Back to Journals » Drug Design, Development and Therapy » Volume 20
Advances in Research on Autophagy in Steroid-Induced Osteonecrosis of the Femoral Head: Dual Regulation of Its Beneficial and Detrimental Effects
Authors Liu M, Huang J, Zhang G, Chen H, Zhao H
Received 8 January 2026
Accepted for publication 28 March 2026
Published 8 July 2026 Volume 2026:20 594555
DOI https://doi.org/10.2147/DDDT.S594555
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Solomon Tadesse Zeleke
Mingqiang Liu1,2,*, Jiwei Huang1,2,*, Guangzhi Zhang3, Haiwei Chen4, Haiyan Zhao1,2
1The First Clinical College of Medicine, Lanzhou University, Lanzhou, 730000, People’s Republic of China; 2Department of Orthopedics, The First Hospital of Lanzhou University, Lanzhou, 730000, People’s Republic of China; 3Department of Orthopedics, The Second Hospital of Lanzhou University, Lanzhou, 730000, People’s Republic of China; 4Emergency Orthopedics Department, The Second Hospital of Lanzhou University, Lanzhou, 730000, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Haiyan Zhao, Email [email protected]
Abstract: Steroid-induced osteonecrosis of the femoral head (SONFH) is a common and refractory clinical condition with a complex pathogenesis that remains incompletely understood. As a highly conserved intracellular degradation and recycling pathway, autophagy plays a dual regulatory role in the pathological process of SONFH: moderate activation of autophagy exerts protective effects by clearing damaged organelles and maintaining cellular homeostasis; conversely, excessive or dysfunctional autophagy aggravates cell injury and bone tissue destruction. This narrative review systematically summarizes the dynamics, mechanisms, and regulatory networks of autophagy in SONFH from the perspectives of both beneficial and detrimental autophagy, providing references for a deeper understanding of SONFH and the development of targeted therapies. Furthermore, we explore therapeutic strategies based on the dual nature of autophagy, including precise pharmacological modulation, restoration of autophagic-lysosomal function, combined treatment approaches, and the application of novel biomaterials and targeted delivery systems. By integrating current mechanistic insights and proposing stage-specific regulatory strategies, this review highlights the translational potential of autophagy modulation in SONFH and provides a theoretical foundation for future clinical interventions.
Keywords: steroid-induced osteonecrosis of the femoral head, autophagy, beneficial autophagy, detrimental autophagy, dichotomous regulation, therapeutic targets
Introduction
Clinical Challenges in Steroid-Induced Osteonecrosis of the Femoral Head
Steroid-induced osteonecrosis of the femoral head is a disease characterized primarily by vascular insufficiency of the femoral head and osteocyte apoptosis, which is caused by long-term or high-dose administration of glucocorticoids. It accounts for 30%–40% of non-traumatic osteonecrosis of the femoral head. The disease presents no specific manifestations in the early stage, followed by femoral head collapse and hip joint dysfunction in the middle stage and beyond. This eventually necessitates total hip arthroplasty in the end stage, imposing a substantial burden on the medical resources of both patients and society. Clinically feasible methods include core decompression and bone grafting, etc. However, they can only cure patients with mild early-stage lesions and cannot restore the physiological state of already necrotic bones. Therefore, to fundamentally change the condition, it is necessary to understand the etiology and pathogenesis of steroid-induced femoral head necrosis.1
The Dual Characteristics of Autophagy and Its Research Significance
Autophagy is an adaptive response of cells to internal and external environmental stimuli, maintaining cellular energy homeostasis by degrading damaged proteins and organelles. Under physiological conditions, autophagy is at a basal level; when cells are subjected to stress such as nutrient deprivation and oxidative stress, autophagy will be activated to resist damage. The role of autophagy is not always absolutely beneficial. Excessive activation or functional defects can both lead to metabolic disorders in cells and subsequently cause cell death.2 In steroid-induced avascular necrosis of the femoral head, the dynamic changes of autophagy are closely related to the progression of the disease. Clarifying the mechanism of the conversion from beneficial autophagy to harmful autophagy can provide new ideas for targeted regulation of autophagy in the treatment of SONFH.3
The Biological Basis and Regulatory Network of Autophagy
The Basic Process and Function of Autophagy
Autophagy is divided into five stages: initiation, nucleation, elongation, fusion and degradation. Under the stimulation of stress signals, the ULK1 complex is activated to initiate autophagy. The Beclin1-Vps34 complex participates in the nucleation of autophagosomes. The Atg12-Atg5-Atg16L1 and LC3-PE systems mediate the elongation of autophagosome membranes. Eventually, autophagosomes fuse with lysosomes to form autolysosomes, achieving substrate degradation and material recycling.4 Under physiological conditions, autophagy participates in the regulation of bone metabolism by eliminating abnormal proteins and maintaining the renewal of organelles, which is very important for osteoblast differentiation, bone cell survival and vascular homeostasis.5,6 (See Figure 1)
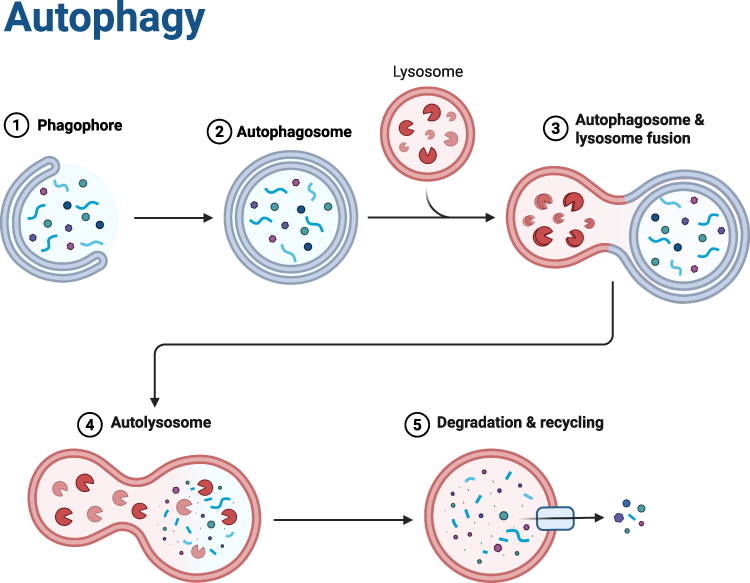 |
Figure 1 This figure illustrates the process of cellular autophagy—a core biological pathway for intracellular substance degradation and recycling: First, a phagophore is initiated to enclose target intracellular materials (such as damaged organelles and macromolecules); the phagophore then closes to form an autophagosome. Next, the autophagosome undergoes membrane fusion with a hydrolase-containing lysosome, forming an autolysosome. Finally, the hydrolases in the autolysosome degrade the enclosed contents, and the resulting degradation products are recycled for reuse by the cell, thus completing the degradation and recycling process. |
The Key Regulatory Signaling Pathways of Autophagy
The initiation, progression and termination of autophagy are precisely regulated by multiple signaling pathways, and the homeostatic balance of each pathway is the basis for maintaining normal autophagic function. mTORC1, as a negative hub for autophagy initiation, inhibits the initiation of autophagy by phosphorylating the Ser757 site of ULK1 when nutrients are abundant or growth signals are activated. Under stress conditions, its activity is suppressed, thereby removing the inhibition. When ULK1 is activated and autophagy is initiated, its functional abnormalities are closely related to diseases such as tumors and metabolic disorders.7 AMPK is a core molecule for cellular energy sensing. It gets activated under energy stress and enhances the activity of ULK1 by directly phosphorylating its Ser317 and Ser777 sites. At the same time, it indirectly phosphorylates TSC2 or Raptor to inhibit the mTORC1 signaling pathway. This constitutes the core axis for regulating autophagy in response to energy status.8 The PI3K-Akt pathway responds to pro-proliferative signals and negatively regulates autophagy by activating mTORC1. Its excessive activation can inhibit the clearance of abnormal cells by autophagy.9 p53 can achieve bidirectional regulation through nuclear-cytoplasmic localization differences. Nuclear p53 transcribes and activates autophagy-related genes, while cytoplasmic p53 inhibits the activity of Beclin1. The balance between them is crucial for autophagy function.10 During endoplasmic reticulum stress, the PERK/eIF2α pathway activates autophagy-related genes by upregulating ATF4, while the IRE1α/XBP1 pathway regulates the expression of autophagy genes by splicing XBP1 to generate XBP1s, thereby alleviating stress-induced damage.11 Abnormalities in the above-mentioned pathways (such as the persistent activation of mTORC1, defects in AMPK, etc.) can lead to an imbalance in autophagy. Both excessive and insufficient autophagy are closely related to the pathological processes of various diseases, including tumors, neurodegenerative diseases, and metabolic syndrome. A deep understanding of the regulatory mechanisms of these pathways is the core to understanding the connection between autophagy’s physiological functions and diseases.12 (See Figure 2)
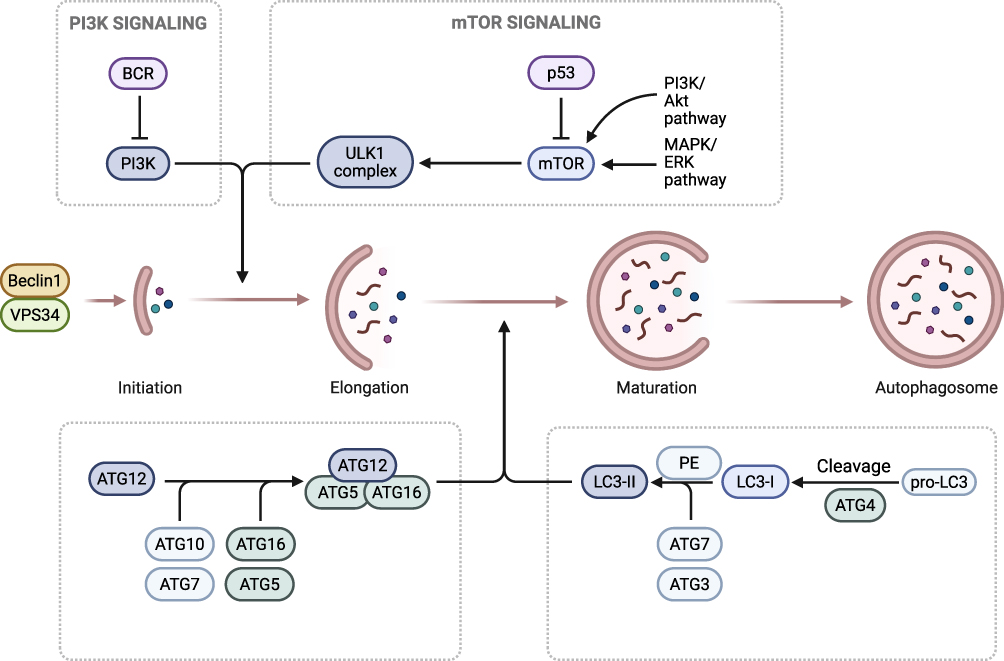 |
Figure 2 This figure depicts the molecular regulatory mechanisms and core progression of autophagy: The upstream PI3K signaling pathway (activated by BCR) and mTOR signaling pathway (regulated by p53, along with the PI3K/Akt and MAPK/ERK pathways) modulate the activity of the ULK1 complex, thereby initiating autophagy. The autophagy process proceeds through sequential stages: Initiation (involving Beclin1 and VPS34), Elongation (ATG12 is conjugated to ATG5 via ATG7/ATG10, then associates with ATG16 to form a complex that mediates membrane elongation), and Maturation (pro-LC3 is cleaved by ATG4 into LC3-I; subsequently, LC3-I is modified by ATG7/ATG3 and conjugated to phosphatidylethanolamine (PE) to form LC3-II, which contributes to membrane maturation). The process ultimately results in autophagosome formation. |
The Manifestations and Mechanisms of Beneficial Autophagy in Steroid-Induced Femoral Head Necrosis
The Definition and Characteristics of Beneficial Autophagy
Beneficial autophagy refers to the protective role of autophagy in cells by eliminating damaged components and maintaining energy homeostasis in the early stage of disease or under moderate stimulation. Its characteristics include a moderate increase in autophagy levels, an elevated LC3-II/LC3-I ratio, increased degradation of p62, normal fusion of autophagosomes with lysosomes, and enhanced cell survival ability.13 In SONFH, beneficial autophagy is mainly manifested as protective effects on osteocytes, osteoblasts and vascular endothelial cells. (See Table 1)
 |
Table 1 Protective Roles and Mechanisms of Beneficial Autophagy in Different Cell Types During Steroid-Induced Osteonecrosis of the Femoral Head |
Protective Autophagy in Osteocytes
