Back to Journals » Drug Design, Development and Therapy » Volume 19
Advances in Pharmacological Activities, Biosynthesis, and Structural Modification of Ursodeoxycholic Acid (UDCA): A Review
Authors Li P, Wang J, Hao H, Chen X, He X, Shao Q
Received 31 July 2025
Accepted for publication 27 November 2025
Published 5 December 2025 Volume 2025:19 Pages 10775—10810
DOI https://doi.org/10.2147/DDDT.S557300
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Anastasios Lymperopoulos
Ping Li,1 Jia Wang,2 Han Hao,2 Xufei Chen,2 Xirui He,3 Qing Shao1
1Pharmacy Department, Xi’an Mental Health Center, Xi’an, Shaanxi, 710061, People’s Republic of China; 2Key Laboratory of Resource Biology and Modern Biotechnology in Western China, College of Life Science, Northwest University, Xi’an, Shaanxi, 710069, People’s Republic of China; 3School of Bioengineering, Zhuhai Campus, Zunyi Medical University, Zhuhai, Guangdong, 519041, People’s Republic of China
Correspondence: Qing Shao, Pharmacy Department, Xi’an Mental Health Center, Chang’an District, Xi’an, Shaanxi, 710061, People’s Republic of China, Tel +86 13891842435, Email [email protected]
Abstract: UDCA is a natural steroid in bear bile, which has important medicinal value. It is used to treat hepatobiliary diseases in clinic. Due to its unique molecular skeleton, UDCA has a wide range of biological activities, special target and low toxicity, and is considered to be a powerful structural mother nucleus, which has attracted more and more researchers’ attention and become the focus of attention again. So far, UDCA derivatives with diverse structures have been designed and their biological activities have been extensively studied. In this paper, the pharmacological activities, biosynthesis and structural modification of UDCA were systematically discussed. It is believed that with the deepening of research, more UDCA derivatives with better medicinal properties will become drugs and better serve human health.
Keywords: ursodeoxycholic acid, pharmacological activities, biosynthesis, structural modification
Introduction
The medicinal value of animals as a natural resource cannot be ignored. Animal-derived remedies constitute a vital component of traditional Chinese medicine. Classified among China’s scarcest medicinal animal products is bear bile, the earliest written record of the use of bear bile to treat diseases can be traced back to the Jin Dynasty’s “Prescriptions for Emergent Reference”, while the earliest summary of its clear efficacy of bear bile can be found in the Tang Dynasty’s “ Theory of Medicinal Property”.1 Bear bile refers to the dried bile of the brown bear (Uesus arctos Linnaeus), the black bear (Selenarctos thibctanus Covier), of the genus Ursus arctos or the genus Ursus, family Ursidae. In traditional Chinese medicine, bear bile exerts pharmacological effects of calming the liver, harmonizing bile, relieving fever, detoxifying, spasmolysis, and protecting the liver.2 Modern medicine shows that bear bile has anti-inflammatory,3,4 anti-apoptotic,5 antioxidant,6 and anti-tumor activities,7 and can treat various liver diseases,8 which is basically in line with the understanding of Chinese medicine. Bear bile is currently applied therapeutically for multiple disorders in paediatrics, gynaecology, internal medicine and surgery.1
Bear bile is primarily composed of bile acids, amino acids, proteins, bile pigments, mineral elements and other chemical components, of which bile acids are the most abundant.9 Bile acids are important active ingredient of bile, and their structures are based on the parent nucleus of cholanic acid, different types of bile acids are formed according to the number and position of their substituents. To date, eight distinct classes of bile acids have been identified in bear bile: tauroursodeoxycholic acid (TUDCA), taurochenodeoxycholic acid (TCDCA), ursodeoxycholic acid (UDCA), chenodeoxycholic acid (CDCA), taurodeoxycholic acid (TDCA), taurolithocholic acid (TLCA), cholic acid (CA), and taurocholic acid (TCA), the structures of these eight bile acids are shown in Figure 1. The percentages of main bile acids in natural bear bile are as follows: TUDCA accounts for 51.25% ± 11.38%, TCDCA for 25.45% ± 8.63%, UDCA for 18.17% ± 0.80%, and CDCA for 15.17% ± 0.42%.1 Bile acids are the main contributors to the pharmacological activity of bear bile; thus, the proportion of bile acids is the core factor in the modern compounding or transformation of bear bile powder.
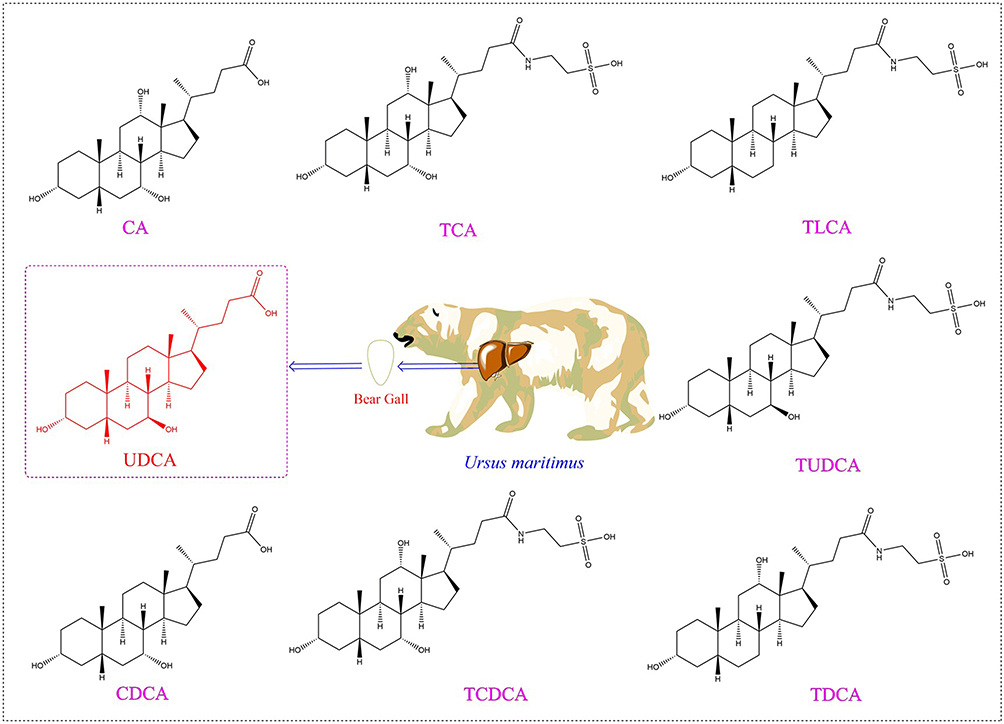 |
Figure 1 Chemical structure of bile acids in bear bile. |
UDCA, also known as 3α,7β-dihydroxy-5β-cholestan-24-acid, is a hydrophilic natural active bile acid first discovered in polar bear bile in 1920. UDCA can serve as a “unique molecular scaffold” and “structural mother nucleus” due to its bifunctional structure that combines a rigid steroid nucleus and a flexible side chain. This structure enables it to both retain biological activity and be chemically modified for expanded functionality. In its molecular structure, the steroid nucleus—formed by the trans-fusion of 3 six-membered rings and 1 five-membered ring—bears 3α-hydroxyl and 7β-hydroxyl groups. These groups can bind to the hydrogen bond sites of targets such as bile acid receptors, exerting basic physiological activities like regulating bile secretion and anti-inflammation. Additionally, the hydroxyl groups on the nucleus can be modified with ether bonds or ester bonds to enhance water solubility or targeting ability. Attached to the C17 position of the steroid nucleus is a linear valeric acid side chain containing a carboxyl group (-COOH). This side chain exhibits good flexibility without disrupting the conformation of the steroid nucleus. The carboxyl group can also undergo esterification or amidation reactions to form amide or ester derivatives, which helps regulate lipid solubility, extend half-life, or conjugate targeting groups (eg, polypeptides, antibodies). It is precisely this structure that endows UDCA with “modifiability” and “target adaptability”. Furthermore, as an endogenous bile acid that can be synthesized in small quantities by the human body, UDCA’s nucleus structure has been validated through biological evolution, featuring low toxicity and a clear metabolic pathway. Derivatives modified based on UDCA are also more easily recognized by the human metabolic system, which helps reduce the risk of off-target toxicity.10,11 UDCA exhibits significant uniqueness compared to other bile acids such as CDCA and TUDCA. Mechanistically, it adopts a mild regulatory mode independent of FXR, exerting its effects by activating GP-BAR1 and regulating endoplasmic reticulum stress, which can reduce disturbances to physiological homeostasis. CDCA strongly activates FXR, easily leading to secondary damage, while TUDCA has a single action pathway. In terms of pharmacological activity, UDCA possesses both broad-spectrum anti-inflammatory and anti-apoptotic properties (eg, inhibiting the NLRP3 inflammasome and balancing Bax/Bcl-2) as well as long-term safety, making it suitable for multiple disease scenarios such as PBC and NAFLD. High concentrations of CDCA can exacerbate inflammation and carry the risk of inducing hepatocyte apoptosis, whereas TUDCA only exerts anti-apoptotic effects in the nervous system and has weak anti-inflammatory effects in hepatobiliary diseases. In terms of clinical value, UDCA is a first-line drug for PBC and is also applicable to multiple hepatobiliary diseases such as ICP and PSC. The clinical application of CDCA has decreased due to side effects caused by high doses, and TUDCA is only suitable for adjuvant treatment of neurological diseases without the potential for cross-field derivation.12–14 In terms of adverse reaction comparison, multiple double-blind studies have shown that UDCA has significant advantages over CDCA. In a 1-year multicenter double-blind trial, the discontinuation rate due to diarrhea in the UDCA group was only 5%, which was significantly lower than that in the CDCA group, and serum transaminases remained within the normal range throughout the trial. Additionally, another double-blind study further confirmed that the incidence of diarrhea in the CDCA group was significantly higher than that in the UDCA group, while the combined use of the two could effectively reduce the incidence of adverse reactions—highlighting the advantages of UDCA in safety and tolerability.15,16 With over a century of research, UDCA has been proven to possess multiple biological and pharmacological activities: it is effective in dissolving gallstones and treating a range of hepatobiliary disorders, including cholestasis, cholestatic pancreatitis, primary biliary cholangitis (formerly primary biliary cirrhosis), non-alcoholic hepatitis, and drug-induced hepatitis, as well as conditions like colitis; it also exerts effects such as immunomodulation, lowering blood lipids and promoting lipid excretion, hypoglycemia, anti-cardiovascular disease activity, anti-inflammation, anti-tumor activity, and neuroprotection.17–24 Clinically, UDCA obtained FDA approval as an original drug for the treatment of primary biliary cirrhosis in 1997 and is now widely used in clinical practice.25 Beyond its applications in hepatobiliary diseases, in the treatment of cystic fibrosis-related disorders, UDCA has been approved in the European Union (EU) as an adjuvant therapy for pancreatic insufficiency in patients with cystic fibrosis. UDCA can regulate the properties of mucus secreted by the pancreas, reduce pancreatic duct obstruction, and thereby assist in improving the digestive and absorptive functions of patients.26 Additionally, it has been shown to reduce the infection rate and severity of COVID-19.27
Biosynthesis is a modern interdisciplinary field combining genetic and metabolic engineering. It has the advantages of high efficiency, economy, stability, green environmental protection and short cycle time, etc. It represents a novel approach to modern drug development, offering distinct advantages and promising potential.28 UDCA and its related preparations are widely used in clinical applications and have high clinical value. However, the source animals of UDCA are now listed as the second class of national protected animals, and it is expected that UDCA obtained through the biosynthetic pathway can partially ease bear bile supply shortages.
The amphiphilicity and acid-base properties of UDCA, as well as its unique steroidal ring molecular structure and intrinsic physiological activity, have drawn widespread of many researchers in the domain of biomedicine and chemistry in recent years.29,30 The introduction of active groups on the carboxyl and hydroxyl groups of UDCA to synthesize new compounds has been a hot topic for researchers in medicinal chemistry, and UDCA derivatives have antibacterial, anticancer, anti-inflammatory, hypocholesterolemic, hepatoprotective and other biological activities.31,32 Structurally modified UDCA derivatives have the advantages of high biological activity and wide range of applications. Therefore, the structural modification of UDCA has become the focus of medical research and application development.
UDCA is a natural steroid found in bear bile, possessing significant medicinal value. It is clinically used for the treatment of hepatobiliary diseases. Endowed with a unique molecular skeleton, UDCA exhibits a broad range of biological activities, specific targeting ability, and low toxicity, making it a highly promising structural parent nucleus. This characteristic has attracted increasing attention from researchers, re-establishing UDCA as a focus of interest. To date, a variety of UDCA derivatives with diverse structures have been designed, and their biological activities have been extensively investigated. This review systematically explores the pharmacological activities, biosynthesis, and structural modification of UDCA, aiming to provide a reference for the subsequent development of more UDCA derivatives with superior pharmaceutical properties, promote their application as clinical drugs, and better serve human health. To support the above research content, this review systematically collects, analyzes, and integrates literature on the pharmacological activities and biosynthesis of UDCA, as well as the mechanism of action of its derivatives. All information is retrieved from globally recognized scientific database systems, including English databases (such as Elsevier, ScienceDirect, and PubMed), Chinese databases (such as China National Knowledge Infrastructure (CNKI) and Wanfang Data), and comprehensive academic search platforms (such as Google Scholar and Baidu Scholar). The literature inclusion covers UDCA-related studies published in all languages, and a final literature pool is formed through a three-step screening process: initial screening based on titles and abstracts, full-text re-screening, and reference tracing. Additionally, content in the review such as the comparison of bile acid molecular structures and the chemical modification sites of UDCA derivatives is illustrated using ChemBioDraw Ultra 14.0 software, ensuring the accuracy and standardization of information presentation.
Biological Activity of UDCA
Protect the Liver and Gallbladder
UDCA exhibits multiple liver-protective effects by altering bile acid composition—reducing toxic hydrophobic acids while promoting nontoxic hydrophilic ones. It also enhances bile flow and demonstrates cytoprotection, anti-apoptosis, and immune regulation. UDCA treatment can significantly improve abnormal liver function in various cholestatic conditions, including primary sclerosing cholangitis, pregnancy-related intrahepatic cholestasis, cystic fibrosis-associated liver disease, hepatic graft-versus-host disease, parenteral nutrition-induced cholestasis, and certain pediatric cholestatic disorders.33–36 Non-alcoholic fatty liver disease (NAFLD), the global most prevalent liver disorder, affects 2.8–24% of people worldwide, progressing from simple steatosis to severe NASH.37 In a 2019 study, it was clarified that UDCA was effective in improving various tissue characteristics or biochemical indices of NASH and had a synergistic effect with vitamin E on hepatoprotection in NASH rats, and this synergistic effect was more pronounced than that of the drug alone in regulating tissue SOD activity and NF-κB expression levels, suggesting that this synergistic protective mechanism may be associated with the inhibition of hepatocellular steatosis and inflammatory cell infiltration, thereby attenuating liver injury and insulin resistance.38 Recent research shows UDCA alleviates liver inflammation in NASH mice via regulating the “gut microbiota-bile acid axis”, with its core focusing on bile acid metabolism control. Specifically, UDCA significantly raises serum levels of itself and its derivative TUDCA. TUDCA exerts dual effects: it activates the G protein-coupled receptor TGR5 to promote energy metabolism, thereby indirectly reducing liver inflammation; it also activates the farnesoid X receptor (FXR) to inhibit the synthesis of endogenous primary bile acids in the liver, mitigating liver injury caused by bile acid accumulation. Notably, this regulatory mechanism only relieves liver inflammation in NASH mice without improving liver fibrosis, providing a research direction for combining UDCA with anti-fibrotic drugs in subsequent NASH treatment.39
Immunomodulatory Activity
UDCA’s effectiveness in autoimmune liver diseases indicates its immunomodulatory effects. Studies show UDCA suppresses IL-2, IL-4, and IFN-γ release from activated T cells and reduces antibody production by B cells. Additionally, it induces glucocorticoid receptor nuclear translocation without ligands, enhancing its DNA-binding capacity. UDCA suppresses IFN-γ-induced MHC class II expression via glucocorticoid receptor signaling.40 In addition, other studies have shown that the immunomodulatory properties of reduced IFN-γ in UDCA are via increased GR in liver lymphocytes, independent of IL-12/18.41 Both bile duct-ligated rats and PBC patients with cholestasis show similar MHC class I overexpression on hepatocytes and bile ducts. This aberrant expression may trigger lymphocyte activation and subsequent immune-mediated liver damage. Studies have shown that UDCA can reduce class I antigen expression in several cholestatic liver diseases.18
Hypolipidemic Activity
In recent years, UDCA has become an effective basic therapeutic agent to prevent and treat hyperlipidemia. Clinical research indicates UDCA can significantly reduce serum cholesterol, triglyceride and LDL-C levels, promote the rise of serum HDL-C level, and at the same time inhibit the accumulation of intrahepatic fat and improve liver function, as well as stabilize the liver cell membrane and inhibit the production of cytokines by mononuclear cells. UDCA exerts dual hepatoprotective and lipid-modulating effects: it safeguards hepatocytes, boosts hepatic metabolism and lipid transport, while concurrently reducing blood lipids by enhancing excretion and limiting absorption.2,42
Hypoglycemic Activity
In a 2004 study, it was shown that UDCA lowered blood glucose in rats by inhibiting the overexpression of SGLT2 and restoring CAT and GPX activity to counteract oxidative stress.20 In addition, Lukivskaya et al observed that UDCA could affect the morphology and function of pancreatic islet β-cells in rats, and the serum insulin level of diabetic rats in the UDCA treatment group exhibited a 2.5-fold increase compared to the model group, and UDCA could also scavenge STZ-generated NO and oxygen free radicals, it prevents pancreatic β-cell apoptosis, thereby lowering blood glucose. UDCA also eliminates STZ-induced NO and oxygen free radicals, prevents pancreatic β-cell apoptosis, and reduces blood glucose.43,44
Preventing and Treating CVD
Cardiovascular disease (CVD) is a chronic circulatory disorder affecting heart and blood vessels, with multifactorial causes including smoking, diabetes, kidney disease, metabolic syndrome, and obesity. CVD has a complex pathogenesis and diverse clinical manifestations, with prolonged progression and limited treatment options. Accounting for one-third of global deaths, it remains the leading cause of worldwide mortality and morbidity.45 UDCA can block calcium influx into cardiomyocytes and control the mitochondrial permeability transition pore (PTP) of cardiomyocytes to protect cardiomyocytes. Rajesh et al showed that intravenous UDCA (40 mg/kg) protected Wistar rats against left coronary artery occlusion-reperfusion injury. UDCA triggered Akt/Bad phosphorylation, blocking phosphorylated Bad mitochondrial translocation, which suppressed Bcl-2 expression, PTP opening, and cytochrome C release, ultimately reducing cardiomyocyte apoptosis.2,21
Anti-Inflammatory Activity
Ulcerative colitis (UC) is a non-specific inflammatory bowel disease with high morbidity and low overall treatment response, characterized by recurrent abdominal pain, diarrhea, and muco-purulent stools. The disease’s pathogenesis remains unclear but thought to be the result of genetic, immune, microbial, and environmental interactions.46 Recent studies highlight UDCA’s therapeutic potential in UC, demonstrating anti-inflammatory, cytoprotective, and complication-reducing effects. Research suggests that the UDCA-bifidobacteria coadministration in UC mice markedly lowers IL-10/TLR4 levels, enriches gut microbiota, and enhances treatment efficacy.47 Small molecules of traditional Chinese medicines can interact under decoction conditions and form supramolecular self-assembly systems induced by weak bonds, resulting in significant changes in bioactivity compared with free monomeric components.48 UDCA and berberine were found to interact to form supramolecular self-assemblies under certain conditions. The supramolecule was found to be effective in ameliorating the loss of body mass and shortening of the colon in mice with colitis, reducing the disease activity index, and significantly reducing the serum levels of TNF-α and IL-6. Flow cytometry revealed the supramolecule markedly decreased neutrophil infiltration in colonic lamina propria versus the mechanical mixture group.49
Anti-Cancer Activity
Cancer, characterized by aberrant cell proliferation, is a major global health burden. WHO projects global cancer deaths to rise from 10 million (2020) to 16.3 million by 2040.50 UDCA demonstrates therapeutic efficacy against multiple malignancies, including gastric, oral, hepatic, ovarian, prostate, pancreatic, colorectal cancers and leukemia. UDCA modulates oncogenic pathway regulation in cancer genomes, such as cell cycle control, PI3K/AKT, P53, TNF-κB, and JAK-STAT, etc. UDCA can also specifically modulate apoptotic thresholds, suppress the growth of cancer cells and induce cell autophagy and apoptosis.25 UDCA demonstrates synergistic potential with established anticancer drugs. For example, it enhances SN38 (irinotecan’s active metabolite)-triggered apoptosis across multiple cancer cell lines through amplified DNA damage induction.51 In a 2015 study, it was found that UDCA can induce different types of cell death depending on the intracellular signaling environment of gastric cancer cells, thereby effectively killing cisplatin-resistant gastric cancer cells. Specifically, for cisplatin-sensitive SNU601/WT cells, UDCA induces apoptosis by upregulating and redistributing TRAIL-R2/DR5 on lipid rafts, while also promoting the localization of CD95/Fas to lipid rafts to downregulate ATG5 and inhibit autophagy. For cisplatin-resistant SNU601/R cells, due to the increased expression of c-FLIP (L) which inhibits the apoptotic pathway, and the failure of CD95/Fas to localize to lipid rafts (resulting in no downregulation of ATG5), UDCA instead induces non-protective autophagic cell death to reduce their survival rate. This dual-pathway mechanism of “apoptosis-autophagy” provides a molecular basis for overcoming chemoresistance in gastric cancer.52
Antiviral Activity
SARS-CoV-2 has had a serious impact on human health worldwide due to its rapid spread, wide reach, rapid mutation rate, and high morbidity and mortality. The most severe clinical manifestation of COVID-19—the novel coronavirus infection triggered by SARS-CoV-2 is respiratory failure and is characterized by high-signal inflammation, which can eventually progress to multi-organ failure.53–55 Angiotensin-converting enzyme II (ACE2) acts as a pivotal molecule involved in regulating vasoconstriction, modulating inflammatory responses, and mitigating oxidative stress. Moreover, ACE2 has a significant function in combating SARS-CoV-2—the virus responsible for the COVID-19 pandemic—given that it is the first receptor in the viral spiking protein host.56 Relevant studies have shown that neocoronaviruses recognize and bind to the ACE2 protein receptor on cells through their spiking proteins, enter into human host cells, and interfere with the normal function of the cells. In this process, the synthesis and expression of ACE2 protein is controlled by the transcriptional regulator FXR. Researchers have shown that UDCA can inhibit FXR signaling in seven organs, including the respiratory tract and intestinal tract, reduce the level of ACE2, decrease the activity of ACE2 protein, and close the channel recognized by the COVID-19 virus, thus preventing its entry into human cells from the root cause. Among patients with chronic liver disease who have contracted the novel coronavirus, studies indicate that those receiving UDCA treatment exhibit lower rates of hospitalization, IC admission and mortality compared to untreated cases. Additionally, individuals undergoing UDCA therapy show an extremely low likelihood of progressing to moderate, severe, or critical stages of the illness.27,57 The various pharmacological activities of UDCA can be seen in Figure 2.
