effectively highlighted in Figure 5, featuring structure-based drug design using the 3D structure of target proteins to enhance efficacy and reduce side effects, de novo drug design for creating novel compounds, predicting adverse drug reactions (ADRs) to ensure safety, and the application of precision nanomedicine to customize treatments based on individual genetic profiles for targeted and effective therapies.
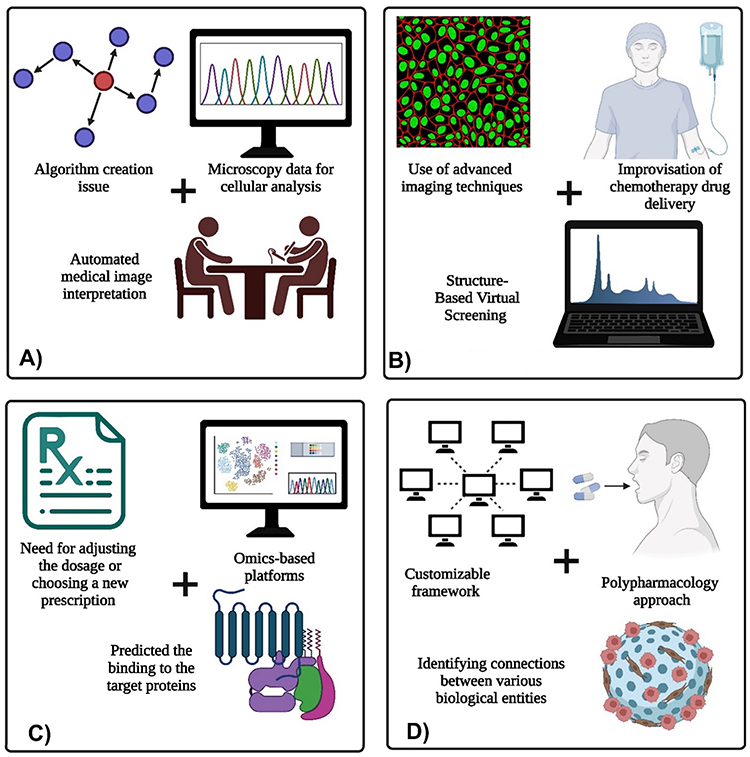 |
Figure 4 Applications of computational and data-driven approaches in nano medicine and drug discovery, including: (A) Image analysis of medical diagnosis, (B) Medical imaging role in drug delivery optimization, (C) Application of genomic data analysis, and (D) Drug repurposing requirement for drug discovery (created using BioRender). |
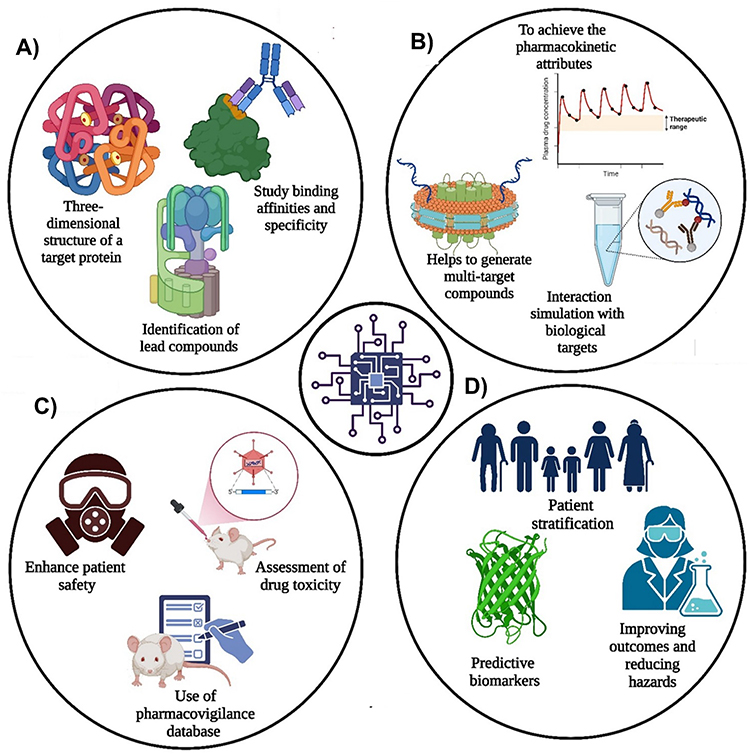 |
Figure 5 Key aspects of modern nano medicine and drug design summarizing: (A) Structure-based drug design, including the three-dimensional structure of the target protein; (B) Importance of de novo drug design; (C) Prediction of adverse drug reactions; and (D) Importance of precision medicine (created using BioRender). |
Virtual Screening
High-Throughput Screening (HTS)
HTS enables scientists to test thousands to millions of compounds to determine whether they interact with specific biological targets of interest. This was achieved using ML models that predict the bioactivity of any compound, thereby significantly reducing the need for rigorous physical screenings. The virtual screening method utilizes technologies such as AutoDock, DeepChem, and Docking Score ML® to examine large compound libraries for potential molecules, including sunitinib, against 155 cancer targets.82 These models can evaluate one or a set of chemical properties and biological information to determine the importance of the compound in solving the problem, thus enhancing and reducing screening costs and time required for screening.
Structure-Based Drug Design (SBDD)
SBDD is a technique that employs the three-dimensional structure of target proteins to develop new drugs. The behavior of a potential drug is predicted using deep learning (DL) models for its interaction with these proteins, including its binding affinity and specificity. SBDD can help identify lead compounds that need to be further improved by building and testing iterations.83
De Novo Drug Design
Generative adversarial networks (GANs) are generative models that are trained to generate new molecules with one or more specific properties inherent to them. This makes it possible for GANs to generate compounds with the required biological activity and pharmacokinetic attributes because the model is built on existing chemical data.84 Reinforcement learning (RL) enhances drug design because the designer cycles through new molecular structures of the drug, making improvements based on the results of the interaction simulation with biological targets. The use of DL can extend enormous chemical spaces, which can help identify drug prospects. De novo drug design tools, such as GANs, RL, simplified molecular input line entry systems (SMILES®), and POLYGON®(a polypharmacology approach based on generative RL), are used to design novel chemical structures and generate multi-target compounds. These methods have been used to develop 32 novel sulfonamide compounds targeting the MEK1 or mTOR pathway using deep generative chemistry.52,85
Drug Target Identification
Genomic Data Analysis
Genomic data analysis employs computational methodologies, including ML models, predictive algorithms, and bioinformatics tools, to systematically analyze large-scale genomic and proteomic datasets associated with various diseases. By integrating genome-wide association studies (GWAS), these approaches can identify critical genetic and molecular signatures linked to disease progression, thereby uncovering novel biomarkers and potential therapeutic targets. Platforms such as DrugBank® and Matador® enhance this process by enabling drug-target interaction prediction, transcriptomic data classification, and the development of drug repurposing strategies. These resources facilitate the prioritization of high-value targets and validation of candidate molecules, thereby bridging genomic insights with actionable therapeutic development.49,58,60–62
Network-Based Approaches
Biological networks can be further analyzed using deep learning models to understand the relevant nodes that can potentially act as drug targets. Knowledge regarding targets can be obtained from the implementation of such interactions, which can provide the highest impact on disease modulation. These approaches utilize tools such as graph theory, ML algorithms, the human disease network (HDN), and the ADTree model. They are employed to understand drug–target interactions, predict disease genes, and explore biological networks to gain insights into disease mechanisms and therapeutic targets.86,87
A major research gap in disease biomarker discovery is the limited ability to accurately infer miRNA–disease associations from complex biological systems. Conventional computational approaches often rely on single-layer networks or similarity-based methods, which fail to capture multilevel interactions and hidden biological relationships. Li et al, have addressed these limitations by developing a gene-mediated tripartite heterogeneous multiplex network model, integrating microRNA, gene, and disease interactions. This framework enables the extraction of latent features and improve representation of biological complexity. The outcomes demonstrated enhanced prediction accuracy, identification of novel miRNA–disease associations, and improved robustness, supporting its application in biomarker discovery and precision medicine.88
Predictive Toxicology
In recent years, ML models have been used to efficiently identify potentially toxic compounds during the early stages of drug development. These predictive models use structural features and biological activity data to evaluate the safety profiles of compounds and reduce the risks and potential adverse effects during clinical trials. Tools and software, such as QSAR, DL models, SVMs, RF, DTs, MeDeBERTa, and RoBERTa (MIMIC), have been used to assess safety, predict potential ADRs, and develop automated ADR detection systems in real-world settings.89–91
A critical research gap in environmental toxicology is the limited mechanistic understanding of bisphenol S (BPS)-induced atherosclerosis at the molecular and systems levels. Conventional toxicological studies often fail because of their reliance on isolated pathways and lack of integrative computational analysis, which limits mechanistic insights. Guo et al, have addressed these limitations by combining network toxicology, molecular docking, and machine learning to systematically identify key targets and pathways involved in BPS-induced vascular damage. The integrated approach revealed interactions with lipid metabolism, inflammation, and oxidative stress pathways. The outcomes demonstrated enhanced mechanistic clarity and predictive capability, supporting risk assessment and targeted intervention strategies for environmental toxin-induced cardiovascular diseases.92
Adverse Drug Reaction Prediction
Large-scale ML platforms consider aspects such as a patient’s EHRs and clinical data to estimate the propensity for ADRs. With the help of this type of model, high-risk medications and patients can be detected by analyzing large numbers, which can enhance patient safety.84 Tools for predicting ADRs include Tox21, which uses ML to assess drug toxicity; DeepADR for predicting ADRs from drug features and protein interactions; MeDeA, which analyzes clinical data to predict ADRs; the FDA’s FAERS, which is integrated with ML for ADR detection; and the pharmacovigilance database VigiBase, which employs data mining for ADR prediction.89–91
Precision Nano Medicine
Patient Stratification
Based on a patient’s genetic, phenotypic, and clinical information, they were divided into different groups using ML approaches. Due to this stratification, biomarkers are used in the formulation of therapies that work precisely on specific groups of patients, improving outcomes and reducing risks.86,87
Predictive Biomarkers
The DL algorithm can learn from other biomarkers to predict a patient’s response to a specific treatment. These biomarkers are useful for developing patient-specific treatment plans, enabling clinicians to select the best treatment modalities for patients.93
A major research gap in nanocomposite-based sensing and computing systems is the lack of multifunctional platforms capable of integrating sensing, logic operations, and data security within a single architecture. Conventional nanomaterials often fail owing to limited functionality, poor scalability, and inability to support complex logic computing tasks. Zhou et al, have addressed these limitations by engineering Au–Ag–Cr multifunctional nanocomposites with tunable optical and electronic properties for multiplex sensing and logic gate implementation. The system demonstrated advanced signal processing, logic computing capability, and secure information encoding. The outcomes highlighted scalable information protection and multifunctional performance, offering promising applications in smart sensing and nano-enabled computing systems.94
Biopharmaceutical Production for Nano Medicine
A significant research gap in regenerative nanomedicine is the lack of biomaterials that can simultaneously support tissue regeneration, controlled drug delivery, and immunomodulation in a single construct. Conventional scaffolds and delivery systems often fail because of poor bioactivity, limited mechanical stability, and inadequate control over therapeutic release. These limitations have led to the exploration of layered double hydroxides (LDHs) as multifunctional nanomaterials with tunable compositions, high loading capacities, and biocompatibility. LDH-based systems enable the sustained release of bioactive agents and enhance the cellular response. The outcomes demonstrated improved tissue regeneration, osteogenesis, and wound healing, emphasizing the potential of LDHs as promising platforms for advanced tissue engineering and regenerative therapies.95
Process Optimization
In biopharmaceutical manufacturing, ML algorithms minimize waste, maximize production, and enhance quality. Some of these models allow for the prediction of process parameters related to process efficiency and product quality, providing insights into the conditions that are most conducive to efficiency and quality.96,97 Tools such as MATLAB, TensorFlow, and Keras allow for defect detection in manufacturing processes, whereas platforms such as IBM Maximo, Uptake, and PTC ThingWorx use ML for predictive maintenance.97 AI algorithms, including 2D-CNNs and the SHAPLEY® technique with Bayesian optimization methods, have been used to optimize biopharmaceutical production processes, particularly in the efficient and continuous manufacturing of monoclonal antibodies.98,99 Aspen Plus and DynoChem optimize production processes, whereas Zenith Technologies and Honeywell UniSim monitor and control batch processes.66 SIMCA and Mettler-Toledo practice PAT to ensure real-time quality; however, advanced tools, such as Llamasoft, SAP IBP, and Microsoft Azure ML, aid in inventory optimization and supply chain management.100,101
Quality Control (QC)
AI-based prognostic maintenance reduces equipment failures and ensures continuous production. DL algorithms can identify other variations because this process ensures customization in the production of biopharmaceuticals. These models use live data from manufacturing processes to detect variations from ideal benchmarks to ensure the integrity of product production.96 Model expert systems (MES), model predictive control (MPC), process analytical technology (PAT), and AI-driven prognostic maintenance (XenoSite, FAME, and SMARTCyp) are key technologies for ensuring the quality and consistency of biopharmaceutical products.100,101 MPC optimizes complex processes, such as bioreactor control, whereas PAT monitors critical parameters in real-time, improving drug quality. XenoSite, FAME, and SMARTCyp aid in understanding drug metabolism and safety, such as that of piroxicam and paracetamol.
Pharmacokinetics for Nano Medicine
ADME Prediction
Artificial ML algorithms have been used for computer-aided modelling of ADMET compound properties. These models are based on chemical structure and physicochemical properties and can filter candidates to rank those with good pharmacokinetic properties, thus minimizing the risk of late-stage failures.102 ADME interpreters, such as SwissADME, TopPreds, and ADMET predictors, provide insights into solubility, permeability, and absorption,103 whereas PKCSM and QikProp use ML and computational chemistry to predict metabolism and drug-likeness.103 Simcyp Simulator and MetaSite focus on simulating human pharmacokinetics and predicting the accuracy of metabolic pathways.90
Dose Optimization
Pharmacokinetic data are used to aid in the development of appropriate dosing regimens using DL models.104 Using different dosing profiles, these models allow for the determination of safe and optimal dosing regimens for specific patients. PK/PD modelling, AI algorithms, RNNs, and the batch-constrained Q-learning algorithm can optimize treatment regimens and support dose decision-making through expert systems (PFES).45 These digital clinical decision support systems have enhanced PK/PD modelling and ML for Warfarin, Apixaban, Rivaroxaban, and Dabigatran, facilitating personalized therapy.104,105
Data Driven Nano Medicine
Data management and integration improve manufacturing competence using real-time analytics, thereby reducing production expenses by 20%.37,65,66,106,107 In nanomedicine, ML and analytics facilitate the design of targeted drug delivery systems, enhancing efficacy while reducing side effects, with studies showing up to a 50% increase in drug bioavailability in preclinical models.37,106,107 In solid pharmaceutical development, data-driven methods optimize formulation and processing, improving drug stability and bioavailability by 30% and 40%, respectively, in oral dosage forms.65,66 Tools such as FormulationAI®, Pycaret, KNIME, TensorFlow, and PyTorch have been used to design innovative formulations and carriers and have been applied to ciprofloxacin and estradiol.37,106 GROMACS and MATLAB have been used to model drug delivery systems in nanomedicine and to aid in simulating nanoparticle behavior.57
A major challenge in prodrug-based chemotherapy is achieving a balance between nanoassembly stability and rapid intracellular drug activation. Conventional doxorubicin prodrugs often fail owing to premature drug leakage, poor self-assembly behavior, and inefficient activation within tumor cells. Feng et al have addressed these limitations by precisely tailoring the molecular structure of doxorubicin prodrugs to enable stable nanoassembly with stimulus-responsive activation. The engineered prodrugs exhibited enhanced structural stability during circulation and rapid drug release in tumor-specific environments. The outcomes demonstrated improved cellular uptake, potent antitumor efficacy, and reduced systemic toxicity, highlighting the potential of rational prodrug design for advanced nanomedicine applications.108
The diverse roles of ML and DL in enhancing pharmaceutical performance by driving the rapid identification and optimal design of lead molecules are listed in Table 3.
 |
Table 3 The Diverse Role of ML and DL Impacting Pharmaceutical Performance |
Case Studies and Reports: Showcasing ML/DL Success Stories in Drug Discovery for Nano Medicine
This section highlights relevant case studies that showcase the successful application of ML and DL in drug discovery. The first case study focuses on the use of DL to predict protein–ligand binding affinities, demonstrating improved predictive accuracy in drug design. The second study examined the use of ML for repurposing existing drugs, illustrating how algorithms can uncover new therapeutic uses, thus accelerating drug development.118 Finally, the third case study explored target identification through biological network analysis and revealed how ML techniques can be used to identify novel drug targets. Collectively, these studies highlight the transformative impact of ML and DL on drug discovery processes.