Osteocytes, as the core for maintaining the homeostasis of bone structure, are of great significance to bone quality in terms of their survival and functional integrity. Under pathological conditions such as glucocorticoid exposure, osteocytes will activate autophagy as a defense mechanism, exerting a protective role by eliminating damaged components and maintaining intracellular homeostasis.14 Short-term or low-dose hormone exposure can specifically activate autophagy in bone cells, which is manifested by the upregulation of autophagy markers such as LC3-II and an increase in the number of autolysosomes. Among them, selective mitochondrial autophagy reduces the accumulation of ROS by eliminating oxidatively damaged mitochondria and blocks the initiation of apoptotic signals.15,16 This protective autophagy reduces the accumulation of cytotoxic substances by degrading damaged components and utilizes autophagic degradation products to replenish energy through gluconeogenesis to maintain cellular metabolism. In vitro experiments have also confirmed that after treating osteoid cells MLO-Y4 with a low concentration of dexamethasone (10−8 mol/L), autophagy activation increases the LC3-II/LC3-I ratio by 30% to 50%, and reduces the apoptosis rate by 20% to 30%. However, autophagy inhibitors will exacerbate hormone-induced apoptosis, which indirectly reflects the protective role of autophagy.17 However, this defense has the characteristic of dynamic balance. In the short term or at low doses of hormones, autophagy protects the survival of bone cells by “clearing damage, maintaining metabolism, and inhibiting apoptosis”. Long-term or high-dose exposure may lead to autophagic function exhaustion or excessive activation, causing autophagic cell death, which instead aggravates bone mass loss. Therefore, the dynamic balance of autophagy in bone cells is the key to exerting a protective effect, which also provides a new idea for the early intervention of hormone-induced osteonecrosis.18,19
Autophagic Regulation of Osteoblast Function
Osteoblasts are the core cells in bone formation, responsible for the synthesis, secretion and mineralization of bone matrix. The core reason for the reduction in bone mass and bone structure damage in glucocorticoid-induced femoral head necrosis may be the functional impairment of osteoblasts. Autophagy, by regulating the material metabolism and functional homeostasis of osteoblasts, has become the core mechanism for protecting them against hormone-induced damage.20 The protective regulation of autophagy on osteoblasts is manifested in two aspects. Firstly, it reduces endoplasmic reticulum stress by selectively degrading misfolded type I collagen and damaged organelles, while maintaining the stability of the osteoblast differentiation transcription factor Runx2, ensuring its transcriptional activation of osteoblast-related genes such as COL1A1 and osteocalcin, and maintaining the osteoblast differentiation potential. On the other hand, by degrading redundant biological macromolecules to produce metabolic intermediates such as amino acids and fatty acids, ATP is generated through gluconeogenesis or beta-oxidation, providing energy for osteoblast proliferation, collagen secretion, and alkaline phosphatase (ALP) activity. Moreover, the inorganic phosphate released can promote the deposition of hydroxyapatite crystals in the bone matrix, thereby accelerating mineralization.21,22 Studies have shown that in the hormone-induced SONFH rat model, activating autophagy in osteoblasts can reduce the decline in femoral head bone density by 15% to 20%, increase trabecular thickness and quantity, and up-regulate the expression of osteogenic markers such as osteocalcin and ALP.23 In vitro experiments have shown that autophagy activation can enhance the stability of Runx2, increase osteogenic differentiation nodules, while autophagy inhibition leads to reduced mineralization capacity and decreased secretion of bone matrix.24,25 In the pathology of SONFH, glucocorticoids inhibit the expression of autophagy-related genes such as Beclin1 and ATG7, leading to a reduction in autophagic function in osteoblasts. Targeted activation of autophagy can partially reverse the inhibitory effect of hormones on osteogenic function and slow down bone mass loss, providing a potential strategy for the intervention of SONFH.26,27
The Autophagic Protective Effect of Vascular Endothelial Cells
Impaired blood supply to the femoral head is also an important mechanism of glucocorticoid-induced femoral head necrosis. As the functional core of the vascular wall, autophagy of vascular endothelial cells plays a key protective role in maintaining vascular homeostasis.28,29 Under physiological conditions, autophagy reduces the accumulation of inactive endothelial nitric oxide synthase (eNOS) by selectively degrading damaged eNOS, and eliminates damaged mitochondria to reduce the impact of reactive oxygen species (ROS) on normal eNOS, maintaining the homeostasis of nitric oxide (NO) synthesis and ensuring vascular dilation function.30,31 Under short-term hormone stimulation, autophagy in vascular endothelial cells is activated. Autophagy alleviates endoplasmic reticulum stress by degrading stress-related abnormal proteins and damaged endoplasmic reticulum structures, eliminates damaged mitochondria that release cytochrome c to block the apoptotic pathway, and also maintains the stability of tight junction proteins occludin and claudin, ensuring the integrity of the vascular endothelial barrier and function, and reducing apoptosis and abnormal vascular permeability.32,33 Studies have shown that in hormone-damaged vascular endothelial cells, the autophagy agonist rapamycin can enhance the migration ability of endothelial cells, significantly increase the length and number of branches of the lumen formed, and autophagy activation can up-regulate the expression of phosphorylated eNOS, increase NO release to improve vascular dilation function, and at the same time reduce the levels of endoplasmic reticulum stress markers GRP78 and apoptotic protein cleaved-caspase3. In the pathology of SONFH, long-term or high-dose hormone use can inhibit autophagy in vascular endothelial cells, leading to abnormal eNOS activity and reduced NO synthesis, as well as vascular dilation dysfunction. Coupled with the increased apoptosis and barrier disruption of endothelial cells caused by the accumulation of endoplasmic reticulum stress, this further aggravates the blood supply disorder of the femoral head.34,35 Short-term autophagy activation, by maintaining eNOS function, alleviating stress injury and enhancing angiogenesis, has become an important defense mechanism for improving the microcirculation of the femoral head. This suggests that targeting the activation of autophagy in vascular endothelial cells may provide a new perspective for the early prevention and treatment of SONFH through improving blood perfusion.36,37
The Manifestations and Mechanisms of Harmful Autophagy in Steroid-Induced Femoral Head Necrosis
The Definition and Triggering Factors of Harmful Autophagy
Harmful autophagy refers to the process in which long-term or excessive activation of autophagy leads to excessive consumption of cellular substances, obstruction of the autophagic flux, and ultimately cell death. Its characteristics include the accumulation of a large number of autophagosomes but with degradation obstacles, abnormal accumulation of p62, and an increase in markers of apoptosis or necrosis.38 In SONFH, the triggers of harmful autophagy include exposure to long-term high-dose hormones, oxidative stress damage, continuous stimulation by inflammatory factors, and dysfunction of the autophagy-lysosome system.29 (See Table 2)
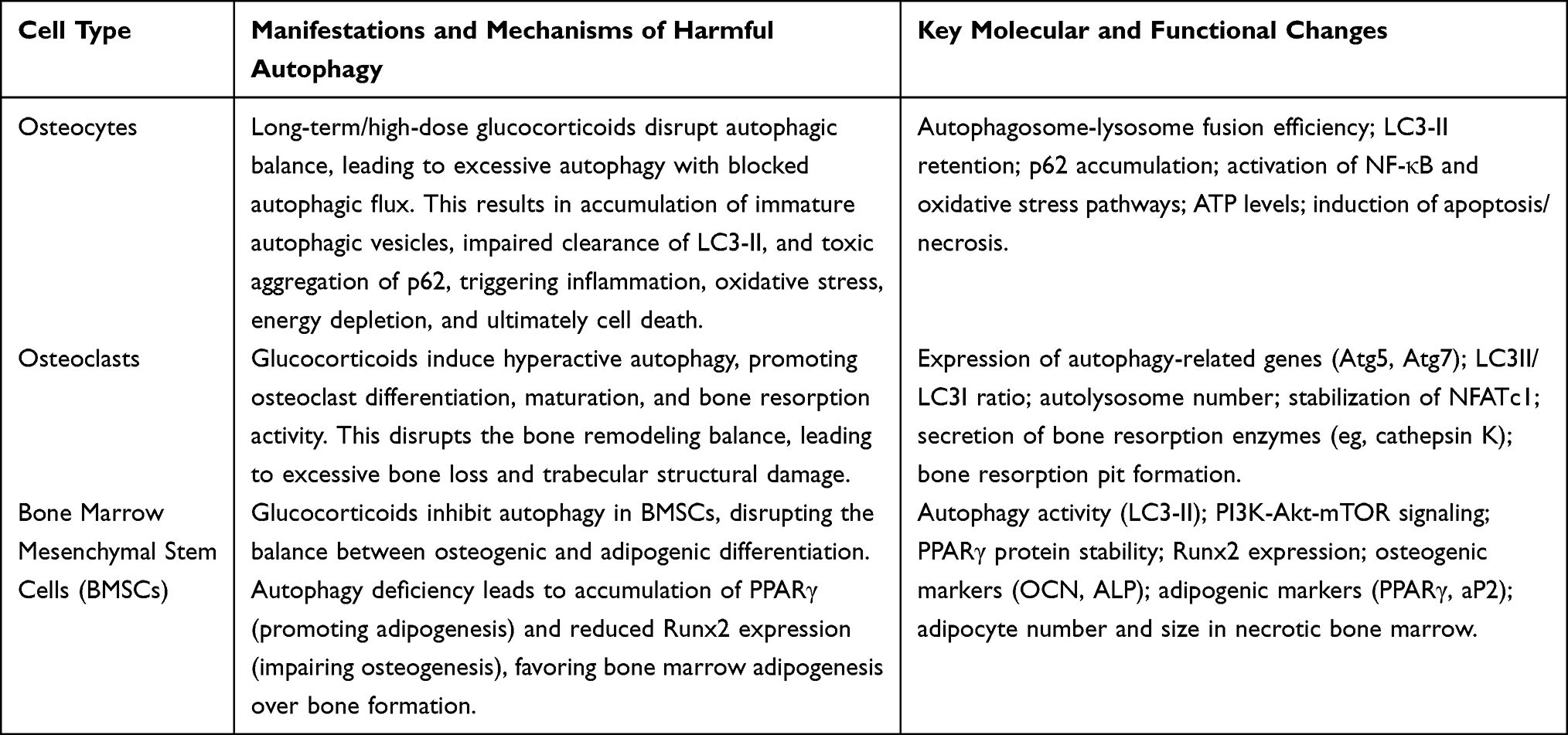 |
Table 2 Cell-Specific Manifestations of Harmful Autophagy in SONFH |
Excessive Autophagy and Apoptosis of Osteocytes
Autophagy’s protective effect on osteocytes is dose and time-dependent. Short-term or low-dose hormone-induced autophagy activation can exert adaptive defense functions. However, long-term and high-dose glucocorticoid effects can disrupt autophagy balance, leading to excessive autophagy activation in osteocytes and a shift towards a harmful phenotype, which becomes a significant factor in osteocyte death and bone structure destruction in the middle and late stages of SONFH.3 Under the influence of long-term and high-dose hormones, excessive autophagy in bone cells is characterized by blocked autophagic flux and functional disorders. The fusion efficiency of autophagosomes and lysosomes is reduced, leading to the accumulation of a large number of immature autophagic vesicles. The clearance of LC3-II is delayed, and p62 protein continuously accumulates to form toxic aggregates, which in turn activates inflammatory pathways such as NF-κB and induces oxidative stress. At the same time, excessive autophagy degrades normal biomolecules and functional organelles, leading to excessive consumption of nutrients, a decline in ATP levels, and energy failure, which inhibits the activity of lysosomal enzymes, forming a vicious cycle of “excessive autophagy, insufficient energy, and degradation disorder”, and then triggers cell death.28,39 During the course of SONFH, excessive autophagy in osteocytes disrupts intracellular homeostasis, depletes energy reserves, and induces toxic accumulation, accelerating osteocyte death. This leads to a decline in the bone tissue’s ability to sense mechanical stimuli, exacerbates the imbalance between bone resorption and formation, and promotes femoral head collapse. This reveals the double-edged sword effect of autophagy. We need to precisely regulate autophagic balance, and targeting the inhibition of abnormal autophagic flux may slow down osteocyte death and delay the occurrence and development of the disease.24,40
Enhanced Autophagy in Osteoclasts and Increased Bone Resorption
Osteoclasts, as the main functional cells for bone resorption, the excessive activation of osteoclasts leading to an overabundance of osteoclasts and hyperactive bone resorption is the core link in the loss of bone mass and the destruction of trabecular structure in the femoral head.41 Relevant studies have indicated that long-term exposure to glucocorticoids can induce excessive enhancement of autophagy in osteoclasts, significantly promoting their differentiation, maturation, and bone resorption function. This disrupts the normal physiological state where the rate of bone formation exceeds that of bone resorption. In the advanced stage of SONFH, a large number of osteoclasts will also undergo apoptosis.42