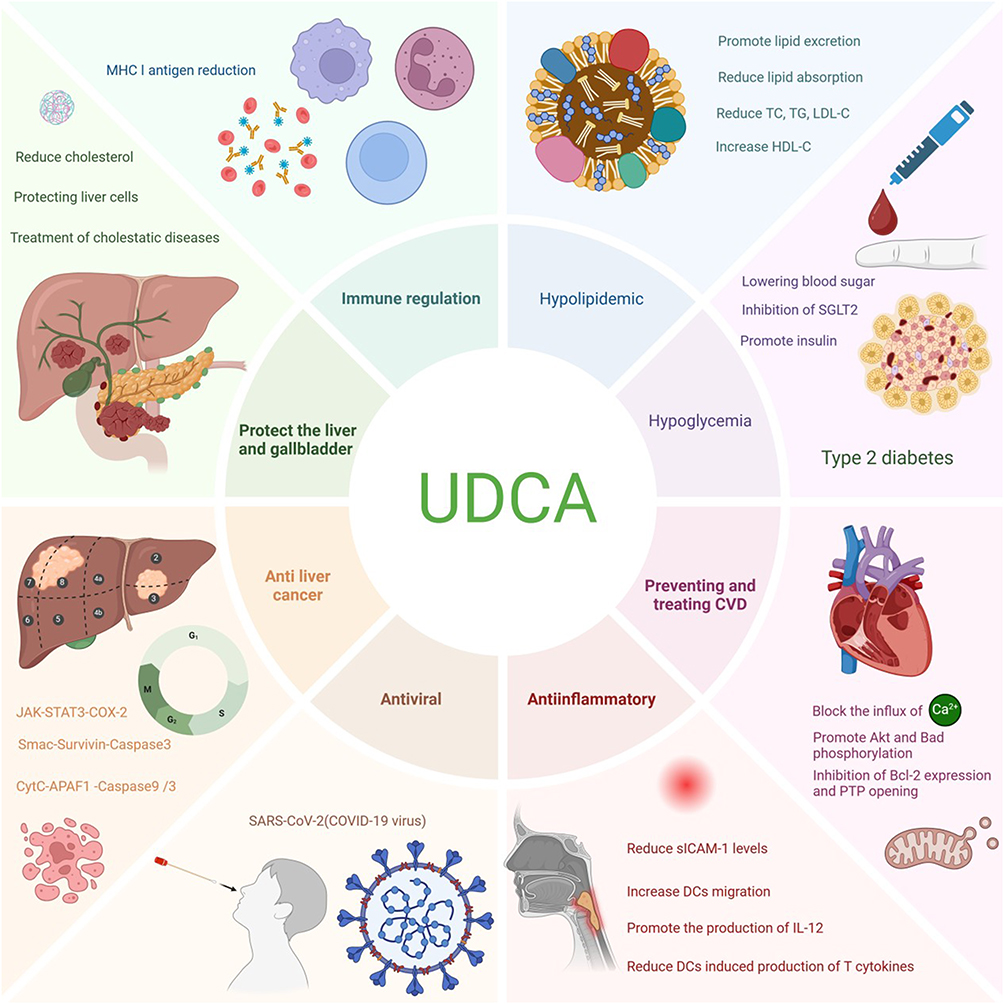 |
Figure 2 Biological Activities of UDCA. |
Despite its numerous pharmacological activities, UDCA still has key limitations in drug development. UDCA itself has low water solubility, which is highly affected by pH; while its derivatives have better solubility, specific data for some of them remain unclear. Its oral bioavailability is unstable, influenced by multiple factors including solubility, formulation differences, gastrointestinal environment, drug-drug interactions, and disease status. Additionally, UDCA may interfere with cellular functions and exhibit genotoxicity; high doses increase liver-related risks, affect adipocyte function, and cause systemic adverse reactions. All these factors pose challenges to its clinical application and drug development.58,59
Biosynthesis of UDCA
Bear bile has been widely used as a precious and potent therapeutic agent in traditional Chinese medicine (TCM) for more than 1300 years. Nevertheless, the present method of acquiring it via bear farming involving bear farming have raised ethical concerns regarding animal welfare issues.60 At first, UDCA was obtained through the drainage of live bear bile, but the source of bear bile was limited and violated the animal protection law. Although a diversity of processes have been designed to produce UDCA from CA, CDCA and HDCA, which are simple and readily available natural steroid compounds, the low yield, cumbersome steps, harsh reaction conditions and the use of a variety of toxic chemical reagents have severely limited the industrial production and application of UDCA. The industrial production of UDCA by chemical method is about 30% of the market share, and the preparation of low purity, far from meeting the market demand.61 Compared with the drawbacks of the chemical synthesis of UDCA, its biosynthesis is relatively green, efficient, and safe, and has thus become a research hotspot and a focus in industrial production. The natural biosynthetic pathway of ursodeoxycholic acid (UDCA) belongs to the bile acid synthesis branch of cholesterol metabolism. First, in the liver, cholesterol undergoes enzymatic reactions involving cholesterol 7α-hydroxylase (CYP7A1) to generate CDCA. Subsequently, CDCA enters the intestine, where it is catalyzed by 7α/β-hydroxysteroid dehydrogenase (7α/β-HSDH) produced by intestinal flora (such as some Clostridium and Bacteroides species). This process first oxidizes the 7α-hydroxyl group of CDCA to a ketone group, forming 7-oxolithocholic acid (7-oxo-LCA), and then reduces the 7-ketone group to a 7β-hydroxyl group, ultimately producing UDCA. The entire process reflects the synergistic interaction between the host’s liver and intestinal flora, with intestinal flora playing a key role in the production of the secondary bile acid UDCA.62 The above research on the natural biosynthesis of UDCA plays a guiding role in its large-scale biosynthesis. UDCA biosynthesis can be catalyzed by either free enzymes or whole-cell systems, utilizing accessible bile acid substrates such as CDCA, CA, or LCA; among these, CDCA is the most commonly employed, given its well-studied catalytic conversion pathway in biosynthesis.11
CDCA can be biosynthesized to UDCA in a two-step reaction (Figure 3A) involving two core enzymes: 7α-hydroxysteroid dehydrogenase (7α-HSDH) and 7β-hydroxysteroid dehydrogenase (7β-HSDH). Specifically, 7α-HSDH mediates the oxidative dehydrogenation of CDCA, generating 7-ketolithocholic acid (7-oxo-CA), while 7β-HSDH subsequently catalyzes the hydrogenation and reduction of 7-oxo-CA to form UDCA.63–65
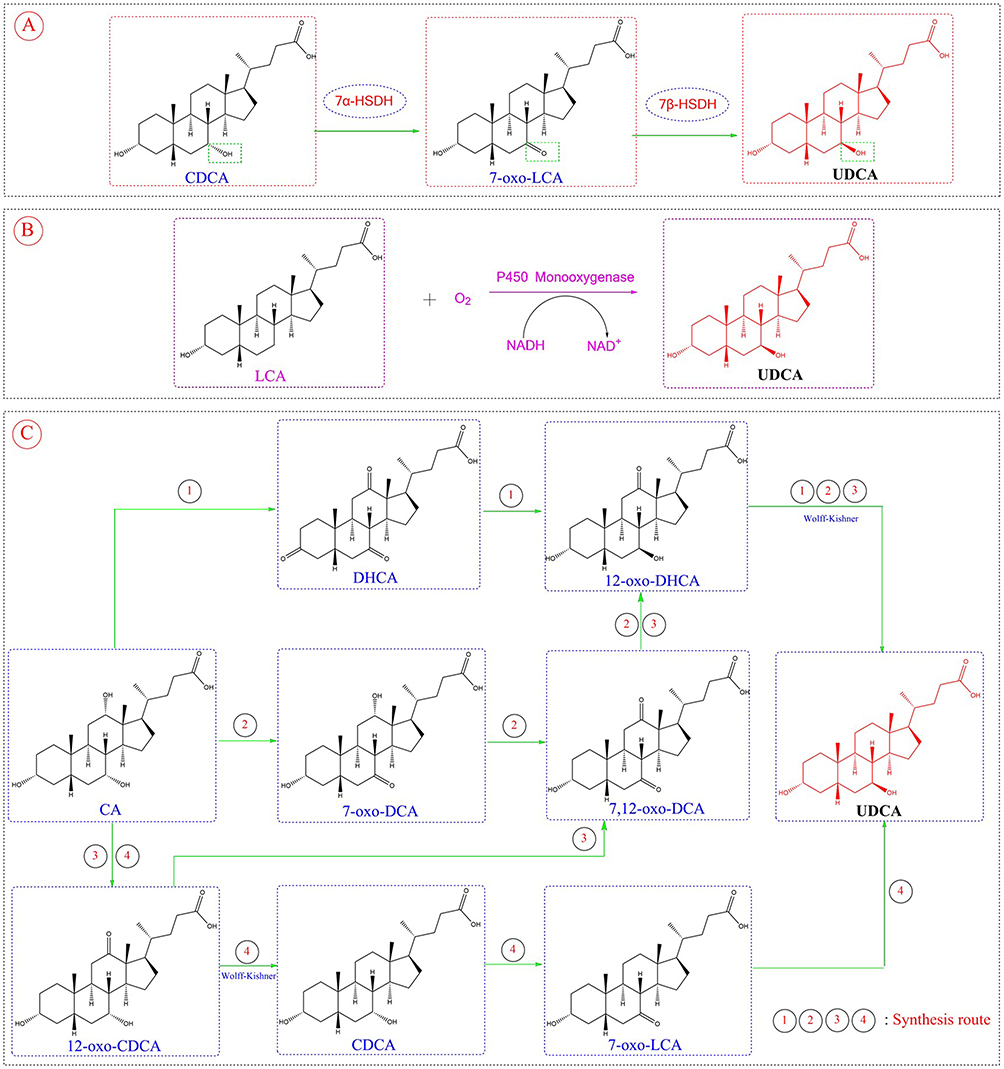 |
Figure 3 The three biosynthetic pathway of UDCA. (A) CDCA is converted to UDCA via 7-oxolithocholic acid (7-oxo-LCA) under the catalysis of enzymes (7α-HSDH, 7β-HSDH). (B) LCA generates UDCA with the action of cytochrome P450 monooxygenase and other substances. (C) Using CA as starting materials, UDCA is synthesized through multiple reactions (including oxidation, Wolff-Kishner reduction, etc.) and a variety of intermediates, where ①②③④ represent different reaction steps. |
LCA is likewise found in the bile of livestock like cattle, sheep, and pigs, serving as a plentiful and low-cost by-product from the meat processing sector. From a structural point of view, synthesizing UDCA with LCA as the substrate only necessitates hydroxylation at the C-7 position of LCA, as illustrated in Figure 3B. Cytochrome P450 monooxygenase is capable of catalyzing this reaction, but it faces numerous challenges in practical applications: its O2-dependent catalytic mechanism strictly requires the involvement of nicotinamide adenine dinucleotide phosphate (NAD(P)H) and its cognate reductase, while the complex structure of this protein complex reduces the operational stability and catalytic efficiency of P450 monooxygenase.66 Notably, a study by Grobe et al showed that the engineered cytochrome P450 monooxygenase OleP (CYP107D1) from Streptomyces antibioticus can achieve regio- and stereo-selective 7β-hydroxylation of lithocholic acid (LCA) to produce ursodeoxycholic acid (UDCA), but this engineered enzyme has extremely low conversion efficiency—only 67 μM UDCA is produced in a reaction system with 2.5 mM LCA.67 However, a breakthrough has been made in recent research in this field: Zhou et al first identified a fungal P450 enzyme (designated as P450FE) from the filamentous fungus Fusarium equiseti HG18, which can catalyze the conversion of LCA to UDCA through 7β-hydroxylation. When heterologously expressed in Pichia pastoris, this enzyme does not require additional coenzymes (relying on the endogenous electron transport system of Pichia pastoris) and achieves a UDCA yield of 5.2%. Subsequent studies using AlphaFold 2 modeling, molecular docking, and site-directed mutagenesis confirmed that Q112, V362, and L363 are key regulatory residues; among them, the V362I mutant exhibits a UDCA yield of 13.6%, which is 2.6 times that of the wild-type enzyme. The discovery of this enzyme also enriches the current enzyme resource library—where only a small number of microorganisms (such as Fusarium equiseti HG18 and Gibberella zeae) are capable of converting LCA to UDCA—and lays a foundation for the industrial production of UDCA.68
As the cheapest and most plentiful bile acid, CA is widely present in bovine, ovine, and porcine bile. Among them, bovine bile has the highest content, with a bile acid content of 44 g in 1000 mL of bovine bile. Four pathways convert CA to UDCA61 as shown in Figure 3C, however, all require the Wolff-Kishner reaction to reduce the intermediate’s C-12 carbonyl to methylene.69 Therefore, it is not currently possible to achieve the full-process biosynthesis of UDCA using bile acids as substrates.
As an advanced production method, the biosynthesis of UDCA promotes the transformation of the production process towards environmental friendliness, reduces costs, increases profits, and improves product quality. Its market and development prospects are broad, and its development potential is enormous. Compared with chemical synthesis, biosynthetic UDCA has shown superiority and has achieved industrial production of UDCA using CDCA as a substrate. Yet, practical production still has limitations. Firstly, The enzymes used—particularly 7β-HSDH—are astable and experience rapid loss of enzyme activity in the reaction system. Secondly, substrate concentrations in enzymatic reactions remain relatively low, while some enzymes suffer severe products or intermediates inhibition. Currently, most published enzyme catalyzed reactions are carried out within the concentration range of 10–20 μmol/L, which is still far from industrial application (generally believed to reach 100 μmol/L). Mechanical shear in production can physically damage enzymes, easily reducing their activity and recovery rate. The recyclability and reuse of enzymes in catalytic reactions still need to be further investigated. It is imperative to develop efficient heterologous expression systems beyond Escherichia coli.