Using DL to Predict Protein-Ligand Binding Affinities for Nano Medicine
The prediction of protein–ligand binding affinity is crucial for drug discovery and nanomedicine, as it helps identify potential drug candidates by estimating the strength of ligand (or drug molecule) binding to target proteins. Traditional methods, such as molecular docking and molecular dynamics simulations, are time-consuming and computationally expensive. Deep learning (DL) offers a powerful alternative by leveraging large datasets and complex models to predict binding affinities with high accuracy.119 In this case study, we explored how a DL model can be used to predict protein–ligand binding affinities. The model employs a neural network architecture trained on a dataset of known protein–ligand complexes with their corresponding binding affinities. The process involves several key steps: (i) data collection: gathering a large dataset of protein–ligand complexes with experimentally determined binding affinities; such as the Protein Data Bank (PDB) and Binding DB; (ii) feature extraction: converting protein and ligand structures into numerical representations, including 3D coordinates, physicochemical properties, and interaction fingerprints;119 (iii) model training: using a DL architecture, such as a convolutional neural network (CNN) or graph neural network (GNN), to learn the relationship between the features and binding affinities- the model is trained on a subset of the data and validated on a separate set to ensure generalization;118 and (iv) prediction: applying the trained model to new protein–ligand pairs to predict their binding affinities and identify promising drug candidates for further experimental validation.89,91,119
Studies have shown that DL models can achieve high accuracy in predicting binding affinities, often outperforming traditional methods. For instance, a CNN model trained on the PDBBind dataset demonstrated improved predictive performance compared with classical docking methods.89,91,119 In silico Medicine and BenevolentAI exploit DL to accelerate the drug development process by predicting binding affinities for cancer and related threats.109 The AtomNet model by Atomwise, which uses CNNs to predict binding affinities, was able to identify potential inhibitors for the Ebola virus in less than a day. Recently, Schrödinger and Exscientia integrated DL with computational chemistry to optimize the binding affinities of cancer therapeutics.65,112 These approaches not only accelerate the drug discovery process but also reduce the cost and time associated with experimental screening.
The prediction of the binding affinity between ligands and proteins is vital for drug screening and optimization. Several models, such as AlphaFold2, are used to predict protein structures; however, they suffer from limitations due to the low quality of the database, inappropriate architecture of the model, and less accurate input representation. Wang et al studied the prediction of protein-ligand binding affinity through DL models. DL models can extract the required features from raw data, followed by feature representation and classification. The binding affinities for all types of biomolecular complexes in the Protein Data Bank were comprehensively collected from the protein data bank (PDBbind) database, where pKa values were used to express the binding affinity values. The PDBbind database consists of three overlapping subsets: core, refined, and general sets (Figure 6A). The number of protein–ligand complexes in the PDBbind database gradually increased with respect to the protein data bank entries (Figure 6B). The DL model can assist in pattern discovery for better prediction of new data and improvement in the generalization ability of the model. The distribution of the protein–ligand binding affinity values in the PDBbind database was balanced. The tertiary structure of the pocket along with the corresponding protein is provided in the Protein Data Bank structure file. The Davis database is one of the largest datasets, including kinase proteins and clinically relevant inhibitors with their respective Kd values (Figure 6C). The Davis database contains information on approximately 442 proteins and 367 kinases. The uniform distribution of 367 human kinases from the human kinome tree is depicted in Figure 6D. The negative protein–ligand complexes were based on lower pKd values. This was influenced by weak binding affinities (Figure 6E). The uniform distribution of 216 human kinases in the human kinome tree is shown in Figure 6F. The uneven distribution of binding affinity is shown in Figure 6G.
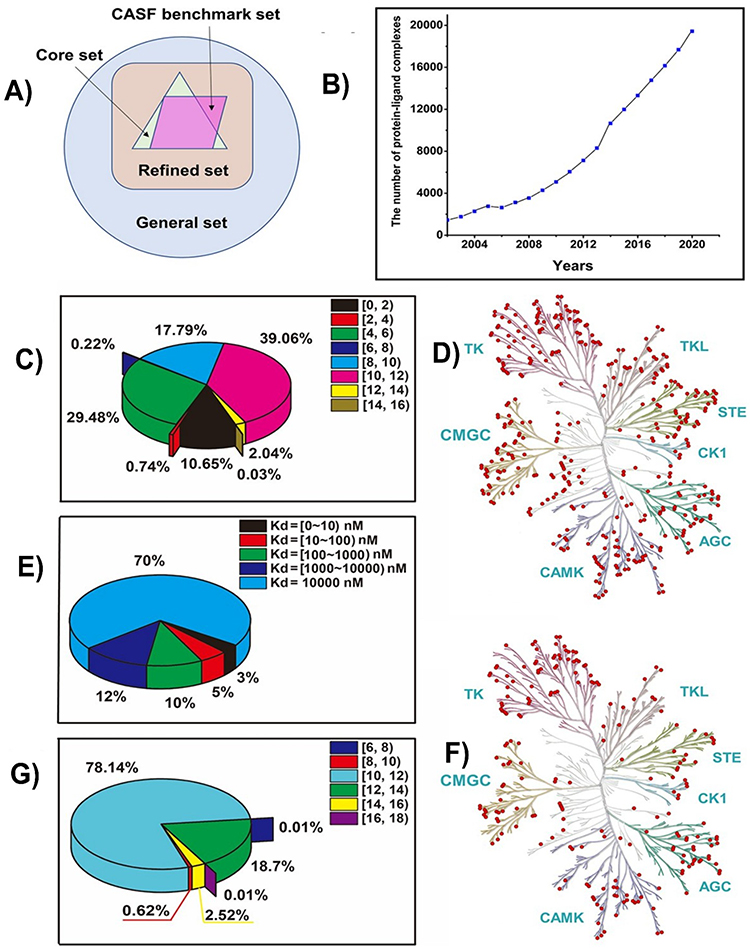 |
Figure 6 Overview of the datasets used for protein-ligand binding affinity analysis. (A) Summary of overlapping subsets in the PDBbind database, including the core, refined, and general sets. (B) Number of the protein-ligand complexes in the PDBbind database from 2002 to 2020. (C) Distribution of the protein-ligand binding affinity values is presented as a pie chart. (D) Distribution of 367 human kinases in the DAVID database on the human kinome tree, with red dots representing each kinase. (E) Distribution of protein-ligand binding affinity values presented as a pie chart. (F) Distribution of 216 human kinases from the KIBA database on the human kinome tree, represented by red dots for each kinase. (G) Distribution of protein-ligand binding affinity values in the KIBA database, represented by a pie chart. Adapted and reprinted from.119 |
The crucial steps involved in predicting protein–ligand binding affinity using deep learning (DL) models are presented in Figure 7. A sufficient number of training samples are required for protein–ligand binding affinity databases. The inputs to the DL models can be provided by the features extracted from the training samples, whereas the selection of the DL model is vital for the accurate prediction of protein–ligand binding affinity.119
 |
Figure 7 Predicted interactions between proteins and ligands based on DL models. The figure illustrates the critical stages involved in binding affinity prediction, including the use of sufficiently large and diverse protein-ligand datasets, feature extraction from training samples as model inputs, and the selection of appropriate DL architectures for accurate affinity estimation. Adapted and reprinted from.119 |
The input features obtained from the protein–ligand interaction model played a crucial role in categorizing the model into interaction-free and interaction-based models, as presented in Figure 8A. Generalized models depend on the extracted information about the interaction-based models from the protein–ligand interactions in the 3D structures of the protein–ligand complexes. In the case of interaction-free models, the prediction of protein–ligand affinity can be performed without considering the information from the protein–ligand interactions. The ligand SMILES strings and protein sequences, along with the ligand protein monomer structures, were used as inputs for interaction-free models (Figure 8B). The extraction of ligand and protein features from the ligand SMILES strings and protein sequences was performed using DeepDTA, which involves two CNN blocks, similar to that described above.113
 |
Figure 8 Conceptual workflows of deep learning models for protein–ligand binding affinity prediction. (A) Interaction-driven DL model conceptual workflow. (B) Interaction-free DL model conceptual workflow, in which the protein-ligand binding affinity is predicted without protein-ligand interaction information in the structure-free models. Ligand SMILES strings and protein sequences, along with ligand protein monomer structures, were used as inputs for the interaction-free models. Adapted and reprinted from.119 |
One of the most preferred input representations for structure-based models involves 3D voxel grids of pocket ligand pairs obtained from CNN models. It also includes the interaction graphs of the protein-ligand pair of the GNN. The physicochemical properties of atoms or residues are influenced by interaction graphs and 3D voxel grids. One of the challenges related to the input representation of structure-based models is the variation in the binding affinity of the protein-ligand pair in different cells because of variations in the cellular environment and protein substrates for the selected target protein, as presented in Figures 9A and 8B. As shown in Figure 9A, approximately 16 of the 68 ligands from the Davis database, especially at the fourth, sixth, and 15th positions, bind to CDK4-CyclinD1 along with CDK4-Cyclin D3 complexes through different binding affinities. The β-sheets from the catalytic domain play a vital role in the different structures of CDK4, as shown in Figure 9B. Figure 9C–H show the interactions of the three ligands targeting CDK4-CyclinD1 with the CDK4-CyclinD3 complex. The results showed stronger interactions of the fourth and 15th ligands with CDK4-CyclinD3 than with CDK4-CyclinD1. Cellular environmental information and substrate binding to target proteins are missing for the inputs used in this study. Changes in atomic positions affect the binding of ligands to proteins; hence, dynamic structural information is vital for predicting protein-ligand binding affinity.
 |
Figure 9 Structural and interaction analysis of ligand binding to CDK4-Cyclin complexes. (A) Binding affinity values for 16 ligands from the Davis database targeting CDK4-CyclinD1 along with the CDK4-CyclinD3 complexes. (B) Structure of the CDK4-CyclinD1 and CDK4-CyclinD3 complexes. (C–H) includes the diagrams of 2D protein-ligand interaction for the 4th, 6th, and 15th ligands targeting the CDK4-CyclinD1 and CDK4-CyclinD3 complexes. The red color indicates the interactions among the hydrophobic residues of the protein and the hydrophobic interactions between the residues and ligands. The green color indicates the hydrogen bonds between the ligands and residues, while the yellow color indicates the hydrogen bond residues. Adapted and reprinted from.119 |
ML for Repurposing Existing Drugs for Nano Medicine
Drug repurposing involves the identification of new therapeutic agents against existing drugs. This approach can significantly reduce the time and cost of drug development because the safety profiles of these drugs are well established. ML techniques are particularly useful for identifying potential repurposing opportunities by analyzing large-scale biological and clinical data.120 In this case study, we examined the application of ML for repurifying existing drugs for new indications. The process involves the following steps: (i) data integration, which involves combining data from various sources, including drug–target interactions, gene expression profiles, disease-associated genes, and clinical trial outcomes;91,119 (ii) feature engineering, which involves extracting relevant features from the integrated data, including molecular fingerprints of drugs, biological pathways, and phenotypic effects; (iii) model building, which involves developing ML models, such as random forests, support vector machines (SVMs), or deep neural networks (DNNs), to predict the likelihood of a drug being effective for a new indication, trained on known drug–disease associations, and validated on held-out data;89,91 and (iv) validation, which involves experimentally testing the top predictions to confirm their efficacy in new disease contexts, including in vitro assays, animal models, and clinical trials.
ML has successfully identified several repurposed candidate drugs. For example, a study using ML models identified the antimalarial drug hydroxychloroquine as a potential treatment for COVID-19, leading to subsequent clinical trials.112,113 Recently, Exscientia and Healx have started using AI for drug repurposing in oncology and other therapeutic areas, including rare diseases.53,54 Although not all predictions lead to successful therapies, ML-driven drug repurposing is a valuable tool for rapidly identifying new treatment options, particularly in response to emerging health crises. In another study, Rodriguez et al investigated the application of ML for the identification of candidates for drug repurposing in Alzheimer’s disease (AD). In this study, an ML framework called Drug Repurposing in Alzheimer ‘sDisease (DRAID) was developed with respect to the pathological aspects of AD severity at the Braak stage and its molecular mechanisms. The developed framework was applied to a list of genes involving FDA-approved and clinically tested drugs, which were inspected for common trends among their respective targets. The association between the gene lists and the disease was identified using ML with the help of mRNA expression profiles from the human brain at different levels of AD progression. Human brain gene expression levels were obtained from official datasets, such as The Mount Sinai/JJ Peters VA Medical Center Brain Bank (MSBB) and The Mayo Clinic Brain Bank (MAYO), as shown in Figure 10B. Braak staging was based on the neuropathological assessment of the accumulation of neurofibrillary tangles in three different stages. The training and evaluation of the predictor can be performed using DRAID to recognize disease categories from mRNA expression levels, as shown in Figure 10A. Background distributions of random gene lists were created with the same length for the evaluation of DGL. The performance of the predictors trained on the gene lists reported in previous studies of AMP-AD datasets is shown in Figure 10C.
 |
Figure 10 ML framework for drug repurposing in Alzheimer’s disease (AD). (A) Overview of the ML framework required for the potential association of the gene lists along with Alzheimer’s disease. Input gene lists derived from experimental data or extracted from database resources in the literature were used in the framework. The framework subsamples a specific gene of interest from the gene expression matrix. Cross-validation was used to train and evaluate the Braak disease stage. (B) The ML framework used AMP-AD datasets, and the schematic highlights the brain regions represented in each dataset of the Mount Sinai/JJ Peters VA Medical Center Brain Bank (MSBB) and Mayo Clinic Brain Bank (MAYO). (C) The performance reported with the respective area under the curve was used to distinguish between disease stages using the predictors. The performance of the predictor related to the gene list presented in the literature is denoted by a vertical line. Adapted and reprinted from.56 |
A significant research gap in liver cancer management is the limited integration of artificial intelligence (AI) for early diagnosis, prognosis, and personalized therapy optimization. Conventional clinical approaches often fail owing to reliance on imaging interpretation variability, late-stage detection, and lack of predictive precision. Grapă et al addressed these limitations by highlighting the role of AI-driven models, including ML and DL, in analyzing imaging, genomics, and clinical data. These approaches improved early detection accuracy, risk stratification, and treatment planning. The outcomes demonstrated enhanced diagnostic precision, improved clinical decision-making, and the potential for personalized medicine, transforming liver cancer management through AI integration.121
Target Identification Through Analysis of Biological Networks for Nano Medicine
Target identification is a critical step in drug discovery that involves identifying molecular targets (eg, proteins and genes) associated with a disease. Analyzing biological networks, such as protein-protein interaction networks and gene regulatory networks, provides insights into the complex interactions within cells and helps identify key nodes that can be targeted by drugs.122 In this case study, we explored how the analysis of biological networks can aid in target identification. The process involved the following steps: (i) network construction: building biological networks from various data sources, such as protein-protein interactions, gene expression data, and signalling pathways, using tools such as STRING and Cytoscape, which are commonly used for network construction and visualization;113,114 (ii) network analysis: applying graph-theoretic algorithms to analyze the networks to identify central nodes (eg, hubs, bottlenecks) that play critical roles in network stability and function (centrality analysis), detect modules or clusters of nodes that are densely connected and may represent functional units (community detection), and identify pathways that are significantly enriched with disease-associated genes or proteins (pathway enrichment); (iii) target prioritization: prioritizing potential targets based on their network properties and biological relevance with high centrality nodes, for instance, may represent critical regulators of disease pathways; and (iv) experimental validation: testing the top-ranked targets in experimental assays to confirm their role in the disease and their potential as drug targets.
