Back to Journals » Therapeutics and Clinical Risk Management » Volume 10
Advanced prostate cancer – patient survival and potential impact of enzalutamide and other emerging therapies
Authors Patel N, Finianos A, Whitaker K, Aragon-Ching J
Received 27 May 2014
Accepted for publication 1 July 2014
Published 16 August 2014 Volume 2014:10 Pages 651—664
DOI https://doi.org/10.2147/TCRM.S57509
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Nihar K Patel, Antoine Finianos, Kristen D Whitaker, Jeanny B Aragon-Ching
Department of Medicine, Division of Hematology and Oncology, George Washington University Medical Center, Washington, DC, USA
Abstract: The advent of exponential growth of novel agents tested and approved for the treatment of patients with metastatic castration-resistant prostate cancer (mCRPC) has brought about a need for understanding of the mechanism of action, side-effects, and clinical efficacy of these drugs as they relate to these patients. This review will provide a synopsis of the treatment landscape in mCRPC as varying agents such as abiraterone acetate, cabazitaxel, sipuleucel-T, radium, and selected emerging agents are presented. A distinct focus on the utilization of enzalutamide, its mechanism of action, key pivotal trials that brought about its US Food and Drug Administration approval, as well as patient-focused perspectives and clinical implications are discussed herein.
Keywords: metastatic castration-resistant prostate cancer, enzalutamide, systemic therapies
Introduction to the management issues in the treatment of advanced prostate cancer
Prostate cancer remains the most common non-cutaneous cancer in the US. In 2014 alone, the projected incidence of prostate cancer is 233,000 cases with deaths occurring in 29,480 men,1 making metastatic prostate cancer therapy truly an unmet medical need. With the increasing availability of new therapies that brought about a significant change in the treatment options for metastatic prostate cancer in the last 5 years alone, issues regarding the optimal sequencing or combination of these agents have arisen. Currently, several guidelines exist that help direct clinicians as to the best sequencing approach and most would evaluate presence or lack of symptoms, performance status, as well as burden of disease to help determine the best sequencing for these agents.2,3 However, therapy failure remains a significant challenge especially since cross-resistance from each agent is increasingly described.4–6 In addition, patients may lose the therapeutic window to gain substantial benefit from each drug that has been proven to provide overall survival gains. Hence, better methods of identifying the target population who have the most potential to benefit remain an important though somewhat elusive goal. This review focuses on the landscape of systemic treatment for prostate cancer with specific attention to enzalutamide, its properties, as well as other current and selected emerging therapies.
Overview of the current and emerging treatment options
The rapid evolution of drug therapies in prostate cancer has vastly improved upon the use of docetaxel since its pivotal US Food and Drug Administration (FDA) approval in 2004 and has brought about a new era where progress has been made beyond the use of androgen deprivation therapy (ADT) with the addition of novel hormonal agents, immunotherapy, second-line chemotherapy as well as radiopharmaceuticals (see Table 1). The choice of sequencing currently relies on patient profiles, whether symptoms of metastatic disease exist or not.7,8 Men who are asymptomatic or minimally symptomatic may benefit from early use of Sipuleucel-T, while treatment using docetaxel is usually reserved for patients with pain. Radium is used predominantly for patients with bony metastases especially in those who are not candidates for aggressive chemotherapy and abiraterone acetate can be given for effects on pain palliation. Agents such as cabazitaxel, abiraterone acetate, enzalutamide or radium can all be offered after progression on docetaxel. While survival outcomes are undeniably improved with the use of these therapies, patients’ disease will ultimately progress on each regimen. The following section gives a brief discussion on recent drug approvals as commonly utilized in the clinic as well as selected emerging therapies.