Hormones can induce hyperactive autophagy in osteoclasts, up-regulate autophagy-related genes Atg5 and Atg7, increase the ratio of LC3II/LC3I, and increase the number of autolysosomes. Studies have shown that after knocking out Atg5 or Atg7, the ability of hormones to promote the differentiation of osteoclast precursors into mature multinucleated osteoclasts is weakened, the number of TRAP-positive multinucleated cells decreases, and the area of bone resorption lacunae reduces. This suggests that hyperactive autophagy is one of the key conditions for hormone-induced osteoclast activation. This process involves downstream enzymes such as NFATc1-related regulatory and stabilizing proteins, cathepsin K, as well as ubiquitination modification and other links.43 On the one hand, excessive autophagy can maintain the stability of NFATc1 protein by selectively eliminating ubiquitination modification enzymes or negative regulatory factors (such as SOCS1) related to NFATc1 binding, and promote its transcriptional activation of downstream target genes related to transcription, thereby ensuring that the osteoclast differentiation program can proceed continuously and orderly.44 On the other hand, autophagic degradation products can serve as a source of amino acids and energy for osteoclasts, and for bone resorption enzymes such as myeloperoxidase (MPO) and cathepsin K (assessed based on the mCATH model). When necessary, they can also utilize amino acids and energy through the endoplasmic reticulum and Golgi apparatus pathways to regulate the misfolded state of bone resorption enzyme-related proteins, promoting the correct folding and secretion of these enzymes and maintaining the integrity of lysosomal function. This helps osteoclasts exert their bone resorption capacity in bone resorption lacunae, enabling them to break down type I collagen and other tissue proteins in the bone matrix.39 During the course of SONFH, excessive autophagy-mediated bone resorption leads to bone structure destruction, characterized by thinning of trabeculae, increased spacing, reduced connectivity, and decreased integrity of bone microstructure.45 Long-term hyperactive autophagy of osteoclasts can cause trabecular fractures, dissolution and absorption, leading to a more significant reduction in bone mass in the weight-bearing area of the femoral head, making the bone’s mechanical properties more vulnerable to damage, and thus making it difficult to bear the load, resulting in femoral head collapse. Therefore, hyperactive autophagy of osteoclasts can play a role in connecting hormone exposure with excessive activation of osteoclasts.28 By targeting the inhibition of excessive autophagy in osteoclasts, it can reduce bone resorption and protect bone microstructure, etc, and is expected to become an effective treatment method for delaying femoral head collapse in SONFH.31
Autophagy Deficiency and Differentiation Imbalance of Bone Marrow Mesenchymal Stem Cells
The normal process of bone marrow mesenchymal stem cells (BMSCs) differentiating into osteoblasts is conducive to maintaining the homeostasis of bone tissue, while the abnormal differentiation of BMSCs into adipocytes, namely bone marrow adipogenesis, is one of the most significant pathological changes in SONFH.46 On the one hand, the imbalance of differentiation weakens the bone formation ability; on the other hand, the continuously accumulated fat cells will also compress the bone marrow sinusoids, disrupt the local blood circulation, cause insufficient blood supply, and thus aggravate the avascular necrosis of the femoral head.2,47
Here, due to the regulation of hormones, autophagy of BMSCs is specifically inhibited by glucocorticoids, which leads to the suppression of the PI3K-Akt-mTOR signaling pathway of autophagy, resulting in a decrease in autophagic activity. During this process, mTOR exerts a negative regulatory effect on autophagy by inhibiting the PI3K-Akt-mTOR signaling pathway, and its activity depends on the phosphorylation and activation mediated by PI3K-Akt.48 When hormones inhibit the PI3K-Akt-mTOR signaling pathway, the classic autophagy initiation signals under the condition of reduced mTOR activity cannot be effectively activated. As a result, there are not sufficient conditions to promote the formation of autophagosomes and enhance autophagic degradation function. Therefore, this situation ultimately leads to an imbalance in two aspects of differentiation: on the one hand, the inhibition of autophagy leads to a decrease in the degradation level of the key transcription factor PPARγ for fat differentiation, resulting in the accumulation of a certain amount of endogenous PPARγ within the cells.24,49 On the other hand, autophagy deficiency leads to the disruption of energy metabolism homeostasis during osteogenic differentiation: that is, the autophagy deficiency results in the lack of the process by which damaged organelles and protein complexes within the cells are decomposed into energy substrates such as amino acids and ATP, reducing the supply of energy substrates (such as ATP and amino acids) needed for osteogenic differentiation, and thus lowering the expression level of osteogenic differentiation-related gene Runx2. Moreover, after differentiation and maturation, osteoblasts still rely on the osteogenic markers (such as OCN and ALP) secreted by themselves to maintain functions such as proliferation and mineralization.50
Detection of BMSCs in the necrotic area of the femoral head in patients with SONFH revealed that the expression level of the autophagy marker protein LC3-II was lower than that in the normal control group, and autophagic activity was impaired. At the same time, histological section counting showed that the number of adipocytes in the lesion area was 1.5 to 2 times that in normal bone marrow tissue, and the volume of adipocytes was increased.33,51 Moreover, in line with our experimental results, the detection of autophagic activity in BMSCs was found to be positively correlated with the mRNA and protein levels of osteogenesis-related markers (Runx2, OCN, ALP), but negatively correlated with adipogenic differentiation-related markers (PPARγ and aP2). Clinically, it has been demonstrated that autophagy deficiency in BMSCs leads to an imbalance in osteogenic and adipogenic differentiation of BMSCs. Restoring autophagy in BMSCs can improve the differentiation balance. This provides molecular evidence that targeted intervention strategies for BMSCs autophagy can be used as one of the treatment methods for SONFH.52
The Conversion Mechanism Between Beneficial Autophagy and Harmful Autophagy
The Critical Regulatory Role of Oxidative Stress
Oxidative stress is the core regulatory hub of autophagic phenotypic conversion. Oxidative stress regulates autophagy in a concentration-dependent manner through ROS, maintaining the dynamic balance between the two.53 ROS generates a certain amount of endogenous ROS (generally considered to be less than 10–20 μmol/L) through mitochondrial respiratory chain, NADPH, oxidase and other pathways. At this concentration, ROS can activate the AMPK-mTOR pathway. The activated ROS can directly oxidize the cysteine residue (such as Cys130) of AMPK, causing the autophagy-related extracellular signal-regulated kinase 6 (ERK6) to be phosphorylated and inhibit its kinase activity, thereby releasing the inhibition of the autophagy initiation complex (ULK1/Atg13/FIP200), and simultaneously up-regulating the expression of autophagy-related molecules such as Atg5-Atg12 complex and LC3, promoting the formation and degradation of autophagosomes and the degradation of substrates. Moderate autophagy can achieve the function of maintaining intracellular homeostasis by removing mitochondrial damage and protein misfolding through mitochondrial autophagy, and can play a protective role in cells under stress conditions such as starvation and mild injury.54 When cells are chronically deprived of oxygen, experience hormonal imbalances, or are exposed to toxins, severe mitochondrial dysfunction leads to a significant release of ROS. If the intracellular ROS level is excessively high (more than twice the normal level), it can cause a shift in the autophagy regulatory mechanism. On one hand, under the influence of ROS, the active sites of autophagy-related proteins (such as Cys) are extensively oxidized and inactivated. For instance, Atg3, a key ligase for LC3 lipidation, when its Cys223 is oxidized, cannot interact with LC3, thereby hindering the synthesis of LC3-PE.55 Oxidation of Cys572 in Atg7 inhibits the E1-like enzyme activity of Atg7, preventing the formation of Atg5 and Atg12, which leads to the obstruction of the construction of the ATG12-Atg16 complex and the subsequent formation of autophagosomes being affected.56 ROS can damage the autophagy pathway through the above-mentioned mechanism, preventing the formation of proteins necessary for the normal operation of the autophagy pathway.57 On the other hand, when high concentrations of ROS can overly enhance autophagic activity by activating the ASK1-MKK3/6-p38MAPK pathway, during this process, the phosphorylated p38MAPK not only directly phosphorylates Beclin-1 Ser90 to strengthen its binding force with Vps34 and thereby trigger autophagy, but also forms an autophagy-inflammation vicious cycle by up-regulating pro-inflammatory cytokines such as IL-6 and TNF-α, making the elimination rate of autophagosomes lower than the generation rate. Excessive immature autophagosomes will remain in the cells and release pro-apoptotic molecules.58 In addition, in human umbilical vein endothelial cells induced by H2O2, the apoptosis rate can be increased through excessive ROS damage. However, this damage promotes the autophagic process to degrade the damaged mitochondria, ultimately reducing the apoptosis rate.59,60 When ROS increased to 42 μmol/L (2.1 times the threshold), although the LC3II/LC3I ratio still rose, the cell survival rate decreased compared to the control group, and the p62 protein could not be normally degraded. In the cardiomyocyte model, ischemia-reperfusion led to a ROS burst. When it exceeded 2.3 times the threshold, it could cause an increase in autophagy-related damage markers (oxidative modification of Atg7, phosphorylation level of p38) and an increase in the area of cardiomyocyte death. Therefore, according to the different characteristics of cells, cells have different physiological thresholds for ROS. For example, in this study, the “2 times threshold” of cardiomyocytes to ROS on the 3rd to 4th day was a non-fixed value obtained through multiple experiments.61,62 However, regardless of the type of stress-induced cell death or injury-related disease, the “2-fold threshold” rule remains largely unchanged. Clinically, it is possible to maintain mitochondrial ROS within the 2-fold threshold through targeted regulation, such as using SOD mimetics or mitochondrial-targeted antioxidants like MitoQ, to keep ROS within a normal range.63 The opposite approach can also be adopted to regulate it. By using inhibitors such as SB203580 to suppress the over-activated p38MAPK, the progression of AUS to DUS can be prevented.64 Future studies are warranted to further clarify the specific thresholds of ROS in regulating the autophagic phenotypic switch in osteocytes, osteoblasts, and osteoclasts. Combined with cell-type-specific oxidative stress response characteristics, the dual regulatory mechanism of ROS on autophagy across different pathological stages of SONFH will be elucidated, so as to provide precise targets for staged targeted intervention. Studies have shown that in the diabetic nephropathy (DN) model, if the ROS content in the activated autophagy of the DN-reduced renal tubular epithelial cells (AECs) is down-regulated to within 1.5 times the threshold, the autophagy of AECs can be restored, effectively reducing proteinuria and renal interstitial fibrosis. Therefore, regulating the ROS content to a certain threshold in clinical practice may also become a therapeutic intervention method.65