Network-based target identification has led to the discovery of several novel drug targets. Celgene (now part of Bristol Myers Squibb), Genentech (Roche), Insilico Medicine, Novartis and Schrödinger practice biological network analysis for target identification, gene expression, and omics data to discover novel drug targets, particularly cancer, autoimmune disorders and aging.113 For instance, network analysis in cancer research has identified the protein BRD4 as a key regulator of oncogenic transcriptional programs, leading to the development of BRD4 inhibitors for cancer therapy.93 This approach provides a systematic framework for uncovering new targets and understanding the molecular mechanisms underlying diseases, ultimately contributing to the development of more effective and targeted therapies.122
Popov et al adopted a DL-based approach to identify potential binding sites for viral drug targets. The DrubBank database was used to analyze draggability for the detection of the binding site with the help of molecular docking to address the issue of selection of antiviral drug targets. This study proposed a structural and DL approach for identifying vulnerable regions of viral proteins with respect to drug-binding sites. The mutability and protein dynamics of the binding site, along with the putative mechanism of drug action, were investigated in this study. The validation of the developed system was done by using severe acute respiratory syndrome coronavirus spike glycoprotein S. The study showed conformation along with the oligomer-specific binding site near the receptor-binding domain which further consists of vital amino acid residues. The complexation of the drug candidate molecules, along with the molecular dynamics simulations of the spike protein, indicated an equilibrium shift towards the inactive conformation compared to that of the drug-free simulations. Inhibition of the interaction between the human angiotensin-converting enzyme 2 receptor and the conformational transition of the spike protein was achieved through small-molecule targeting of the binding site. Drug-like molecules with high docking scores that could form interactions were selected and screened using different factors, such as partition coefficient, number of hydrogen bond donors, topological polar surface areas, and number of rotatable bonds, as presented in Figure 11A and B. Many polar contacts had high scores in molecular docking studies, as shown in Figure 11B. The highly similar compound nature and potential artifacts were used to select 20 drug-like molecules, as shown in Figure 11C. Figure 11C shows the superimposed docking poses of these compounds in the context of the BiteNet-predicted binding site.123
 |
Figure 11 Insights from molecular docking studies for identifying drug-like molecules. (A) Distribution pattern of the molecular properties of top-scoring compounds, including the topological polar surface area, molecular weight, number of potential polar contacts, predicted octanol-water partition coefficient, and number of hydrogen bond donors, and number of rotatable bonds. (B) It includes examples of five different docked compounds with three-dimensional conformers and polar interactions contributing to high docking scores. (C) Superimposed docking poses for selected compounds are shown, with the BiteNet-predicted binding site represented by a mesh. Adapted and reprinted from.123 |
Figure 12 illustrates the structure-driven topological importance and sequence-based conservation of a vulnerable binding site in the coronavirus spike protein. Briefly, the topological importance of amino acid residues is vital for the mutational tolerance of viral proteins to predict vulnerable epitopes. The topological importance of the spike trimers was studied using structure-based network analysis. The receptor-binding domain-related regions were compared with those of common drug targets to detect the binding site (Figure 12A-D). The detected binding site had lower mutation tolerance than that of the receptor-binding domain. Valdar conservation scores were calculated, and the detected binding site was more conserved than the coronavirus compared to the receptor-binding domain. The results demonstrated that the identified binding site corresponded to a vulnerable region of the coronavirus spike structure.
 |
Figure 12 Structure-driven topological importance and sequence-based conservation of a vulnerable binding site in the coronavirus spike protein. (A and B) Spike trimer structure colored in the context of structure-driven topological importance, along with the Valdar conservation score of the spike amino acid residues. (C and D) Box plots of the structure-driven topological importance along with Valdar’s conservation score which was calculated based on the exposed amino acid residues of the spike, receptor-binding domain, and the identified binding site, highlighting the lower mutation tolerance and higher conservation of the predicted vulnerable region. Adapted and reprinted from.123 |
ML-enabled virtual screening was implemented to evaluate antituberculosis activity using molecular dynamics simulations. Zheng et al studied the application of ML with virtual screening for the evaluation of antituberculosis activity of aldoxorubicin and quarfloxin to address drug resistance issues of Mycobacterium tuberculosis on a global platform. In this study, a virtual screening workflow was developed using multiple ML and DL models. The DrugBank database was used, and approximately 11576 compounds were extracted and screened for Mycobacterium tuberculosis (Mtb) activity. Three data-splitting settings were used to predict antituberculosis bioactive compounds. Preliminary screening revealed that aldoxorubicin and quarfloxin potently inhibited the Mycobacterium tuberculosis strain H37Rv. The screened compounds also exhibited antibacterial activity against multidrug-resistant tuberculosis (MDR-TB) isolates. The direct binding of selected compounds to Mycobacterium tuberculosis DNA gyrase was validated using molecular docking, molecular dynamics simulations, and surface plasmon resonance experiments. The three-dimensional structure of DNA gyrase and the binding poses and interaction profiles of the two selected antibacterial agents with DNA gyrase are shown in Figure 13A and C. The binding poses showed that quarfloxacin and aldoxorubicin docked into the same hydrophobic binding pocket as evybactin (Figure 13A and B). The residues PRO353 (A) and ARG354 (A) formed hydrogen bonds with aldoxorubicin, whereas the residue PRO353 (A) formed hydrogen bonds with quarfloxin. The results showed that quarfloxin and aldoxorubicin may bind to the same allosteric sites targeted by thiophenes in Mycobacterium tuberculosis DNA gyrase, thus ensuring their antituberculosis activity.124
 |
Figure 13 Protein–ligand binding modes of aldoxorubicin, quarfloxin, and the reference ligand evybactin with Mycobacterium tuberculosis DNA gyrase. (A) Complexes of Mtb DNA and ligands in the form of a protein-ligand complex. (B) Binding modes of Mtb DNA gyrase along with the three ligands with that of the protein hydrophobic surface along with the binding residues and protein ligand complexes. (C) Ligand-focused binding mode profiles of the three ligands (aldoxorubicin, quarfloxin, and evybactin). Adapted and reprinted from.124 |
Recent Innovations in ML for Nano-Enabled Drug Discovery and Targeting
MISATO is a comprehensive dataset developed by Siebenmorgen et al with the goal of revolutionizing structure-based drug discovery (DD) by combining molecular dynamics (MD) and quantum mechanical (QM) data for approximately 20,000 experimental protein–ligand complexes. In addition to containing more than 170 μs of MD simulations in explicit water that captured binding affinities and conformational landscapes, this dataset includes quantum-refined ligand structures that eliminate crystallographic errors. The study demonstrated how MISATO could improve the precision of ML models in DD by validating AI baseline models, MD trajectories, and QM computations. Additionally, the study offers a user-friendly Python interface for preprocessing and template notebooks, making it easy for researchers to use. MISATO is a potent tool for next-generation AI models, combining quantum and Newtonian insights to reveal hidden factors in protein–ligand interactions, despite drawbacks such as QM calculations limited to ligands and the requirement for longer MD timeframes. By addressing important DD difficulties with unprecedented accuracy and efficiency, our study lays the groundwork for creative AI applications. Training models on datasets is essential for ensuring consistency and accuracy. Several challenges are associated with the PDBbind database, depending on their quantum chemical protocol, including the total number of available structures being lower than the training sizes of the AI targets, as well as the complex ligand association involved in molecular recognition, as shown in Figure 14.125
 |
Figure 14 Impact of an optimized quantum-mechanical protocol on structural consistency and electronic properties in protein-ligand datasets. (A) Overview of the statistical changes induced by the developed optimized protocol. (B) Comparison of the partial charge with D4 polarizability for all halogens in the database. The wrong atom assignment was analyzed with the help of the outliers. (C) Representative examples of inconsistent structures involving 1WUG with elongated NO bonds. The 4MDN included nitrogen in the angular violation compared with that of the VSEPR expectations. The protonation state problem is represented by 5GTR. (D and E) Calculated electronic densities of ketamine and tramadol. The electronic density is represented by a dashed circle. (F and G) Deviations among the amino acids with the respective adaptability of the residues to the protein pocket. The blue color indicates the targets, whereas the red color indicates the AI model prediction. The developed model identified small sphere right residues along with amino acids with high flexibility. Reproduced with permission from reference.125 Copyright 2024, Nature. |
Madushanka et al presented the QM40 dataset and studied the urgent problem of a lack of high-quality datasets for ML and DL applications in quantum mechanical (QM) predictions. This collection includes 162,954 compounds with 10–40 atoms, including common drug-related components such as C, O, N, S, F, and Cl. It covers 88% of the chemical space of FDA-approved drugs. The B3LYP/6–31G (2df,p) level of theory in Gaussian16 was used for the calculations, guaranteeing compatibility with well-known datasets such as QM9 and Alchemy, and enabling the concatenation of datasets. Numerous resources are available in QM40, such as 16 QM parameters, Cartesian coordinates, Mulliken charges, and bond-specific information, such as force constants for local vibrational modes. Owing to these characteristics, QM40 is a valuable standard for assessing ML and DL techniques in QM predictions. This dataset fills important gaps in the incorporation of artificial intelligence into quantum chemistry by allowing researchers to develop predictive models and guarantee their alignment with current frameworks.126
To overcome the difficulties in the long-term administration of eye medications, especially for chronic diseases such as glaucoma, Hsueh et al devised a novel approach. While polymer-based implants carry risks such as corneal injury, traditional treatments such as eye drops often suffer from poor patient adherence. As shown in Figure 15, multifunctional peptides for ocular nanodrug delivery were evaluated using a peptide microarray. Scientists have used super-learning-based ML techniques to create multifunctional peptides that circumvent these restrictions. Peptides with optimal melanin-binding, cell-penetrating, and minimal cytotoxic properties were identified using this technology. HR97, the main peptide, was coupled with brimonidine, a medication that is usually administered several times daily. The HR97-brimonidine compound considerably outperformed free brimonidine in rabbit models, reducing intraocular pressure (IOP) for up to 18 days with a single intracameral injection. This demonstrates the potential of computational peptide design for the development of effective implant-free drug delivery methods. Notwithstanding the drawbacks, including poor conjugation yields and the requirement for additional drug release rate adjustment, this work shows promise as a platform for long-term eye treatment and other biomedical uses.127
 |
Figure 15 Peptide microarray based evaluation of multifunctional peptides for ocular nano medicine and drug delivery. (A and B) Illustration of the first peptide microarray involving the anchoring of peptides to the microarray. Binding event characterization was performed using melanin nanoparticles along with surface biotinylation, and the quantification of melanin binding with the peptide was detected using DyLight 680-conjugated streptavidin. (C) In vitro melanin-binding assay using melanin nanoparticles and biotin quantification kit. (D) In vitro relationship between predicted melanin binding and experimentally measured binding, with melanin binding predictions on the x-axis and experimental binding values on the y-axis. (E and F) Comparison of melanin binding and cell penetration into melanin-induced human adult retinal pigment epithelial cells. The predicted non-cell-penetrating peptides are represented by blue triangles, while the predicted cell-penetrating peptides are represented by magenta dots. (G) Summary of the intracellular concentrations of cell-penetrating and non-cell-penetrating peptides. Reproduced with permission from.127 Copyright 2023, Nature. |
RosettaVS, a sophisticated structure-based virtual screening tool developed by Zhou et al, performed better than other state-of-the-art methods in predicting docking poses and binding affinities for drug development. Their method, which combines AI acceleration with receptor flexibility, was used to screen multi-billion chemical libraries against two unrelated targets: the voltage-gated sodium channel NaV1.7 and ubiquitin ligase KLHDC2. In less than seven days, the screening procedure identified four hit compounds for NaV1.7 and seven hit compounds for KLHDC2, all of which had single-digit micromolar binding affinities. X-ray crystallographic validation confirmed the accuracy of the expected binding postures of KLHDC2. This study highlights the strong performance of RosettaVS across various protein pockets, including polar and hydrophobic areas, proving its effectiveness in lead discovery. To further increase the precision and effectiveness of virtual screening campaigns, the authors propose that future developments should use deep learning models, GPU acceleration, and improved active learning strategies. A schematic representation of the DL-guided virtual screening protocol and experimental findings is shown in Figure 16.128
 |
Figure 16 Performance and application of the RosettaVS DL-guided virtual screening platform. (A) Schematic representation of DL-supervised structure-based virtual screening protocol, integrating receptor flexibility and AI acceleration. (B and C) Illustration of C29 binding to ubiquitin ligase KLHDC2. (D) Comparative study of the computationally predicted binding pose and the experimentally resolved binding pose of C29. The yellow color indicates the high-resolution X-ray crystal structure which was superimposed on that of the predicted docked pose in magenta. (E) Inhibition of the inactivated state of Nav1.7 by compounds Z8739902234 and Z8739905023. (F) Docking structure of Z8739902234 and Z8739905023 within the NaV1.7 binding site. Reproduced with permission from.128 Copyright 2024, Nature. |
Similar to the word2vec technique in natural language processing, Chen et al developed a unique ML method called functional representation of gene signatures (FRoGS) to improve bioinformatics analysis by incorporating gene function knowledge. FRoGS considerably outperformed conventional models based solely on gene IDs by embedding genes into vectors that reflect their biological functions and integrating information from gene ontology (GO) and empirical functions. When applied to the L1000 datasets from the Broad Institute, FRoGS enhanced compound–target predictions and provided a more comprehensive understanding of the underlying mechanisms of action. By incorporating more pharmacological data sources, the model performance was further enhanced, and more accurate predictions were produced, supported by experimental data. This study showed that superior transfer learning and application to a range of bioinformatics tasks, including therapeutic target identification and disease gene discovery, are made possible by embedding gene functions into vector representations. Furthermore, FRoGS is a useful tool for various biomedical applications because it can model gene functions using pre-existing knowledge, which enables a more effective analysis with smaller training datasets. A scheme of the neural network for the prediction of the compound binding probability to that of the target and other experimental findings is presented in Figure 17.129
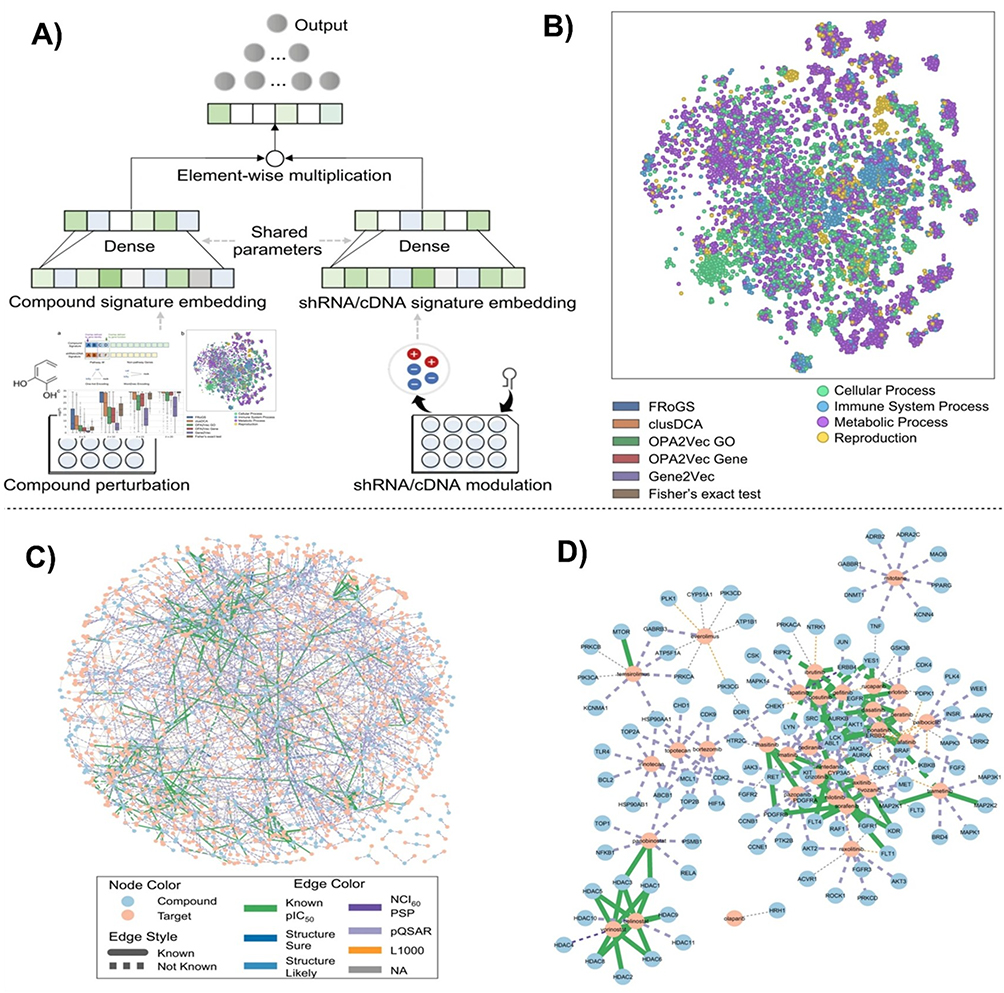 |
Figure 17 Functional representation of gene signatures (FRoGS) for gene function aware compound-target prediction. (A) Scheme of the neural network for predicting the compound binding probability to that of the target, namely, L1000 gene set signature embeddings. (B) Projection of t-distributed stochastic neighbor embedding (t-SNE) for learned gene embedding vectors. (C) Network simplification by retaining less than or equal to five best-scoring targets per compound to less than or equal to ten best-scoring compounds per target (≤5 targets per compound and ≤10 compounds per target) to enhance interpretability and prediction robustness. (D) Illustration of other antineoplastic agents (L01X), demonstrating the model’s capability to uncover pharmacologically relevant compound–target relationships. Reproduced with permission from.129 Copyright 2024, Nature. |