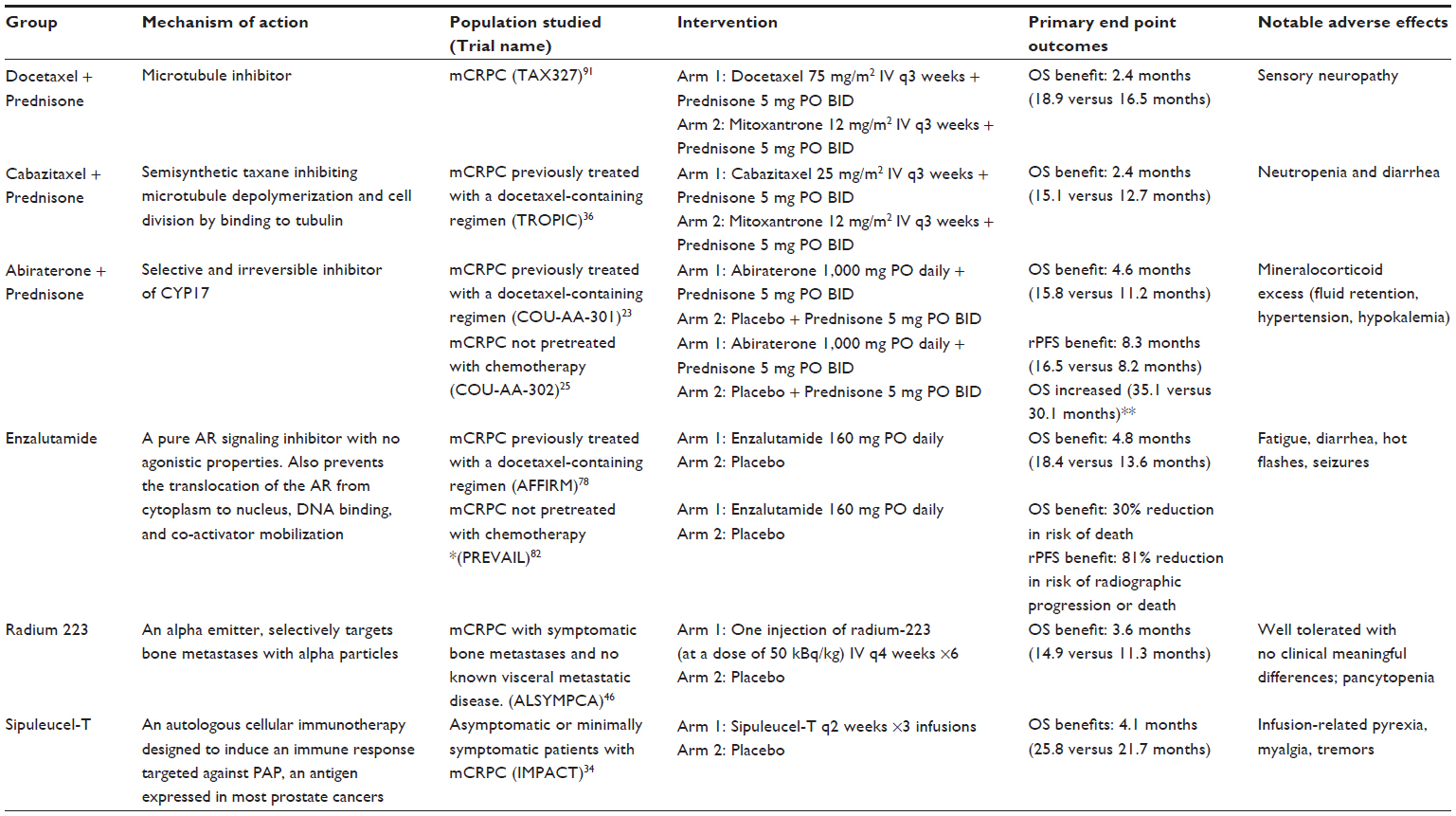 | Table 1 FDA approved therapies for advanced prostate cancer* |
Background on hormonal therapy
Androgens in the form of testosterone or the more potent dihydrotestosterone (DHT) have been well-defined drivers of progression of prostate cancer and differentiation of the prostate gland. As such, the backbone of treatment for advanced prostate cancers was established decades ago when castration in the form of surgical orchiectomy achieved significant prostate tumor regression.9 Since then, substitution to chemical castration has been employed mostly due to patient preference.10 ADT has therefore become the standard systemic treatment for locally advanced or metastatic prostate cancer.11 While ADT is almost always effective in most patients, disease progression to castration resistance inevitably occurs.12 It is now increasingly recognized that the androgen receptor (AR) remains overexpressed despite seemingly castrate levels of testosterone, since alternative receptors may activate the AR or other target genes may help perpetuate the castrate-resistant phenotype,13,14 hence the term “castration-resistance” has become widely adopted in the literature. The enhanced understanding of the role of these androgens in stimulating the growth of prostate cancer has led to the development and approval of both abiraterone and enzalutamide, the latter of which will be the main focus of this review.