The Bidirectional Influence of Inflammatory Signals
Inflammatory signals act as crucial regulatory mediators for the transformation of autophagic phenotypes. Inflammatory factors precisely regulate the function of autophagy through concentration gradients at different concentrations and cross-interactions via distinct signaling pathways. This significant role in the occurrence and development of bone metabolic diseases, ischemic injuries, and other conditions cannot be overlooked.66 During the physiological compensatory period, low-concentration inflammatory factors (such as IL-6) play a protective role. When cells are slightly damaged, IL-6 secreted by surrounding immune cells (generally maintained at a concentration of about 5–10 pg/mL) binds to the IL-6R/gp130 complex on the cell membrane via its ligand IL-6, stimulating Janus kinase (JAK) within the cell. This leads to the phosphorylation of STAT3, which then enters the nucleus and directly binds to the TTCCCGTAA sequence in the promoter region of the Beclin1 gene, activating the transcriptional expression of Beclin1. As a key regulatory factor for initiating autophagy, Beclin1 can form a complex with Vps34 histone, activating the activity of PI3K and accelerating the formation of autophagosomes.67–70 Studies have shown that in osteoblast models, treatment with 10 pg/mL IL-6 can increase the Beclin1 protein level by 2.1 times and the accumulation of LC3-II by 1.8 times, improving the effect of autophagic clearance of damaged organelles (with a gain efficiency of 40%), and to a certain extent, counteract the cellular damage caused by oxidative stress, which is beneficial for the protection of osteocytes during the bone tissue repair period.71 Autophagy in the inflammatory response has a transformation into a harmful state, that is, from the primary cell protection/adaptation to secondary tissue damage, which is mainly characterized by excessive production of pro-inflammatory factors, and is accompanied by the autophagy activation state changing from the initial primary regulator to a negative factor directly regulating the development of inflammation.72 When the inflammatory response is uncontrollable, it leads to the production of a large amount of pro-inflammatory factors (>50 pg/mL), mainly TNF-α (>50 pg/mL) and IL-1β (>20 pg/mL), which activate the downstream signaling cascade through transmembrane receptors TNFR1/IL-1R. This process activates the IkB kinase (IKK) complex via the TNF-α-TRADD/RIPK1/TNFRSF1A signaling pathway, causing the degradation of the NF-κB inhibitory protein IκBα through phosphorylation. As a result, the NF-κB (p65/p50) dimer enters the nucleus to function, increasing the expression of autophagy-related genes such as ULK1 and Atg5, and thus activating the initial signal of autophagy at an excessively high level.73 In addition, NF-κB also transcribes pro-apoptotic genes such as Bax, intensifying the interaction between autophagy and apoptosis to promote damage. Most importantly, high concentrations of IL-1β activate p38MAPK-Hsp27, which inhibits the activity of v-ATPase on the lysosomal membrane, causing lysosomal acidification disorders and reducing the fusion rate of autophagosomes with lysosomes. This stops the cycle of autophagic substrate degradation. At this point, the imbalance between the hyperactive initiation of autophagy and the obstruction of autophagic clearance leads to the accumulation of damaged proteins and organelles in the cytoplasm, exacerbating tissue damage.74 Given the high spatiotemporal correlation between the dynamic evolution of the inflammatory microenvironment and the autophagy phenotype changes during the pathological process of non-traumatic avascular necrosis of the SONFH, the authors speculate that there may be directional changes in the regulation of inflammatory or autophagy signals at different stages of the disease course of SONFH, which leads to the changes at the gene and molecular levels in this direction appearing as marker molecules at a certain period.75 Studies have shown that in the early stage of SONFH (1 to 3 months after onset), the concentration of IL-6 in the core tissue of the femoral head increases. Under immunofluorescence detection, it can be observed that the co-localization of Beclin1 and LC3 in bone cells increases, autophagosomes are scattered and evenly distributed, and the autophagic positive feedback loop is activated.76 However, as the disease progresses, that is, in the middle stage (4 to 6 months), a significant change in the composition of the inflammatory factor spectrum can be observed: the concentration of TNF-α increases, the concentration of IL-1β rises, the level of IL-6 decreases and tends to normal, accompanied by a marked enrichment of p62 protein in osteocytes, an increase of 70% in the nuclear translocation rate of NF-κB p65, and a decrease in the expression of lysosomal marker LAMP2, indicating that the molecular characteristics of autophagy have converted to pathogenic autophagy and are currently within the effective time window.77 In addition, some scholars established a mouse model of ONFH and found that after blocking the NF-κB signaling pathway in the ONFH model mice, the excessive activation of autophagy mediated by TNF-α was alleviated, and the survival rate of bone cells in the femoral head of the model mice increased.78 The above-mentioned research indicates that the changes in the concentration of inflammatory factors and the transformation of signaling pathways are both key targets for regulating the phenotypic variations of autophagy. Therefore, the bidirectional regulation of inflammatory signals can provide stage-specific strategies for disease intervention.79–81 For instance, in the early stage of inflammation, a mild enhancement of the IL-6/STAT3 signaling pathway can be utilized to boost the beneficial autophagy effect; however, in the stage where inflammation is out of control, targeted intervention in the TNF-α/NF-κB signaling pathway or improvement of lysosomal function (such as through the use of chloroquine derivatives, etc.) can be adopted to inhibit autophagic damage, thereby providing new ideas for the clinical treatment of inflammation-related bone diseases such as SONFH.
Autophagy-Lysosome System Dysfunction
The autophagy-lysosome system is an important degradation and recycling system within cells, and its functional integrity is the basis for maintaining cellular homeostasis.82 The normalization of lysosomal function is a prerequisite for ensuring the orderly and efficient process of autophagy and is the basis for the efficient degradation of intracellular macromolecules and some damaged organelles. Under pathophysiological conditions, hormones can disrupt the working efficiency of the autophagy-lysosome system, mainly through two pathways: on the one hand, hormones can directly disrupt the acidic microenvironment within lysosomes, causing an increase in the pH value within lysosomes. The imbalance of acid-base ratio will affect the activity of various acidic hydrolases (such as cathepsins, glycosidases, etc.) within lysosomes, reducing the degradation efficiency of autophagic substrates.83,84 On the other hand, hormones can induce the disruption of the structural stability of lysosomal membranes, increasing their permeability. As a result, active substances such as cathepsins, which are originally contained within lysosomes, leak out in large quantities into the cytosol, activating the caspases family of proteases in a cascade reaction, ultimately leading to the occurrence of apoptosis.85,86 When hormones continuously act on the body, the formation rate of autophagosomes is higher than the degradation rate of lysosomes, and the autophagic flux will be blocked. During this period, since autophagosomes are not decomposed in time, they accumulate in the cells and fail to exert their autophagic functions. Instead, it can lead to aggravated intracellular stress responses and accumulation of oxidative damage, turning effective autophagy into harm. That is to say, if autophagy-soluble lysosome system balance is disrupted due to long-term hormone interference, it can cause cell damage and tissue lesions.23,87 In addition, systematic detection of lysosome function‑related indicators should be performed in bone tissue from patients with SONFH, including lysosomal pH, lysosomal‑associated membrane protein 2 expression levels, cathepsin activity, and changes in lysosomal membrane permeability. This will help clarify the specific role of lysosomal dysfunction in autophagic imbalance in SONFH and provide a theoretical basis for therapeutic strategies targeting lysosomal repair.
Currently, several potential molecular switches involved in the phenotypic transition of autophagy have been reported. p53 exerts bidirectional regulation through differential nuclear-cytoplasmic localization: nuclear p53 transcriptionally activates autophagy-related genes, whereas cytoplasmic p53 inhibits Beclin1 activity. mTORC1 and AMPK constitute the core axis of energy metabolism sensing, negatively and positively regulating autophagy initiation, respectively. ROS modulate autophagic activity in a concentration‑dependent manner, promoting protective autophagy at low concentrations and inducing detrimental autophagy at high concentrations. NF-κB mediates the crosstalk between inflammatory signaling and autophagy, participating in phenotypic transition through transcriptional regulation of autophagy‑related genes. Beclin1, as a key initiator of autophagy, is also involved in the fine‑tuning of autophagic activity through its post‑translational modifications (eg, phosphorylation, ubiquitination). The synergistic interplay of these molecular switches determines whether autophagy exerts protective or detrimental effects in SONFH, and further elucidation of their regulatory network will provide a theoretical basis for stage‑specific targeted intervention.