By incorporating biological pathways into neural network topologies, Hartman et al developed biologically informed neural networks (BINNs) to improve the interpretability of ML models used in proteomics. Using plasma proteomic data, this novel method was used to categorize COVID-19 and septic acute kidney injury (AKI) sub-phenotypes. In several situations, BINNs have outperformed traditional techniques by successfully identifying biomarkers and pathways pertinent to each disease, such as metabolic proteins in AKI and immune-related proteins in COVID-19. Researchers have been able to pinpoint the impact of particular proteins and pathways because of the sparse structure of BINNs, which allows introspection. The complementary nature of these techniques was highlighted when feature attribution revealed biomarkers that were missed by differential expression analysis. In the test sets, the BINNs achieved excellent accuracy and showed strong generalization to unseen data. With their adaptability to genomic and metabolomic datasets, this study demonstrated the promise of BINNs for biomarker discovery, pathway analysis, and sub-phenotype classification. This study provides a strong framework for combining proteomics and ML, creating opportunities for cutting-edge biomedical research. The scheme for data collection, analysis, and interpretation is shown in Figure 18.130
 |
Figure 18 Biologically informed neural networks (BINNs) for plasma proteome-based sub-phenotype classification and biomarker discovery. Scheme for data collection and analysis of the plasma proteome obtained from patients with COVID 19 and AKI. The collected data were re-analyzed and used to develop datasets for the selected disease conditions. BINN is generated for each dataset by subsetting the pathway database, such as Reactome, through the proteomic content of the dataset of interest, followed by layerization through a sequential neural network-like structure. Differentiation among the sub-phenotypes was performed using trained BINNs, whereas SHAP was used for network interpretation to provide pathway analysis and biomarker identification. Reproduced with permission from reference.130 Copyright 2023, Nature. |
To identify broadly neutralizing antibodies (bNAbs) against HIV-1 from immunological repertoires, Foglierini et al developed RAIN, a computational pipeline that combines machine learning and single-cell B cell receptor (BCR) sequencing data. RAIN uses specific sequence-based properties to differentiate bNAbs from non-bNAbs, in contrast to conventional techniques that depend on sequence alignment or one-hot encoding. Using structural analysis by cryo-electron microscopy and functional neutralization testing, this study confirmed RAIN’s predictions of RAIN and identified high-affinity bNAbs that target the HIV-1 envelope glycoprotein’s CD4-binding region. Unconventional mutations in antibody frameworks and conserved feature weights across antigenic locations are important discoveries that suggest possible immune escape pathways. RAIN sheds light on HIV-1 immune responses and escape pathways, while achieving exceptional precision, offering a creative and effective technique for accelerating bNAb development. This method has wide-ranging implications for immune repertoire analysis and antibody development. The Compile, Analyze, and Tally Nab Panels (CATNAP) database was used in this study. CATNAP was primarily used to analyze data related to HIV neutralizing antibodies to study the potential genetic signatures related to these antibody interactions with proteins. The study scheme and 3D construction of the Fab4251-SOSIP complex are presented in Figures 19 and 20, respectively.131
 |
Figure 19 RAIN computational pipeline for the identification of broadly neutralizing antibodies (bNAbs) against HIV-1. Schematic illustration of the data collection from the CATNAP database along with the healthy donor repertoires which were later converted for the training along with the validation of ML models such as anomaly detection, decision tree, random forest, and super learner, were trained and validated to distinguish bNAbs from non-bNAbs. The workflow was used for processing of BCR sequences while the predicted bNAbs were produced and tested in neutralization along with binding assays. Reproduced with permission from.131 Copyright 2024, Nature. |
 |
Figure 20 Structural characterization of the Fab4251–HIV-1 Env SOSIP complex. (A and B) 3D reconstruction of Fab4251-SOSIP complex and Fab4251-DS-SOSIP through nsEM. (C) Cryo-EM density map of the Fab4251-DS-SOSIP complex. (D) Atomic model of Fab4251-DS-SOSIP. (E) Footprint illustration of light- and heavy-chain binding surfaces on DS-SOSIP. (F) Binding of SOSIP and Fab4251 to the CD4 receptor. (G) Close-up view of VH H54 from Fab4251, along with F43 in CD4 receptor. (H) Comparative representation of VRC01 class antibodies on SOSIP, along with Fab4251, VRC01, PG04, and 3BNC60, illustrating shared and distinct epitope recognition. Reproduced with permission from.131 Copyright 2024, Nature. |
A groundbreaking assessment of ML models for forecasting the absorption, distribution, metabolism, and excretion (ADME) characteristics of targeted protein degraders (TPDs), such as molecular glues and hetero bifunctionals, was conducted by Peteani et al. According to the study, molecular glues produced the lowest prediction errors, such as 0.11 for CYP3A4 inhibition and 0.28 for mouse metabolic clearance, and ML-based quantitative structure-property relationship (QSPR) models achieved prediction accuracy for TPDs comparable to that of traditional modalities. Transfer learning techniques, such as fine-tuning multitask graph neural networks (MT-GNNs), greatly enhance prediction performance; however, hetero bifunctionals present more difficulties. Using a surrogate dataset of more than 270,000 chemical structures, the scientists confirmed that these models are applicable to TPD-centric initiatives and can be scaled up. This study demonstrated how ML, especially for new modalities such as TPDs, can accelerate the design-make-test-analyze (DMTA) cycle in drug discovery by providing accurate property predictions. Their results support the wider use of ML in pharmaceutical research to expedite the creation of TPDs with advantageous ADME profiles.132
HiDDEN, a computational technique developed by Goeva et al to improve case-control labels in single-cell RNA-seq investigations, enables the detection of minute perturbation effects that are frequently missed by conventional procedures. Through simulations, HiDDEN outperformed current techniques in precisely labelling cells by detecting modest disturbance signals and limited fractions of damaged cells. When used on human multiple myeloma precursor conditions, HiDDEN was able to detect malignancy in previously undiagnosed early-stage samples. Similarly, an endothelial subpopulation implicated in early blood-brain barrier failure was discovered in a rat model of demyelination. In heterogeneous datasets, this approach also demonstrated resilience to batch effects and fluctuating cell counts, making it easier to identify marker genes through differential expression testing. HiDDEN has potential for wider applications, such as multi-omics data and multi-stage disease progressions, although it requires cell-type-specific applications and further downstream analysis to handle heterogeneity perturbation responses. This method offers important insights into genomic and translational research and represents a major breakthrough in the detection of subtle transcriptional alterations. The developed scheme for the HiDDEN label refinement framework is shown in Figure 21.133
 |
Figure 21 HiDDEN framework for refining case–control labels in single-cell RNA-sequencing studies. (A) Setup of single-cell experiments for the case-control study. (B) Overview of standard clustering containing cells with mixed case-control sample-level labels. (C) Average log-normalized expression of perturbation markers. (D) Illustration of the HiDDEN label refinement framework involving a dimensionality reduction method for the analysis of gene expression profiles. (E) Scatterplot of adjusted p-values per gene computed through differential expression using case-control sample labels and HiDDEN-refined binary labels, highlighting improved detection of marker genes. (F and G) tSNE embeddings for the gene expression of Naïve B cells along with Memory B cells, following label refinement. (H) Representation of the dataset, including human plasma cells from healthy donors, patients with multiple myeloma, and two precursor disease states. Reproduced with permission from reference.133 Copyright 2024, Nature. |
One of the key research gaps in the field of liposomal nanomedicine is the lack of reproducible and scalable optimization strategies for clinically relevant formulations. Traditional microfluidic and trial-and-error-based methodologies are constrained by a lack of control over the parameters, low throughput, and inability to address complex formulation–process interactions. These limitations have been overcome by Buttitta et al, who combined machine learning with microfluidic systems to optimize liposome synthesis parameters in a data-driven manner. This strategy allowed the size, polydispersity, and encapsulation efficiency to be accurately controlled. The results showed improved formulation reproducibility, increased predictive power, and faster optimization, indicating the promise of ML-driven microfluidics to simplify the process of translational nanomedicine creation and clinical-scale production, as presented in Figure 22. The panels (A–F) in Figure 22 depict the design and therapeutic analysis of selenium nanoparticle-based nanoplatforms for targeted cancer therapy. Panel A shows the synthesis and functionalization of nanoparticles, and surface engineering resulted in better stability and tumor specificity than unmodified systems. Panel B exhibits cellular uptake and intracellular trafficking via receptor-mediated pathways, which facilitated effective drug delivery. Panel C showed the generation of ROS and redox imbalance with the result of mitochondrial dysfunction and apoptosis. Panels D and E show increased cytotoxicity and tumor growth inhibition using in vitro and in vivo experiments. Panel F shows immune modulation and regulation of signaling pathways, demonstrating that the nanoplatform had synergistic anticancer activity based on combined oxidative and molecular actions.134
 |
Figure 22 Nanoplatform of targeted anticancer therapy based on selenium nanoparticles. (A) Diagram of nanoparticle production and functionalization of surfaces. (B) Cellular uptake and intracellular trafficking pathways. (C) Mechanistic description of ROS production, mitochondrial dysfunction and induction of apoptosis. (D and E) In vitro and in vivo cytotoxicity/tumor inhibition. (F) Signaling pathways and immune response modulation that help to increase therapeutic efficacy.134 |
Nanomedicine translation has failed to precisely predict nanoparticle biodistribution and tumor delivery in biological systems. The simplified assumptions, absence of nanoparticle-specific parameters, and limited predictive capabilities in complex tumor environments are limitations of conventional physiologically based pharmacokinetic (PBPK) models. Chou et al have overcome these shortcomings by incorporating artificial intelligence in PBPK modeling, which facilitates the prediction of nanoparticle transportation, accumulation, and clearance using data as illustrated in Figure 23. In Figure 23, panels A–F illustrate an AI-aided PBPKQSAR composite model to predict nanoparticle biodistribution and tumor delivery. Panel A depicts the concept of nanomedicine modeling using databases, which confirmed the view that the integration of data enhanced predictive modeling relative to conventional empirical models. Panel B demonstrated AI–QSAR modeling with RF and DNN to predict tumor-related parameters, addressing the drawback of traditional models. Panel C provided evidence of PBPK organ-level distribution simulation. Panels D through F verified the performance of the model with a high correlation of the prediction and experimental results. Taken together, the results indicated that AI-enhanced PBPK modeling outperformed standard PBPK modeling in terms of prediction accuracy and the ability to rationally design nanomedicines to treat cancer with respect to their targeting. Collectively, the findings suggested that AI-enhanced PBPK modeling significantly improved prediction accuracy and facilitated the rational design of nanomedicines for targeted cancer therapy. The hybrid model enhanced the estimation of the parameters and nonlinear biological interactions. These results illustrated better predictive efficiency on tumor delivery efficiency, which justified rational nanocarrier design and facilitated the clinical translation of nanoparticle-based therapeutics.135
 |
Figure 23 PBPK-QSAR AI-based modelling of nanoparticle biodistribution. (A) Database-based framework between nanoparticle physicochemical characteristics and tumor targeting strategies. (B) Prediction of important tumor-related parameters with RF and DNN algorithms by the AI-QSAR model. (C) AI-based PBPK model that models tissue biodistribution and tumor delivery efficiency. (D–F) Correlation plots of the predicted versus experimental values (DE24, DE168, DEmax), which show the model accuracy and validation performance.135 |
Nanomedicine has been shown to be unable to efficiently predict the delivery of nanoparticles to tumors, which hinders its clinical translation. Classical physiological-based pharmacokinetic (PBPK) models are limited by simplistic assumptions, few specific descriptors of nanoparticles, and insufficient biological variability. These limitations decrease the predictive accuracy of tumor accumulation. This has been done by Imanparast et al who combined AI with PBPK modeling, allowing optimization of parameters by data and complex nonlinear interactions, as presented in Figure 24. Panels A–L in Figure 24 illustrate the design and validation of a microfluidic biochip system that can be used to control drug delivery and analyze biomolecular interactions. Figure 24A–D demonstrate chip fabrication and microchannel architecture, which contributed to the view that the microfluidic design facilitated the ability to accurately handle fluids in contrast to traditional static systems. Figure 24E–G demonstrate the generation of gradients and the reproducible distribution of the flow with the help of a peristaltic pump, which guaranteed reproducible experimental conditions. Figure 24H–J show flow simulations that ensured effective mixing and velocity control. Figure 24K and L demonstrated molecular docking interactions, validating drug–protein binding. The results showed that the microfluidic platform with its integrated approach enhanced the accuracy of drug screening and analysis of biomolecular interactions. The hybrid system increased nanoparticle biodistribution and tumor uptake prediction to a great extent. The results showed improved model accuracy and strength, which is a potent weapon in rational nanocarrier design and expedites the translation of nanomedicine.136
 |
Figure 24 Microfluidic biochip system of controlled drug delivery and interaction analysis. (A–D) Design and manufacture of microchip and bioreceptor channels. (E) Peristaltic pump experimental with controlled flow of fluids. (F and G) Creation of concentration gradients between microchannels. (H–J) Simulation of velocity distributions of various flow rates using computational fluid dynamics simulations. (K and L) Molecular docking experiments that demonstrate the interactions of drug molecules and target proteins in support of biochip applications in drug screening and biomolecular analysis.136 |
One of the key gaps in nanomedicine research is that high-spatial-resolution mapping and the prediction of intratumor nanoparticle distributions remain unachievable. Previous imaging and computer models have been limited by low resolution, failure to capture heterogeneity, and poor predictive ability generalization across tumor types. Tang et al have overcome these shortcomings by designing GANDA, a conditional deep generative adversarial network, that can predict nanoparticle distribution by pixel-to-pixel with the input of biological imaging as presented in Figure 25. The panels A–D in Figure 25 show a deep learning model of GAN to predict the distribution of quantum dots (QDs) in the tumor microenvironment. Panels A–B demonstrate patch-wise image decomposition and reconstruction, which allows the viewpoint that generative modeling enhanced the accuracy of spatial prediction in contrast to the traditional analysis of images. The model combined both DAPI and CD31 channels to produce synthetic QD distributions through loss optimization of adversarial and pixel-wise losses. Panel C emphasized its use in screening EPR enhancers, which can be used to implement better nanoparticle delivery approaches. Panel D showed patient stratification of nanomedicine suitability. The research suggested that predictive modeling based on AI increased the accuracy in nanomedicine design and tailored therapeutic decision-making. The model was able to incorporate microenvironmental aspects to produce realistic distribution maps. The results provided evidence of better spatial precision, increased ability to predict nanoparticle deposition profiles, and the possibility of stratifying patients, alleviating more specific and targeted nanomedicine design.137
 |
Figure 25 GAN-based model to predict the distribution of nanoparticles in tumor tissues. (A) Training process of image decomposition into patches and creation of QD distribution through adversarial learning. (B) Reconstruction of full tumor images from generated patches. (C) Application in screening EPR-enhancing strategies for improved nanoparticle delivery. (D) Predicted nanoparticle distribution-patient stratification, which can be used to identify candidates to be used in nanomedicine-based therapies.137 |
A summary of ML applications in drug discovery and targeting is presented in Table 4.
 |
Table 4 Summary of ML for Drug Discovery and Drug Targeting |
Table 5 presents an overview of the chosen clinical trials (identified by NCT numbers on ClinicalTrials.gov) that have demonstrated the application of AI-driven solutions to diagnostics, risk prediction, patient stratification, treatment optimization, and clinical decision support systems. The studies included interventional and observational designs and utilized a broad range of AI methodologies, such as machine learning (ML), deep learning (DL), predictive analytics, and algorithm validation systems.
 |
Table 5 Representative Clinical Case Studies Highlighting the Integration of Artificial Intelligence (AI) Across Diverse Healthcare Applications |
The column “AI Technique/Model” defines the main method of computation that was used, and the column “Application Area” defines the clinical use of the AI system. All these case studies demonstrate the growing use of AI in augmenting precision medicine, clinical outcomes, and data-driven healthcare decision-making. Despite the main emphasis on AI, a number of studies have also introduced nanomedicine with possible translational importance, especially in the fields of imaging, optimization of targeted therapy, and advanced therapeutic monitoring.