Recent Progress in the Medicinal Chemistry of UDCA Derivatives
Cholesterol Lowering Activity
Bile acids, a major component of bile, evolved from cholesterol and are the largest organic acids secreted by bile into the liver. The liver tissue of the body can efficiently lower hepatic cholesterol buildup through bile acid metabolism, thereby reducing the damage of cholesterol to liver cells.34 As a minor human bile constituent, UDCA decreases biliary cholesterol saturation by limiting intestinal cholesterol reabsorption and hepatic cholesterol secretion.70
UDCA has been successfully used to dissolve cholesterol gallstones, but its low efficacy and long treatment time are due to the easy occurrence of 7-dehydroxylation reaction, which generates hepatotoxic lithocholic acid. A series of new UDCA phosphonic acid derivatives were synthesized using UDCA as raw material, and their in vitro cholesterol dissolving efficiencies were evaluated. Compound 1 is more effective in dissolving cholesterol than Compound 2 and natural bile salts (Figure 4). The ability to dissolve cholesterol is influenced by its structure and decreases significantly with increasing body Ph.71
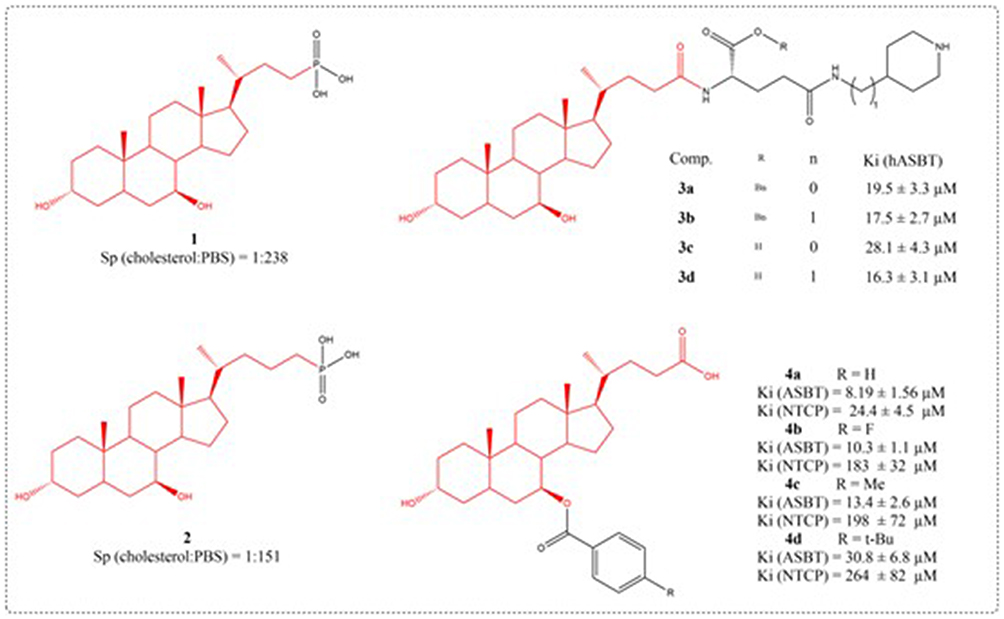 |
Figure 4 Structures of UDCA derivatives 1–4. |
The human apical sodium-dependent bile acid transporter (hASBT), a 348-amino acid intestinal transporter, is crucial for bile acid enterohepatic cycling and cholesterol regulation.72 A total of 29 aminopiperidine-based conjugates were prepared, all showing hASBT inhibition (Ki: 0.95 to 31.8 μM). The Ki values of UDCA derivatives 3a-3d are 19.5, 17.5, 28.1, and 16.3 μM, respectively (Figure 4). Piperidine nitrogen amidation slightly reduced activity, whereas carbon replacement enhanced potency. Esterifying the glutamic acid linker had little effect, indicating no need for a negative charge near C-24 for binding.73 ASBT actively absorbs intestinal bile acids. After absorption by ASBT, bile acids are mainly extracted into liver cells through the sodium taurocholate cotransporter peptide (NTCP) on the outer surface of the liver cell base. The ASBT and NTCP are potential prodrug targets. Three compounds had higher affinity for ASBT than NTCP: compounds 4b, 4c, and 4d (Figure 4). While prior literature indicates NTCP has broader substrate specificity, these compounds strongly bind ASBT and weakly or not at all inhibit NTCP. Compounds 4b, 4c, and 4d are similar, with larger substituents reducing their ASBT and NTCP affinity (F < CH3 < t-butyl). Impact was more significant in NTCP (Ki ≥200 μM) (Figure 5). Compound 10 potently inhibited ASBT and NTCP, with greater affinity for NTCP. Its Ki values resembled UDCA’s. Unlike UDCA, compound 10’s C-24 methyl esterification maintained affinity, particularly with free 3-/7-hydroxyl groups.74
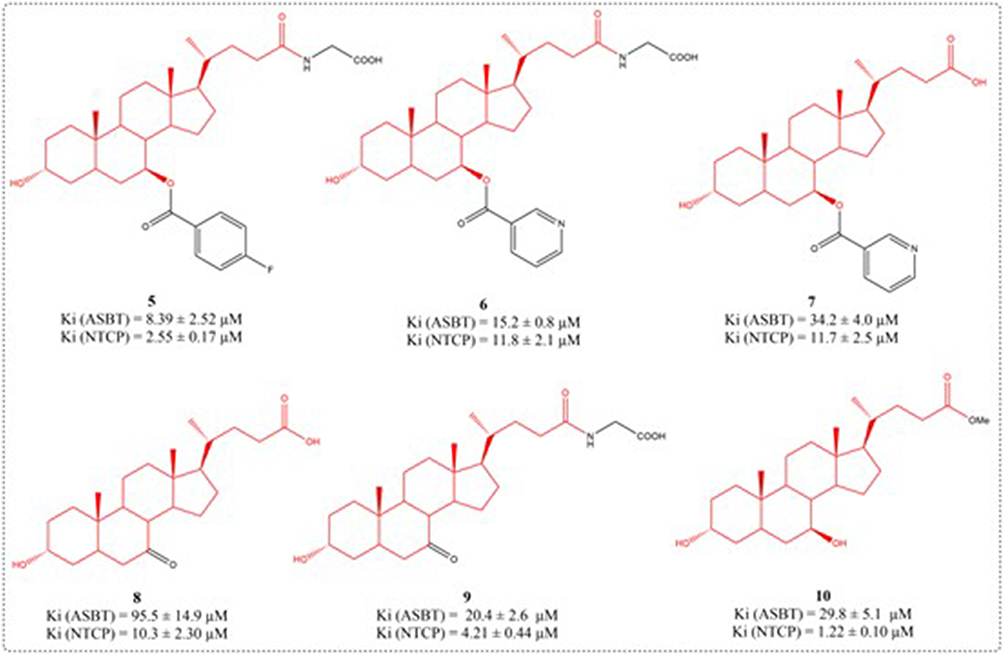 |
Figure 5 Structures of UDCA derivatives 5–10. |
Bile acids are signaling molecules that interact with the nuclear receptor FXR and the G-protein coupled receptor 1 (GP-BAR1/TGR5).75,76 GP-BAR1 is a promising pharmacological target for the treatment of steatohepatitis, type 2 diabetes, and obesity. UDCA is the 7β-hydroxy epimer of CDCA; though its pharmacology resembles FXR modulation, it is not a FXR agonist. Sepe et al designed and synthesized 27 compounds using UDCA as a scaffold. In vivo and in vitro pharmacological tests showed that compound 12 administration selectively upregulates small intestinal proglucagon 1 (a GP-BAR1 target) without affecting hepatic FXR target genes. Compounds 12 dose-dependently activated the cAMP response element in GP-BAR1-transfected HEK-293T cells, with an EC50 of 17.2 μM (Figure 6). Furthermore, compound 12 induces a notable remodelling of the bile acid pool in a rodent model of cholestasis.77
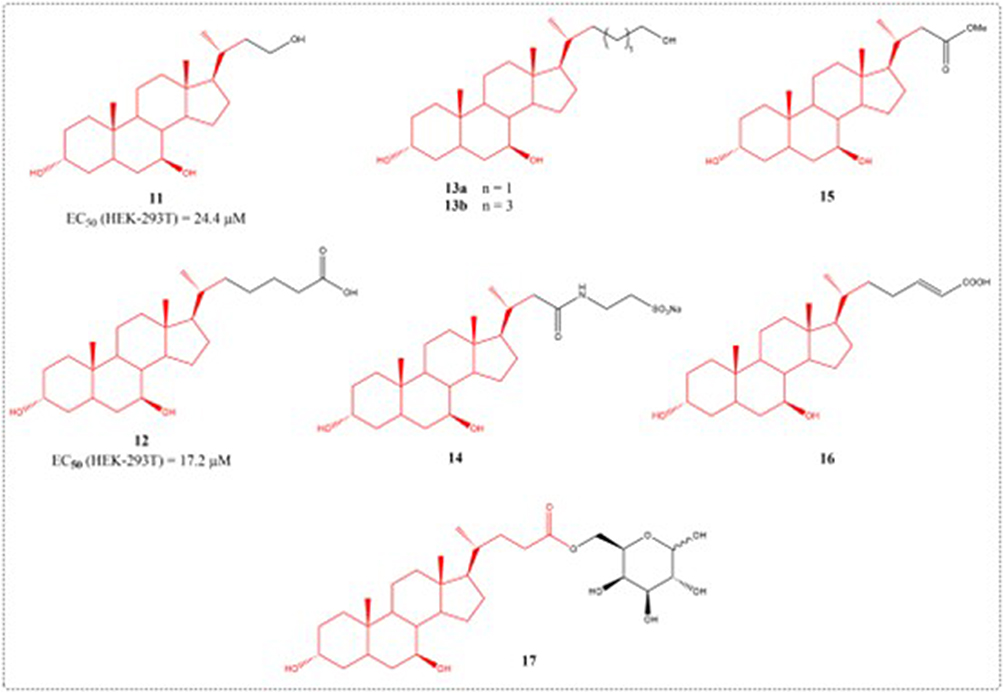 |
Figure 6 Structures of UDCA derivatives 11–17. |
A common liver disorder in susceptible women, estrogen-induced cholestasis arises during pregnancy, oral contraceptive use, or postmenopausal therapy, yet lacks effective treatments.78 UDCA is currently employed to treat estrogen-related cholestasis, including pregnancy-associated intrahepatic cholestasis.79 Though clinically effective, UDCA has poor solubility in gastro-duodeno-jejunal contents, and its pharmacological doses hardly dissolve in the healthy male stomach and intestine.80 To enhance solubility and target hepatic transporters, Guida et al developed a novel UDCA galactosyl prodrug (compound 17). Compound 17 exhibited stability at pH 7.4 and 1.2, releasing the parent compound in human plasma. In cholestatic rats, compound 17 was more potent than UDCA’s role in lowering serum biomarkers (AST, ALT, ALP) and cytokines (TNF-α, IL-1β).81
Liver Protective Activity
Liver disorders including viral hepatitis, fibrosis, cirrhosis, and hepatocellular carcinoma exhibit high prevalence and therapeutic challenges, significantly endangering public health. NO has various effects such as improving hepatic hemodynamic properties, repairing liver injury, resisting portal hypertension, and improving liver fibrosis However, currently available nitroglycerin in the market can cause serious systemic side effects such as hypotension when used to treat liver diseases. The development of liver specific NO releasing drugs is of great significance.82,83 Li et al used UDCA as a drug targeting carrier, amino acids as linkers, and simulated the negative charge of the 24th carboxyl group of UDCA molecule with the alpha carboxyl group of amino acids. The carrier was coupled with NO donor nitrate ester through amide bonds, and a series of novel liver targeted nitric oxide release complexes were designed and synthesized. The pharmacological evaluation of acute hepatic damage triggered by carbon tetrachloride and acetaminophen in mice showed that compounds 18a, 18b, 18c, 18d, and 19 could reduce serum ALT and AST concentrations by 50% to 70% (Figure 7). 18a has good liver targeting ability, and its liver targeting ability is better than that of the positive drug NCX-1000.84 NCX-1000, also known as compound 20, was confirmed in a 2004 study to selectively release NO in the liver, reduce the accumulation of IFN-g, TNF-a, Fas/Fas ligands, as well as inducible nitric oxide synthase (iNOS) messenger RNA induced by acetaminophen, lower mortality rates, and improve liver histopathology.85
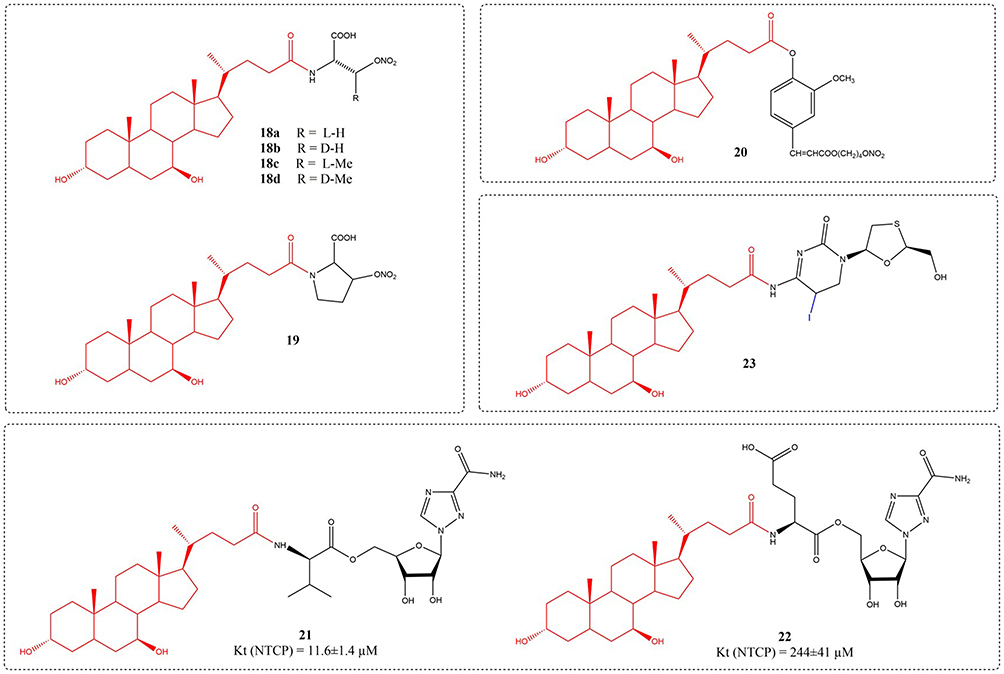 |
Figure 7 Structures of UDCA derivatives 18–23. |
Ribavirin, a guanosine analog, is used in the treatment of hepatitis C.86 Yet, the applicability of this agent is constrained by dose-dependent hemolytic anemia, with roughly 20% and 5% of treated patients requiring dose reduction or discontinuation, respectively.87 Dong et al synthesized six prodrugs that bind to ribavirin at bile acids C-3 or C-24. This strategic modification was intended to facilitate liver-targeted drug delivery while minimizing off-target impacts on red blood cells (RBCs). Among them, compound 21 exhibits the greatest affinity for NTCP, with its Kt value measuring 11.6 Μm.88 The nucleoside analog lamivudine (3TC), an oral antiviral agent, is approved for hepatitis B treatment, demonstrating efficacy in suppressing viral reverse transcriptase and improving virological, biochemical, and histological outcomes in chronic HBV patients. Due to its high-water solubility, liver uptake is reduced and there are side effects such as cephalalgia, vertigo, sleep disturbances, exhaustion, GI upset, and ALT elevation, etc. Prolonged utilization of 3TC for treatment may result in the emergence of 3TC resistant mutants. Motaleb et al designed and synthesized a novel coenzyme drug 23 using 3TC and UDCA as raw materials, which has the ability to increase targeted tissue drug uptake by 3TC and is labelled with125I.89
Chronic hepatitis B virus (HBV) infection persists as a global health challenge, affecting around 240 million individuals and causing 686,000 yearly deaths worldwide.90 Thus, the creation of potent medications for HBV treatment has emerged as a key healthcare priority. At present, six nucleos(t)ide analogues—lamivudine, adefovir dipivoxil, entecavir, telbivudine, along with tenofovir derivatives (disoproxil and alafenamide), have received approval for HBV.91 Li et al developed and created a range of adefovir mono-L-amino acid and mono-cholic acid drug conjugates. Compound 24c emerged as the most promising molecular candidate, exhibiting the strongest antiviral activity (EC50= 0.42 μM, SI 1063.07) along with the greatest cellular uptake in primary hepatocytes and NTCP-HEK293 cells (Figure 8). Its pharmacokinetic characteristics and tissue distribution indicate that it has good drug properties and pharmacokinetic properties.92,93 As regulators of lipid metabolism and energy homeostasis, bile acids serve as endocrine signaling molecules with therapeutic potential for NASH. Wang et al synthesized new bile acid analog compounds using UDCA as a substrate in 2020 and evaluated their potential anti NASH effects in vitro and in vivo. Compound 25 had three pharmacological effects on NASH. First, reduced the Cd36 expression in hepatocytes, suppressing FA uptake into the liver. Second, stimulated the phosphorylation of AMPKα and ACC and enhanced the expression of Cpt-1α, promoting FA oxidation in hepatocytes. Third, it activated TGR5 on Kupffer cells, elevated intracellular cAMP concentrations and suppressed IκBα phosphorylation, ultimately resulting in diminished inflammatory responses.94
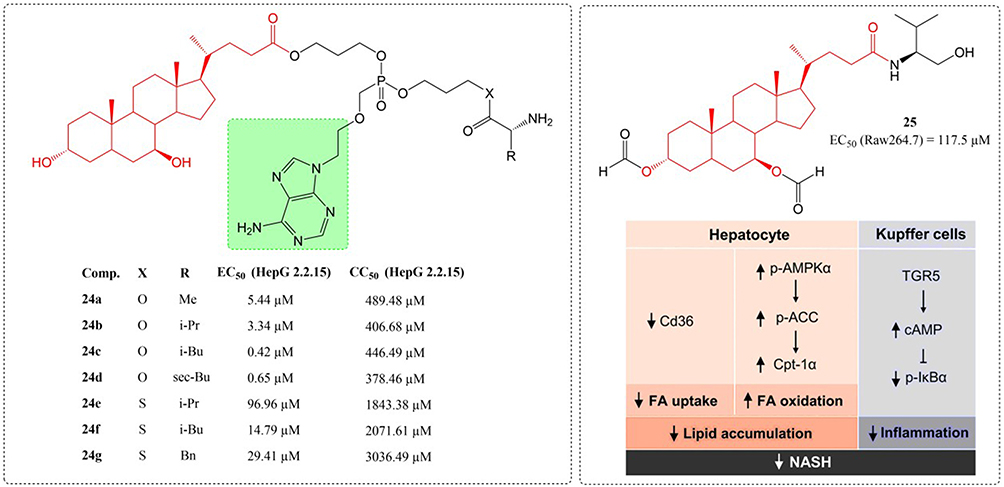 |
Figure 8 Structures of UDCA derivatives 24–25. EC50: Half Maximal Effective Concentration; CC50: Half Maximal Cytotoxic Concentration. ↓: inhibited; ↑: promoting. |
The term IR injury refers to cellular damage occurring upon reperfusion of tissues that were rendered hypoxic during a period of ischemia caused by diminished blood supply.95 Liver IR injury results from complex interactions between various elements including hypoxic metabolism, mitochondrial dysfunction, oxidative damage, Kupffer cell, and cytokines/chemokines, where oxidative stress emerges as a particularly critical component.96 UDCA enhances hepatic blood circulation to stimulate metabolic activity and boost glutathione (GSH) synthesis. Furthermore, it is capable of generating homeostatic substances that can clear ROS produced in the course of IR injury. Although UDCA demonstrates significant potential for preventing and treating IR injury, its effectiveness is limited by a brief plasma half-life.97 To overcome the limitations of UDCA’s insufficient solubility in aqueous media and diminished bioavailability. A team under Hong’s direction synthesized a dimeric UDCA derivative with a disulfide bridge (ssUDCA) as a potential IR injury treatment. This compound demonstrated the ability to spontaneously form stable nanospheres in aqueous environments while exhibiting hydrogen peroxide scavenging capacity along with anti-inflammatory and antiapoptotic properties (Figure 9). In a murine hepatic IR injury model, administration of ssUDCA (5 mg/kg) markedly attenuated tissue damage by reducing ROS generation and downregulating proinflammatory cytokine expression.98
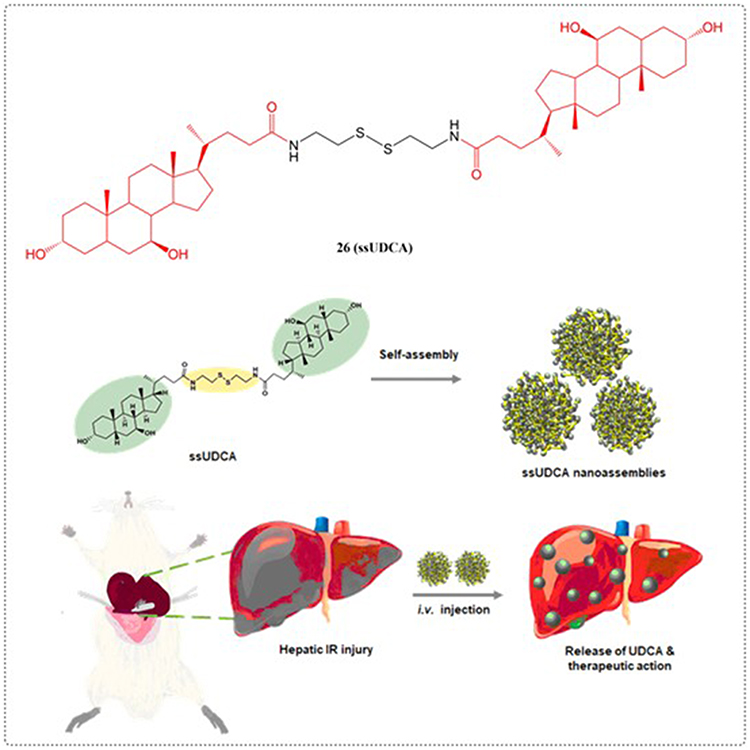 |
Figure 9 Structures of UDCA derivatives 26. |
Polycystic liver diseases (PLDs) represent an inherited condition marked by the gradual formation of symptomatic bile duct cysts, with liver transplantation remaining the sole definitive treatment.99 Both UDCA and inhibitors of histone deacetylase 6 (HDAC6) have shown considerable promise as potential treatment. Structural modification of UDCA yielded novel conjugates with selective HDAC6 inhibition, as demonstrated through systematic design, synthesis, and functional analysis by Caballero-Camino et al (27a-e). In vivo studies revealed that 27a effectively attenuated cyst development in liver and kidney tissues while restoring normal ciliogenesis and controlling abnormal proliferation in cholangiocytes. These UDCA-HDAC6i conjugates open a therapeutic avenue for PLDs (Figure 10).