Therapeutic Strategies Based on the Dual Nature of Autophagy
Drug Intervention for Precise Regulation of Autophagic Activity
Based on the duality of autophagy, the treatment of autophagy should also be precisely regulated in a phased and targeted manner: In the early stage of the disease course, the beneficial aspect of autophagy should be promoted to exert a protective effect on osteocytes and osteoblasts. At this stage, rapamycin-like mTOR inhibitors can be used to activate autophagy.88 In the middle and later stages, it is necessary to suppress excessive autophagy and apply a small amount of autophagy inhibitors regionally to effectively alleviate the excessive bone resorption caused by overly high autophagy in osteoclasts.89 Additionally, curcumin can exert the dual characteristics of autophagy by regulating the AMPK pathway, inducing beneficial autophagy at low concentrations and inhibiting excessive autophagy at high concentrations, which holds significant clinical significance.90
Repair Strategies Targeting the Autophagy-Lysosome System
To improve the function of lysosomes and restore autophagic flux while avoiding harmful autophagy, one approach is to apply lysosomal acidifiers (such as chloroquine derivatives) to enhance lysosomal activity and increase the degradation of autophagic substrates. On the other hand, gene therapy can be used to upregulate the expression of lysosome-associated membrane protein LAMP2, thereby enhancing the fusion ability of autophagosomes and lysosomes.91 Studies have shown that restoring lysosomal function can increase the efficiency of autophagic flux in bone cells damaged by hormones and reduce the rate of cell apoptosis.92
The Synergistic Effect of Combined Treatment Strategies
The combined application of autophagy regulation and traditional treatment methods can achieve an effect greater than the sum of their individual effects. For instance, core decompression combined with local injection of autophagy agonists can effectively improve blood circulation and simultaneously activate autophagy. Clinical observations have shown that it can increase the Harris hip score of early-stage SONFH patients by 10 to 15 points.41 The concurrent use of autophagy regulators with anti-osteoporosis drugs (such as bisphosphonates) can inhibit excessive autophagy and bone resorption of osteoclasts and delay femoral head collapse.16
A pH-Responsive Rapamycin Liposome Delivery System Based on Macrophage Membrane Biomimicry for Macrophage Autophagy Regulation
In the course of SONFH development, the imbalance of autophagy in macrophages is a crucial factor. Excessive autophagy can accelerate osteocyte apoptosis and bone mass reduction, while moderate activation of autophagy can clear damaged organelles and inhibit the release of inflammatory factors (such as TNF-α, IL-1β, etc), thereby improving the local microenvironment and promoting bone repair. Moreover, how to precisely regulate autophagy in macrophages also determines the therapeutic effect of SONFH, which is also a key challenge to be solved by an efficient targeted delivery system. However, traditional drug delivery systems have certain deficiencies in the regulation of macrophage autophagy: First, they lack good targeting ability and need to achieve macrophage targeting through chemical modification ligands such as mannose or RGD. However, the synthesis methods are complex, not stable enough, have low specificity, and are not easy to accumulate at the lesion sites of macrophages.93 Secondly, the hydrophobic drug rapamycin (an autophagy activator) is affected by the carrier itself and has a “lag” during the process of entering cells. Traditional drug delivery systems cannot respond to the acidic environment of lysosomes, and the drug is easily trapped in endosomes or lysosomes, failing to exert its effect in a timely manner.94 Thirdly, temporal and spatial inconsistency cannot simultaneously and efficiently connect itself and the connection points of the autophagy pathway.95 The novel rapamycin delivery system based on “cell bionic camouflage and intracellular environment response” can precisely regulate autophagy in macrophages in SONFH. Its innovation lies in the construction of a “phagocytosis-acidolysis-autophagy” full-pathway intracellular single-cell regulation model.96 This is mainly reflected in the three-layer intervention approach: The first layer is the “zero modification” targeted phagocytosis mimicking the macrophage membrane. By wrapping the liposome with the macrophage membrane, the inherent surface marker proteins (CD11b, CD45, CD47, MHC-II, etc.) of the liposome derived from macrophages are retained. Through “homotypic targeting”, the “self-recognition camouflage” of the macrophage membrane-labeled particles is achieved, which enables the macrophages to have a higher phagocytic rate for their own membrane-labeled particles. No other chemical modification ligands are needed to make the target-sized particles specifically taken up by macrophages. This not only avoids the cumbersome process of traditional ligand modification but also retains the natural membrane proteins, enhancing the system’s biocompatibility and blood stability, and improving the phagocytic effect of macrophages. As a result, the system can be used for precise targeting of local macrophages in SONFH.97 The second layer is the “instantaneous release” in response to lysosomal pH. That is, DOPE (conical lipid) and CHEMS (weak acid-sensitive lipid) are assembled to form the core structure of the liposome. The bilayer structure maintained by DOPE in the neutral environment of blood (pH 7.4) ensures the stability of the system during circulation. When liposomes enter macrophages, in the acidic environment of lysosomes (pH 5.0), DOPE undergoes a structural transformation to the “inverted hexagonal phase (HII phase)”, which prompts the rapid rupture of liposomes and triggers the “instantaneous release” of rapamycin within lysosomes. This “extracellular stability and intracellular triggering” response mode overcomes the characteristics of traditional drug delivery systems, which have slow release rates and do not respond to intracellular signals. Under this response mode, rapamycin can “take effect at specific sites” within lysosomes, achieving precise drug release at key sites for autophagy regulation.98 The third layer is the regulation of full-pathway activation, which achieves the full-process control of the entire process by tracking a series of events that occur at different stages of rapamycin within macrophages. Its characteristic lies in reaching the target site in the shortest time after drug release. The mTOR pathway instantaneously activates LC3-II and promotes the degradation of p62, thereby regulating the operation of the autophagy pathway. This represents a “phagocytic acidolysis autophagy” regulatory mode, achieving a seamless regulation path. It aims to precisely adjust autophagy function at different stages of the SONFH disease course, that is, to dynamically regulate the intervention intensity in response to the “double-edged sword” effect of autophagy during the course of SONFH.88,99 For the first time, it has integrated the biomimetic targeting of macrophage membranes, pH-responsive release, and rapamycin autophagy intervention, achieving synchronous optimization in targeting efficiency, drug release time and space, and pathway activation. Macrophages were extracted and purified from the lesion tissue of SONFH to improve the autophagy effect of macrophages in SONFH, realizing the design of an innovative dual-regulation delivery system. The scientific value of this system lies not only in breaking through the technical difficulties of traditional drug delivery systems, but also in establishing a “coordinated design strategy of cell natural characteristics, intracellular environment response, functional pathway activation”, which can provide new ideas for the research of “intracellular response type immune regulation drugs” and “cell subtype precise targeting platform”, and ultimately achieve the therapeutic effect of “strengthening beneficial effects and weakening harmful effects” in the regulation of autophagy balance in SONFH. (See Figure 3)
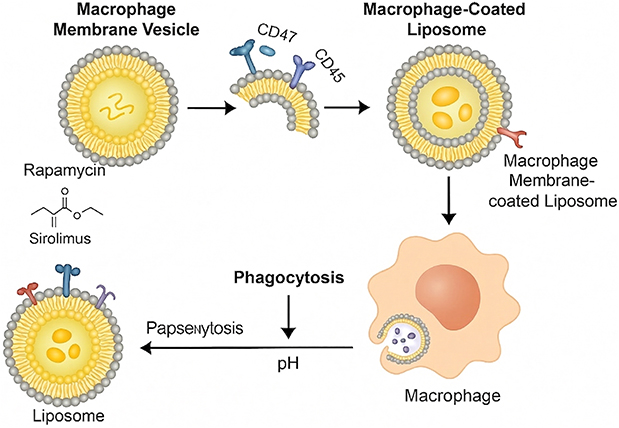 |
Figure 3 This figure illustrates the preparation and intracellular delivery process of macrophage membrane-coated liposomes: First, macrophage membrane vesicles carrying CD47 and CD45 molecules are isolated. Subsequently, these vesicles are combined with liposomes loaded with rapamycin (generic name: sirolimus) to construct macrophage membrane-coated liposomes. These coated liposomes can be taken up by macrophages via phagocytosis; afterward, with the involvement of pH regulation and intracellular related processes, the intracellular delivery of the liposomes is completed. |
Temporal-Synergistic Material System: Dynamic Integration Strategy of Autophagy Regulation and Bone Repair in SONFH