Abiraterone acetate
The ability to synthesize androgens at multiple sites and intratumorally brought about the discovery of steroidogenesis inhibition as a drug development approach to treat metastatic castration-resistant prostate cancer (mCRPC).13 Abiraterone inhibits the microsomal enzyme cytochrome P450 isoform-17 (CYP17) that catalyzes two sequential reactions of 17-α-hydroxylase and 17,20 lyase.15 Abiraterone acetate was developed as a candidate oral small molecule inhibitor of CYP17.16–18 The loss of CYP17 as a result of abiraterone therapy results in augmented cortisol activity which interrupts negative feedback control of adrenocorticotropic hormone (ACTH), thereby resulting in increased upstream effectors with a syndrome of mineralocorticoid excess characterized by hypokalemia, hypertension and fluid retention,17 which can be mitigated by addition of low-dose steroids such as prednisone.19 The subsequent early phase clinical trials demonstrated feasibility and safety of the use of abiraterone.20–22 The phase III multicenter, randomized trial known as the COU-AA-30123 ultimately led to US FDA approval for abiraterone. One thousand one hundred and ninety-five men with mCRPC who were previously treated and failed docetaxel were randomized 2:1 to either abiraterone plus prednisone versus prednisone with placebo.23 The primary end point of the trial was overall survival (OS), while secondary end points included time to prostate-specific antigen (PSA) progression, progression-free survival (PFS), and PSA response rates. The results of the trial marked the first time in years that an oral, well-tolerated androgen-signaling inhibitor yielded promising survival benefit. The OS was superior in the abiraterone acetate arm compared with the prednisone/ placebo arm (14.6 versus 10.9 months; hazard ratio [HR] 0.65; P<0.001). Similarly, all secondary end points were statistically significant in favor of abiraterone, including time to PSA progression, PFS, and PSA response rates. Further OS analysis showed maintained improvement in OS at the time of reporting (15.8 versus 11.2 months, P<0.0001).24 In COU-AA-302, the role of abiraterone in the chemotherapy naive population was subsequently studied. This phase III randomized, placebo-controlled trial enrolled 1,088 patients with mildly symptomatic or asymptomatic mCRPC and assigned to receive prednisone (5 mg twice daily) with 1,000 mg of abiraterone or placebo.25 The co-primary end points of this trial were radiographic progression-free survival (rPFS) and OS, which was met for rPFS, showing benefit in the abiraterone group (16.5 months versus 8.2 months, P<0.0001) but not for OS, though a strong trend toward improved survival was seen, but did not cross the O’Brien-Fleming efficacy boundary for statistical significance or the pre-specified statistical efficacy boundary (alpha-level: 0.0035).26 Similar to the earlier study, unblinding was carried out after interim analysis and cross-over was allowed. There was a notable delay in chemotherapy administration for the abiraterone arm at 25.2 months versus 16.2 months in the prednisone alone arm. These favorable results led to ultimate approval by the FDA of abiraterone for pre-chemotherapy mCRPC patients.