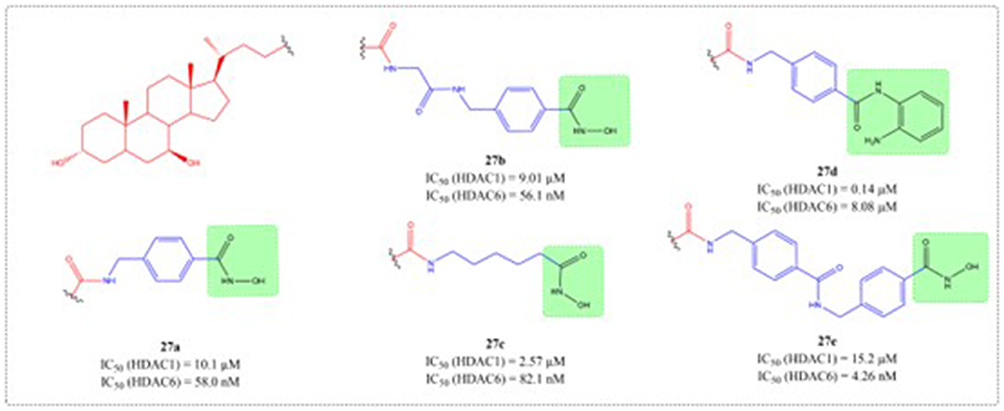 |
Figure 10 Structures of UDCA derivatives 27a-e. IC50: Half Maximal Inhibitory Concentration. |
Through systematic structure modification involving various bifunctional linkers, Liu et al generated dimerized bile acid compounds (DBADs) and characterized their capacity to block HBV infection in NTCP-receptor expressing HepG2 cell systems (Figures 11–14). DBADs exhibit potent and sustained NTCP inhibition, with different linkers and compositions exhibiting different inhibitory properties. The cyclized DBAD derivative (compound 37) demonstrated strong binding affinity for human NTCP (hNTCP). When administered intraperitoneally to hNTCP-transgenic mice, DBA-41 significantly elevated total bile acid levels in serum. These DBAD represent an innovative pharmacological approach for developing therapeutics that block both HBV entry and substrate transport via NTCP inhibition.100
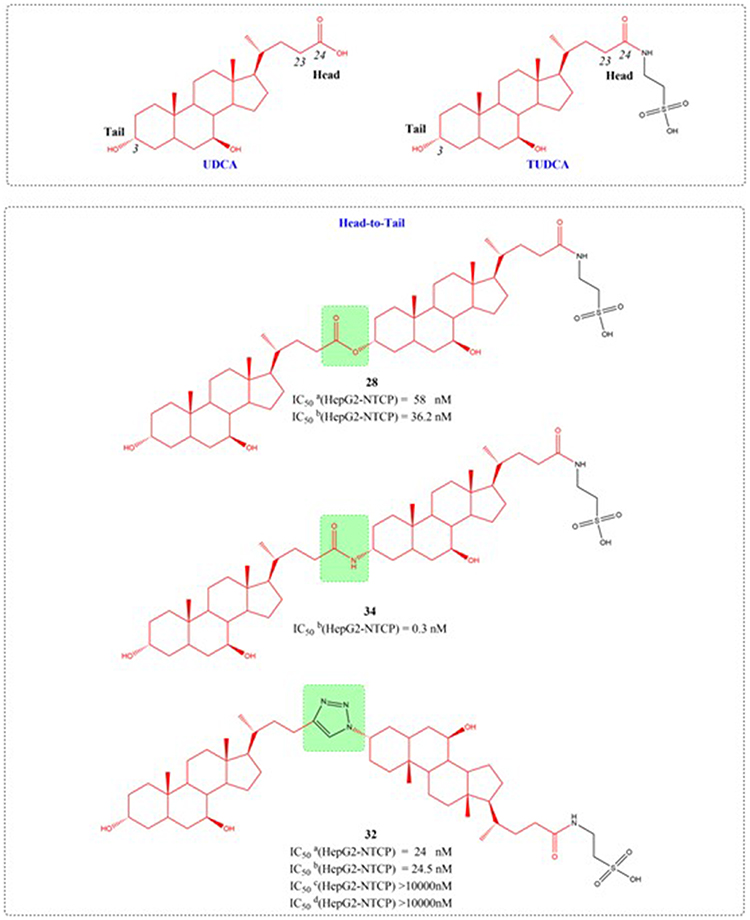 |
Figure 11 Structures of UDCA derivatives (28, 32, and 34). IC50: Half Maximal Inhibitory Concentration.The group in the green box is a linker. |
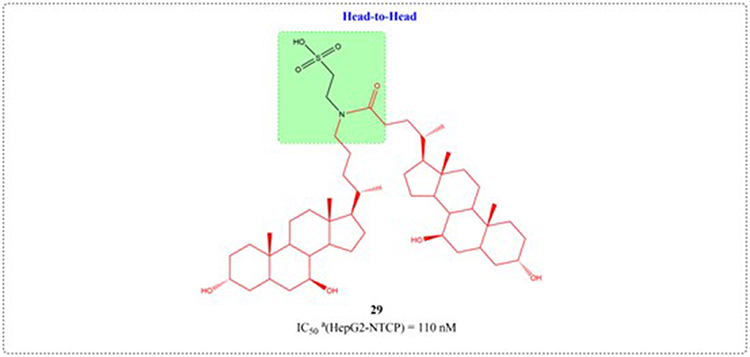 |
Figure 12 Structures of UDCA derivatives 29. IC50: Half Maximal Inhibitory Concentration. The group in the green box is a linker. |
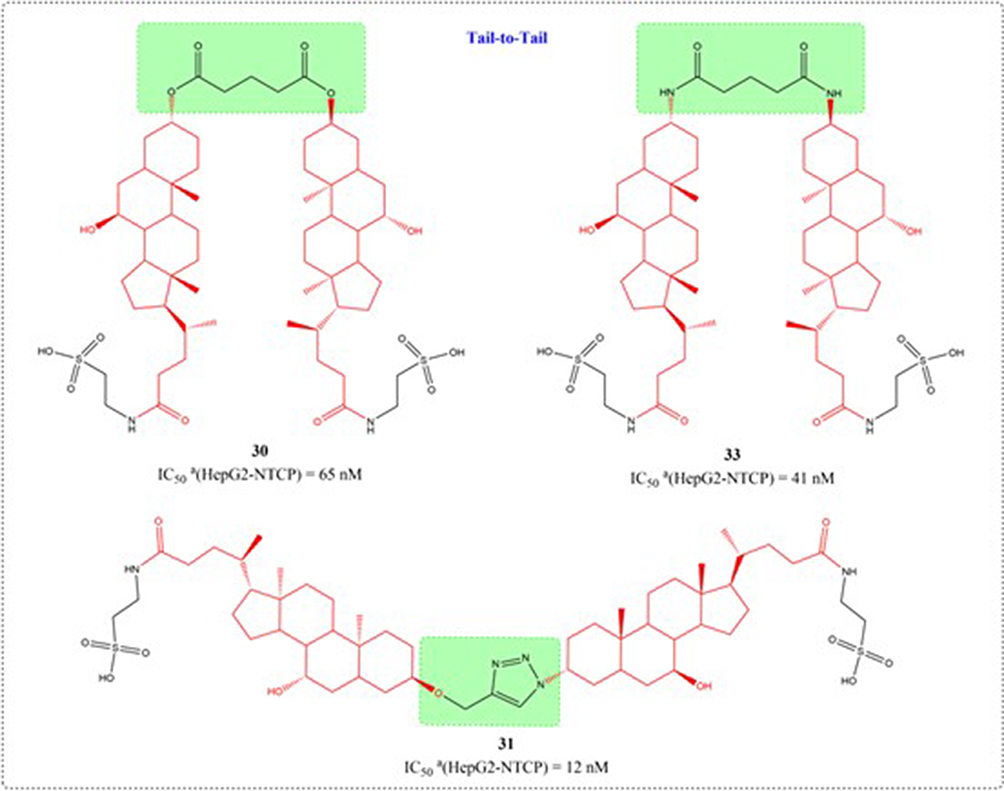 |
Figure 13 Structures of UDCA derivatives (30, 31, and 33). IC50: Half Maximal Inhibitory Concentration. The group in the green box is a linker. |
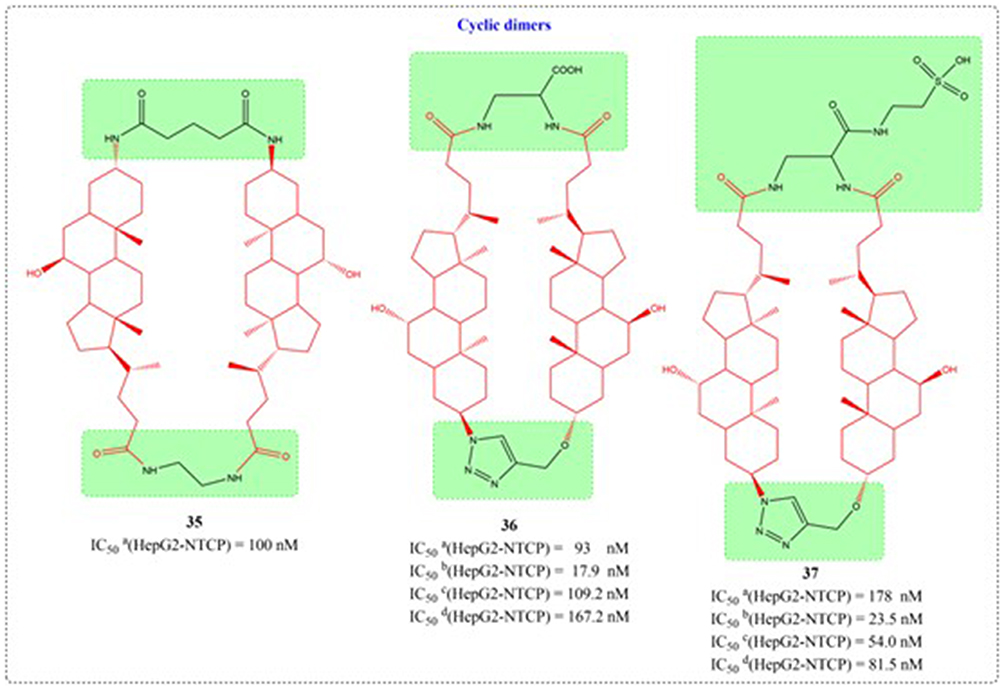 |
Figure 14 Structures of UDCA derivatives 35–37. IC50: Half Maximal Inhibitory Concentration. |
Leveraging UDCA’s ability to direct the cellular activity of therapeutic compounds, Criado et al engineered a collection of innovative platinum complexes (38a-38d). The complexes underwent in vitro cytotoxicity profiling against an array of cancer cell lines, including human colorectal adenocarcinoma, murine hepatoma, human hepatocellular carcinoma, and murine leukemia cellular systems. Several synthesized compounds demonstrated antitumor activity comparable to cisplatin in experimental evaluations (Figure 15).101
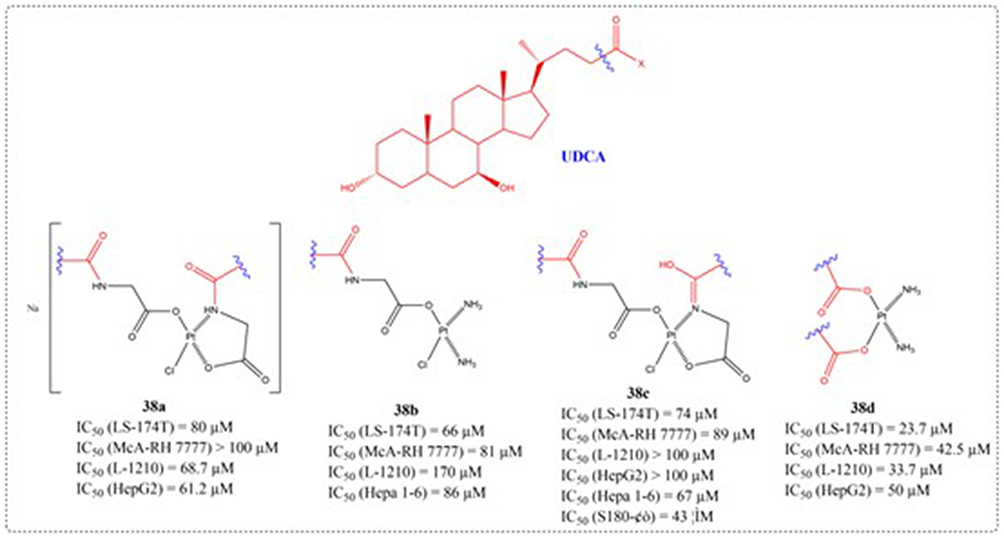 |
Figure 15 Structures of UDCA derivatives 38a-d. IC50: Half Maximal Inhibitory Concentration. |
Drug targeting can help overcome chemoresistance mitigation while adverse reactions. Vallejo et al designed the coupling of a nitrogen-fixing base (NB) onto the lateral chain of UDCA (39–40). Cell viability was quantified via formazan assay following 6-hour or 72-hour drug treatment across multiple cell models, including HepG2, LS 174T, Caco-2, Hepa 1–6, McA-RH7777, CCRF S-180 II, and CHO cell lines (Figure 16). Compound 40 shows potent cell-inhibitory activity, with efficacy even against cisplatin-resistant Hepa 1–6/R cells, and is capable of extending the survival time of nude mice bearing tumors formed by the in situ implantation of Hepa1-6/R cells. Targeting the hepatic-intestinal circuit with structurally modified UDCA analogs presents an innovative pharmacological strategy for managing associated malignancies.102 As a major secondary bile acid, UDCA plays a preventive role in gastrointestinal disorders among cancer patients, particularly in cases of stomach, colon, lung, breast, and liver cancers. Brossard et al introduced piperazine onto the C-7 hydroxyl group of UDCA to generate novel piperazine-UDCA derivatives and discussed their cytotoxic inhibitory effects on human tumor cell lines (41–42). This investigation highlights hybrid heterocyclic steroids as potential next-generation anticancer therapeutics, featuring increased bioactivity and synthetic simplicity.103
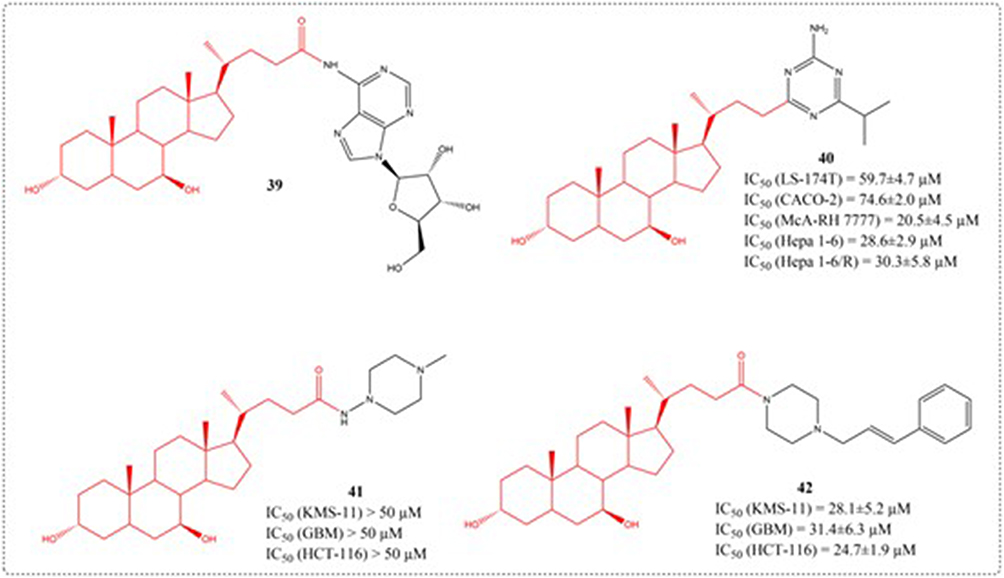 |
Figure 16 Structures of UDCA derivatives 39–42. IC50: Half Maximal Inhibitory Concentration. |
As an important category of medications, sulfonamides demonstrate extensive antimicrobial activity against various pathogens. Recently, some sulfonamide derivatives have been reported to have potential antitumor effects.104 Ren et al used UDCA as a starting material to design and synthesize a series of N-sulfonyl-3,7-dioxo-5β-cholan-24-amides, UDCA derivatives (43–45). Compounds 43b, 44a and 44b show high inhibitory activity on these human cancer cell lines (IC50: 2.39–9.34 μM), the structure and activity of related compounds can be seen in Figure 17.105
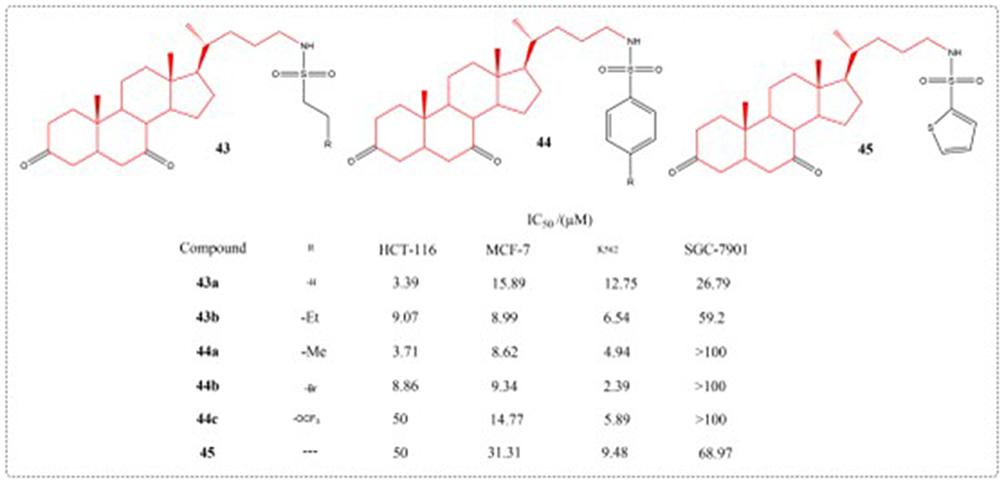 |
Figure 17 Structures of UDCA derivatives 43–45. IC50: Half Maximal Inhibitory Concentration. |
Research evidence confirms the anti-apoptotic effects of the bile acid UDCA across both cellular and animal model systems. Nevertheless, its clinical application is constrained by low water solubility. Dosa et al designed and synthesized several phosphate prodrugs. Compound 46 – a covalently bound phosphate ester – exhibits anti-apoptotic activity comparable to UDCA, functioning independently of alkaline phosphatase activation. In contrast, although highly effective in these tests, the POMC prodrug designated as compound 47 required enzymatic activation through supplementation with alkaline phosphatase, and the structures of compounds 46–47 are shown in Figure 18.106
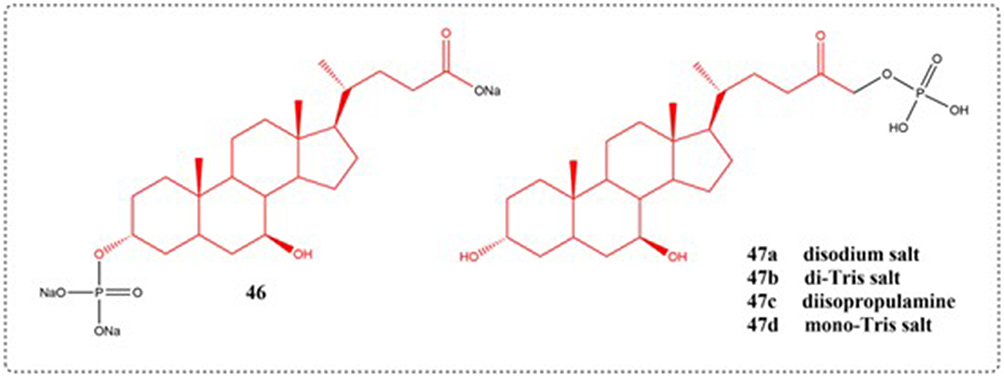 |
Figure 18 Structures of UDCA derivatives 46–47. |
The 2014 study by Li et al reported the design of CPT analogs incorporating bile acid groups at positions C-10 or C-20, which demonstrated reduced camptothecin-related toxicity while showing enhanced selectivity for hepatoma and colon carcinoma cells (Figure 19). The anti-tumor activity results showed that the introduction of bile acid groups at position 20 of CPT can reduce in vivo toxicity and enhance specificity for liver cancer cells.107
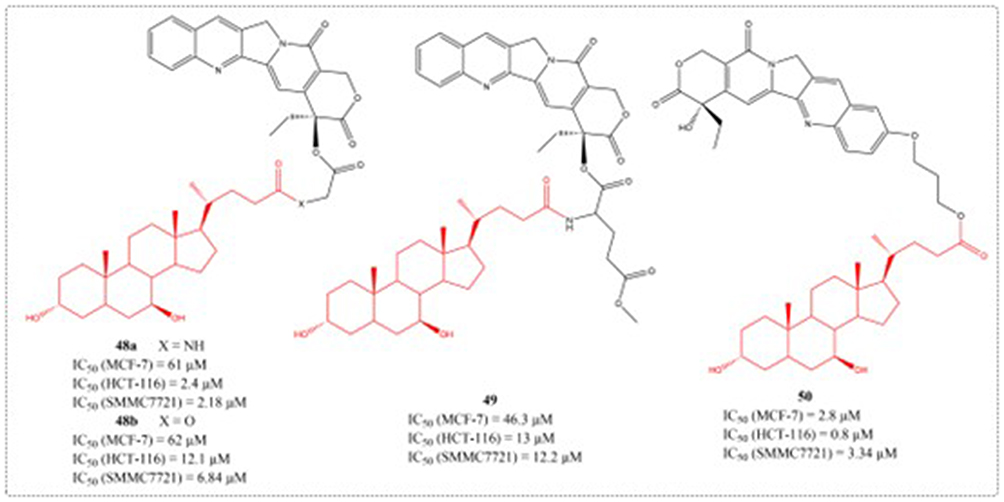 |
Figure 19 Structures of UDCA derivatives 48–50. IC50: Half Maximal Inhibitory Concentration. |
Brossard et al. Multiple new bile acid-piperazine carboxamide conjugates (prepared from cholic, ursodeoxycholic, chenodeoxycholic, deoxycholic and lithocholic acids) were developed and screened for their ability to induce apoptosis in DLD-1, HCT-116, and HT-29 colorectal carcinoma cells. It contains UDCA derivative 51, but its anti-tumor activity is poor.108 Samadi et al synthesized UDCA maleic anhydride derivatives (52) in 2017, but their IC50 values for C6 rat glioma cells exceeded 180 μM, the structures of compounds 51–52 are shown in Figure 20.109
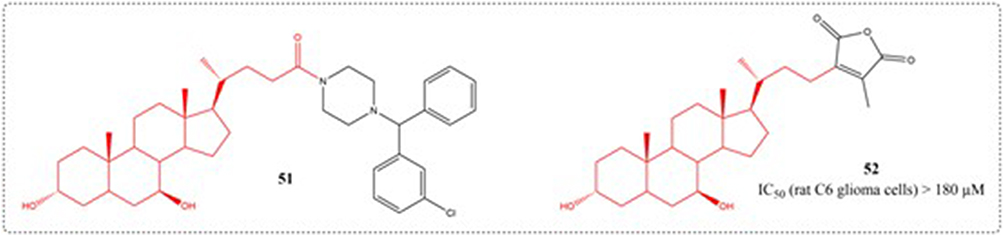 |
Figure 20 Structures of UDCA derivatives 51–52. IC50: Half Maximal Inhibitory Concentration. |