When treating SONFH, it is necessary to address issues such as microcirculation disorders within the bone, functional impairment of BMSCs, imbalance between osteogenesis and osteoclastogenesis, as well as persistent activation of oxidative stress and inflammation. Additionally, the double-edged sword nature of autophagy (lack of protective autophagy and excessive autophagy) is also one of the difficulties in its treatment.1,31 Based on methacrylated gelatin (Gel-MA), magnesium oxide-tannic acid complex (MgO2-TA), and copper/bioactive glass (CuBG), a temporally coordinated material system was constructed to improve the early microenvironment, regulate autophagy in the middle stage, and promote osteogenic repair in the later stage, aiming to provide a new strategy for the treatment of steroid-induced osteonecrosis of the femoral head (SONFH): Gel-MA, as a 3D scaffold substrate, has excellent biocompatibility and sustained-release ability, providing a favorable platform for cell adhesion and autophagy signal transmission. MgO2-TA serves as a microenvironment-responsive unit. In the hypoxic and acidic local microenvironment of SONFH, it can release oxygen and magnesium ions to generate oxygen, release magnesium ions to alleviate hypoxia stress and reduce excessive autophagy mediated by HIF-1α, lower oxidative stress and eliminate reactive oxygen species (ROS) to prevent abnormal activation of autophagy, and promote angiogenesis. CuBG, through its porous structure and sustained release of copper ions, induces protective autophagy (upregulation of LC3 and Beclin-1 expression) in BMSCs, promotes osteogenic differentiation (activation of Wnt/β-catenin and other pathways), and regulates macrophage reprogramming, thereby preventing autophagic disorders caused by inflammation. The temporal coordination mechanism of this system is highly consistent with the pathological development process of SONFH. It exerts an anti-infection effect through photothermal sterilization in the 0–4 weeks before surgery. The use of MgO2-TA to repair hypoxia and oxidative stress conditions, improve tissue autophagy and induce its initiation of angiogenesis. From 4 to 8 weeks after the operation, CuBG activates protective autophagy and osteogenic signals to enhance the function of BMSCs. From 8 to 12 weeks after the operation, the continuous release of material ions during the degradation process promotes bone matrix mineralization and the balance of osteogenesis and osteoclastogenesis, ultimately maintaining autophagic homeostasis. Therefore, through this temporal coordination system, effective regulation of the pathway of “microenvironment improvement, autophagy balance, and bone reconstruction” can be achieved. Finally, it is necessary to further improve the performance of the system materials and clarify the related issues such as its molecular mechanism, in order to provide a complete solution for the precise treatment of SONFH. This system may provide a strong support for the clinical application of SONFH. (See Figure 4)
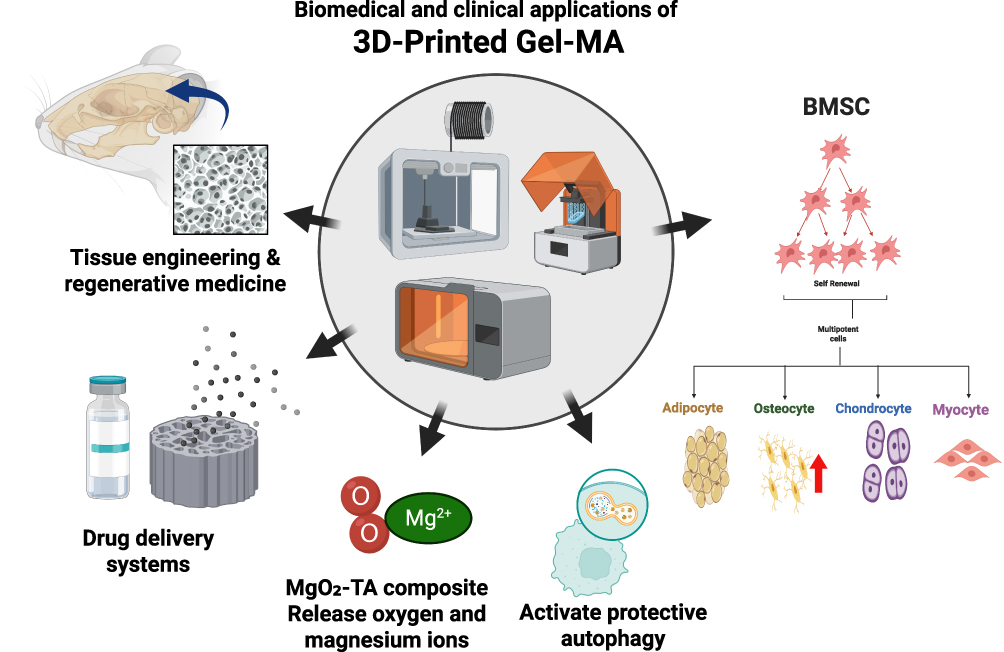 |
Figure 4 This figure illustrates the multi-domain applications and related functional mechanisms of 3D-printed Gel-MA (gelatin methacryloyl) materials: In the center of the figure are 3D printing devices, which are used to fabricate Gel-MA-based materials; its application areas cover tissue engineering and regenerative medicine, drug delivery systems, and other fields. Meanwhile, this material can combine with MgO2-TA composites to release oxygen and magnesium ions, and activate protective autophagy. In addition, it can support the survival of bone marrow mesenchymal stem cells (BMSCs) and promote their differentiation into multiple cell types including adipocytes, osteocytes, chondrocytes, and myocytes. Note: The red upward arrow denotes enhancement. |
Summary and Outlook
At the early stage or under mild stimulation, autophagy exerts a protective effect on osteocytes, osteoblasts and vascular endothelial cells, eliminating damaged organelles, maintaining energy homeostasis and inhibiting apoptosis. Under long-term and high-dose hormone stimulation, autophagy function is disrupted, manifested as excessive activation of autophagy in osteocytes, hyperactive autophagy in osteoclasts, and autophagy dysfunction in bone marrow mesenchymal stem cells, leading to abnormal bone metabolism, bone marrow adipogenesis, and aggravated blood supply insufficiency. This transformation from “autophagy balance of cell communities and subgroups” to “autophagy imbalance of cell communities and subgroups” is influenced by multiple factors, including oxidative stress, inflammatory signaling pathways, and dysfunction of the autophagy-lysosome system. At present, the above-mentioned imbalanced state can be intervened through phased drug administration, autophagy, lysosomal function repair, and combined drug administration methods. With the help of biomimetic macrophage membrane delivery systems or temporally coordinated materials, new paths are opened up for the dynamic regulation of autophagy. Further research is needed to identify the molecular switches that regulate the phenotypic transition of autophagy, clarify the regulatory mechanisms of key autophagy proteins mediated by ROS and inflammatory factors, and draw the spatiotemporal expression maps of autophagy-related genes in specific intervals through single-cell sequencing technology combined with spatial transcriptomics and other techniques. This will help determine the autophagy patterns of different cell types in various cell interactions and construct the autophagy-mediated signaling network among cell populations or subpopulations, thereby explaining the role of “autophagy balance in cell communities and subpopulations” in maintaining bone homeostasis. Clinically speaking, it is also very necessary to apply autophagy activity detection to the research of preclinical diagnosis, staged treatment and other aspects. In addition, efforts should be made to actively optimize the research and development of autophagy regulators such as rapamycin derivatives and curcuminoids, accelerate the transformation of related drugs to clinical research, and give full play to their roles. In addition, when promoting multi-disciplinary exchanges, it is possible to achieve simultaneous structural repair and functional regulation by constructing 3D printed bone organoids loaded with autophagy regulators; and to establish a large database composed of genes, phenotypes, therapeutic effects, etc. through artificial intelligence, and build prediction models for genes, phenotypes, and therapeutic effects to promote the implementation of individualized treatment in clinical applications. Explore the regulatory targets of active components in traditional Chinese medicine such as tanshinone and icaritin, and realize a comprehensive treatment strategy combining traditional Chinese and Western medicine. Based on the above, we have fully recognized the dialectical relationship of “precise regulation of autophagic balance”, which provides new ideas for analyzing the mechanism of occurrence and development of such diseases. Ultimately, it aims to precisely regulate “autophagic balance precise regulation”, transforming the treatment strategy from symptomatic treatment to etiological intervention, laying a foundation for improving the quality of life of patients.
In summary, future research should focus on the following key directions. First, it is essential to identify the molecular switches that regulate the phenotypic transition of autophagy and elucidate their mechanisms of action at different pathological stages of SONFH. Second, leveraging cutting-edge technologies such as single-cell sequencing and spatial transcriptomics will enable the construction of spatiotemporal expression maps of autophagy-related genes across distinct cell subpopulations in bone tissue, revealing the heterogeneity and network interactions of autophagy regulation among cells. Third, integrating artificial intelligence to develop multidimensional prediction models—combining genomic, phenotypic, and treatment response data—will provide decision-making support for individualized and dynamic precision regulation of autophagy. Furthermore, active efforts should be made to advance the clinical translation of autophagy modulators (eg, rapamycin derivatives, active components of traditional Chinese medicine) and to explore the optimal timing and administration strategies for stage-specific intervention in SONFH. Through these efforts, the regulation of autophagy is expected to evolve from “symptom-based intervention” to “etiology-based treatment”, ultimately offering more precise and effective clinical solutions for patients with SONFH. In the future, cutting-edge technologies such as single-cell sequencing and spatial transcriptomics should be employed to map the spatiotemporal expression profiles of autophagy-related genes across distinct cell subpopulations in bone tissue, revealing the heterogeneity and network interactions of autophagic regulation among cells. Meanwhile, artificial intelligence should be integrated to construct multidimensional prediction models that combine genomic, phenotypic, and treatment response data, providing decision-making support for individualized and dynamic precision regulation of autophagy.100–107
Data Sharing Statement
All data in this article are available from the corresponding author Haiyan Zhao upon reasonable request.
Acknowledgments
This publication is funded in part by the National Natural Science Foundation of China (Project Approval number: 82060394); National Natural Science Foundation of China (Project Approval number: 82560433).
Disclosure
The authors report no conflicts of interest in this work.
References
1. Huang C, Wen Z, Niu J, et al. Steroid-induced osteonecrosis of the femoral head: novel insight into the roles of bone endothelial cells in pathogenesis and treatment. Front Cell Dev Biol. 2021;9:777697. doi:10.3389/fcell.2021.777697
2. Zhong C, Xu H, Chen J, et al. Human umbilical cord mesenchymal stem cells prevent steroid-induced avascular necrosis of the femoral head by modulating cellular autophagy. Biomedicines. 2024;12(12):2817. doi:10.3390/biomedicines12122817
3. Liang XZ, Luo D, Chen YR, et al. Identification of potential autophagy-related genes in steroid-induced osteonecrosis of the femoral head via bioinformatics analysis and experimental verification. J Orthop Surg Res. 2022;17(1):86. doi:10.1186/s13018-022-02977-x
4. Guo YF, Su T, Yang M, et al. The role of autophagy in bone homeostasis. J Cell Physiol. 2021;236(6):4152–19. doi:10.1002/jcp.30111
5. Zhu C, Shen S, Zhang S, et al. Autophagy in bone remodeling: a regulator of oxidative stress. Front Endocrinol. 2022;13:898634. doi:10.3389/fendo.2022.898634
6. Gomez-Puerto MC, Verhagen LP, Braat AK, et al. Activation of autophagy by FOXO3 regulates redox homeostasis during osteogenic differentiation. Autophagy. 2016;12(10):1804–1816. doi:10.1080/15548627.2016.1203484
7. Fitzwalter BE, Towers CG, Sullivan KD, et al. Autophagy inhibition mediates apoptosis sensitization in cancer therapy by relieving FOXO3a turnover. Dev Cell. 2018;44(5):555–565e553. doi:10.1016/j.devcel.2018.02.014