Ethical and Regulatory Considerations for Nano Medicine
Addressing Challenges Related to Data Privacy and Security for Nano Medicine
Appropriate measures must be taken to protect patient autonomy and privacy. Competing objectives may impact private data custodians; thus, they should be structurally supported to maintain data protection and prevent unauthorized use.186 Concerns about the external risks of privacy violations caused by AI-driven techniques are another set of issues.187,188 Emerging technologies that can efficiently reidentify such data may be undermined or rendered useless for de-identifying or hiding patient health information. The danger to patient data stored by private custodians can arise as a result of this. The European Commission has implemented legislation with standardized AI standards that outline an organizational accountability principle for privacy and data, which is similar to the European General Data Protection Regulation.189
Strong privacy protection is achievable when institutions are fundamentally incentivized to work together to secure data protection through their design. Although commercial healthcare AI systems can be controlled to preserve privacy, these systems have conflicting objectives.190 As we have seen, businesses might not be sufficiently motivated to uphold privacy protection if they profit from the data or use them in other ways, and if punishments for breaking the law are not severe enough to deter such behavior. The use of commercial AI consumes large amounts of data, which raises additional issues concerning the external threat of privacy violations caused by extremely complex algorithmic systems.191,192 Numerous countries, including the United States, Canada, and Europe, have experienced an increase in healthcare data breaches. Healthcare AI is currently in an exciting stage of development and adoption, and the health gains these technologies produce for the patients whose data they utilize should be large, if not enormous.193 However, significant privacy issues are associated with the use of commercial healthcare AI.
Ensuring the Transparency and Interpretability of ML Models for Nano Medicine
In ML projects, it is crucial to choose characteristics that are pertinent, significant, and easy to comprehend for the system and task. Features are characteristics or attributes that characterize information and affect the conclusions or predictions made by the models. The extent to which an individual can accurately predict a model’s outcome is known as its interpretability.194 The more interpretable an ML model, the simpler it is to understand why specific judgments or predictions are made using it. Interpretability and explanations are essential for promoting learning and satisfying the desire to understand why particular predictions or behaviors are produced by computers.
Regulatory Implications of Using AI in Drug Discovery for Nano Medicine
New regulatory problems, such as certification, personal use, data sharing management, privacy security, and responsibility, are caused by rapidly evolving medical AI scenarios. The Food and Drug Administration (FDA) in the United States declared that it had no plans to enforce the necessity of certain lower-risk device software functions, including symptom checkers. During the outbreak, the British Medicines and Healthcare Products Regulatory Agency authorized the fast-track approval of medical devices. According to a new assessment by the FDA and the International Medical Device Regulators Forum (IMDRF), AI technologies are distinct from conventional medical devices. The IMDRF and FDA have collaborated to create a new classification called “Software as a Medical Device” (SaMD),192 highlighting the need for an updated regulatory framework that considers the safety challenges that AI systems must address in complex environments; that is, learning periods that may cause major discrepancies in system performance.195,196 The current rule makes it challenging to obtain consent for data processing. The analysis of data from thousands of patients makes it challenging to deploy AI technologies for medical purposes because consent is required for the collection and use of personal data.
Challenges and Future Directions
The integration of ML and DL techniques into drug discovery and targeting has led to a remarkable period of invention and rapid advancement.47,197 From target identification and validation to predictive modeling and personalized medicine, these cutting-edge technologies have proven their ability to drastically accelerate and improve several aspects of pharmaceutical research. Despite significant accomplishments and encouraging results, the path to realizing the full potential of ML and DL in this sector is distinguished by various intricate hurdles and warrants further exploration and improvement.
The creation of novel pharmaceuticals that can successfully treat diseases is a difficult and resource-intensive process in drug discovery. The selection of appropriate pharmacological targets, design and screening of candidate compounds, and rigorous testing to ensure safety and efficacy are frequently performed in this regard. Traditionally, this procedure has depended significantly on expensive and time-consuming procedures. ML and DL have become formidable tools for analyzing large datasets, identifying intricate patterns, and creating predictive models that aid in the selection of new drug candidates and accelerate the discovery process. ML and DL have accelerated the process of finding novel medications, reducing the time and expense involved in bringing them to the market.197 Additionally, they have opened the door for personalized medicine, which involves the development of customized treatment programs for patients based on their genetic profiles and medical histories.195–198 These results demonstrate the considerable promise of ML and DL in revolutionizing the pharmaceutical sector. Despite their potential, the incorporation of ML and DL into drug development and targeting is not without difficulties. The availability and quality of data are among the greatest challenges. To train and produce precise predictions, ML and DL models primarily depend on large, high-quality datasets.197,199 However, obtaining such data is difficult in the pharmaceutical industry. Data-related problems, such as data bias, data shortages, and privacy concerns, frequently occur, highlighting the need for solutions to address these issues.
Data Availability and Quality
The availability and quality of data are major issues faced by academics and pharmaceutical companies in the rapidly changing field of drug discovery and targeting.197 In this section, we explore these issues and examine various strategies and advancements that have the potential to improve data accessibility and quality when applied to ML and DL applications in drug development and targeting. The ability of ML and DL approaches to glean insightful information from enormous datasets has been established; however, their usefulness is largely dependent on the data on which they are trained.200 The availability of extensive high-quality datasets is crucial for the successful application of ML and DL techniques in drug development.47 Data-related difficulties have several aspects. Data scarcity is a significant challenge, particularly when addressing specific disease regions or rare illnesses, where datasets may be limited in terms of size and scope. Second, data quality is a major concern.201 For ML and DL models to be reliable, biomedical data must be precise, error-free, and consistent, whether they are genomic, clinical, or molecular interaction data.199 Furthermore, the risk of data bias from skewed or underrepresented datasets is high and can result in biased model predictions and unfavorable results.198 Finally, to preserve sensitive patient information by changing data protection legislation, strong protections are required owing to ethical and data privacy concerns.202 Potential initiatives and steps to enhance data accessibility and quality in the context of ML are as follows: (i) data sharing initiatives: encouraging pharmaceutical companies and research institutions to share their data can significantly enhance the pool of available data for ML and DL applications; (ii) data augmentation: employing techniques such as data augmentation, transfer learning, and domain adaptation to make the most of the existing data and mitigate issues related to data scarcity; and (iii) QC standards: to improve data quality, QC standards and best practices for data collection, curation, and annotation should be established.
Interpretability and Transparency
The creation of exact predictive models is frequently recognized as a notable accomplishment in ML and DL applications for nanomedicine, drug discovery, and targeting. However, a significant problem arises when these models are expected to make choices that directly affect patients’ health and well-being.203 The interpretability and transparency of ML and DL models are central to this dilemma; they are two crucial characteristics necessary for establishing confidence, promoting collaboration, and ensuring the proper integration of these technologies into the pharmaceutical sector.75,76 Transparency refers to the model’s inner workings, which are transparent and easy to understand, including the aspects considered in the model. Challenges and future directions in this aspect include the following: (i) model explainability: developing methods for explaining the predictions of complex DL models, making it easier for researchers and regulators to understand why a particular decision was made; (ii) regulatory compliance: aligning ML and DL models with regulatory requirements, ensuring that they meet standards for transparency and interpretability; and (iii) ethical considerations: addressing ethical dilemmas related to the black-box nature of DL models, particularly in cases where models may influence clinical decisions.
Ethical and Regulatory Considerations
The integration of ML and DL in drug discovery raises several ethical and regulatory issues. It is essential to navigate these challenges to ensure the responsible and safe use of these technologies.204 Future directions include the following:
- Data privacy and security: Numerous countries and continents around the world, including the United States, Canada, and Europe, have faced an increase in healthcare data breaches,193 necessitating the strengthening of data privacy measures to protect sensitive patient information and confidential research data.205
- Bias and fairness: Developing techniques to detect and mitigate bias in ML and DL models to ensure fair and equitable outcomes.190 Data bias arises when certain populations or experimental conditions are underrepresented in datasets, leading to models that may not generalize well to diverse groups, as highlighted by Queshi et al for clinical algorithms.206
- Regulatory frameworks: Collaborating with regulatory bodies to establish guidelines and standards for validating ML and DL models for drug discovery and targeting.191 Privacy concerns also pose a significant challenge, with regulations such as the General Data Protection Regulation (GDPR) in Europe and the Health Insurance Portability and Accountability Act (HIPAA) in the United States aiming to protect sensitive medical information; however, issues related to data security and confidentiality persist.207
Collaborative Efforts Between ML and Domain Experts
The synergy between ML and domain experts in fields such as pharmacology and biology is critical for the success of ML and DL applications in drug discovery.103 Key challenges include (i) interdisciplinary collaboration, which involves encouraging collaborative efforts between ML and domain experts to bridge the gap between technological capabilities and pharmaceutical domain knowledge, and (ii) training and education, which involves establishing programs and initiatives to train domain experts in ML and vice versa, fostering a shared understanding of both fields.
Integration of Multimodal Data
In drug discovery, information is obtained from various sources, including genomic data, medical imaging, and scientific literature.206 The integration of multimodal data sources is vital for obtaining a comprehensive understanding of the subject. Challenges and future directions include the following: (i) data fusion techniques: developing advanced techniques for integrating data from different modalities, such as combining genomic data with medical imaging or textual information; (ii) cross-domain learning: exploring methods for transferring knowledge and insights across different data modalities to improve drug discovery outcomes; and (iii) semantic integration: developing methods to harmonize and standardize data from diverse sources, enabling more effective analysis and interpretation.
Addressing these challenges and charting future directions for the integration of ML and DL in drug discovery and targeting will not only advance research but also contribute to the development of safer and more effective drugs.109 By addressing these issues, the pharmaceutical industry can harness the full potential of these technologies while ensuring ethical and regulatory compliance in their use.
Challenges and Constraints in ML Enabled Research for Nano Medicine
The use of ML and DL methodologies in nanomedicine and drug development and targeting has spawned a new wave of innovation and revolutionized pharmaceutical research. As these technologies become increasingly involved in the search for novel drug candidates, it is critical to understand that, like any scientific enterprise, they have limitations and unexplored areas that require further research.203,208 To advance the profession, it is essential to recognize these constraints and identify areas for future research. In this section, we examine the current limitations that prevent ML and DL from realizing their full potential in drug discovery. These limitations address a variety of issues, such as technical challenges, ethical and legal concerns, and difficulty in comprehending the outcomes of complex ML models. Although these limitations pose challenges, they can serve as starting points for identifying significant research gaps. Finding solutions to these issues provides stimulating research opportunities that move the discipline closer to a more efficient, open, and ethically upright future.209 Several limitations and further research opportunities should be considered when using ML and DL for drug discovery (Figure 26).
 |
Figure 26 Overview of existing constraints and challenges limiting the application of ML and DL in nano medicine and drug discovery. (A) Data fusion and interpretability, (B) regulatory compliance, (C) ethical concerns and algorithmic bias, and (D) data quality and availability (created using BioRender). |
The key constraints of ML and DL models in nanotechnology and drug discovery are as follows:
- Data quality and availability: The creation and effectiveness of ML and DL models may be hampered by a lack of high-quality, complete datasets.210
- Ethical concerns: Working with sensitive medical and genomic data presents substantial ethical problems in terms of protecting patient privacy and data security.73
- Interpretability: The reasoning behind the predictions made by complex deep learning models may be challenging to comprehend because they frequently lack interpretability.211
- Fairness and bias: ML and DL algorithms may unintentionally reinforce biases found in the training data, producing unfair outcomes.8
- Regulatory compliance: Collaboration with regulatory agencies may be necessary because existing regulatory frameworks may not completely consider the intricacies of AI technology in drug research.9
- Data fusion: Challenges in data fusion, cross-domain learning212 and semantic integration arise when data from several sources, such as genomes, medical imaging, and scientific literature, are combined.4
Conclusion
The combination of machine learning and deep learning with nanomedicine is transforming drug discovery by enabling the development of therapeutics focused on data and precision. In addition to increasing the speed of traditional processes, AI-based solutions are enabling the rational design and optimization of nanocarrier systems, enhancing drug bioavailability, targeting efficiency, and safety profiles. ML-guided nanoformulations have shown great potential to improve delivery efficiency and therapeutic outcomes, highlighting their importance in promoting precision nanomedicine. Moreover, the capacity of AI models to deconstruct intricate nano–bio phenomena, such as protein corona formation and biodistribution profiles, offers a crucial basis for the creation of next-generation nanotherapeutics. Although these advances have been made, there is limited clinical translation. There is still a major disparity between preclinical achievements and human validation, especially because standardized and large-scale clinical trials of AI-optimized nanotherapeutics are not available. To overcome this, AI–nanomedicine-combined clinical trials, including adaptive designs, biomarker-based stratification, and real-time data analytics, are urgently needed. These experiments will be necessary to confirm predictive models, assess safety and effectiveness in heterogeneous patient groups, and build trust in AI-enabled nanomedicine systems. In the future, explainable AI convergences with multi-omics integration and standardized nanomedicine databases have the potential to further promote model reliability and clinical applicability. Most importantly, although AI-guided nanomedicine has colossal potential to enhance precision therapy, its effective translation will require closing the gap between computational innovation and clinical validation by providing specific AI–nano clinical trial systems. Although ML and DL have made massive contributions to drug discovery and nanomedicine design, their use is limited by the heterogeneity of the data, their low interpretability, and lack of clinical validation. Most of the reported successes rely on preclinical or retrospective research, and little real-life translation. Issues related to nano–bio interactions, reproducibility, and regulatory acceptance continue to exist. Thus, in addition to the development of technologies, more efforts should be devoted to critical assessment, standardized data, and properly structured AI–nanomedicine clinical trials to guarantee reliable, safe, and clinically significant results.
Data Sharing Statement
All data are presented in this manuscript and no new data was generated in this review article.
Acknowledgments
The authors acknowledge their respective institutions for providing the necessary support for the completion of this review article.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
This study received no external funding.