Navacchia et al synthesized a novel library of fully biomimetic conjugated compounds (53–58) combining nucleosides (A, G, and U) with UDCA using the conjugate method of click chemistry. Toxicity was tested on K562, HCT116, and normal fibroblasts. Paving the way for the coupling of nucleosides and bile acids to become new molecular entities for cancer therapy (Figure 21).110
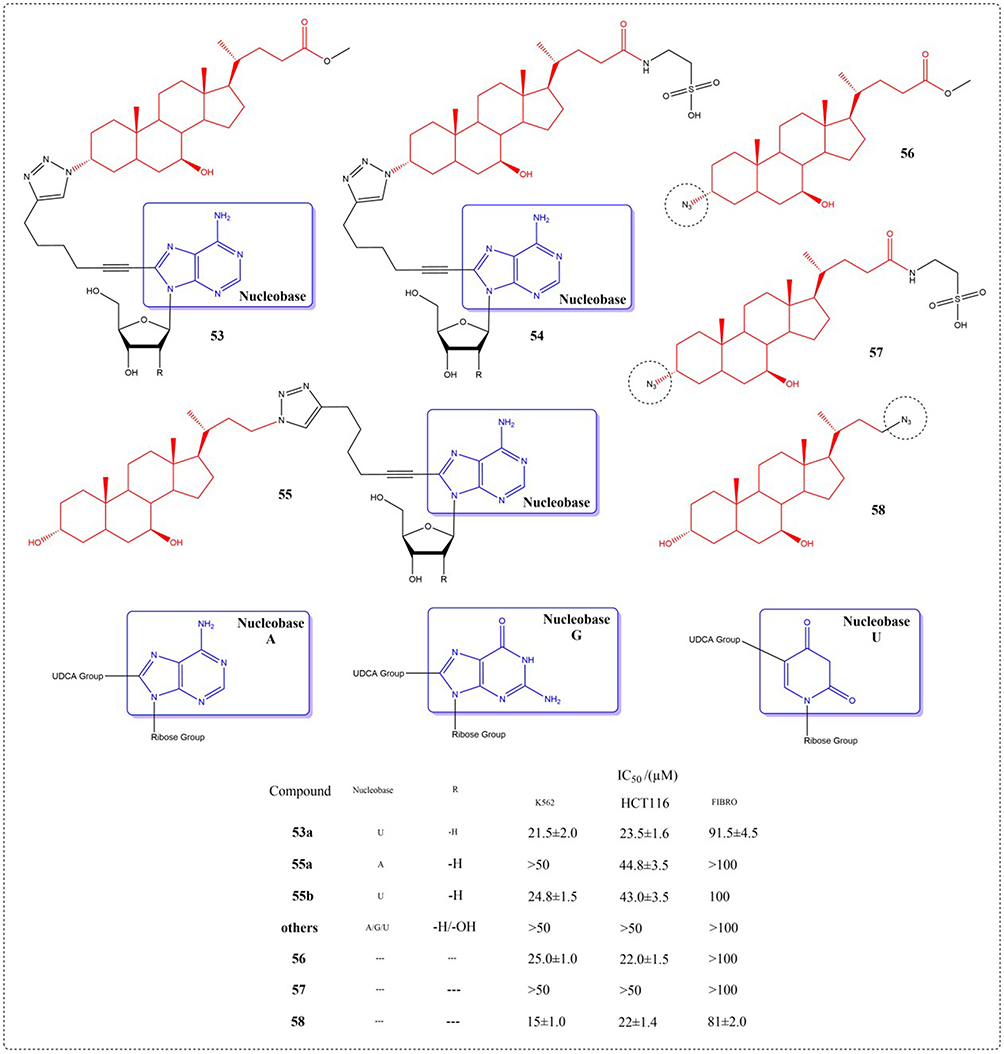 |
Figure 21 Structures of UDCA derivatives 53–58. IC50: Half Maximal Inhibitory Concentration. The group in the blue box is a nucleobase. |
Over the past three decades, multiple artemisinin derivatives have exhibited potent tumor-suppressive effects. However, no anticancer drug based on artemisinin has been approved for marketing so far.111 Among various dihydroartemisinin derivatives synthesized by Zou et al, the ursodeoxycholic acid hybrid (59) exhibited the highest biological activity. Compound 59 demonstrated potent cytotoxicity with sub-micromolar IC50 values across fifteen cancer cell lines. In vivo studies confirmed its antitumor efficacy in both A549 xenograft and Lewis lung carcinoma transplantation murine models. All of the results demonstrated that compound 59 is a potential anticancer agent that warrants further investigation.112 Subsequently, in 2019, the anti-tumor activity of UDCA-DHA hybrid compounds (61–63) synthesized by Marchesi’s team also confirmed that UDCA is a suitable partner for the preparation of novel DHA hybrids.113 The compound structures related to the previous text are shown in Figure 22.
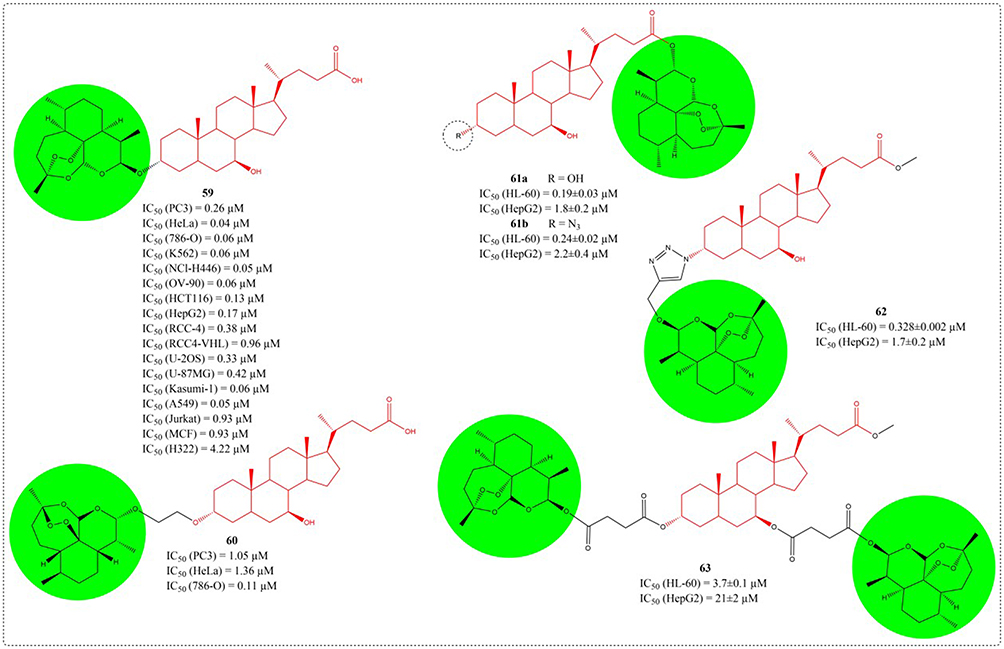 |
Figure 22 Structures of UDCA derivatives 59–63. IC50: Half Maximal Inhibitory Concentration. The group in the green circle is dihydroartemisinin. |
As an endogenous bile acid, UDCA has low toxicity and is continuously reabsorbed in the intestinal system and remains in the bile circulation. Research has indicated that UDCA can to some extent restrain tissue overgrowth and tumor development in inflamed intestines, playing an important role in the intestinal health. In a 2019 study, researchers used UDCA as raw material to optimize the structure of the 11th, 12th, 7th, and 24th substituents of UDCA. A total of 37 novel UDCA derivatives were s chemically prepared and subsequently assessed for their ability to inhibit colon cancer cell proliferation in vitro. Among them, compounds 64–66 showed the best activity, and compound 66c had the best anti colon cancer activity (Figure 23). At the same time, preliminary structure-activity relationships of this series of UDCA derivatives were obtained, providing important support for further in-depth research and the discovery of candidate compounds.114
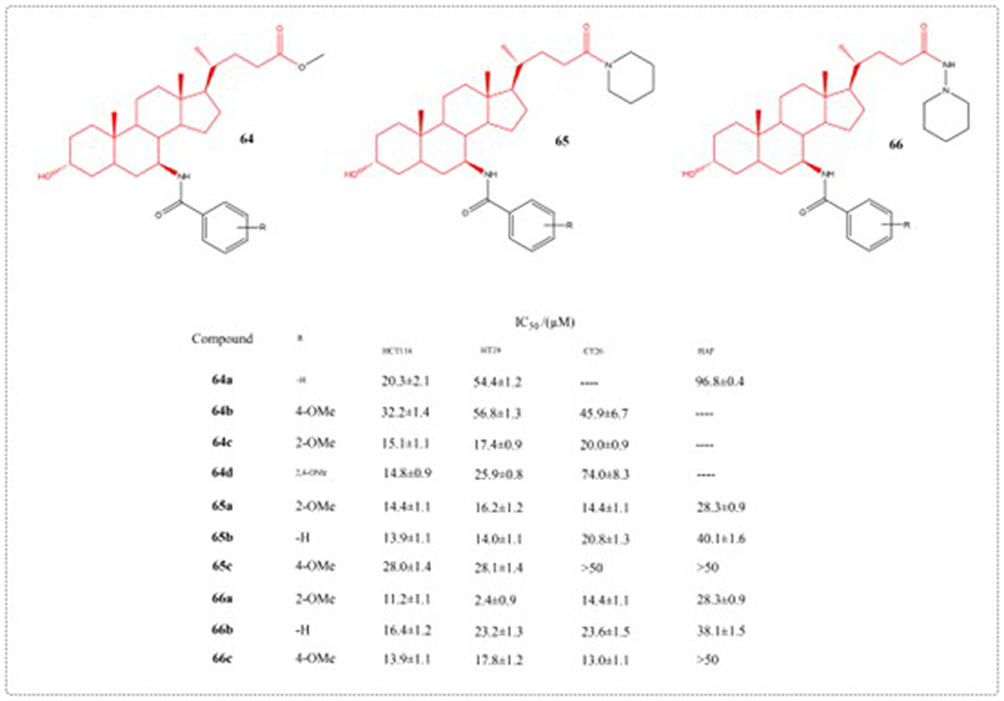 |
Figure 23 Structures of UDCA derivatives 64–66. IC50: Half Maximal Inhibitory Concentration. |
In 2014, Chen et al developed twenty derivatives using UDCA, identifying compound 67 as an agent capable of inhibiting both hepatocellular carcinoma cells (SMMC-7721 and HepG2) and shielding healthy hepatocytes from deoxycholic acid-induced injury. In vivo studies showed that compound 67 not only had superior antitumor efficacy to UDCA but also exhibited fewer adverse effects than 5-fluorouracil. Proteomics confirms that mTOR/S6K1, cyclinD1/CDK2/4, and caspase-dependent apoptotic signaling pathways are the mechanisms by which it exerts anti-tumor activity.115 In 2022, Chen et al. Optimized the structure of 67, and compound 68 had the strongest anti hepatoma activity (Figure 24). The mechanism study showed that compound 68 inhibited the proliferation of HepG2 cells by inhibiting G0/G1 phase, inhibited the activation of PI3K/Akt/mTOR pathway, and triggers the apoptosis of HepG2 cell by activating caspase signaling pathway.116
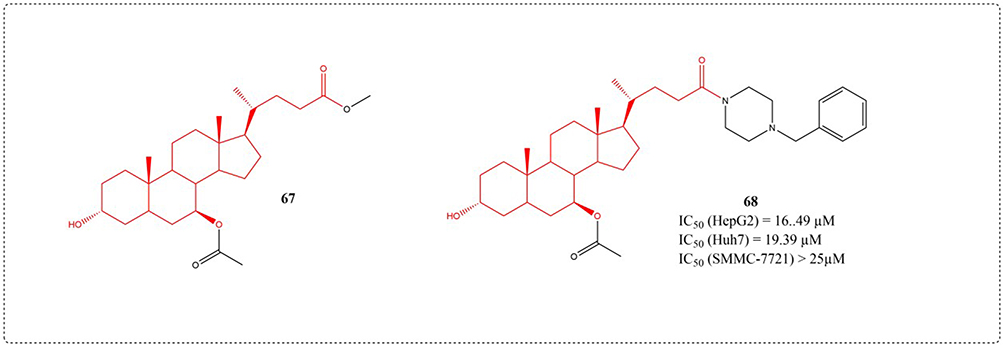 |
Figure 24 Structures of UDCA derivatives 67–68. IC50: Half Maximal Inhibitory Concentration. |
The enzyme autotaxin (ATX) catalyzes the conversion of lysophosphatidylcholine to the biologically active lipid mediator lysophosphatidic acid (LPA). Abnormal LPA expression is capable of driving the advancement of illnesses like cancer and fibrosis. TUDCA and UDCA are partial non-competitive modulators with micromolar affinity for the ATX tunnel.117 Clark designed and synthesized type V ATX inhibitors based on endogenous steroid regulator UDCA (Figure 25). Biochemical profiling of compounds 73b and 73c confirmed the therapeutic potential of novel type V ATX inhibitors, while derivative 23 demonstrated dual efficacy in suppressing both LPA biosynthesis and downstream receptor signaling.118
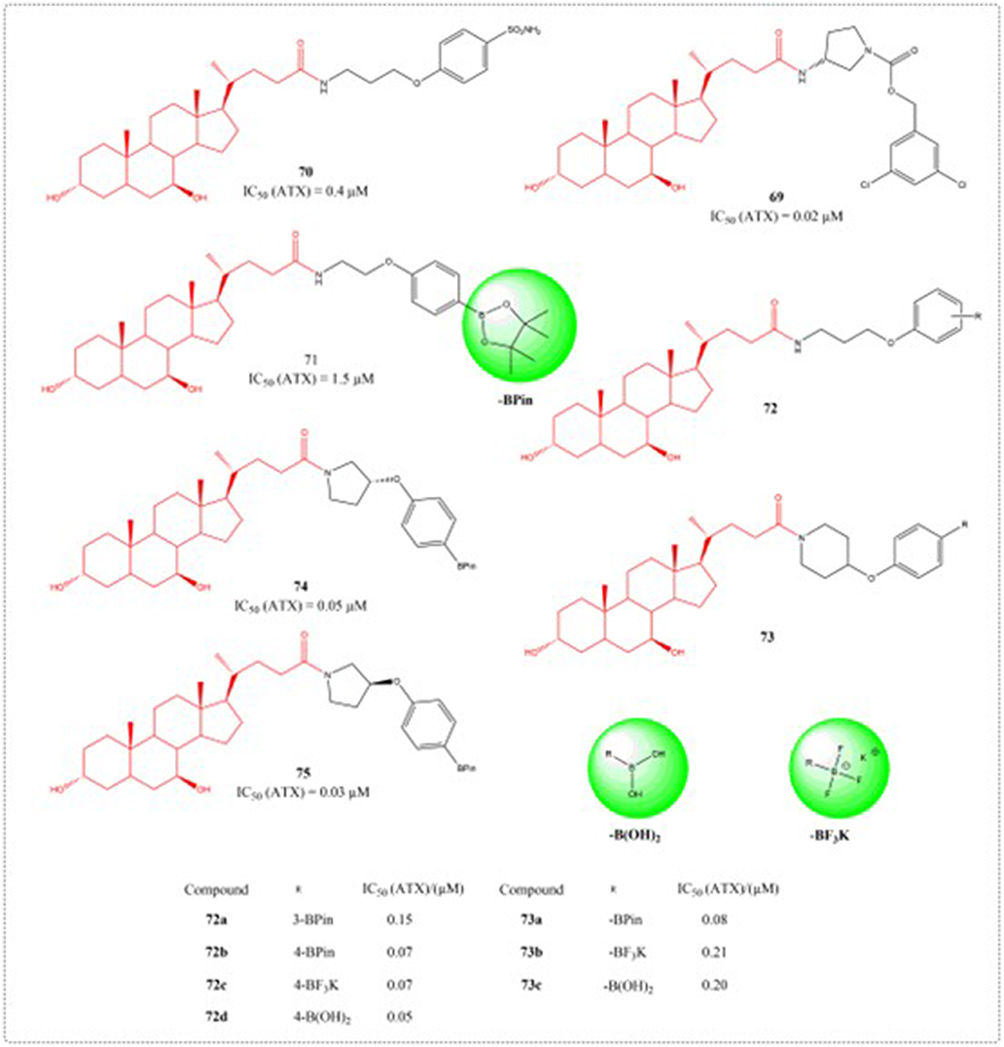 |
Figure 25 Structures of UDCA derivatives 69–75. IC50: Half Maximal Inhibitory Concentration. The groups in the green circles are boron-containing groups. |
Antibacterial Activity
In recent years, with the long-term overuse of antibiotics, the clinical emergence of multidrug-resistant bacteria is often closely related to high morbidity and mortality. The effectiveness of traditional antibiotics is decreasing, the development of new antibiotics is slow and bacteria are easy to obtain drug resistance, which increases the difficulty of clinical anti infection treatment. Therefore, it is urgent to find potential antibiotic alternatives.119 Currently, numerous bile acid-based derivatives have demonstrated significant antibacterial properties in recent studies.120 Huang et al developed and prepared eight novel steroid derivatives in 2009, including UDCA derivatives (76a and 76b). The strain’s inhibitory effects against Escherichia coli and Staphylococcus aureus was evaluated. Compounds 76a and 76b displayed markedly distinct growth inhibition profiles, attributable to variations in their side-chain lengths (Figure 26).121 Berberine has antibacterial activity against various bacteria and is is among the frequently employed medications for the treatment of treating intestinal infections. Due to its quaternary ammonium structure and high water solubility, this compound demonstrates limited intestinal absorption following oral dosing. Affects its biological availability. Li et al conceived and fabricated 15 berberine bile acid analogues in 2012, and evaluated their antibacterial activity against Gram positive bacteria S. aureus ATCC 25923, S. aureus ATCC 8799, Bacillus subtilis AS 1.398, and E. coli ATCC 31343 using MIC and MBC. Among them, compound 77 showed good inhibitory activity against S. aureus ATCC 25923 and S. aureus ATCC 8799.