Sipuleucel-T
Therapeutic advances for prostate cancer entered a new realm of treatment possibilities with the advent of immunotherapy. Identification of certain tumor-associated antigens such as PSA and prostatic acid phosphatase (PAP) that are overexpressed in prostate cancers provided an attractive target for immune-based therapies.27,28
Sipuleucel-T is an activated autologous dendritic cellular vaccine designed to target PAP. The vaccine is manufactured through ex-vivo stimulation of antigen presenting cells (APCs) which in turn activate cytotoxic T lymphocytes. It is an intensive process which entails harvesting a patient’s own peripheral cells via apheresis. The cells are then exposed to a fusion peptide consisting of PAP and granulocyte-macrophage colony stimulating factor which serves as an immune adjuvant to enhance immune activation,29 undergo quality assurance for a minimum of cluster of differentiation (CD)54 expression threshold for adequate cell collection, and are subsequently shipped back to an infusion facility where they can be administered intravenously to the patient every 2 weeks for three infusions.30 With the most common adverse reactions being transient fever and flu-like symptoms, early trials of sipuleucel-T demonstrated the safety of this form of therapy, with no increased risk of autoimmunity.31
Two initial small phase III trials evaluated the ability of sipuleucel-T to treat minimally symptomatic mCRPC.32,33 By using the conventionally established time to progression (TTP) end points, the first of these trials failed to meet the primary end point and resulted in the premature discontinuation of the second small phase III trial. Ultimately, improvement in the secondary end point of OS was shown, leading to a second, larger phase III study, with OS as the end point, coined the IMPACT trial (NCT00065442), which definitively addressed the clinical benefit of sipuleucel-T in mCRPC.34 Five hundred and twelve patients with minimally symptomatic or asymptomatic mCRPC were enrolled, randomizing 2:1 in favor of sipuleucel-T relative to the placebo. As noted in the previous studies, there was no significant difference in TTP based on objective parameters (3.7 months in the sipuleucel-T arm versus 3.6 months in the placebo arm; HR 0.95; P=0.63). Nonetheless, the findings for OS showed a significant advantage for patients treated with sipuleucel-T at 25.8 versus 21.7 months for placebo, with a 3 year survival probability of 31.7% versus 23.0% (HR 0.77; P=0.02). These findings prompted FDA approval of sipuleucel-T for the treatment of minimally symptomatic mCRPC, ushering in a modern age of immunotherapy for the treatment of cancer. An ongoing, phase III trial is evaluating the treatment of sipuleucel-T in patients with hormone sensitive prostate cancer (PROTECT), via NCT00779402.
Cabazitaxel
Cabazitaxel is a semisynthetic taxane tested in taxane-resistant cell lines which inhibits microtubule depolymerization and cell division by binding to tubulin, resulting in cell cycle arrest.35 Despite 4 years since its original approval, cabazitaxel remains the first and only chemotherapy to have shown a survival benefit as second-line treatment for patients whose disease has progressed beyond docetaxel. The approval was based on data from TROPIC,36 a Phase III trial (NCT00417079) conducted in 755 patients with mCRPC previously treated with docetaxel; participants were randomized to receive either cabazitaxel or mitoxantrone, both in combination with prednisone. The primary end point of the study was OS which was met with patients receiving cabazitaxel achieving a median OS of 15.1 months versus 12.7 months in the mitoxantrone plus prednisone arm; P=0.0001. The median PFS was also significantly better with cabazitaxel at 2.8 months versus 1.4 months in the mitoxantrone arm (HR 0.74; P<0.0001). Other secondary end points such as PSA response, time to PSA progression and tumor response were all superior in the cabazitaxel over mitoxantrone arms. However, treatment-related deaths were more frequent in the 30 days after last treatment for cabazitaxel compared with mitoxantrone (4.9% versus 2.4%). Other hematologic toxicities, such as neutropenia (82% versus 58%) occurring as a grade 3 event or higher, were much more often seen in the cabazitaxel arm than mitoxantrone, as was the non-hematologic toxicity diarrhea (6% versus 1%). In addition, febrile neutropenia occurred in 8% of patients in the cabazitaxel group compared with 1% in the mitoxantrone group. Based on the safety profile, the product label of cabazitaxel had clear safety warnings with regard to monitoring for neutropenia, and the product label recommended use of growth factors in high-risk patients, such as those who are >65 years of age, with extensive prior radiation, or with serious comorbidities and poor nutritional status.37 Conversely, the rates of neuropathy were certainly favorable and the pain responses were comparable though not superior to mitoxantrone.38 The perceived hematologic toxicity of cabazitaxel has limited its clinical use in patients with progressive mCRPC. Given its place in sequencing in the “post-docetaxel” space, few patients are deemed fit enough for further chemotherapy after being heavily pre-treated.