8. Rosenfeldt MT, O’prey J, Morton JP, et al. p53 status determines the role of autophagy in pancreatic tumour development. Nature. 2013;504(7479):296–300. doi:10.1038/nature12865
9. Jung S, Jeong H, Yu SW. Autophagy as a decisive process for cell death. Exp Mol Med. 2020;52(6):921–930. doi:10.1038/s12276-020-0455-4
10. Doherty J, Baehrecke EH. Life, death and autophagy. Nat Cell Biol. 2018;20(10):1110–1117. doi:10.1038/s41556-018-0201-5
11. Cooper KF. Till death do us part: the marriage of autophagy and apoptosis. Oxid Med Cell Longev. 2018;2018(1):4701275. doi:10.1155/2018/4701275
12. Cabrera S, Marino G, Fernandez AF, et al. Autophagy, proteases and the sense of balance. Autophagy. 2010;6(7):961–963. doi:10.4161/auto.6.7.13065
13. Wong PM, Feng Y, Wang J, et al. Regulation of autophagy by coordinated action of mTORC1 and protein phosphatase 2A. Nat Commun. 2015;6(1):8048. doi:10.1038/ncomms9048
14. Yao W, Dai W, Jiang JX, et al. Glucocorticoids and osteocyte autophagy. Bone. 2013;54(2):279–284. doi:10.1016/j.bone.2013.01.034
15. Jia J, Yao W, Guan M, et al. Glucocorticoid dose determines osteocyte cell fate. FASEB J. 2011;25(10):3366–3376. doi:10.1096/fj.11-182519
16. Wang J, Zhang Y, Cao J, et al. The role of autophagy in bone metabolism and clinical significance. Autophagy. 2023;19(9):2409–2427. doi:10.1080/15548627.2023.2186112
17. Xia X, Kar R, Gluhak-Heinrich J, et al. Glucocorticoid-induced autophagy in osteocytes. J Bone Miner Res. 2010;25(11):2479–2488. doi:10.1002/jbmr.160
18. Kar R, Riquelme MA, Hua R, et al. Glucocorticoid-induced autophagy protects osteocytes against oxidative stress through activation of MAPK/ERK signaling. JBMR Plus. 2019;3(4):e10077. doi:10.1002/jbm4.10077
19. Wei L, Chai S, Yue C, et al. Resveratrol protects osteocytes against oxidative stress in ovariectomized rats through AMPK/JNK1-dependent pathway leading to promotion of autophagy and inhibition of apoptosis. Cell Death Discov. 2023;9(1):16. doi:10.1038/s41420-023-01331-2
20. Liang X, Ma Q, Wang L, et al. Geraniin promotes osteoblast proliferation, bone formation, and autophagy by regulating the PI3K/Akt/mTOR cascade to improve glucocorticoid-induced osteoporosis. Calcif Tissue Int. 2025;116(1):77. doi:10.1007/s00223-025-01387-5
21. Han Y, Zhang L, Xing Y, et al. Autophagy relieves the function inhibition and apoptosis‑promoting effects on osteoblast induced by glucocorticoid. Int J Mol Med. 2018;41(2):800–808. doi:10.3892/ijmm.2017.3270
22. Liu S, Fang T, Yang L, et al. Gastrodin protects MC3T3-E1osteoblasts from dexamethasone-induced cellular dysfunction and promotes bone formation via induction of the NRF2 signaling pathway. Int J Mol Med. 2018;41(4):2059–2069. doi:10.3892/ijmm.2018.3414
23. Wang T, Liu X, He C. Glucocorticoid-induced autophagy and apoptosis in bone. Apoptosis. 2020;25(3–4):157–168. doi:10.1007/s10495-020-01599-0
24. Zhou M, Liu L, Xu Y, et al. Effects of osteoblast autophagy on glucocorticoid-induced femoral head necrosis. Jt Dis Relat Surg. 2020;31(3):411–418. doi:10.5606/ehc.2020.73036
25. Li MY, Shen HH, Cao XY, et al. Targeting a mTOR/autophagy axis: a double-edged sword of rapamycin in spontaneous miscarriage. Biomed Pharmacother. 2024;177:116976. doi:10.1016/j.biopha.2024.116976
26. Yuan J, Gao YS, Liu DL, et al. PINK1-mediated mitophagy contributes to glucocorticoid-induced cathepsin K production in osteocytes. J Orthop Translat. 2023;38:229–240. doi:10.1016/j.jot.2022.11.003
27. Piemontese M, Onal M, Xiong J, et al. Suppression of autophagy in osteocytes does not modify the adverse effects of glucocorticoids on cortical bone. Bone. 2015;75:18–26. doi:10.1016/j.bone.2015.02.005
28. Li X, Li YS, Li LJ, et al. Overactivated autophagy contributes to steroid-induced avascular necrosis of the femoral head. Exp Ther Med. 2017;14(1):367–372. doi:10.3892/etm.2017.4508
29. Peng P, Nie Z, Sun F, et al. Glucocorticoids induce femoral head necrosis in rats through the ROS/JNK/c-Jun pathway. FEBS Open Bio. 2021;11(1):312–321. doi:10.1002/2211-5463.13037
30. Yin Z, Wu L, Huang J, et al. Immunological mechanisms in steroid-induced osteonecrosis of the femoral head. Front Immunol. 2025;16:1626617. doi:10.3389/fimmu.2025.1626617
31. Chen K, Liu Y, He J, et al. Steroid-induced osteonecrosis of the femoral head reveals enhanced reactive oxygen species and hyperactive osteoclasts. Int J Biol Sci. 2020;16(11):1888–1900. doi:10.7150/ijbs.40917
32. Tang H, Yuan L, Xu Z, et al. Glucocorticoids induce femoral head necrosis in rats through the HIF-1alpha/VEGF signaling pathway. Sci Rep. 2025;15(1):29205. doi:10.1038/s41598-025-15018-4
33. Luo H, Wei J, Wu S, et al. Elucidating the role of the GC/GR/GLUT1 axis in steroid-induced osteonecrosis of the femoral head: a proteomic approach. Bone. 2024;183:117074. doi:10.1016/j.bone.2024.117074
34. Su Q, Chen J. Regulatory effect and mechanism of tanshinone I on cell apoptosis in steroid-induced osteonecrosis of the femoral head. Kaohsiung J Med Sci. 2025;41(12):e70086. doi:10.1002/kjm2.70086
35. Shao W, Wang B, Wang P, et al. Inhibition of sympathetic tone via hypothalamic descending pathway propagates glucocorticoid-induced endothelial impairment and osteonecrosis of the femoral head. Bone Res. 2024;12(1):64. doi:10.1038/s41413-024-00371-3
36. Yao X, Yu S, Jing X, et al. PTEN inhibitor VO-OHpic attenuates GC-associated endothelial progenitor cell dysfunction and osteonecrosis of the femoral head via activating Nrf2 signaling and inhibiting mitochondrial apoptosis pathway. Stem Cell Res Ther. 2020;11(1):140. doi:10.1186/s13287-020-01658-y
37. Cui Y, Alken A, Wang W, et al. YTHDF3-associated m6A regulation and cuproptosis-related gene expression in steroid-induced osteonecrosis of the femoral head. J Mol Histol. 2025;56(5):279. doi:10.1007/s10735-025-10544-x
38. Luo P, Gao F, Han J, et al. The role of autophagy in steroid necrosis of the femoral head: a comprehensive research review. Int Orthop. 2018;42(7):1747–1753. doi:10.1007/s00264-018-3994-8
39. Zhang J, Cao J, Liu Y, et al. Advances in the pathogenesis of steroid-associated osteonecrosis of the femoral head. Biomolecules. 2024;14(6):667. doi:10.3390/biom14060667
40. Wang Y, Zhang Y, Lu H, et al. Integrative analysis of cellular autophagy-related genes in steroid-induced osteonecrosis of the femoral head. Am J Transl Res. 2023;15(9):5850–5872.
41. Liu B, Gao F, Xiu X, et al. Denosumab can prevent collapse in patients with early-stage steroid-induced osteonecrosis of the femoral head by inhibiting osteoclasts and autophagy. Orthop Surg. 2023;15(1):256–265. doi:10.1111/os.13584
42. Wang Q, Yang Z, Li Q, et al. Lithium prevents glucocorticoid-induced osteonecrosis of the femoral head by regulating autophagy. J Cell Mol Med. 2024;28(10):e18385. doi:10.1111/jcmm.18385
43. Chang C, Greenspan A, Gershwin ME. The pathogenesis, diagnosis and clinical manifestations of steroid-induced osteonecrosis. J Autoimmun. 2020;110:102460. doi:10.1016/j.jaut.2020.102460
44. Liu S, Huang Y, Wang C, et al. Epimedium protects steroid-induced avascular necrosis of femoral head in rats by inhibiting autophagy. Exp Ther Med. 2018;16(6):5047–5052. doi:10.3892/etm.2018.6827
45. Huang Z, Wang Q, Zhang T, et al. Hyper-activated platelet lysates prevent glucocorticoid-associated femoral head necrosis by regulating autophagy. Biomed Pharmacother. 2021;139:111711. doi:10.1016/j.biopha.2021.111711
46. Gao Y, Chen C, Liu R, et al. Research progress of connexin 43 mediated gap junction communication regulating bone metabolism in glucocorticoid-induced osteonecrosis of the femoral head. Exp Cell Res. 2025;449(1):114598. doi:10.1016/j.yexcr.2025.114598
47. Zhang Y, Ma L, Lu E, et al. Atorvastatin upregulates microRNA-186 and inhibits the TLR4-mediated MAPKs/NF-kappaB pathway to relieve steroid-induced avascular necrosis of the femoral head. Front Pharmacol. 2021;12:583975. doi:10.3389/fphar.2021.583975
48. Zhang S, Wang C, Shi L, et al. Beware of steroid-induced avascular necrosis of the femoral head in the treatment of COVID-19-experience and lessons from the SARS epidemic. Drug Des Devel Ther. 2021;15:983–995. doi:10.2147/DDDT.S298691
49. Tang B, Chen Y, Zhao P, et al. MiR-601-induced BMSCs senescence accelerates steroid-induced osteonecrosis of the femoral head progression by targeting SIRT1. Cell Mol Life Sci. 2023;80(9):261. doi:10.1007/s00018-023-04903-8
50. Li L, Zhao S, Leng Z, et al. Pathological mechanisms and related markers of steroid-induced osteonecrosis of the femoral head. Ann Med. 2024;56(1):2416070. doi:10.1080/07853890.2024.2416070
51. Huang C, Qing L, Xiao Y, et al. Insight into steroid-induced ONFH: the molecular mechanism and function of epigenetic modification in mesenchymal stem cells. Biomolecules. 2023;14(1):4. doi:10.3390/biom14010004
52. Cao R, Li H, Liu G, et al. Aging and autophagic phenotypic changes in bone marrow mesenchymal stem cells in glucocorticoid-induced osteonecrosis. Int Immunopharmacol. 2025;152:114389. doi:10.1016/j.intimp.2025.114389
53. Rahman MA, Ahmed KR, Haque F, et al. Recent advances in cellular signaling interplay between redox metabolism and autophagy modulation in cancer: an overview of molecular mechanisms and therapeutic interventions. Antioxidants. 2023;12(2):428. doi:10.3390/antiox12020428
54. Medvedev R, Ploen D, Spengler C, et al. HCV-induced oxidative stress by inhibition of Nrf2 triggers autophagy and favors release of viral particles. Free Radic Biol Med. 2017;110:300–315. doi:10.1016/j.freeradbiomed.2017.06.021
55. Zhou J, Li XY, Liu YJ, et al. Full-coverage regulations of autophagy by ROS: from induction to maturation. Autophagy. 2022;18(6):1240–1255. doi:10.1080/15548627.2021.1984656
56. Azad MB, Chen Y, Gibson SB. Regulation of autophagy by reactive oxygen species (ROS): implications for cancer progression and treatment. Antioxid Redox Signal. 2009;11(4):777–790. doi:10.1089/ars.2008.2270
57. Kroemer G, Marino G, Levine B. Autophagy and the integrated stress response. Mol Cell. 2010;40(2):280–293. doi:10.1016/j.molcel.2010.09.023