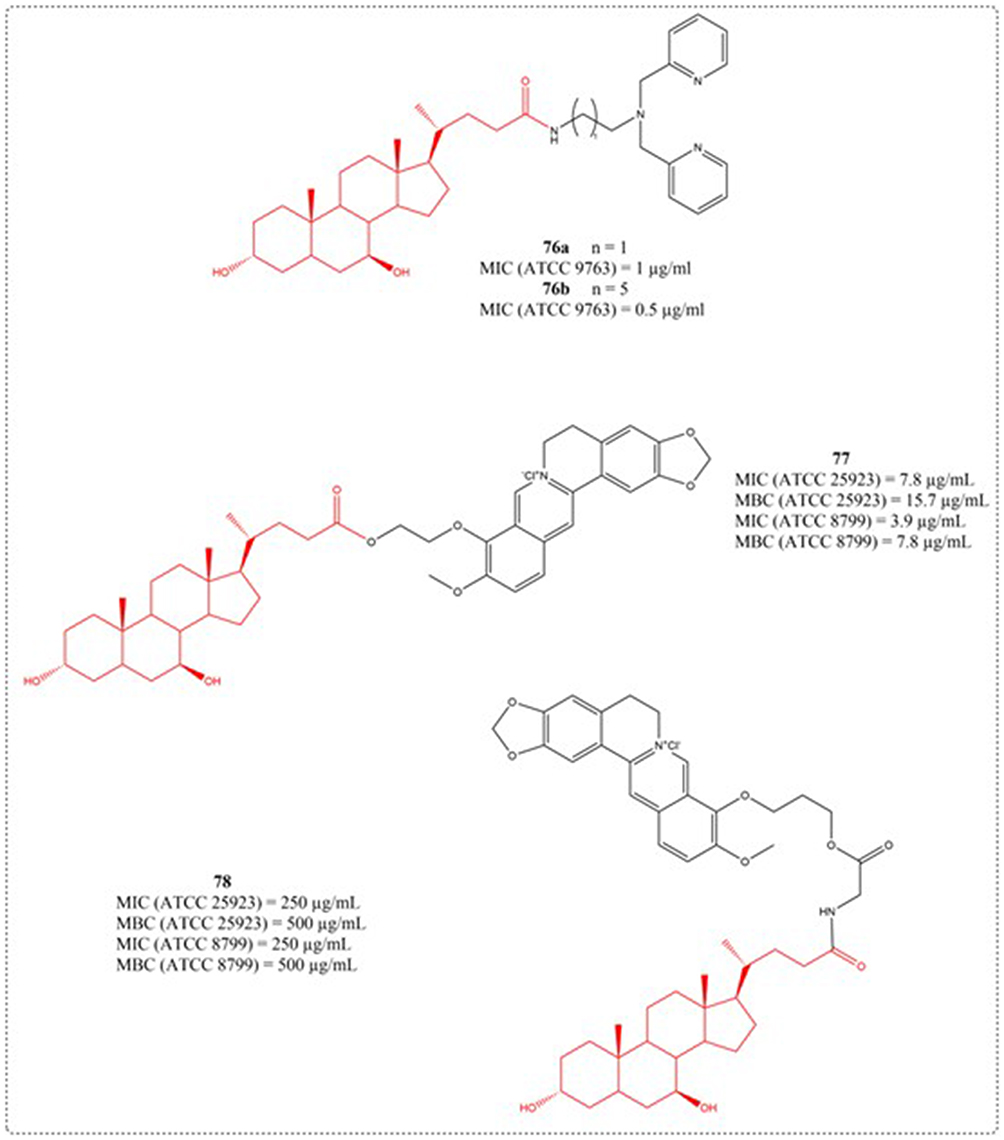 |
Figure 26 Structures of UDCA derivatives 76–78. Abbreviations: MIC, Minimum Inhibitory Concentration; MBC, Minimum Bactericidal Concentration. |
Menthol, a straightforward monoterpene, possesses marked biological functionalities ranging from anticancer, antibacterial, antifungal, and antiviral actions to anti-inflammatory, antipruritic, analgesic, and antitussive properties.122 In a study conducted in 2016, menthyl 1, 4-disubstituted 1, 2, 3-triazole conjugates incorporating hydroxybenzaldehydes, phenols and bile acids were prepared using click chemistry approaches. Compound 79 has a potent inhibitory action on Enterococcus faecium, with a MIC value of 7 µM, exceeding its positive drug cefixime (MIC=35 µM).123 Cateni et al prepared new derivatives with guanidine groups on their side chains using UDCA and trametenolic acid (TMA) as starting materials. The derived compounds 80 and 81 were very active against the Gram-positives Bacillus subtilis and Staphylococcus aureus, even at low MICs, and also showed an activity also against Mycobacterium smegmatis.124
Clostridium difficile, a Gram-positive, anaerobic bacterium capable of forming spores, induces colon infections that can be fatal. Traditional antibiotic-driven methods for addressing Clostridium difficile infection perturb the commensal microbiota and tend to be ineffective in eradicating bacterial spores—two major contributors to the recurrence of the infection.125 In 2017, Stoltz et al a number of bile acid analogues were discovered to function as powerful suppressors of spore germination in the highly virulent NAP1 strain of C. difficile. Activity profiling identified C24-tetrazole and -amide UDCA derivatives as top performers. The C3 position accommodated various functional groups (compounds 82–89), with derivative 85c (Figure 27) emerging as a dual-function agent - potently inhibiting C. difficile spore formation while demonstrating minimal absorption in Caco-2 intestinal barrier models, indicative of gut-selective activity.126
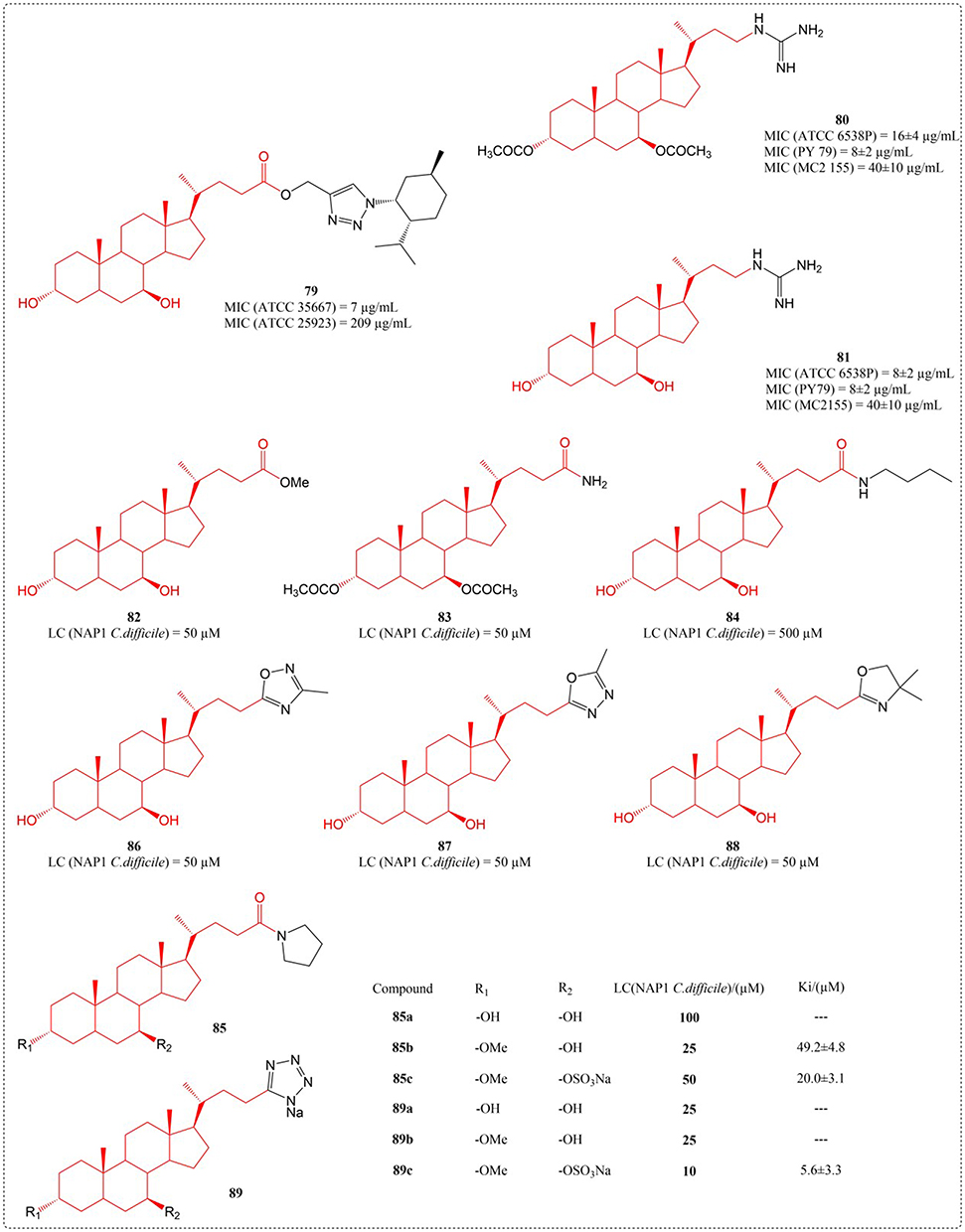 |
Figure 27 Structures of UDCA derivatives 79–89. Abbreviations: MIC, Minimum Inhibitory Concentration; LC, Lethal Concentration; Ki, Inhibition Constant. |
Anti-AIDS Activity
3′-Azido-3′-deoxythymidine (zidovudine or AZT) became the first medication sanctioned by the FDA for managing acquired immunodeficiency syndrome (AIDS), a condition resulting from human immunodeficiency virus (HIV) infection.127 Due to its short half-life, high concentrations in clinical use can easily lead to side effects such as anemia.128 Dalpiaz et al proposed a novel prodrug 90 (Figure 28) obtained by combining AZT with UDCA, to avoid the poor permeability of AZT within the central neural system or intracellular compartments. Marked species differences were observed in UDCA-AZT stability: instantaneous decomposition in rat plasma/blood (t½ <10 s) contrasted with prolonged hydrolysis in human specimens (plasma: 7.53 ± 0.44 h; whole blood: 3.71 ± 0.16 h), facilitating timed AZT liberation in human physiology.32
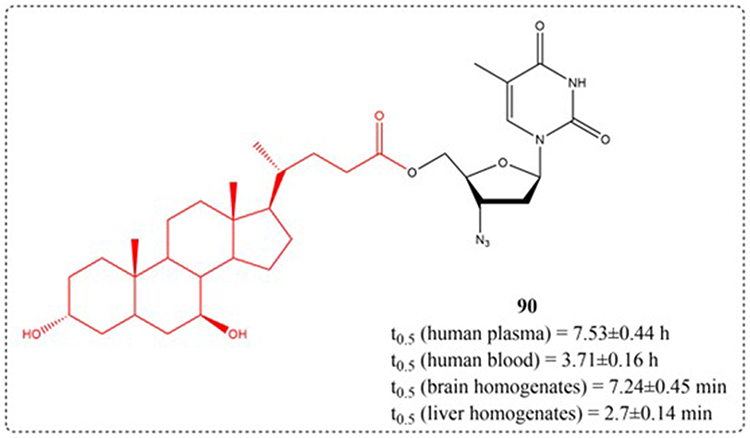 |
Figure 28 Structures of UDCA derivatives 90. t0.5: Half-life. |
Antiviral Activity
Although some progress has been made in managing chronic hepatitis B virus (HBV) infection, current first-line drugs have not been able to fully eradicate the covalently closed circular DNA (cccDNA) of HBV so as to completely eliminate HBV. Targeted therapy can specifically bind to the target sites, thereby blocking virus invasion, virus transcription, and protein translation, achieving precise treatment.129 Bile acid is currently the only orally effective small molecule liver targeted drug carrier, and its biggest advantage as a drug delivery carrier is its high efficiency in hepatic and intestinal circulation and good organ specificity. Upon conjugation with bile acids, drugs become substrates for bile acid transporters and enter the enterohepatic circulation of bile acids, thereby increasing drug absorption and concentration in the liver, endowing drugs with liver selectivity. Su et al designed and synthesized six novel structures of single L-amino acid ester and single bile acid ester derivatives of adefovir using adefovir as raw material. Pharmacological activity confirmed that the introduction of bile acid ester structural fragments on the phosphonic acid group of adefovir can significantly improve the ability of compounds to be taken up by liver cells and enhance their antiviral activity. The antiviral activity and liver cell uptake ability of compound 91c are the best, and it is worth further investigation (Figure 29).130
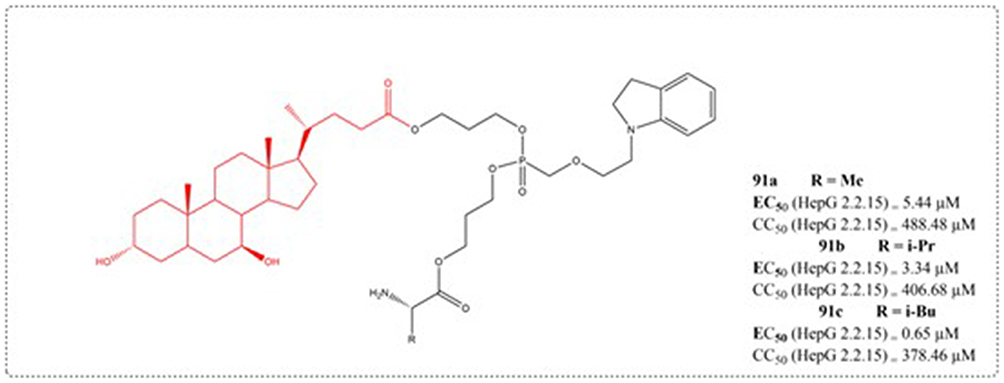 |
Figure 29 Structures of UDCA derivatives 91. EC50: Half Maximal Effective Concentration; CC50: Half Maximal Cytotoxic Concentration. |
Antidiabetic Activity
Type 2 diabetes (T2DM) is a metabolic disorder characterised by glucose toxicity, lipotoxicity, insulin resistance, and other pathological manifestations, and represents an urgent global health problem.131 GP-BAR1 (also known as TGR5), a G-protein-coupled receptor mediating bile acid-induced non-genomic responses, has gained attention as a potential therapeutic target for metabolic diseases such as obesity and type II diabetes. Yu et al designed GP-BAR1 agonists based on bile acids and conducted in vitro activity screening. The study confirmed that compound 92 is an effective GP-BAR1 ligand with specificity on GPBAR1 and does not activate FXR. Novel, highly potent GP-BAR1 modulators represent a promising therapeutic class with enhanced efficacy and safety for metabolic diseases.132 Additionally, Berberine Ursodeoxycholate (HTD1801), developed by HighTide Therapeutics, is an oral anti-inflammatory metabolic modulator targeting the gut-liver axis. Its core mechanism of action lies in achieving dual intervention of “metabolic regulation + inflammation inhibition” through synergistic effects of dual targets: on one hand, it activates the adenosine monophosphate-activated protein kinase (AMPK) pathway, a key regulator of cellular energy homeostasis, which improves insulin sensitivity, promotes fatty acid oxidation, and enhances cellular metabolic stress capacity; on the other hand, it potently inhibits the activation of the NLRP3 inflammasome, reduces the release of inflammatory factors such as IL-1β, thereby blocking inflammation-driven metabolic disorders and simultaneously improving blood glucose, lipid profiles, and organ inflammatory status. This unique mechanism enabled it to meet the primary efficacy endpoints in two Phase 3 clinical trials (SYMPHONY 1 and SYMPHONY 2) involving Chinese patients with type 2 diabetes mellitus (T2DM), not only significantly reducing glycated hemoglobin (HbA1c) but also improving lipid indices and inflammatory markers. Based on the above positive data, the company plans to submit a New Drug Application (NDA) to the Center for Drug Evaluation (CDE) of the National Medical Products Administration (NMPA) in 2025 for the treatment of T2DM.133
Insecticidal Activity
Malaria, along with various neglected conditions like human African trypanosomiasis (HAT, sleeping sickness), Chagas disease, and leishmaniasis—all triggered by protozoan parasites—endanger the well-being of millions worldwide, with a particular impact on developing regions in Asia, Africa, and Latin America.134 The available treatments have significant drawbacks and high costs and also, emerging parasite resistance is reducing the efficacy of these treatment strategies. For all these reasons, the development of new antiprotozoal agents is urgently needed.135 In a study by Harikandei et al in 2020, novel isothiocyanate derivatives were synthesized starting from noscapine, bile acids, amino acids, and some aromatic compounds. The synthesized compounds were evaluated for antiprotozoal activity against four pathogenic parasites: Trypanosoma brucei rhodesiense, Trypanosoma cruzi, Leishmania donovani, and Plasmodium falciparum. The IC50 value of compound 95 against T.b. rhodesiense is 1.6 uM.136
Anti-Inflammatory Activity
Inflammation represents the body’s physiological immune reaction to infectious agents or tissue damage, and its primary function is to clear harmful pathogens and reestablish tissue homeostasis.137 Li et al engineered and fabricated a group of UDCA-cinnamic acid hybrids in 2023. The capacity of synthesized derivatives to attenuate inflammatory responses was examined by monitoring LPS-induced NO generation in RAW264.7 murine macrophage cultures. Compound 96d exhibited the strongest inhibitory effect on NO (IC50 = 7.70 μM), and considerably diminished the levels of TNF-α, IL-1β, IL-6, and PGE2, downregulated the level of iNOS and COX-2, which may involve the Akt/NF-κB and MAPK signaling cascade. This substance holds potential as a novel lead compound for the further advancement of anti-inflammatory medications.138 In addition, the study by Sharma et al confirmed that the compound 97 can inhibit TNF-α-induced NF-κB activity and induce reverse transcription activation of GRE.31 The structure of a series of compounds (92–100) with anti diabetes activity, insecticidal activity and anti-inflammatory activity synthesized in the above literature is shown in Figure 30.