Disclosure
The authors declare no conflict of interest.
References
1. Hughes J, Rees S, Kalindjian S, Philpott K. Principles of early drug discovery. Br J Pharmacol. 2011;162(6):1239–62. doi:10.1111/j.1476-5381.2010.01127.x
2. Leeson PD, Springthorpe B. The influence of drug-like concepts on decision-making in medicinal chemistry. Nat Rev Drug Discov. 2007;6(11):881–890. doi:10.1038/nrd2445
3. DiMasi JA, Hansen RW, Grabowski HG. The price of innovation: new estimates of drug development costs. J Health Econ. 2003;22(2):151–185. doi:10.1016/S0167-6296(02)00126-1
4. Donadon MF, Martin-Santos R, Osório FDL. The associations between oxytocin and trauma in humans: a systematic review. Front Pharmacol. 2018;9. doi:10.3389/fphar.2018.00154
5. Zhang B, Wang J, Wang X, et al. Proteogenomic characterization of human colon and rectal cancer. Nature. 2014;513(7518):382–387. doi:10.1038/nature13438
6. Fang Y, Pan X, Shen H-B, Valencia A. De novo drug design by iterative multiobjective deep reinforcement learning with graph-based molecular quality assessment. Bioinformatics. 2023;39(4). doi:10.1093/bioinformatics/btad157
7. Bzdok D, Meyer-Lindenberg A. Machine learning for precision psychiatry: opportunities and challenges. Biol Psychiatry Cogn Neurosci Neuroimaging. 2018;3(3):223–230. doi:10.1016/j.bpsc.2017.11.007
8. Mehrabi N, Morstatter F, Saxena N, Lerman K, Galstyan A. A survey on bias and fairness in machine learning. ACM Comput Surv. 2022;54(6):1–35. doi:10.1145/3457607
9. Saria S, Butte A, Sheikh A. Better medicine through machine learning: what’s real, and what’s artificial? PLOS Med. 2018;15(12):e1002721. doi:10.1371/journal.pmed.1002721
10. Mittelstadt B. Principles alone cannot guarantee ethical AI. Nat Mach Intell. 2019;1(11):501–507. doi:10.1038/s42256-019-0114-4
11. Guan C, Mendes BB, Conniot J, et al. Accelerating discoveries in cancer nanomedicine using AI. Cell Biomater. 2025;1(11):100279. doi:10.1016/j.celbio.2025.100279
12. Morla-Folch J, Ranzenigo A, Fayad ZA, Teunissen AJP. Nanotherapeutic heterogeneity: sources, effects, and solutions. Small. 2024;20(17). doi:10.1002/smll.202307502
13. Joseph T, Kar Mahapatra D, Esmaeili A, et al. Nanoparticles: taking a unique position in medicine. Nanomaterials. 2023;13(3):574. doi:10.3390/nano13030574
14. Subramanian NA, Palaniappan A. NanoTox: development of a parsimonious in silico model for toxicity assessment of metal-oxide nanoparticles using physicochemical features. ACS Omega. 2021;6(17):11729–11739. doi:10.1021/acsomega.1c01076
15. Kumar R, Kumar A, Bhardwaj S, et al. Nanotoxicity unveiled: evaluating exposure risks and assessing the impact of nanoparticles on human health. J Trace Elem Miner. 2025;13:100252. doi:10.1016/j.jtemin.2025.100252
16. Ivanova ML, Nicholls M, Russo N, Mihaylov G, Nikolic K. Toward predictable nanomedicine: current forecasting frameworks for nanoparticle–biology interactions. Adv Intell Discov. 2026. doi:10.1002/aidi.202500205
17. Mohr AE, Ortega-Santos CP, Whisner CM, Klein-Seetharaman J, Jasbi P. Navigating challenges and opportunities in multi-omics integration for personalized healthcare. Biomedicines. 2024;12(7):1496. doi:10.3390/biomedicines12071496
18. Hemme CL, Atoyan J, Cai A, Liu C. Challenges and opportunities in multi-omics data acquisition and analysis: toward integrative solutions. Biomolecules. 2026;16(2):271. doi:10.3390/biom16020271
19. Mahmoudi M, Landry MP, Moore A, Coreas R. The protein Corona from nanomedicine to environmental science. Nat Rev Mater. 2023;8(7):422–438. doi:10.1038/s41578-023-00552-2
20. Li T, Wang Y, Zhou D. Manipulation of protein Corona for nanomedicines. WIREs Nanomed Nanobiotechnol. 2024;16(4). doi:10.1002/wnan.1982
21. Li H, Wang Y, Tang Q, et al. The protein Corona and its effects on nanoparticle-based drug delivery systems. Acta Biomater. 2021;129:57–72. doi:10.1016/j.actbio.2021.05.019
22. Huzar J, Coreas R, Landry MP, Tikhomirov G. AI-based prediction of protein corona composition on DNA nanostructures. ACS Nano. 2025;19(4):4333–4345. doi:10.1021/acsnano.4c12259
23. Kara A, Ongoren B, Anaya BJ, Lalatsa A, Serrano DR. Continuous manufacturing of nanomedicines using 3D-printed microfluidic devices. Appl Mater Today. 2025;43:102672. doi:10.1016/j.apmt.2025.102672
24. VandenBerg MA, Dong X, Smith WC, et al. Learning from the future: towards continuous manufacturing of nanomaterials. AAPS Open. 2025;11(1):7. doi:10.1186/s41120-025-00111-9
25. Halwani AA. Development of pharmaceutical nanomedicines: from the bench to the market. Pharmaceutics. 2022;14(1):106. doi:10.3390/pharmaceutics14010106
26. Đorđević S, Gonzalez MM, Conejos-Sánchez I, et al. Current hurdles to the translation of nanomedicines from bench to the clinic. Drug Deliv Transl Res. 2022;12(3):500–525. doi:10.1007/s13346-021-01024-2
27. Barua S, Balaji B, Balaji S. AI/ML-based computational models for toxicity prediction. Environ Sci Pollut Res. 2026. doi:10.1007/s11356-025-37354-8
28. Zhang R, Wen H, Lin Z, Li B, Zhou X. Artificial intelligence-driven drug toxicity prediction: advances, challenges, and future directions. Toxics. 2025;13(7):525. doi:10.3390/toxics13070525
29. Younas A, Wang S, Asad M, et al. Recent advances in cancer nanomedicine: from smart targeting to personalized therapeutics - pioneering a new era in precision oncology. Mater Today Bio. 2026;36:102660. doi:10.1016/j.mtbio.2025.102660
30. Zhao L, Liu X, Deng X. AI-engineered multifunctional nanoplatforms: synergistically bridging precision diagnosis and intelligent therapy in next-generation oncology. J Nanobiotechnology. 2025;24(1):73. doi:10.1186/s12951-025-03947-1
31. Bergström CAS, Strafford M, Lazorova L, Avdeef A, Luthman K, Artursson P. Absorption classification of oral drugs based on molecular surface properties. J Med Chem. 2003;46(4):558–570. doi:10.1021/jm020986i
32. Ren D, Wang C, Wei X, Zhang Y, Han S, Xu W. Harmonizing physical and deep learning modeling: a computationally efficient and interpretable approach for property prediction. Scr Mater. 2025;255:116350. doi:10.1016/j.scriptamat.2024.116350
33. Sheng Y, Wang J, Liu S, Jiang Y. IMN4NPD: an integrated molecular networking workflow for natural product dereplication. Anal Chem. 2024. doi:10.1021/acs.analchem.3c04746
34. Ashique S, Faiyazuddin M, Afzal O, et al. Advanced nanoparticles, the hallmark of targeted drug delivery for osteosarcoma-an updated review. J Drug Deliv Sci Technol. 2023;87:104753. doi:10.1016/j.jddst.2023.104753
35. Du X, Li Y, Xia Y-L, et al. Insights into protein–ligand interactions: mechanisms, models, and methods. Int J Mol Sci. 2016;17(2):144. doi:10.3390/ijms17020144
36. Hatvate NT, Khuspe PR, Mandhare TA, Kashid P, Gaikwad VD. Proteomics in oncology: retrospect and prospects. In: K S, editor. Novel Technologies in Biosystems, Biomedical & Drug Delivery. Singapore: Springer Nature Singapore; 2023:243–269.
37. Gholap AD, Uddin MJ, Faiyazuddin M, Omri A, Gowri S, Khalid M. Advances in artificial intelligence for drug delivery and development: a comprehensive review. Comput Biol Med. 2024;178:108702. doi:10.1016/j.compbiomed.2024.108702
38. Cai L, Gao J, Zhao D. A review of the application of deep learning in medical image classification and segmentation. Ann Transl Med. 2020;8(11):713. doi:10.21037/atm.2020.02.44
39. Dubois C, Eigen D, Delmas E, et al. Deep learning in medical image analysis: introduction to underlying principles and reviewer guide using diagnostic case studies in paediatrics. BMJ. 2024;387:e076703. doi:10.1136/bmj-2023-076703
40. Li M, Jiang Y, Zhang Y, Zhu H. Medical image analysis using deep learning algorithms. Front Public Health. 2023;11. doi:10.3389/fpubh.2023.1273253
41. Noorbakhsh J, Farahmand S, Foroughi Pour A, et al. Deep learning-based cross-classifications reveal conserved spatial behaviors within tumor histological images. Nat Commun. 2020;11(1):6367. doi:10.1038/s41467-020-20030-5
42. Cireşan DC, Giusti A, Gambardella LM, Schmidhuber J. Mitosis Detection in Breast Cancer Histology Images with Deep Neural Networks. Springer; 2013:411–418.
43. Boztepe C, Künkül A, Yüceer M. Application of artificial intelligence in modeling of the doxorubicin release behavior of pH and temperature responsive poly(NIPAAm-co-AAc)-PEG IPN hydrogel. J Drug Deliv Sci Technol. 2020;57:101603. doi:10.1016/j.jddst.2020.101603
44. He K, Zhang X, Ren S, Sun J. Deep residual learning for image recognition.
45. Nilius H, Tsouka S, Nagler M, Masoodi M. Machine learning applications in precision medicine: overcoming challenges and unlocking potential. TRAC-Trends Anal Chem. 2024;179:117872. doi:10.1016/j.trac.2024.117872
46. Cammarota G, Ianiro G, Ahern A, et al. Gut microbiome, big data and machine learning to promote precision medicine for cancer. Nat Rev Gastroenterol Hepatol. 2020;17(10):635–648. doi:10.1038/s41575-020-0327-3
47. Senior AW, Evans R, Jumper J, et al. Improved protein structure prediction using potentials from deep learning. Nature. 2020;577(7792):706–710. doi:10.1038/s41586-019-1923-7
48. Jumper J, Evans R, Pritzel A, et al. Highly accurate protein structure prediction with AlphaFold. Nature. 2021;596(7873):583–589. doi:10.1038/s41586-021-03819-2
49. Jin Q, Leaman R, Lu Z. PubMed and beyond: biomedical literature search in the age of artificial intelligence. EBioMedicine. 2024;100:104988. doi:10.1016/j.ebiom.2024.104988
50. Zheng S, Dharssi S, Wu M, Li J, Lu Z. Text mining for drug discovery. In: Bioinformatics and Drug Discovery. Springer; 2019:231–252.
51. Hao Y, Romano JD, Moore JH. Knowledge-guided deep learning models of drug toxicity improve interpretation. Patterns. 2022;3(9):100565. doi:10.1016/j.patter.2022.100565
52. Munson BP, Chen M, Bogosian A, et al. De novo generation of multi-target compounds using deep generative chemistry. Nat Commun. 2024;15(1):3636. doi:10.1038/s41467-024-47120-y
53. Cruz-Burgos M, Losada-Garcia A, Cruz-Hernández CD, et al. New approaches in oncology for repositioning drugs: the case of PDE5 inhibitor sildenafil. Front Oncol. 2021;11. doi:10.3389/fonc.2021.627229
54. Zong N, Wen A, Moon S, et al. Computational drug repurposing based on electronic health records: a scoping review. Npj Digit Med. 2022;5(1):77. doi:10.1038/s41746-022-00617-6
55. Nikidis E, Kyriakopoulos N, Tohid R, Kachrimanis K, Kioseoglou J. Harnessing machine learning for efficient large-scale interatomic potential for sildenafil and pharmaceuticals containing. Nanoscale. 2024;16(38):18014–18026. doi:10.1039/D4NR00929K
56. Rodriguez S, Hug C, Todorov P, et al. Machine learning identifies candidates for drug repurposing in Alzheimer’s disease. Nat Commun. 2021;12(1):1033. doi:10.1038/s41467-021-21330-0
57. Liu R, Wei L, Zhang P. A deep learning framework for drug repurposing via emulating clinical trials on real-world patient data. Nat Mach Intell. 2021;3(1):68–75. doi:10.1038/s42256-020-00276-w
58. Gulshan V, Peng L, Coram M, et al. Development and validation of a deep learning algorithm for detection of diabetic retinopathy in retinal fundus photographs. JAMA. 2016;316(22):2402. doi:10.1001/jama.2016.17216
59. Islam MM, Yang H-C, Poly TN, Jian W-S, Jack Li Y-C. Deep learning algorithms for detection of diabetic retinopathy in retinal fundus photographs: a systematic review and meta-analysis. Comput Methods Programs Biomed. 2020;191:105320. doi:10.1016/j.cmpb.2020.105320
60. Esteva A, Kuprel B, Novoa RA, et al. Dermatologist-level classification of skin cancer with deep neural networks. Nature. 2017;542(7639):115–118. doi:10.1038/nature21056
61. Redmon J, Divvala S, Girshick R, Farhadi A. You only look once: unified, real-time object detection. In:
62. MacEachern SJ, Forkert ND. Machine learning for precision medicine. Genome. 2021;64(4):416–425. doi:10.1139/gen-2020-0131
63. Pakhrin SC, Shrestha B, Adhikari B, Kc DB. Deep learning-based advances in protein structure prediction. Int J Mol Sci. 2021;22(11):5553. doi:10.3390/ijms22115553
64. Pearce R, Li Y, Omenn GS, Zhang Y, Ouzounis CA. Fast and accurate Ab Initio Protein structure prediction using deep learning potentials. PLOS Comput Biol. 2022;18(9):e1010539. doi:10.1371/journal.pcbi.1010539
65. Bannigan P, Aldeghi M, Bao Z, Häse F, Aspuru-Guzik A, Allen C. Machine learning directed drug formulation development. Adv Drug Deliv Rev. 2021;175:113806. doi:10.1016/j.addr.2021.05.016
66. Dawoud MHS, Mannaa IS, Abdel-Daim A, Sweed NM. Integrating artificial intelligence with quality by design in the formulation of lecithin/chitosan nanoparticles of a poorly water-soluble drug. AAPS Pharm Sci Tech. 2023;24(6):169. doi:10.1208/s12249-023-02609-5
67. Puttagunta M, Ravi S. Medical image analysis based on deep learning approach. Multimed Tools Appl. 2021;80(16):24365–24398. doi:10.1007/s11042-021-10707-4
68. Castelvecchi D. Can we open the black box of AI? Nature. 2016;538(7623):20–23. doi:10.1038/538020a
69. Quazi S. Artificial intelligence and machine learning in precision and genomic medicine. Med Oncol. 2022;39(8):120. doi:10.1007/s12032-022-01711-1
70. Zhou R, Lu Z, Luo H, Xiang J, Zeng M, Li M. NEDD: a network embedding based method for predicting drug-disease associations. BMC Bioinf. 2020;21(S13):387. doi:10.1186/s12859-020-03682-4
71. Schork NJ. Artificial intelligence and personalized medicine. In: Cancer Treatment and Research. Springer International Publishing; 2019:265–283.