58. Khan AQ, Rashid K, Alamodi AA, et al. Reactive oxygen species (ROS) in cancer pathogenesis and therapy: an update on the role of ROS in anticancer action of benzophenanthridine alkaloids. Biomed Pharmacother. 2021;143:112142. doi:10.1016/j.biopha.2021.112142
59. Gupta U, Ghosh S, Wallace CT, et al. Increased LCN2 (lipocalin 2) in the RPE decreases autophagy and activates inflammasome-ferroptosis processes in a mouse model of dry AMD. Autophagy. 2023;19(1):92–111. doi:10.1080/15548627.2022.2062887
60. Scherz-Shouval R, Elazar Z. Regulation of autophagy by ROS: physiology and pathology. Trends Biochem Sci. 2011;36(1):30–38. doi:10.1016/j.tibs.2010.07.007
61. Baehrecke EH. Autophagy: dual roles in life and death? Nat Rev Mol Cell Biol. 2005;6(6):505–510. doi:10.1038/nrm1666
62. Mendonca LS, Moreira R, Henriques D, et al. Autophagy- and oxidative stress-related protein deregulation mediated by extracellular vesicles of human MJD/SCA3 iPSC-derived neuroepithelial stem cells and differentiated neural cultures. Cell Death Dis. 2025;16(1):383. doi:10.1038/s41419-025-07659-0
63. Grujicic J, Allen AR. MnSOD mimetics in therapy: exploring their role in combating oxidative stress-related diseases. Antioxidants. 2024;13(12). doi:10.3390/antiox13121444
64. Gong G, Wan W, Zhang X, et al. Management of ROS and regulatory cell death in myocardial ischemia-reperfusion injury. Mol Biotechnol. 2025;67(5):1765–1783. doi:10.1007/s12033-024-01173-y
65. Manojlovic M, Zafirovic S, Tomic Naglic D, et al. Mitochondrial dysfunction, reactive oxygen species, and diabetes mellitus - A triangular relationship: a review. Biomol Biomed. 2025;26(4):547–558. doi:10.17305/bb.2025.13145
66. Monkkonen T, Debnath J. Inflammatory signaling cascades and autophagy in cancer. Autophagy. 2018;14(2):190–198. doi:10.1080/15548627.2017.1345412
67. Cadwell K. Crosstalk between autophagy and inflammatory signalling pathways: balancing defence and homeostasis. Nat Rev Immunol. 2016;16(11):661–675. doi:10.1038/nri.2016.100
68. Matsuzawa-Ishimoto Y, Hwang S, Cadwell K. Autophagy and inflammation. Annu Rev Immunol. 2018;36(1):73–101. doi:10.1146/annurev-immunol-042617-053253
69. Deretic V. Autophagy in inflammation, infection, and immunometabolism. Immunity. 2021;54(3):437–453. doi:10.1016/j.immuni.2021.01.018
70. Lim J, Murthy A. Controlling inflammation by selective autophagy. Cell Death Differ. 2018;25(5):825–827. doi:10.1038/s41418-018-0096-5
71. Tang H, Zhu S, Chen K, et al. IL-17A regulates autophagy and promotes osteoclast differentiation through the ERK/mTOR/Beclin1 pathway. PLoS One. 2023;18(2):e0281845. doi:10.1371/journal.pone.0281845
72. Ge Y, Huang M, Yao YM. Autophagy and proinflammatory cytokines: interactions and clinical implications. Cytokine Growth Factor Rev. 2018;43:38–46. doi:10.1016/j.cytogfr.2018.07.001
73. Tong X, Ganta RR, Liu Z. AMP-activated protein kinase (AMPK) regulates autophagy, inflammation and immunity and contributes to osteoclast differentiation and functionabs. Biol Cell. 2020;112(9):251–264. doi:10.1111/boc.202000008
74. Lyu Q, Wawrzyniuk M, Rutten V, et al. Hsp70 and NF-kB mediated control of innate inflammatory responses in a canine macrophage cell line. Int J Mol Sci. 2020;21(18):6464. doi:10.3390/ijms21186464
75. Shao J, Ding Z, Peng J, et al. MiR-146a-5p promotes IL-1beta-induced chondrocyte apoptosis through the TRAF6-mediated NF-kB pathway. Inflamm Res. 2020;69(6):619–630. doi:10.1007/s00011-020-01346-w
76. Tu J, Li W, Zhang Y, et al. Simvastatin inhibits IL-1beta-induced apoptosis and extracellular matrix degradation by suppressing the NF-kB and MAPK pathways in nucleus pulposus cells. Inflammation. 2017;40(3):725–734. doi:10.1007/s10753-017-0516-6
77. Zheng LW, Wang WC, Mao XZ, et al. TNF-alpha regulates the early development of avascular necrosis of the femoral head by mediating osteoblast autophagy and apoptosis via the p38 MAPK/NF-kappaB signaling pathway. Cell Biol Int. 2020;44(9):1881–1889. doi:10.1002/cbin.11394
78. Zhang XY, Li HN, Chen F, et al. Icariin regulates miR-23a-3p-mediated osteogenic differentiation of BMSCs via BMP-2/Smad5/Runx2 and WNT/beta-catenin pathways in osteonecrosis of the femoral head. Saudi Pharm J. 2021;29(12):1405–1415. doi:10.1016/j.jsps.2021.10.009
79. Martinez GP, Zabaleta ME, Di giulio C, et al. The role of chloroquine and hydroxychloroquine in immune regulation and diseases. Curr Pharm Des. 2020;26(35):4467–4485. doi:10.2174/1381612826666200707132920
80. Shams S, Martinez JM, Dawson JRD, et al. The therapeutic landscape of rheumatoid arthritis: current state and future directions. Front Pharmacol. 2021;12:680043. doi:10.3389/fphar.2021.680043
81. Huang J, Wu L, Zhao Y, et al. Programmed cell death of chondrocytes, synovial cells, osteoclasts, and subchondral bone cells in osteoarthritis. J Inflamm Res. 2025;18:12323–12360. doi:10.2147/JIR.S514309
82. Cid-Diaz T, Leal-Lopez S, Fernandez-Barreiro F, et al. Obestatin signalling counteracts glucocorticoid-induced skeletal muscle atrophy via NEDD4/KLF15 axis. J Cachexia Sarcopenia Muscle. 2021;12(2):493–505. doi:10.1002/jcsm.12677
83. Kudriaeva AA, Sokolov AV, Belogurov AAJ. Stochastics of degradation: the autophagic-lysosomal system of the cell. Acta Naturae. 2020;12(1):18–32. doi:10.32607/actanaturae.10936
84. He Y, Xu Y, Zhang C, et al. Identification of a lysosomal pathway that modulates glucocorticoid signaling and the inflammatory response. Sci Signal. 2011;4(180):ra44. doi:10.1126/scisignal.2001450
85. Sato M, Ueda E, Konno A, et al. Glucocorticoids negatively regulates chaperone mediated autophagy and microautophagy. Biochem Biophys Res Commun. 2020;528(1):199–205. doi:10.1016/j.bbrc.2020.04.132
86. Ma RD, Zhou GJ, Qu M, et al. Corticosterone induces neurotoxicity in PC12 cells via disrupting autophagy flux mediated by AMPK/mTOR signaling. CNS Neurosci Ther. 2020;26(2):167–176. doi:10.1111/cns.13212
87. Tang Q, Zheng G, Feng Z, et al. Trehalose ameliorates oxidative stress-mediated mitochondrial dysfunction and ER stress via selective autophagy stimulation and autophagic flux restoration in osteoarthritis development. Cell Death Dis. 2017;8(10):e3081. doi:10.1038/cddis.2017.453
88. Chen R, Yang C, Yang F, et al. Targeting the mTOR-autophagy axis: unveiling therapeutic potentials in osteoporosis. Biomolecules. 2024;14(11):1452. doi:10.3390/biom14111452
89. Wang K. The potential therapeutic role of curcumin in osteoporosis treatment: based on multiple signaling pathways. Front Pharmacol. 2024;15:1446536. doi:10.3389/fphar.2024.1446536
90. Pantovic A, Krstic A, Janjetovic K, et al. Coordinated time-dependent modulation of AMPK/Akt/mTOR signaling and autophagy controls osteogenic differentiation of human mesenchymal stem cells. Bone. 2013;52(1):524–531. doi:10.1016/j.bone.2012.10.024
91. Mauthe M, Orhon I, Rocchi C, et al. Chloroquine inhibits autophagic flux by decreasing autophagosome-lysosome fusion. Autophagy. 2018;14(8):1435–1455. doi:10.1080/15548627.2018.1474314
92. Hao Y, Fan X, Huang X, et al. Recovery of lysosomal acidification and autophagy flux by attapulgite nanorods: therapeutic potential for lysosomal disorders. Biomolecules. 2025;15(5):728. doi:10.3390/biom15050728
93. Dalle Vedove E, Costabile G, Merkel OM. Mannose and mannose-6-phosphate receptor-targeted drug delivery systems and their application in cancer therapy. Adv Healthc Mater. 2018;7(14):e1701398. doi:10.1002/adhm.201701398
94. Tavakol S, Ashrafizadeh M, Deng S, et al. Autophagy modulators: mechanistic aspects and drug delivery systems. Biomolecules. 2019;9(10). doi:10.3390/biom9100530
95. Silva VR, Neves SP, Santos LS, et al. Challenges and therapeutic opportunities of autophagy in cancer therapy. Cancers. 2020;12(11). doi:10.3390/cancers12113461
96. Liu B, Yan W, Luo L, et al. Macrophage membrane camouflaged reactive oxygen species responsive nanomedicine for efficiently inhibiting the vascular intimal hyperplasia. J Nanobiotechnology. 2021;19(1):374. doi:10.1186/s12951-021-01119-5
97. Khan M, Ullah R, Wang G, et al. Macrophage membrane-functionalized nanotherapeutics for tumor targeted therapy. Theranostics. 2025;15(10):4823–4847. doi:10.7150/thno.108875
98. Hu F, Yue H, Lu T, et al. Cytosolic delivery of HBsAg and enhanced cellular immunity by pH-responsive liposome. J Control Release. 2020;324:460–470. doi:10.1016/j.jconrel.2020.05.042
99. Wang S, Deng Z, Ma Y, et al. The role of autophagy and mitophagy in bone metabolic disorders. Int J Biol Sci. 2020;16(14):2675–2691. doi:10.7150/ijbs.46627
100. Jin J, Cen Q, Wang H, et al. New perspective on neurodegenerative diseases: focusing on the interaction between autophagy and oxidative stress and targeted strategies. Neurobiol Dis. 2026;220:107283. doi:10.1016/j.nbd.2026.107283
101. Ryu JI, Jung J, Kim JY. Sir2 regulates selective autophagy in stationary-phase yeast cells. Microb Cell. 2026;13:1–12. doi:10.15698/mic2026.01.864
102. Yu JX, Zhang WX, Li PY, et al. Curcumin rescues oxidative stress-induced impairment of PINK1/Parkin pathway-mediated mitophagy in APOE4-expressing astrocytes. Mol Neurobiol. 2026;63(1):454. doi:10.1007/s12035-026-05744-9
103. Fine N, Rockel JS, Kapoor M. Transcriptomics in the study of bone and cartilage. Curr Osteoporos Rep. 2026;24(1):5. doi:10.1007/s11914-026-00951-8
104. Ren C, Sun K, Wu R, et al. Single-cell RNA sequencing revealed cell heterogeneity in sagittal suture mesenchyme. Front Cell Dev Biol. 2026;14:1725375. doi:10.3389/fcell.2026.1725375
105. Xia T, Yang C, Li X, et al. Suppression of exosomal miR-199a-3p, a differentially expressed miRNA in steroid-induced osteonecrosis of the femoral head, promotes cell proliferation, osteogenesis, and angiogenesis while inhibiting dexamethasone-induced apoptosis. Front Bioeng Biotechnol. 2026;14:1709739. doi:10.3389/fbioe.2026.1709739
106. Xu H, Huang H, Zou K, et al. From pathogenesis to treatment: the role of autophagic cell death in GONFH and its potential mitigation by naringenin. Theranostics. 2026;16(9):4804–4820. doi:10.7150/thno.129809
107. Quan H, He Y, Xiao P, et al. Scaffold-mediated microenvironmental modulation targeting osteoclasts for ONFH niche reprogramming. Research. 2025;8:1027. doi:10.34133/research.1027
 © 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.