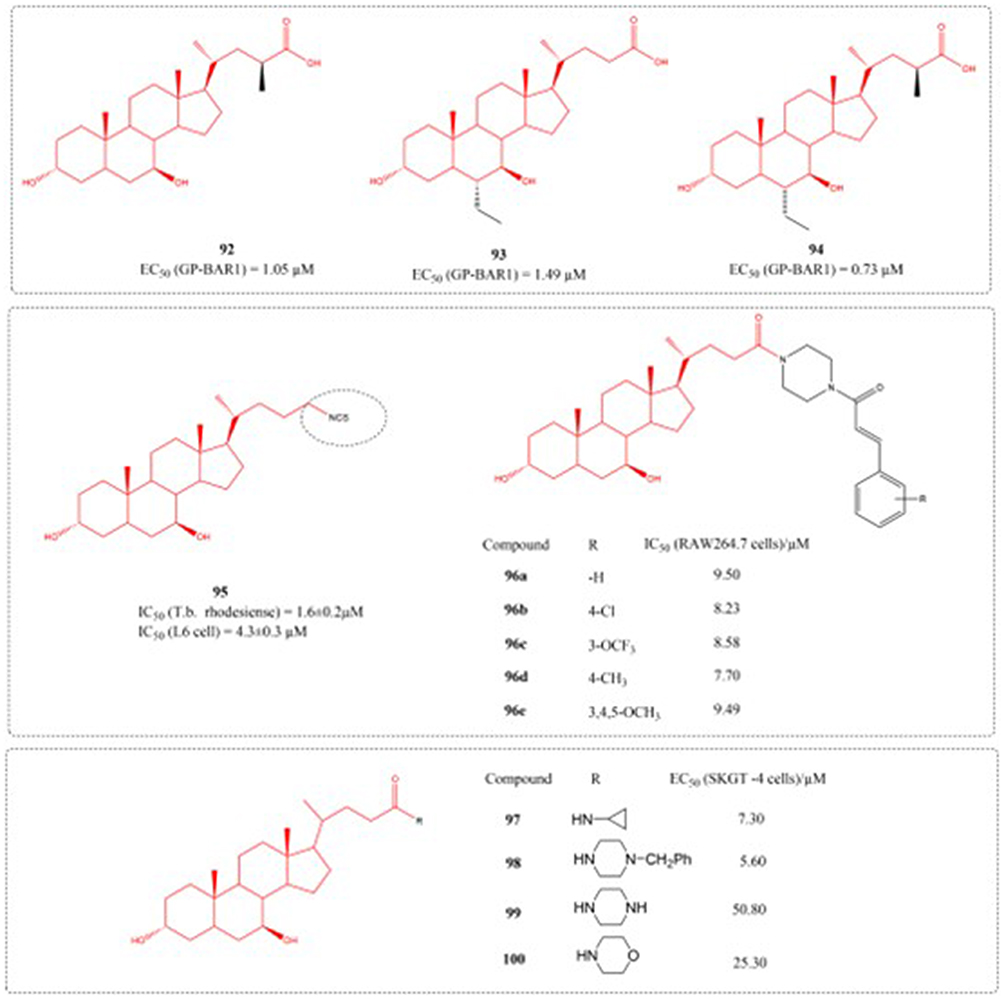 |
Figure 30 Structures of UDCA derivatives 92–100. EC50: Half Maximal Effective Concentration; IC50: Half Maximal Inhibitory Concentration. |
Conclusion
UDCA is a steroid compound found in bear bile and is a drug used to treat gallstones and liver diseases. UDCA has received widespread attention due to its numerous pharmacological activities, such as protecting the liver and gallbladder, regulating immunity, lowering blood lipids, lowering blood sugar, protecting against cardiovascular and cerebrovascular diseases, anti-inflammatory, anti-tumor, antiviral, and other pharmacological activities. In this review, we attempt to comprehensively introduce the pharmacological mechanisms, biosynthesis, and structural modifications of UDCA. Due to its unique targeting properties, it can also be used as a drug carrier. Multiple positions on the hydroxyl, carboxyl, and steroid rings of UDCA molecules can be chemically modified. Currently, drug developers typically modify UDCA by attaching amino acids, peptides, nucleoside analogues, and heterocyclic amines to the carboxyl or hydroxyl groups of UDCA to synthesize derivatives with different biological activities. And the derivatives of UDCA were classified based on drug activity and different modification sites. From the above discussion, it is evident that UDCA and its derivatives demonstrate diverse biological actions and enormous potential in the field of medicinal chemistry. Research has shown that UDCA derivatives have been targeted for their liver protection, cholesterol lowering, anti-tumor, insecticidal, hypoglycemic, and anti-inflammatory effects. Derivatives of UDCA have shown potential application value in both metabolic diseases and oncology. In metabolic diseases, they can exert antidiabetic activity by targeting key pathways or axes. For instance, GP-BAR1 agonists (such as compound 92) designed based on bile acids can specifically bind to GP-BAR1 without activating FXR, providing a safe and efficient direction for disease treatment. Additionally, HTD1801 improves insulin sensitivity by activating the AMPK pathway while inhibiting the NLRP3 inflammasome to reduce inflammatory factors. It has significantly reduced HbA1c and improved lipid profiles as well as inflammatory markers in Phase 3 clinical trials for type 2 diabetes mellitus. In the field of oncology, UDCA can assist in enhancing anticancer activity by constructing innovative platinum complexes (38a-38d). In in vitro tests on cancer cell lines such as human colorectal adenocarcinoma, hepatocellular carcinoma, and murine leukemia, some of these complexes have demonstrated antitumor activity comparable to that of cisplatin, providing new potential candidate drugs for cancer treatment. This effective UDCA derivative has a wide range of effects on other therapeutic targets, and future research on this scaffold may yield some more encouraging results in the field of medicinal chemistry. To better serve human health and enhance the application potential of UDCA derivatives, future research can focus on three core directions: first, developing nanodelivery systems to improve the solubility and bioavailability of UDCA and its derivatives, and reduce dosage by using carriers such as nanoparticles and liposomes; second, optimizing conjugation strategies by conjugating UDCA with ligands like liver-targeting molecules and inflammation site-targeting groups to enhance drug targeting and reduce off-target effects; third, exploring personalized medicine approaches by formulating differentiated medication regimens based on patients’ disease types, genotypes, and metabolic characteristics to improve the safety and efficacy of treatment.
Data Sharing Statement
No datasets were created or analyzed as part of the present study.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
This work was supported by the Shaanxi Provincial Natural Science Basic Research Program (2022JQ-895).
Disclosure
The authors report no conflicts of interest in this work.
References
1. Li X, Su F, Jiang C, et al. Efficacy evolution of bear bile and related research on components. Zhongguo Zhongyao Zazhi. 2022;47(18):4846–4853. doi:10.19540/j.cnki.cjcmm.20220527.601
2. Zhu L, Liao J, Lu X, et al. Research progress in pharmacological effectsand mechanism of Fel Ursi against cardiovascular and cerebrovascular diseases. Zhongguo Zhongyao Zazhi. 2023;48(23):6307–6314. doi:10.19540/j.cnki.cjcmm.20230823.601
3. Zhu H, Wang G, Bai Y, et al. Natural bear bile powder suppresses neuroinflammation in lipopolysaccharide-treated mice via regulating tgr5/akt/nf-κb signaling pathway. J Ethnopharmacol. 2022;291:115063. doi:10.1016/j.jep.2022.115193
4. Yanguas-Casas N, Asuncion Barreda-Manso M, Nieto-Sampedro M, et al. Tauroursodeoxycholic acid reduces glial cell activation in an animal model of acute neuroinflammation. J Neuroinflammation. 2014;11(1):50. doi:10.1186/1742-2094-11-50
5. Rivard A, Steer C, Kren B, et al. Administration of tauroursodeoxycholic acid (tudca) reduces apoptosis following myocardial infarction in rat. Am J Chin Med. 2007;35(2):279–295. doi:10.1142/s0192415x07004813
6. Maruhashi T, Kihara Y, Higashi Y. Bilirubin and endothelial function. J Atheroscler Thromb. 2019;26(8):688–696. doi:10.5551/jat.RV17035
7. Chen H, Shen A, Liu L, et al. Bear bile powder inhibits growth of hepatocellular carcinoma via suppressing stat3 signaling pathway in mice. Chin J Integr Med. 2020;26(5):370–374. doi:10.1007/s11655-019-3010-1
8. Zheng M, Li Y, Wang G, et al. Protective effect of cultured bear bile powder against dimethylnitrosamine-induced hepatic fibrosis in rats. Biomed Pharmacother. 2019;112:108701. doi:10.1016/j.biopha.2019.108701
9. Yuan B, Ren Y, Ma L, et al. Analysis on replacement of traditional Chinese medicine bear bile with bile acids based on drug properties. Zhongguo Zhongyao Zazhi. 2014;39(4):738–743.
10. Guo M, Zhang Y, Wang H, et al. Progress in synthesis of ursodeoxycholic acid from chenodeoxycholic acid. Chin J New Drugs. 2020;29(20):2358–2363.
11. Eggert T, Bakonyi D, Hummel W. Enzymatic routes for the synthesis of ursodeoxycholic acid. J Biotechnol. 2014;191:11–21. doi:10.1016/j.jbiotec.2014.08.006
12. Bouscarel B, Fromm H, Ceryak S, et al. Ursodeoxycholic acid increases low-density lipoprotein binding, uptake and degradation in isolated hamster hepatocytes. Biochem J. 1992;280(3):589–598. doi:10.1042/bj2800589
13. Fiorucci S, Biagioli M, Zampella A, et al. Bile acids activated receptors regulate innate immunity. Front Immunol. 2018;13(9):1853. doi:10.3389/fimmu.2018.01853
14. Zangerolamo L, Vettorazzi JF, Rosa LRO, et al. The bile acid tudca and neurodegenerative disorders: an overview. Life Sci. 2021;272(7):119252. doi:10.1016/j.lfs.2021.119252
15. Erlinger S, Le Go A, Husson J-M, Fevery J. Franco-Belgian cooperative study of ursodeoxycholic acid in the medical dissolution of gallstones: a double-blind, randomized, dose-response study, and comparison with chenodeoxycholic acid. Hepatology. 1984;4(2):308–314. doi:10.1002/hep.1840040222
16. Podda M, Zuin M, Dioguardi ML, et al. A combination of chenodeoxycholic acid and ursodeoxycholic acid is more effective than either alone in reducing biliary cholesterol saturation. Hepatology. 1982;2(3):334–339. doi:10.1002/hep.1840020308
17. Song P, Zhang X, Feng W, et al. Biological synthesis of ursodeoxycholic acid. Front Microbiol. 2023;14:1140662. doi:10.3389/fmicb.2023.1140662
18. Hillaire S, Boucher E, Calmus Y, et al. Effects of bile acids and cholestasis on major histocompatibility complex class i in human and rat hepatocytes. Gastroenterology. 1994;107(3):781–788. doi:10.1016/0016-5085(94)90127-9
19. Zhou C, Gao G, Liu Y. Advances in studies on bear bile powder. Zhongguo Zhongyao Zazhi. 2015;40(7):1252–1258.
20. Lukivskaya O, Lis R, Egorov A, et al. The protective effect of ursodeoxycholic acid in alloxan-induced diabetes. Cell Biochem Funct. 2004;22(2):97–103. doi:10.1002/cbf.1063
21. Rajesh K, Suzuki R, Maeda H, et al. Hydrophilic bile salt ursodeoxycholic acid protects myocardium against reperfusion injury in a pi3k/akt dependent pathway. J Mol Cell Cardiol. 2005;39(5):766–776. doi:10.1016/j.yjmcc.2005.07.014
22. Willart M, van Nimwegen M, Grefhorst A, et al. Ursodeoxycholic acid suppresses eosinophilic airway inflammation by inhibiting the function of dendritic cells through the nuclear farnesoid x receptor. Allergy. 2012;67(12):1501–1510. doi:10.1111/all.12019
23. Chen W, Feng Z, Sun Q. A novel ursodeoxycholic acid-chitosan-folate conjugates for the delivery of calcitriol for cancer therapy. J Drug Delivery Sci Technol. 2022;73. doi:10.1016/j.jddst.2022.103410
24. Huang F. Ursodeoxycholic acid as a potential alternative therapeutic approach for neurodegenerative disorders: effects on cell apoptosis, oxidative stress and inflammation in the brain. Brain Behav Immun. 2021;21(18):100348. doi:10.1016/j.bbih.2021.100348
25. Goossens J, Bailly C. Ursodeoxycholic acid and cancer: from chemoprevention to chemotherapy. Pharmacol Ther. 2019;203:107396. doi:10.1016/j.pharmthera.2019.107396
26. Drzymała-Czyż S, Jończyk-Potoczna K, Lisowska A, et al. Supplementation of ursodeoxycholic acid improves fat digestion and absorption in cystic fibrosis patients with mild liver involvement. Eur J Gastroenterol Hepatol. 2016;28(6):645–649. doi:10.1097/MEG.0000000000000593
27. Brevini T, Maes M, Webb G, et al. Fxr inhibition may protect from sars-cov-2 infection by reducing ace2. Nature. 2023;615(7950):134–142. doi:10.1038/s41586-022-05594-0
28. Chen X, Wei C, Zhao J, et al. Carnosic acid: an effective phenolic diterpenoid for prevention and management of cancers via targeting multiple signaling pathways. Pharmacol Res. 2024;206:107288. doi:10.1016/j.phrs.2024.107288
29. Kitani K. Ursodeoxycholic acid for cholestatic diseases. Lancet. 1988;2(8601):49. doi:10.1016/S0140-6736(88)92983-2
30. Poupon RE, Balkau B, Eschwege E, et al. A multicenter, controlled trial of ursodiol for the treatment of primary biliary cirrhosis. Udca-pbc study group. New Engl J Med. 1991;324(22):1548–1554. doi:10.1056/nejm199105303242204
31. Sharma R, Prichard D, Majer F, et al. Ursodeoxycholic acid amides as novel glucocorticoid receptor modulators. J Med Chem. 2011;54(1):122–130. doi:10.1021/jm100860s
32. Dalpiaz A, Paganetto G, Pavan B, et al. Zidovudine and ursodeoxycholic acid conjugation: design of a new prodrug potentially able to bypass the active efflux transport systems of the central nervous system. Mol Pharm. 2012;9(4):957–968. doi:10.1021/mp200565g
33. Angulo P. Use of ursodeoxycholic acid in patients with liver disease. Curr Gastroenterol Rep. 2002;4(1):37–44. doi:10.1007/s11894-002-0036-9
34. Hall L, Halle Smith J, Evans R, et al. Ursodeoxycholic acid in the management of symptomatic gallstone disease: systematic review and clinician survey. BJS Open. 2023;7(2):zrac152. doi:10.1093/bjsopen/zrac152
35. Zhu G, Shi K, Huang S, et al. Network meta-analysis of randomized controlled trials: efficacy and safety of udca-based therapies in primary biliary cirrhosis. Medicine. 2015;94(11):e609. doi:10.1097/md.0000000000000609
36. Cazzagon N, Floreani A. Primary biliary cholangitis: treatment. Curr Opin Gastroenterol. 2021;37(2):99–104. doi:10.1097/mog.0000000000000708
37. Koehler E, Plompen E, Schouten J, et al. Presence of diabetes mellitus and steatosis is associated with liver stiffness in a general population: the rotterdam study. Hepatology. 2016;63(1):138–147. doi:10.1002/hep.27981
38. Li W, Gao Q, Ma L. Synergistic protective mechanism of vitamin e and ursodeoxycholic acid on nonalcoholic fatty liver in rats. Genom Appl Biol. 2019;38(12):5713–5718.
39. Jiménez-González C, Alonso-Pea M, Argos Vélez P, et al. Unraveling masld: the role of gut microbiota, dietary modulation, and ai-driven lifestyle interventions. Nutrients. 2025;17(9):1580. doi:10.3390/nu17091580
40. Tanaka H, Makino Y, Miura T, et al. Ligand-independent activation of the glucocorticoid receptor by ursodeoxycholic acid. Repression of ifn-gamma-induced mhc class ii gene expression via a glucocorticoid receptor-dependent pathway. J Immunol. 1996;156(4):1601–1608. doi:10.4049/jimmunol.156.4.1601
41. Takigawa T, Miyazaki H, Kinoshita M, et al. Glucocorticoid receptor-dependent immunomodulatory effect of ursodeoxycholic acid on liver lymphocytes in mice. Am J Physiol Gastrointest Liver Physiol. 2013;305(6):G427–G438. doi:10.1152/ajpgi.00205.2012
42. Cheng J, Ren J, Wu Q, et al. Observation on the therapeutic effect of acupuncture combined with herbal medicine on treating fatty liver. Lishizhen Med Materia Medica Res. 2016;27(3):635–636.
43. Lukivskaya O, Patsenker E, Buko VU. Protective effect of ursodeoxycholic acid on liver mitochondrial function in rats with alloxan-induced diabetes: link with oxidative stress. Life Sci. 2007;80(26):2397–2402. doi:10.1016/j.lfs.2007.02.042
44. Zhu M, Liu Q, Guo M, et al. The protective effect of ursodeoxycholic acid on pancreatic beta-cell in stz-induced diabetic rats. Acta Universitatis Medicinalis Nanjing. 2008;28(3):286–290.
45. Zhang S, Yan F, Luan F, et al. The pathological mechanisms and potential therapeutic drugs for myocardial ischemia reperfusion injury. Phytomedicine. 2024;129:155649. doi:10.1016/j.phymed.2024.155649
46. Ning H, Yi M, Huang X, et al. Research progress of traditional Chinese medicine intervention for ulcerative colitis based on notch1 signaling pathway. Zhongguo Zhongyao Zazhi. 2024;49(7):1741–1748. doi:10.19540/j.cnki.cjcmm.20231127.405
47. Jiao J, Jiang X, Zhao Y, et al. Effects of ursodeoxycholic acid combined with bifidobacterium on inflammatory factors and intestinal flora in mice with colitis. Chin J Microecol. 2021;33(5):524–530.