Alpharadin
Radiopharmaceuticals in the form of strontium and samarium have been available since their FDA approval in the 1990s, but while early clinical trials with Strontium-89 (Amersham Healthcare, Arlington Heights, IL, USA) and Samarium-153 (Bayer Healthcare, Berlin, Germany) have shown pain palliation from skeletal metastases,39–42 the absence of significant survival benefit and potential for myelosuppression has relegated their use for predominantly palliative end-of-life care.43–45 Alpharadin or radium-223 is a unique α-emitting radiation agent that served to improve upon the range of β emitters and was the first in its class of radiopharmaceuticals to exhibit OS in mCRPC patients with symptomatic bone metastases.46 Radium-223 emits high-energy alpha particles of short range (<100 μm), inducing mainly double stranded DNA breaks that result in a potent and highly localized cytotoxic effect in the target areas.47,48 The shorter travel distance also means fewer toxic effects on adjacent tissue as well as less myelosuppression.49 Early clinical trials showed safety and feasibility in prostate cancer patients,50 eventually leading to the phase III ALSYMPCA study (NCT00699751) which evaluated 921 men with mCRPC and bone metastases, comparing radium-223 at 6 monthly injections with placebo, in addition to the best standard of care.46 The primary end point was met with a significant improvement in survival for patients treated with radium-223, with a median OS of 14.9 months versus 11.3 months for placebo, maintained through the updated analysis.46 All main secondary end points, including time to first skeletal-related events (SREs) and time to increase in PSA level showed a benefit of radium-223 as compared with placebo. This trial ultimately led to the FDA approval of radium for clinical use in 2013 for predominantly symptomatic men with mCRPC who are post-docetaxel treatment or intolerant of or ineligible for docetaxel with no known visceral metastases. The potential for myelosuppression was low (at 2%) but patients had to have adequate hematologic parameters prior to starting with radium-223.
Emerging therapies
Several novel agents and treatments have been shown to have promising effects in earlier phases of prostate cancer trials. The challenge continues with how to sequence these current and emerging therapies,51 especially since many of the drug approvals have been tested against prior standards utilizing either placebo, prednisone or mitoxantrone. Hence, there is increasing difficulty to compare contemporary drugs with the currently approved landscape of treatment for mCRPC. We discuss herein a select group of trials that are in phase III or nearing completion of phase III (see selected agents in Table 2).