72. Sistaninejhad B, Rasi H, Nayeri P, Moraru L. A review paper about deep learning for medical image analysis. Comput Math Methods Med. 2023;2023(1). doi:10.1155/2023/7091301
73. Murdoch B. Privacy and artificial intelligence: challenges for protecting health information in a new era. BMC Med Ethics. 2021;22(1):122. doi:10.1186/s12910-021-00687-3
74. Ghosal S, Bag S, Chinnadurai RK, Mukherjee M, Pramanik G, Bhowmik S. Investigating the preferential interaction between imatinib mesylate and VEGF G-quadruplex DNA as therapeutic strategies for cancer treatment: biophysical and molecular modelling approaches. Comput Biol Med. 2024;177:108683. doi:10.1016/j.compbiomed.2024.108683
75. Cai L, Chu J, Xu J, et al. Machine learning for drug repositioning: recent advances and challenges. Curr Res Chem Biol. 2023;3:100042. doi:10.1016/j.crchbi.2023.100042
76. Caruana R, Lou Y, Gehrke J, Koch P, Sturm M, Elhadad N. Intelligible models for healthcare. In:
77. Yang F, Zhang Q, Ji X, et al. Machine learning applications in drug repurposing. Interdiscip Sci Comput Life Sci. 2022;14(1):15–21. doi:10.1007/s12539-021-00487-8
78. Parvathaneni V, Kulkarni NS, Muth A, Gupta V. Drug repurposing: a promising tool to accelerate the drug discovery process. Drug Discov Today. 2019;24(10):2076–2085. doi:10.1016/j.drudis.2019.06.014
79. Ma S, Liu J, Li W, et al. Machine learning in TCM with natural products and molecules: current status and future perspectives. Chin Med. 2023;18(1):43. doi:10.1186/s13020-023-00741-9
80. Chen H, Engkvist O, Wang Y, Olivecrona M, Blaschke T. The rise of deep learning in drug discovery. Drug Discov Today. 2018;23(6):1241–1250. doi:10.1016/j.drudis.2018.01.039
81. Zeng M, Guo D, Fernández-Varo G, et al. The integration of nanomedicine with traditional chinese medicine: drug delivery of natural products and other opportunities. Mol Pharm. 2023;20(2):886–904. doi:10.1021/acs.molpharmaceut.2c00882
82. Liu H, B H, Chen P, et al. Docking score ML: target-specific machine learning models improving docking-based virtual screening in 155 targets. J Chem Inf Model. 2024;64(14):5413–5426. doi:10.1021/acs.jcim.4c00072
83. van Montfort RLM, Workman P. Structure-based drug design: aiming for a perfect fit. Essays Biochem. 2017;61(5):431–437. doi:10.1042/EBC20170052
84. Zhang Z, Liu Q, Lee C-K, Hsieh C-Y, Chen E. An equivariant generative framework for molecular graph-structure Co-design. Chem Sci. 2023;14(31):8380–8392. doi:10.1039/D3SC02538A
85. Grisoni F, Huisman BJH, Button AL, et al. Combining generative artificial intelligence and on-chip synthesis for de novo drug design. Sci Adv. 2021;7(24). doi:10.1126/sciadv.abg3338
86. Barabási A-L, Gulbahce N, Loscalzo J. Network medicine: a network-based approach to human disease. Nat Rev Genet. 2011;12(1):56–68. doi:10.1038/nrg2918
87. You Y, Lai X, Pan Y, et al. Artificial intelligence in cancer target identification and drug discovery. Signal Transduct Target Ther. 2022;7(1). doi:10.1038/s41392-022-00994-0
88. Li W, Wang S, Xu J, Xiang J. Inferring latent microRNA-disease associations on a gene-mediated tripartite heterogeneous multiplexing network. IEEE/ACM Trans Comput Biol Bioinforma. 2022;19(6):3190–3201. doi:10.1109/TCBB.2022.3143770
89. Elhag IY. Role of AI in ADME/Tox toward formulation optimization and delivery. In: A Handbook of Artificial Intelligence in Drug Delivery. Elsevier; 2023.
90. Wang MWH, Goodman JM, Allen TEH. Machine learning in predictive toxicology: recent applications and future directions for classification models. Chem Res Toxicol. 2021;34(2):217–239. doi:10.1021/acs.chemrestox.0c00316
91. McMaster C, Chan J, Liew DF, et al. Developing a deep learning natural language processing algorithm for automated reporting of adverse drug reactions. J Biomed Inform. 2023;137:104265. doi:10.1016/j.jbi.2022.104265
92. Guo B, He X. The mechanism of bisphenol s-induced atherosclerosis elucidated based on network toxicology, molecular docking, and machine learning. J Appl Toxicol. 2025;45(6):1043–1055. doi:10.1002/jat.4768
93. Kourou K, Exarchos TP, Exarchos KP, Karamouzis MV, Fotiadis DI. Machine learning applications in cancer prognosis and prediction. Comput Struct Biotechnol J. 2015;13:8–17. doi:10.1016/j.csbj.2014.11.005
94. Zhou J, Lu JY, Xie ZX, Wang DH, He BS, Huang WT. Multifunctional Au–Ag–Cr nanocomposites: from multiplex sensing and advanced logic computing to scalable information protection. Research. 2025;8. doi:10.34133/research.0763
95. Luo J, Cui Y, Xu L, et al. Layered double hydroxides for regenerative nanomedicine and tissue engineering: recent advances and future perspectives. J Nanobiotechnology. 2025;23(1):370. doi:10.1186/s12951-025-03448-1
96. André C, Guerra PJG. Machine learning in biopharmaceutical manufacturing. Eur Pharm J. 2018;23(4):62–65.
97. Maharjan R, Lee JC, Lee K, Han H-K, Kim KH, Jeong SH. Recent trends and perspectives of artificial intelligence-based machine learning from discovery to manufacturing in biopharmaceutical industry. J Pharm Investig. 2023;53(6):803–826. doi:10.1007/s40005-023-00637-8
98. Nimmagadda VSP. AI in pharmaceutical manufacturing: optimizing production processes and ensuring quality control. J AI-Assisted Sci Discov. 2024;4(1):338–379.
99. Alam MN, Anupa A, Kodamana H, Rathore AS. A deep learning-aided multi-objective optimization of a downstream process for production of monoclonal antibody products. Biochem Eng J. 2024;208:109357. doi:10.1016/j.bej.2024.109357
100. Ali KA, Mohin SK, Mondal P, Goswami S, Ghosh S, Choudhuri S. Influence of artificial intelligence in modern pharmaceutical formulation and drug development. Futur J Pharm Sci. 2024;10(1). doi:10.1186/s43094-024-00625-1
101. Eslami T, Jungbauer A. Control strategy for biopharmaceutical production by model predictive control. Biotechnol Prog. 2024;40(2). doi:10.1002/btpr.3426
102. Tao L, Zhang P, Qin C, et al. Recent progresses in the exploration of machine learning methods as in-silico ADME prediction tools. Adv Drug Deliv Rev. 2015;86:83–100. doi:10.1016/j.addr.2015.03.014
103. Mak -K-K, Wong Y-H, Pichika MR. Artificial intelligence in drug discovery and development. In: Drug Discovery and Evaluation: Safety and Pharmacokinetic Assays. Cham: Springer International Publishing; 2023:1–38.
104. Immonen E, Wong J, Nieminen M, et al. The use of deep learning towards dose optimization in low-dose computed tomography: a scoping review. Radiography. 2022;28(1):208–214. doi:10.1016/j.radi.2021.07.010
105. Lee H, Kim HJ, Chang HW, Kim DJ, Mo J, Kim J-E. Development of a system to support warfarin dose decisions using deep neural networks. Sci Rep. 2021;11(1):14745. doi:10.1038/s41598-021-94305-2
106. Dong J, Z W, Xu H, Ouyang D. FormulationAI: a novel web-based platform for drug formulation design driven by artificial intelligence. Brief Bioinform. 2023;25(1). doi:10.1093/bib/bbad419
107. El-Naggar NE-A, Dalal SR, Zweil AM, Eltarahony M. Artificial intelligence-based optimization for chitosan nanoparticles biosynthesis, characterization and in‑vitro assessment of its anti-biofilm potentiality. Sci Rep. 2023;13(1):4401. doi:10.1038/s41598-023-30911-6
108. Feng C, Wang Y, Xu J, et al. Precisely tailoring molecular structure of doxorubicin prodrugs to enable stable nanoassembly, rapid activation, and potent antitumor effect. Pharmaceutics. 2024;16(12):1582. doi:10.3390/pharmaceutics16121582
109. Boldini D, Friedrich L, Kuhn D, Sieber SA. Machine learning assisted hit prioritization for high throughput screening in drug discovery. ACS Cent Sci. 2024. doi:10.1021/acscentsci.3c01517
110. Lynch C, Sakamuru S, Ooka M, et al. High-throughput screening to advance in vitro toxicology: accomplishments, challenges, and future directions. Annu Rev Pharmacol Toxicol. 2024;64(1):191–209. doi:10.1146/annurev-pharmtox-112122-104310
111. Shi M, Wang L, Li P, Liu J, Chen L, Xu D. Dasatinib–SIK2 binding elucidated by homology modeling, molecular docking, and dynamics simulations. ACS Omega. 2021;6(16):11025–11038. doi:10.1021/acsomega.1c00947
112. Hukerikar N, Hingorani AD, Asselbergs FW, Finan C, Schmidt AF. Prioritising genetic findings for drug target identification and validation. Atherosclerosis. 2024;390:117462. doi:10.1016/j.atherosclerosis.2024.117462
113. Pun FW, Ozerov IV, Zhavoronkov A. AI-powered therapeutic target discovery. Trends Pharmacol Sci. 2023;44(9):561–572. doi:10.1016/j.tips.2023.06.010
114. Xie L, He S, Song X, X B, Zhang Z. Deep learning-based transcriptome data classification for drug-target interaction prediction. BMC Genomics. 2018;19(S7):667. doi:10.1186/s12864-018-5031-0
115. Facco P, Zomer S, Rowland-Jones RC, et al. Using data analytics to accelerate biopharmaceutical process scale-up. Biochem Eng J. 2020;164:107791. doi:10.1016/j.bej.2020.107791
116. Gholap AD, Omri A. Advances in artificial intelligence-envisioned technologies for protein and nucleic acid research. Drug Discov Today. 2025;30(5):104362. doi:10.1016/j.drudis.2025.104362
117. Khuspe PR, Mane DV. Artificial intelligence driven discovery of anticancer phytochemicals: from plants to lead compounds. In: Emerging Trends in Phytotherapy of Cancer. Elsevier; 2026:441–460.
118. Chakraborty C, Bhattacharya M, Lee -S-S, Wen Z-H, Lo Y-H. The changing scenario of drug discovery using AI to deep learning: recent advancement, success stories, collaborations, and challenges. Mol Ther Nucleic Acids. 2024;35(3):102295. doi:10.1016/j.omtn.2024.102295
119. Wang H. Prediction of protein–ligand binding affinity via deep learning models. Brief Bioinform. 2024;25(2). doi:10.1093/bib/bbae081
120. Choudhury C, Arul Murugan N, Priyakumar UD. Structure-based drug repurposing: traditional and advanced AI/ML-aided methods. Drug Discov Today. 2022;27(7):1847–1861. doi:10.1016/j.drudis.2022.03.006
121. Grapă C, Mocan T, Mocan LP, et al. Turning the Tide—Artificial Intelligence in the Evolving Landscape of Liver Cancer. Cancers. 2025;17(18):3003. doi:10.3390/cancers17183003
122. Muzio G, O’Bray L, Borgwardt K. Biological network analysis with deep learning. Brief Bioinform. 2021;22(2):1515–1530. doi:10.1093/bib/bbaa257
123. Popov P, Kalinin R, Buslaev P, et al. Unraveling viral drug targets: a deep learning-based approach for the identification of potential binding sites. Brief Bioinform. 2023;25(1). doi:10.1093/bib/bbad459
124. Zheng S, Y G, Y G, et al. Machine learning–enabled virtual screening indicates the anti-tuberculosis activity of aldoxorubicin and quarfloxin with verification by molecular docking, molecular dynamics simulations, and biological evaluations. Brief Bioinform. 2024;26(1). doi:10.1093/bib/bbae696
125. Siebenmorgen T, Menezes F, Benassou S, et al. MISATO: machine learning dataset of protein–ligand complexes for structure-based drug discovery. Nat Comput Sci. 2024;4(5):367–378. doi:10.1038/s43588-024-00627-2
126. Madushanka A, Moura RT, Kraka E. QM40, realistic quantum mechanical dataset for machine learning in molecular science. Sci Data. 2024;11(1):1376. doi:10.1038/s41597-024-04206-y
127. Hsueh HT, Chou RT, Rai U, et al. Machine learning-driven multifunctional peptide engineering for sustained ocular drug delivery. Nat Commun. 2023;14(1):2509. doi:10.1038/s41467-023-38056-w
128. Zhou G, Rusnac D-V, Park H, et al. An artificial intelligence accelerated virtual screening platform for drug discovery. Nat Commun. 2024;15(1):7761. doi:10.1038/s41467-024-52061-7
129. Chen H, King FJ, Zhou B, et al. Drug target prediction through deep learning functional representation of gene signatures. Nat Commun. 2024;15(1):1853. doi:10.1038/s41467-024-46089-y
130. Hartman E, Scott AM, Karlsson C, et al. Interpreting biologically informed neural networks for enhanced proteomic biomarker discovery and pathway analysis. Nat Commun. 2023;14(1):5359. doi:10.1038/s41467-023-41146-4
131. Foglierini M, Nortier P, Schelling R, et al. RAIN: machine learning-based identification for HIV-1 bNAbs. Nat Commun. 2024;15(1):5339. doi:10.1038/s41467-024-49676-1
132. Peteani G, Huynh MTD, Gerebtzoff G, Rodríguez-Pérez R. Application of machine learning models for property prediction to targeted protein degraders. Nat Commun. 2024;15(1):5764. doi:10.1038/s41467-024-49979-3
133. Goeva A, Dolan M-J, Luu J, et al. HiDDEN: a machine learning method for detection of disease-relevant populations in case-control single-cell transcriptomics data. Nat Commun. 2024;15(1):9468. doi:10.1038/s41467-024-53666-8
134. Buttitta G, Lavagna L, Bonacorsi S, et al. Machine Learning-Guided microfluidic optimization of clinically inspired liposomes for nanomedicine applications. Int J Pharm. 2025;686:126362. doi:10.1016/j.ijpharm.2025.126362
135. Chou W-C, Chen Q, Yuan L, et al. An artificial intelligence-assisted physiologically-based pharmacokinetic model to predict nanoparticle delivery to tumors in mice. J Control Release. 2023;361:53–63. doi:10.1016/j.jconrel.2023.07.040
136. Imanparast A, Ameri AR, Attaran N, et al. A preclinical design approach for translation of biohybrid photosensitive nanoplatform for photodynamic therapy of breast cancer. J Control Release. 2025;378:543–558. doi:10.1016/j.jconrel.2024.12.021
137. Tang Y, Zhang J, He D, et al. GANDA: a deep generative adversarial network conditionally generates intratumoral nanoparticles distribution pixels-to-pixels. J Control Release. 2021;336:336–343. doi:10.1016/j.jconrel.2021.06.039
138. Zhang Y, Ye T, Xi H, Juhas M, Li J. Deep Learning Driven Drug Discovery: tackling Severe Acute Respiratory Syndrome Coronavirus 2. Front Microbiol. 2021;12. doi:10.3389/fmicb.2021.739684
139. Serrano DR, Luciano FC, Anaya BJ, et al. Artificial Intelligence (AI) applications in drug discovery and drug delivery: revolutionizing personalized medicine. Pharmaceutics. 2024;16(10):1328. doi:10.3390/pharmaceutics16101328
140. Dara S, Dhamercherla S, Jadav SS, Babu CM, Ahsan MJ. Machine learning in drug discovery: a review. Artif Intell Rev. 2022;55(3):1947–1999. doi:10.1007/s10462-021-10058-4
141. Vatansever S, Schlessinger A, Wacker D, et al. Artificial intelligence and machine learning-aided drug discovery in central nervous system diseases: state-of-the-arts and future directions. Med Res Rev. 2021;41(3):1427–1473. doi:10.1002/med.21764
142. Sumathi S, Suganya K, Swathi K, et al. A review on deep learning-driven drug discovery: strategies, tools and applications. Curr Pharm Des. 2023;29(13):1013–1025. doi:10.2174/1381612829666230412084137
143. Vamathevan J, Clark D, Czodrowski P, et al. Applications of machine learning in drug discovery and development. Nat Rev Drug Discov. 2019;18(6):463–477. doi:10.1038/s41573-019-0024-5
144. Staszak M, Staszak K, Wieszczycka K, Bajek A, Roszkowski K, Tylkowski B. Machine learning in drug design: use of artificial intelligence to explore the chemical structure–biological activity relationship. Wiley Interdiscip Rev Comput Mol Sci. 2022;12(2). doi:10.1002/wcms.1568
145. Vora LK, Gholap A, Jetha K, Thakur RRS, Solanki HK, Chavda VP. Artificial intelligence in pharmaceutical technology and drug delivery design. Pharmaceutics. 2023;15(7):1916. doi:10.3390/pharmaceutics15071916
146. Chang C-H, Lin C-H, Lane H-Y. Machine learning and novel biomarkers for the diagnosis of Alzheimer’s disease. Int J Mol Sci. 2021;22(5):2761. doi:10.3390/ijms22052761
147. Pirruccello JP, Chaffin MD, Chou EL, et al. Deep learning enables genetic analysis of the human thoracic aorta. Nat Genet. 2022;54(1):40–51. doi:10.1038/s41588-021-00962-4
148. Abdallah S, Sharifa M, Almadhoun MKIK, et al. The impact of artificial intelligence on optimizing diagnosis and treatment plans for rare genetic disorders. Cureus. 2023. doi:10.7759/cureus.46860
149. Yang R, Yu Y. Artificial convolutional neural network in object detection and semantic segmentation for medical imaging analysis. Front Oncol. 2021;11. doi:10.3389/fonc.2021.638182
150. Pinto R, Valentim R, Fernandes da Silva L, et al. Use of interrupted time series analysis in understanding the course of the congenital syphilis epidemic in Brazil. Lancet Reg Heal Am. 2022;7:100163. doi:10.1016/j.lana.2021.100163
151. Rahman T, Uddin MK, Bhattacharjee B, et al. Blockchain applications in business operations and supply chain management by machine learning. Int J Comput Sci Inf Syst. 2024;09(11):17–30. doi:10.55640/ijcsis/Volume09Issue11-03
152. Smith TB, Vacca R, Mantegazza L, Capua I. Natural language processing and network analysis provide novel insights on policy and scientific discourse around Sustainable Development Goals. Sci Rep. 2021;11(1):22427. doi:10.1038/s41598-021-01801-6
153. Raparthi M, Reddy Gayam S, Prasad Kasaraneni B, et al. AI-driven decision support systems for precision medicine: examining the development and implementation of AI-driven decision support systems in precision medicine. J Artif Intell Res. 2021;1(1 SE–Articles):11–20.