48. Li W, Wang Z, Liu X, et al. Based on weak bond chemistry, the interaction mechanism between glycyrrhiza protein and berberine in water decocting process of rhizoma coptidis and liquorice was investigated. Yaoxue Xuebao. 2021;56(8):2119–2126. doi:10.16438/j.0513-4870.2021-0258
49. Gao S, Gao F, Kong J, et al. Therapeutic effect of ursodeoxycholic acid-berberine supramolecular nanoparticles on ulcerative colitis based on supramolecular system induced by weak bond. Zhongguo Zhongyao Zazhi. 2023;48(10):2739–2748. doi:10.19540/j.cnki.cjcmm.20230223.301
50. Ansari MF, Khan HY, Tabassum S, et al. Advances in anticancer alkaloid-derived metallo-chemotherapeutic agents in the last decade: mechanism of action and future prospects. Pharmacol Ther. 2023;241:108335. doi:10.1016/j.pharmthera.2022.108335
51. Ikegami T, Matsuzaki Y, Al Rashid M, et al. Enhancement of DNA topoisomerase i inhibitor-induced apoptosis by ursodeoxycholic acid. Mol Cancer Ther. 2006;5(1):68–79. doi:10.1158/1535-7163.Mct-05-0107
52. Lim SC, Han SI. Ursodeoxycholic acid effectively kills drug-resistant gastric cancer cells through induction of autophagic death. Oncol Rep. 2015;34(3):1261. doi:10.3892/or.2015.4076
53. Chen JY, Huang TR, Hsu SY, et al. Effect and mechanism of quercetin or quercetin-containing formulas against COVID −19: from bench to bedside. Phytother Res. 2024;38(6):2597–2618. doi:10.1002/ptr.8175
54. Latarissa IR, Meiliana A, Sormin IP, et al. The efficacy of herbal medicines on the length of stay and negative conversion time/rate outcomes in patients with covid-19: a systematic review. Front Pharmacol. 2024;15:1383359. doi:10.3389/fphar.2024.1383359
55. Zhang W, Ma L, Xie W, et al. Advances in the application of traditional Chinese medicine during the covid-19 recovery period: a review. Medicine. 2024;103(14):e37683. doi:10.1097/md.0000000000037683
56. Fiorillo B, Marchiano S, Moraca F, et al. Discovery of bile acid derivatives as potent ace2 activators by virtual screening and essential dynamics. J Chem Inf Model. 2022;62(1):196–209. doi:10.1021/acs.jcim.1c01126
57. Pang J, Feng JN, Ling W, et al. Commentary: can fxr serve as a potential target for covid-19 prevention? Acta Pharmaceutica Sinica B. 2023;13(4):1786–1788. doi:10.1016/j.apsb.2023.01.023
58. Simoni P, Cerrè C, Cipolla A, et al. Bioavailability study of a new, sinking, enteric-coated ursodeoxycholic acid formulation. Pharmacol Res. 1995;31(2):115–119. doi:10.1016/1043-6618(95)80056-5
59. Kotb MA. Molecular mechanisms of ursodeoxycholic acid toxicity & side effects: ursodeoxycholic acid freezes regeneration & induces hibernation mode. Int J Mol Sci. 2012;13(7):8882–8914. doi:10.3390/ijms13078882
60. Li Y, Huang Y, Feng N, et al. Artificial bear bile: a novel approach to balancing medical requirements and animal welfare. Engineering. 2024;38:100–112. doi:10.1016/j.eng.2023.09.017
61. Tonin F, Arends IWCE. Latest development in the synthesis of ursodeoxycholic acid (udca): a critical review. Beilstein J Org Chem. 2018;14:470–483. doi:10.3762/bjoc.14.33
62. Zhang B, Wang Z, Wang Y, Yang Y, Song J, Zhang B. Studies on the synthesis process of plant-derived ursodeoxycholic acid intermediates. Molecules. 2025;30(7):1454. doi:10.3390/molecules30071454
63. Zhang X, Fan D, Hua X, et al. Large-scale production of ursodeoxycholic acid from chenodeoxycholic acid by engineering 7α- and 7β-hydroxysteroid dehydrogenase. Bioprocess Biosyst Eng. 2019;42(9):1537–1545. doi:10.1007/s00449-019-02151-4
64. Zheng M, Chen K, Wang R, et al. Engineering 7β-hydroxysteroid dehydrogenase for enhanced ursodeoxycholic acid production by multiobjective directed evolution. J Agric Food Chem. 2017;65(6):1178–1185. doi:10.1021/acs.jafc.6b05428
65. Zheng M, Wang R, Li C, et al. Two-step enzymatic synthesis of ursodeoxycholic acid with a new 7β-hydroxysteroid dehydrogenase from ruminococcus torques. Process Biochem. 2015;50(4):598–604. doi:10.1016/j.procbio.2014.12.026
66. Grobe S, Badenhorst CPS, Bayer T, et al. Engineering regioselectivity of a p450 monooxygenase enables the synthesis of ursodeoxycholic acid via 7β-hydroxylation of lithocholic acid. Angew Chem Int Edit. 2021;60(2):753–757. doi:10.1002/anie.202012675
67. Kollerov V, Monti D, Deshcherevskaya N, et al. Hydroxylation of lithocholic acid by selected actinobacteria and filamentous fungi. Steroids. 2013;78(3):370–378. doi:10.1016/j.steroids.2012.12.010
68. Zhou ZR, Liu F, Li S, et al. A fungal p450 enzyme from fusarium equiseti hg18 with 7β-hydroxylase activity in biosynthesis of ursodeoxycholic acid. J Steroid Biochem Mol Biol. 2024;240:106507. doi:10.1016/j.jsbmb.2024.106507
69. Newman S, Gu L, Lesniak C, et al. Rapid wolff-kishner reductions in a silicon carbide microreactor. Green Chem. 2014;16(1):176–180. doi:10.1039/c3gc41942h
70. Zappaterra F, Costa S, Summa D, et al. Glyceric prodrug of ursodeoxycholic acid (udca): novozym 435-catalyzed synthesis of udca-monoglyceride. Molecules. 2021;26(19):5966. doi:10.3390/molecules26195966
71. Babu P, Maitra U. Synthesis and in vitro cholesterol dissolution by 23-and 24-phosphonobile acids. Steroids. 2005;70(10):681–689. doi:10.1016/j.steroids.2005.03.008
72. Pauli-Magnus C, Stieger B, Meier Y, et al. Enterohepatic transport of bile salts and genetics of cholestasis. J Hepatol. 2005;43(2):342–357. doi:10.1016/j.jhep.2005.03.017
73. Gonzalez P, Acharya C, MacKerell A, et al. Inhibition requirements of the human apical sodium-dependent bile acid transporter (hasbt) using aminopiperidine conjugates of glutamyl-bile acids. Pharm Res. 2009;26(7):1665–1678. doi:10.1007/s11095-009-9877-3
74. Kolhatkar V, Polli J. Structural requirements of bile acid transporters: c-3 and c-7 modifications of steroidal hydroxyl groups. Eur J Pharm Sci. 2012;46(1–2):86–99. doi:10.1016/j.ejps.2012.02.012
75. Watanabe M, Houten S, Mataki C, et al. Bile acids induce energy expenditure by promoting intracellular thyroid hormone activation. Nature. 2006;439(7075):484–489. doi:10.1038/nature04330
76. Thomas C, Gioiello A, Noriega L, et al. Tgr5-mediated bile acid sensing controls glucose homeostasis. Cell Metab. 2009;10(3):167–177. doi:10.1016/j.cmet.2009.08.001
77. Sepe V, Renga B, Festa C, et al. Modification on ursodeoxycholic acid (udca) scaffold. Discovery of bile acid derivatives as selective agonists of cell-surface g-protein coupled bile acid receptor 1 (gp-bar1). J Med Chem. 2014;57(18):7687–7701. doi:10.1021/jm500889f
78. Williamson C, Miragoli M, Kadir S, et al. Bile acid signaling in fetal tissues: implications for intrahepatic cholestasis of pregnancy. Dig Dis. 2011;29(1):58–61. doi:10.1159/000324130
79. Lazaridis K, Gores G, Lindor K. Ursodeoxycholic acid ‘mechanisms of action and clinical use in hepatobiliary disorders’. J Hepatol. 2001;35(1):134–146. doi:10.1016/s0168-8278(01)00092-7
80. Parquet M, Metman E, Raizman A, et al. Bioavailability, gastrointestinal transit, solubilization and faecal excretion of ursodeoxycholic acid in man. Eur J Clin Invest. 1985;15(4):171–178. doi:10.1111/j.1365-2362.1985.tb00164.x
81. Di Guida F, Pirozzi C, Magliocca S, et al. Galactosylated pro-drug of ursodeoxycholic acid: design, synthesis, characterization, and pharmacological effects in a rat model of estrogen-induced cholestasis. Mol Pharm. 2018;15(1):21–30. doi:10.1021/acs.molpharmaceut.7b00626
82. Li D, Lu Z, Hui J, et al. Therapeutic effects of zk_(14), a novel nitric oxide donating biphenyldicarboxylate derivative, on hepatic fibrosis in rats. J China Pharm Univ. 2009;40(3):254–257.
83. Liang G, Cao P, Yang X, et al. Synthesis and anti-hepatitis b virus activities of nitric oxide-releasing derivatives of matijin-su. Chin J Org Chem. 2014;34(5):973–979. doi:10.6023/cjoc201312003
84. Li M, He X, Tao L, et al. Design and synthesis of liver targeted no-releasing drugs with bile acids as carriers. Chin J Org Chem. 2008;28(12):2170–2174.
85. Fiorucci S, Antonelli E, Distrutti E, et al. Liver delivery of no by ncx-1000 protects against acute liver failure and mitochondrial dysfunction induced by APAP in mice. Br J Pharmacol. 2004;156(1):33–42. doi:10.1111/j.1476-5381.2009.00273.x
86. Latt N, Alachkar N, Gurakar A. Hepatitis c virus and its renal manifestations: a review and update. Gastroenterol Hepatol. 2012;8(7):434–445.
87. De Franceschi L, Fattovich G, Turrini F, et al. Hemolytic anemia induced by ribavirin therapy in patients with chronic hepatitis c virus infection: role of membrane oxidative damage. Hepatology. 2000;31(4):997–1004. doi:10.1053/he.2000.5789
88. Dong Z, Li Q, Guo D, et al. Synthesis and evaluation of bile acid-ribavirin conjugates as prodrugs to target the liver. J Pharmaceut Sci. 2015;104(9):2864–2876. doi:10.1002/jps.24375
89. Motaleb M, Abo-kul M, Ibrahim S, et al. Synthesis of 125 I-lamivudine and 125 I-lamivudine-ursodeoxycholic acid codrug. J Labelled Compd Rad. 2016;59(11):451–453. doi:10.1002/jlcr.3434
90. Zhang S, Gao S, Zhao M, et al. Anti-hbv drugs suppress the growth of hbv-related hepatoma cells via down-regulation of hepatitis b virus x protein. Cancer Lett. 2017;392:94–104. doi:10.1016/j.canlet.2017.02.003
91. Jia H, Rai D, Zhan P, et al. Recent advance of the hepatitis b virus inhibitors: a medicinal chemistry overview. Fut Med Chemist. 2015;7(5):587–607. doi:10.4155/fmc.15.19
92. Chen Y, Zhang WZ, Li J, et al. Novel Adefovir mono l-amino acid ester, mono bile acid ester derivatives: design, synthesis, biological evaluation, and molecular docking study. Med Chem Res. 2017;26(8):1812–1821. doi:10.1007/s00044-017-1892-z
93. Li T, Li J, Yang Y, et al. Synthesis, pharmacological evaluation, and mechanistic study of Adefovir mixed phosphonate derivatives bearing cholic acid and l-amino acid moieties for the treatment of hbv. Bioorgan Med Chem. 2019;27(16):3707–3721. doi:10.1016/j.bmc.2019.07.012
94. Wang Y, Zhu Y, Niu J, et al. A novel bile acid analog, a17, ameliorated non-alcoholic steatohepatitis in high-fat diet-fed hamsters. Toxicol Appl Pharmacol. 2020;404:115169. doi:10.1016/j.taap.2020.115169
95. Nita M, Grzybowski A. The role of the reactive oxygen species and oxidative stress in the pathomechanism of the age-related ocular diseases and other pathologies of the anterior and posterior eye segments in adults. Oxid Med Cell Longev. 2016;2016:3164734. doi:10.1155/2016/3164734
96. Gao L, Li Y, Zhao J, et al. Mechanism of reactive oxygen/nitrogen species in liver ischemia-reperfusion injury and preventive effect of Chinese medicine. Chin J Integr Med. 2024;31:462–473. doi:10.1007/s11655-024-3810-9
97. Danic M, Stanimirov B, Pavlovic N, et al. Pharmacological applications of bile acids and their derivatives in the treatment of metabolic syndrome. Front Pharmacol. 2018;9:1382. doi:10.3389/fphar.2018.01382
98. Hong S, Lee Y, Shin H, et al. Nanoassemblies of disulfide-bridged bile acid dimers as therapeutics agents for hepatic ischemia/reperfusion injury. ACS Appl Bio Mater. 2021;4(4):3145–3154. doi:10.1021/acsabm.0c01554
99. Limaiem F, Hajri M. Polycystic liver disease: an uncommon genetic condition. Clin Case Rep. 2024;12(5):e8892. doi:10.1002/ccr3.8892
100. Liu Y, Zhang L, Yan H, et al. Design of dimeric bile acid derivatives as potent and selective human ntcp inhibitors. J Med Chem. 2021;64(9):5973–6007. doi:10.1021/acs.jmedchem.1c00078
101. Criado J, Manzano J, Rodríguez-Fernández E. New organotropic compounds -: synthesis, characterization and reactivity of pt(ii) and au(iii) complexes with bile acids: DNA interactions and ‘in vitro’ anticancer activity. J Inorg Biochem. 2003;96(2–3):311–320. doi:10.1016/s0162-0134(03)00240-x
102. Vallejo M, Castro MA, Medarde M, et al. Novel bile acid derivatives (BANBs) with cytostatic activity obtained by conjugation of their side chain with nitrogenated bases. Biochem Pharmacol. 2007;73(9):1394–1404. doi:10.1016/j.bcp.2006.12.027
103. Brossard D, El Kihel L, Clement M, et al. Synthesis of bile acid derivatives and in vitro cytotoxic activity with pro-apoptotic process on multiple myeloma (kms-11), glioblastoma multiforme (gbm), and colonic carcinoma (hct-116) human cell lines. Eur J Med Chem. 2010;45(7):2912–2918. doi:10.1016/j.ejmech.2010.03.016
104. El-Deeb I, Bayoumi S, El-Sherbeny M, et al. Synthesis and antitumor evaluation of novel cyclic arylsulfonylureas: adme-t and pharmacophore prediction. Eur J Med Chem. 2010;45(6):2516–2530. doi:10.1016/j.ejmech.2010.02.038
105. Ren J, Wang Y, Wang J, et al. Synthesis and antitumor activity of n-sulfonyl-3,7-dioxo-5β-cholan-24-amides, ursodeoxycholic acid derivatives. Steroids. 2013;78(1):53–58. doi:10.1016/j.steroids.2012.09.009
106. Dosa P, Ward T, Castro R, et al. Synthesis and evaluation of water-soluble prodrugs of ursodeoxycholic acid (udca), an anti-apoptotic bile acid. Chemmedchem. 2013;8(6):1002–1011. doi:10.1002/cmdc.201300059
107. Li X, Zhao T, Cheng D, et al. Synthesis and biological activity of some bile acid-based camptothecin analogues. Molecules. 2014;19(3):3761–3776. doi:10.3390/molecules19033761
108. Brossard D, Lechevrel M, El Kihel L, et al. Synthesis and biological evaluation of bile carboxamide derivatives with pro-apoptotic effect on human colon adenocarcinoma cell lines. Eur J Med Chem. 2014;86:279–290. doi:10.1016/j.ejmech.2014.07.080
109. Samadi M, Nury T, KhalafiNezhad A, et al. Protecting group-free radical decarboxylation of bile acids: synthesis of novel steroidal substituted maleic anhydrides and maleimides and evaluation of their cytotoxicity on c6 rat glioma cells. Steroids. 2017;125:124–130. doi:10.1016/j.steroids.2017.07.004
110. Navacchia M, Marchesi E, Mari L, et al. Rational design of nucleoside-bile acid conjugates incorporating a triazole moiety for anticancer evaluation and sar exploration. Molecules. 2017;22(10):1710. doi:10.3390/molecules22101710
111. Taleghani A, Emami SA, Tayarani-Najaran Z. Artemisia: a promising plant for the treatment of cancer. Bioorgan Med Chem. 2020;28(1):115180. doi:10.1016/j.bmc.2019.115180
112. Zou X, Liu C, Li C, et al. Study on the structure-activity relationship of dihydroartemisinin derivatives: discovery, synthesis, and biological evaluation of dihydroartemisinin-bile acid conjugates as potential anticancer agents. Eur J Med Chem. 2021;225:113754. doi:10.1016/j.ejmech.2021.113754
113. Marchesi E, Chinaglia N, Capobianco ML, et al. Dihydroartemisinin-bile acid hybridization as an effective approach to enhance dihydroartemisinin anticancer activity. Chemmedchem. 2019;14(7):779–787. doi:10.1002/cmdc.201800756
114. Jiexin X. Synthesis and bioactivity evaluation of ursodeoxycholic acid and its derivatives [Master]. Shanghai: East China Normal University; 2019.
115. Xu Y, Luo Q, Lin T, et al. U12, a udca derivative, acts as an anti-hepatoma drug lead and inhibits the mtor/s6k1 and cyclin/cdk complex pathways. PLoS One. 2014;9(12):e113479. doi:10.1371/journal.pone.0113479
116. Yang R, Du C, Cao T, et al. Synthesis and anti-hepatoma activities of u12 derivatives arresting g0/g1 phase and inducing apoptosis by pi3k/akt/mtor pathway. Pharmaceuticals. 2022;15(1):107. doi:10.3390/ph15010107
117. Keune W, Hausmann J, Bolier R, et al. Steroid binding to autotaxin links bile salts and lysophosphatidic acid signalling. Nat Commun. 2016;7:11248. doi:10.1038/ncomms11248
118. Clark J, Salgado-Polo F, Macdonald S, et al. Structure-based design of a novel class of autotaxin inhibitorsbased on endogenous allosteric modulators. J Med Chem. 2022;65(8):6338–6351. doi:10.1021/acs.jmedchem.2c00368
119. Li Y, Li Q, Chen X, et al. Advances in research on the antibacterial effects and mechanism of chlorogenic acid. Chin J Antibiot. 2024;49(2):141–150.
120. Willemen H, de Smet L, Koudijs A, et al. Micelle formation and antimicrobial activity of cholic acid derivatives with three permanent ionic head groups. Angew Chem Int Edit. 2002;41(22):4275–4277. doi:10.1002/1521-3773(20021115)41:22<4275::Aid-anie4275>3.0.Co;2-u
121. Huang L, Sun Y, Zhu H, et al. Synthesis and antimicrobial evaluation of bile acid tridentate conjugates. Steroids. 2009;74(8):701–706. doi:10.1016/j.steroids.2009.03.005
122. Chen C, Liu Z, Wang S, et al. Research progress in the role of menthol and its receptor trpm8 in tumor. Chin Pharmacol Bull. 2015;31(3):312–314. doi:10.3969/j.issn.1001-1978.2015.03.004
123. Khaligh P, Salehi P, Bararjanian M, et al. Synthesis and in vitro antibacterial evaluation of novel 4-substituted 1-menthyl-1,2,3-triazoles. Chem Pharm Bull. 2016;64(11):1589–1596. doi:10.1248/cpb.c16-00463
124. Cateni F, Zacchigna M, Procida G, et al. Cholane and lanostane derivatives: antimicrobial evaluation. Chemistryselect. 2016;1(15):4856–4860. doi:10.1002/slct.201600556
125. Khanna S, Baddour L, Huskins WC, et al. The epidemiology of clostridium difficile infection in children: a population-based study. Gastroenterology. 2012;142(5):S131. doi:10.1016/S0016-5085(12)60495-8
126. Stoltz K, Erickson R, Staley C, et al. Synthesis and biological evaluation of bile acid analogues inhibitory to clostridium difficile spore germination. J Med Chem. 2017;60(8):3451–3471. doi:10.1021/acs.jmedchem.7b00295
127. De Clercq E. Anti-hiv drugs: 25 compounds approved within 25 years after the discovery of hiv. Int J Antimicrob Agents. 2009;33(4):307–320. doi:10.1016/j.ijantimicag.2008.10.010
128. Walker R, Parker R, Kovacs J, et al. Anemia and erythropoiesis in patients with the acquired immunodeficiency syndrome (aids) and kaposi sarcoma treated with zidovudine. Ann Intern Med. 1988;108(3):372–376. doi:10.7326/0003-4819-108-3-372
129. Wang D, Zhang C, Tang J, et al. Advancements in targeted therapy for chronic hepatitis b virus infection. Chin J New Drugs Clin Remedies. 2024;43(4):241–246.
130. Su H, Chen Y, Fu X, et al. Synthesis, antiviral activity and primary hepatocyte uptake evaluation of Adefovir l-amino acid and bile acid ester derivatives. Chin J Pharm. 2016;47(3):264–268.
131. Li J, Qiu L, Ren Y, et al. Research progress of traditional Chinese medicines and active ingredients targeting m1/m2 macrophage polarization balance in intervening obese with type 2 diabetes. Zhongguo Zhongyao Zazhi. 2024;49(13):3441–3451. doi:10.19540/j.cnki.cjcmm.20240205.703
132. Yu D, Sousa K, Mattern D, et al. Stereoselective synthesis, biological evaluation, and modeling of novel bile acid-derived g-protein coupled bile acid receptor 1 (gp-bar1, tgr5) agonists. Bioorgan Med Chem. 2015;23(7):1613–1628. doi:10.1016/j.bmc.2015.01.048
133. Ji L, Ma J, Ma Y, et al. 847-p: berberine ursodeoxycholate (htd1801) improves key glycemic and cardiometabolic parameters across the type 2 diabetes mellitus disease spectrum. Diabetes. 2024;73(Sup1):7. doi:10.2337/db24-847-P
134. Graebin C, Madeira M, Yokoyama-Yasunaka JK, et al. Synthesis and in vitro activity of limonene derivatives against leishmania and trypanosoma. Eur J Med Chem. 2010;45(4):1524–1528. doi:10.1016/j.ejmech.2009.12.061
135. Orhan I, Sener B, Kaiser M, et al. Inhibitory activity of marine sponge-derived natural products against parasitic protozoa. Mar Drugs. 2010;8(1):47–58. doi:10.3390/md8010047
136. Harikandei KB, Salehi P, Ebrahimi SN, Bararjanian M, Kaiser M, Al-Harrasi A. Synthesis, in-vitro antiprotozoal activity and molecular docking study of isothiocyanate derivatives. Bioorgan Med Chem. 2020;28(1):115185.
137. Medzhitov R. Origin and physiological roles of inflammation. Nature. 2008;454(7203):428–435. doi:10.1038/nature07201
138. Li X, Hu Y, He B, et al. Design, synthesis and evaluation of ursodeoxycholic acid-cinnamic acid hybrids as potential anti-inflammatory agents by inhibiting Akt/NF-rB and MAPK signaling pathways. Eur J Med Chem. 2023;260:115785. doi:10.1016/j.ejmech.2023.115785
 © 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.