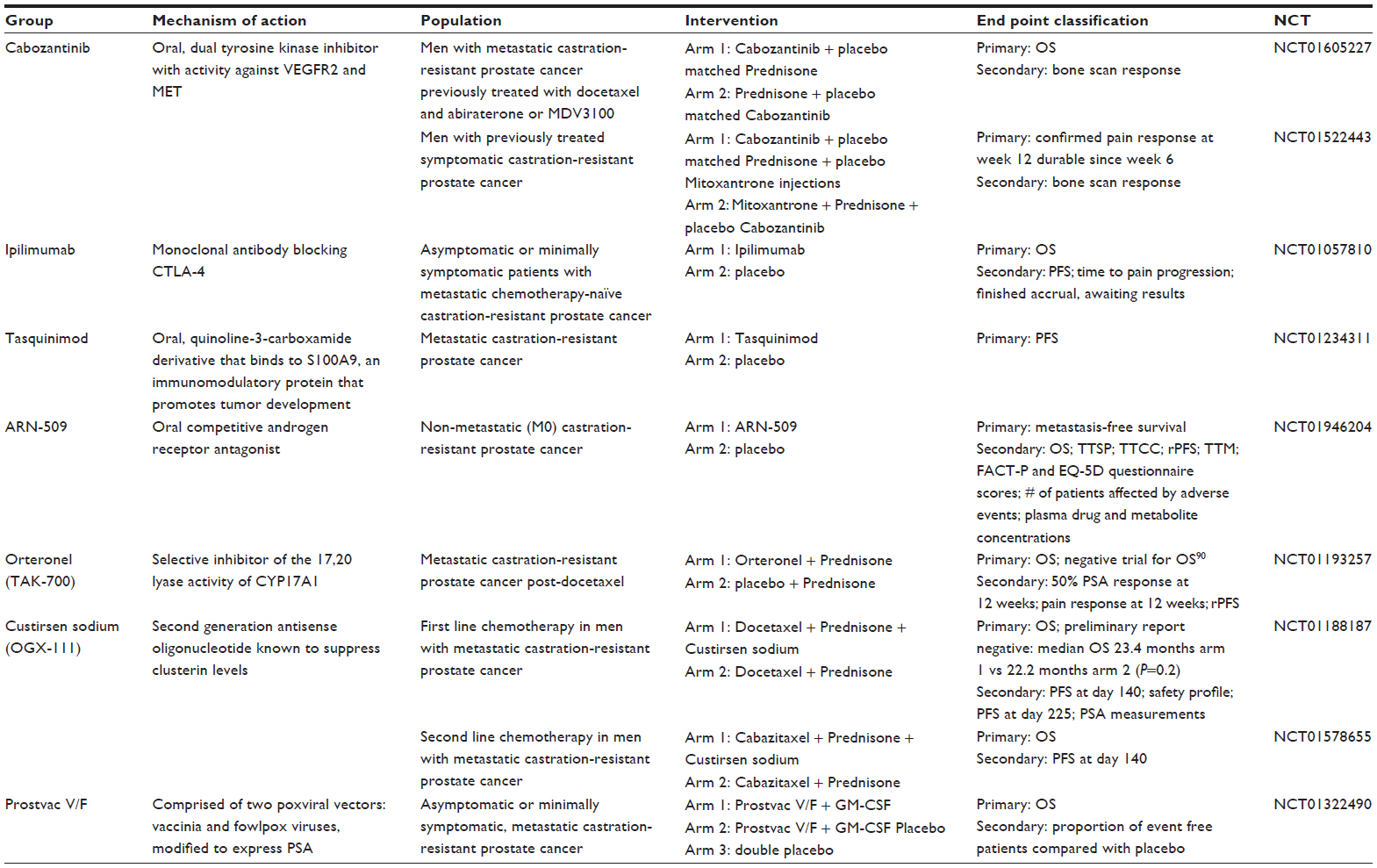 | Table 2 Selected emerging therapies currently in phase III clinical trials |
Cabozantinib
Cabozantinib is a novel, orally bioavailable, dual tyrosine kinase inhibitor with activity against vascular endothelial growth factor receptor 2 (VEGFR2) and the mesenchymal-epithelial transition factor (MET), also known as hepatocyte growth factor receptor, among others. Both MET and VEGFR2 have been implicated in the progression of prostate cancer, particularly with bone involvement.52,53 One hundred and seventy-one CRPC patients were included in a phase II randomized discontinuation trial and were treated initially with cabozantinib for 12 weeks followed by continuation of the drug versus placebo for patients who achieved stable disease. Median PFS for the 31 patients randomized after 12 weeks was 23.9 weeks, versus 5.9 weeks for those on cabozantinib and placebo.54 After 12 weeks of the lead-in therapy, 5% had partial responses and 75% had stable disease, with 72% demonstrating reduction of soft tissue lesions and 68% with improvements on bone scan. Sixty-seven percent of available patients reported an improvement in pain control with a decrease in, or discontinuation use of narcotics by 56%. In a subsequent dose-finding study, cabozantinib at a lower dose, 40 mg compared with 100 mg, was found to be better tolerated with similar clinical effect.55
These encouraging results have prompted two phase III trials. COMET-1 (NCT01605227), with a primary end point of OS is a placebo-controlled trial enrolling 960 men with mCRPC and additional prior therapy, randomly assigned to prednisone or cabozantinib. COMET-2 (NCT01522443) is a placebo-controlled phase III trial evaluating cabozantinib versus mitoxantrone and prednisone in patients with previously treated symptomatic mCRPC, with an emphasis on the effect on pain and bone disease.