154. Li S, Ding Q, Wang X. “Network Target” theory and network pharmacology. In: Network Pharmacology. Singapore: Springer Singapore; 2021:1–34.
155. Zheng W, Yang M, Huang D, Jin M. A deep learning approach for optimizing monoclonal antibody production process parameters. J Adv Comput Syst. 2024;4(12):28–42. doi:10.69987/JACS.2024.41203
156. Liu F, Panagiotakos D. Real-world data: a brief review of the methods, applications, challenges and opportunities. BMC Med Res Methodol. 2022;22(1):287. doi:10.1186/s12874-022-01768-6
157. Gupta R, Srivastava D, Sahu M, Tiwari S, Ambasta RK, Kumar P. Artificial intelligence to deep learning: machine intelligence approach for drug discovery. Mol Divers. 2021;25(3):1315–1360. doi:10.1007/s11030-021-10217-3
158. Noviandy TR, Idroes GM, Hardi I. Machine learning approach to predict AXL kinase inhibitor activity for cancer drug discovery using bayesian optimization-XGBoost. J Soft Comput Data Min. 2024;5(1). doi:10.30880/jscdm.2024.05.01.004
159. Simončič M, Lukšič M, Druchok M. Machine learning assessment of the binding region as a tool for more efficient computational receptor-ligand docking. J Mol Liq. 2022;353:118759. doi:10.1016/j.molliq.2022.118759
160. Hosna A, Merry E, Gyalmo J, Alom Z, Aung Z, Azim MA. Transfer learning: a friendly introduction. J Big Data. 2022;9(1):102. doi:10.1186/s40537-022-00652-w
161. Yang S, Kar S. Application of artificial intelligence and machine learning in early detection of adverse drug reactions (ADRs) and drug-induced toxicity. Artif Intell Chem. 2023;1(2):100011. doi:10.1016/j.aichem.2023.100011
162. Wei Q, Ramsey SA. Predicting chemotherapy response using a variational autoencoder approach. BMC Bioinf. 2021;22(1):453. doi:10.1186/s12859-021-04339-6
163. Porokhin V, Liu L-P, Hassoun S, Martelli PL. Using graph neural networks for site-of-metabolism prediction and its applications to ranking promiscuous enzymatic products. Bioinformatics. 2023;39(3). doi:10.1093/bioinformatics/btad089
164. Ashburner JM, Chang Y, Wang X, et al. Natural language processing to improve prediction of incident atrial fibrillation using electronic health records. J Am Heart Assoc. 2022;11(15). doi:10.1161/JAHA.122.026014
165. Lamani MR, Padmaja K, Asha V, Somesha M, Shilpa BL, Anand M. Leveraging natural language processing for enhanced pharmacovigilance in reproductive health. In: Data-Driven Reproductive Health. Singapore: Springer Nature Singapore; 2024:143–155.
166. Kang M, Ko E, Mersha TB. A roadmap for multi-omics data integration using deep learning. Brief Bioinform. 2022;23(1). doi:10.1093/bib/bbab454
167. Abbasi M, Santos BP, Pereira TC, et al. Designing optimized drug candidates with Generative Adversarial Network. J Cheminform. 2022;14(1):40. doi:10.1186/s13321-022-00623-6
168. Sucharitha P, Ramesh Reddy K, Satyanarayana SV, Garg T. Absorption, distribution, metabolism, excretion, and toxicity assessment of drugs using computational tools. In: Computational Approaches for Novel Therapeutic and Diagnostic Designing to Mitigate SARS-Cov-2 Infection. Elsevier; 2022:335–355.
169. Abbasi K, Razzaghi P, Poso A, Ghanbari-Ara S, Masoudi-Nejad A. Deep learning in drug target interaction prediction: current and future perspectives. Curr Med Chem. 2021;28(11):2100–2113. doi:10.2174/0929867327666200907141016
170. Greenaway RL, Jelfs KE. Integrating computational and experimental workflows for accelerated organic materials discovery. Adv Mater. 2021;33(11). doi:10.1002/adma.202004831
171. Noor F, Tahir Ul Qamar M, Ashfaq UA, Albutti A, Alwashmi ASS, Aljasir MA. Network pharmacology approach for medicinal plants: review and assessment. Pharmaceuticals. 2022;15(5):572. doi:10.3390/ph15050572
172. Alharbi F, Vakanski A. Machine learning methods for cancer classification using gene expression data: a review. Bioengineering. 2023;10(2):173. doi:10.3390/bioengineering10020173
173. Dhakal A, McKay C, Tanner JJ, Cheng J. Artificial intelligence in the prediction of protein–ligand interactions: recent advances and future directions. Brief Bioinform. 2022;23(1). doi:10.1093/bib/bbab476
174. Saldívar-González FI, Medina-Franco JL. Approaches for enhancing the analysis of chemical space for drug discovery. Expert Opin Drug Discov. 2022;17(7):789–798. doi:10.1080/17460441.2022.2084608
175. Andaur Navarro CL, Damen JAA, Takada T, et al. Risk of bias in studies on prediction models developed using supervised machine learning techniques: systematic review. BMJ. 2021:n2281. doi:10.1136/bmj.n2281
176. Sadique Z, Grieve R, Diaz-Ordaz K, Mouncey P, Lamontagne F, O’Neill S. A machine-learning approach for estimating subgroup- and individual-level treatment effects: an illustration using the 65 trial. Med Decis Mak. 2022;42(7):923–936. doi:10.1177/0272989X221100717
177. Mei S, Zhang K. A machine learning framework for predicting drug–drug interactions. Sci Rep. 2021;11(1):17619. doi:10.1038/s41598-021-97193-8
178. Perron Q, Mirguet O, Tajmouati H, et al. Deep generative models for ligand-based de novo design applied to multi-parametric optimization. J Comput Chem. 2022;43(10):692–703. doi:10.1002/jcc.26826
179. Kosvyra A, Ntzioni E, Chouvarda I. Network analysis with biological data of cancer patients: a scoping review. J Biomed Inform. 2021;120:103873. doi:10.1016/j.jbi.2021.103873
180. Chirico N, Sangion A, Gramatica P, Bertato L, Casartelli I, Papa E. QSARINS -Chem standalone version: a new platform-independent software to profile chemicals for physico-chemical properties, fate, and toxicity. J Comput Chem. 2021;42(20):1452–1460. doi:10.1002/jcc.26551
181. Noorain SV, Parveen B, Parveen R, Parveen R. Artificial intelligence in drug formulation and development: applications and future prospects. Curr Drug Metab. 2023;24(9):622–634. doi:10.2174/0113892002265786230921062205
182. Gautam N, Ghanta S, Mueller J, et al. Artificial intelligence, wearables and remote monitoring for heart failure: current and future applications. Diagnostics. 2022;12(12):2964. doi:10.3390/diagnostics12122964
183. Wörheide MA, Krumsiek J, Kastenmüller G, Arnold M. Multi-omics integration in biomedical research – a metabolomics-centric review. Anal Chim Acta. 2021;1141:144–162. doi:10.1016/j.aca.2020.10.038
184. Rothman JE. Starting at Go: protein structure prediction succumbs to machine learning. Proc Natl Acad Sci. 2023;120(39). doi:10.1073/pnas.2311128120
185. Abouarab B, Bazarian C, Ben Chaouch Z, et al. Financing repurposed drugs for rare diseases: a case study of Unravel Biosciences. Orphanet J Rare Dis. 2023;18(1):287. doi:10.1186/s13023-023-02753-y
186. Scheibner J, Ienca M, Kechagia S, et al. Data protection and ethics requirements for multisite research with health data: a comparative examination of legislative governance frameworks and the role of data protection technologies†. J Law Biosci. 2020;7(1). doi:10.1093/jlb/lsaa010
187. Challen R, Denny J, Pitt M, Gompels L, Edwards T, Tsaneva-Atanasova K. Artificial intelligence, bias and clinical safety. BMJ Qual Saf. 2019;28(3):231–237. doi:10.1136/bmjqs-2018-008370
188. Zeng X, Wang F, Luo Y, et al. Deep generative molecular design reshapes drug discovery. Cell Reports Med. 2022;3(12):100794. doi:10.1016/j.xcrm.2022.100794
189. Zhuo R, Huffaker B, Claffy KC, Greenstein S. The impact of the General Data Protection Regulation on internet interconnection. Telecommun Policy. 2021;45(2):102083.
190. Vasey B, Nagendran M, Campbell B, et al. Reporting guideline for the early stage clinical evaluation of decision support systems driven by artificial intelligence: DECIDE-AI. BMJ. 2022;377:e070904. doi:10.1136/bmj-2022-070904
191. Pesapane F, Bracchi DA, Mulligan JF, et al. Legal and regulatory framework for AI solutions in healthcare in EU, US, China, and Russia: new scenarios after a pandemic. Radiation. 2021;1(4):261–276. doi:10.3390/radiation1040022
192. Lippi ME, Morello F, Di Pietro L, De Maria C, Calderai V. Legislation for open-source medical devices: current scenario, risks and possibilities. In: Engineering Open-Source Medical Devices. Cham: Springer International Publishing; 2022:145–166.
193. Azzi S, Gagnon S, Ramirez A, Richards G. Healthcare applications of artificial intelligence and analytics: a review and proposed framework. Appl Sci. 2020;10(18):6553. doi:10.3390/app10186553
194. Naik N, Hameed BMZ, Shetty DK, et al. Legal and Ethical consideration in Artificial Intelligence in healthcare: who takes responsibility? Front Surg. 2022;9. doi:10.3389/fsurg.2022.862322
195. Tiwari PC, Pal R, Chaudhary MJ, Nath R. Artificial intelligence revolutionizing drug development: exploring opportunities and challenges. Drug Dev Res. 2023;84(8):1652–1663. doi:10.1002/ddr.22115
196. Bai Q, Tan S, Xu T, Liu H, Huang J, Yao X. MolAICal: a soft tool for 3D drug design of protein targets by artificial intelligence and classical algorithm. Brief Bioinform. 2021;22(3). doi:10.1093/bib/bbaa161
197. Stokes JM, Yang K, Swanson K, et al. A Deep Learning Approach to Antibiotic Discovery. Cell. 2020;180(4):688–702.e13. doi:10.1016/j.cell.2020.01.021
198. Goldstein BA, Navar AM, Pencina MJ. Risk prediction with electronic health records. JAMA Cardiol. 2016;1(9):976. doi:10.1001/jamacardio.2016.3826
199. Topol EJ. High-performance medicine: the convergence of human and artificial intelligence. Nat Med. 2019;25(1):44–56. doi:10.1038/s41591-018-0300-7
200. Prajapati JB, Paliwal H, Saikia S, et al. Impact of AI on drug delivery and pharmacokinetics: the present scenario and future prospects. In: A Handbook of Artificial Intelligence in Drug Delivery. Elsevier; 2023:443–465.
201. Philip AK, Faiyazuddin M. An overview of artificial intelligence in drug development. In: A Handbook of Artificial Intelligence in Drug Delivery. Elsevier; 2023:1–8.
202. European Union. Regulation (EU) 2016/679 of the European Parliament and of the Council of 27 April 2016 on the protection of natural persons with regard to the processing of personal data and on the free movement of such data, and repealing Directive 95/46/EC (General Da). European Union; 2016.
203. O’Driscoll A, Daugelaite J, Sleator RD. “Big data”, Hadoop and cloud computing in genomics. J Biomed Inform. 2013;46(5):774–781. doi:10.1016/j.jbi.2013.07.001
204. Niazi S. The coming of age of AI/ML in drug discovery, development, clinical testing, and manufacturing: the FDA perspectives. Drug Des Devel Ther. 2023;17:2691–2725. doi:10.2147/dddt.s424991
205. Sartor J, Lagioia F. The impact of the General Data Protection Regulation (GDPR) on artificial intelligence; 2020.
206. Qureshi R, Irfan M, Gondal TM, et al. AI in drug discovery and its clinical relevance. Heliyon. 2023;9(7):e17575. doi:10.1016/j.heliyon.2023.e17575
207. Lagioia F, Sartor G. AI systems under criminal law: a legal analysis and a regulatory perspective. Philos Technol. 2020;33(3):433–465. doi:10.1007/s13347-019-00362-x
208. Golas SB, Shibahara T, Agboola S, et al. A machine learning model to predict the risk of 30-day readmissions in patients with heart failure: a retrospective analysis of electronic medical records data. BMC Med Inform Decis Mak. 2018;18(1):44. doi:10.1186/s12911-018-0620-z
209. Ienca M, Vayena E. On the responsible use of digital data to tackle the COVID-19 pandemic. Nat Med. 2020;26(4):463–464. doi:10.1038/s41591-020-0832-5
210. Behrooz H, Hayeri YM. Machine learning applications in surface transportation systems: a literature review. Appl Sci. 2022;12(18):9156. doi:10.3390/app12189156
211. Lundberg SM, Erion G, Chen H, et al. Explainable AI for trees: from local explanations to global understanding; 2019.
212. Mazumdar H, Khondakar KR, Das S, Halder A, Kaushik A. Artificial intelligence for personalized nanomedicine; from material selection to patient outcomes. Expert Opin Drug Deliv. 2025;22(1):85–108. doi:10.1080/17425247.2024.2440618
 © 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.