Tasquinimod
Tasquinimod, a quinolone-3-carboxamide, with antiangiogenic, immunomodulatory, and anti-metastatic properties has previously demonstrated preclinical evidence of antitumor activity in prostate cancer.56,57 A phase I trial of tasquinimod in patients with chemotherapy-naïve CRPC showed common adverse events including inflammation, nausea, and fatigue.58 Rare but serious adverse events included sinus tachycardia, cerebral infarction, and hyperamylasemia. More recently, an international, double-blinded, phase II trial enrolled 201 men who were randomized in a 2:1 fashion for therapy with tasquinimod or placebo. Although there were no significant changes in PSA progression in the two arms of the trial, rPFS favored the tasquinimod arm (7.6 versus 3.3 months; P=0.0042).59 Patients whose cancer had already metastasized to their bones and who took tasquinimod remained progression-free for even longer – 8.8 months, compared with 3.4 months for placebo. Currently, a phase III study (NCT01234311) of tasquinimod versus placebo is enrolling mCRPC patients in order to determine its true efficacy in mCRPC.
Ipilimumab
Ipilimumab is a monoclonal antibody that binds to cytotoxic T lymphocyte antigen 4 (CTLA-4), a molecule expressed on cytotoxic T lymphocytes after activation by antigen-presenting cells.60 Ipilimumab serves as a checkpoint inhibitor that overcomes immune activity T cell suppression to enable a more enhanced anti-tumor immune response.
CA184-043 was a phase III trial comparing ipilimumab given at 10 mg/kg to placebo, following radiotherapy to stimulate immune priming in patients with mCRPC, who have received and failed prior treatment with docetaxel.61 The study’s primary end point of OS did not reach statistical significance with a median OS of 11.2 months (95% CI 9.5–12.7) for ipilimumab and 10.0 months (95% CI 8.3–11.0) for placebo (HR 0.85; 95% CI 0.72–1.00; P=0.053). However, benefit was observed across some efficacy end points in a post-hoc assessment of predefined subgroups of patients with favorable features, such as those without visceral metastases, alkaline phosphatase < 1.5× normal, or a hemoglobin of 110 g/L or higher.62 Ipilimumab was also tested in asymptomatic or minimally symptomatic patients with chemotherapy-naïve mCRPC (NCT01057810). The trial has finished accrual and awaiting results.
Review of pharmacology, mode of action, and pharmacokinetics of enzalutamide
Enzalutamide, a small molecule AR antagonist is a once daily second generation AR signaling inhibitor that was approved by the FDA in 2012 for the treatment of mCRPC post-chemotherapy with docetaxel.63 Given that the AR transcription program remains active despite emergence of castration resistance,64 this property has been exploited to screen for anti-androgens with specific ability to bind and inhibit the AR without the agonistic effects.65
Enzalutamide was selected for development based on research done at Sawyers’ laboratory66 (University of California, Los Angeles, CA, USA) where RD162 and MDV3100 were identified as nonsteroidal diarylthiohydantoin agonists, from amongst 200 derivatives, from the activity matched with the chemical structure. Enzalutamide was further selected as the lead candidate due to its high affinity and selectivity for the ligand binding domain of the AR, despite increased AR expression, and used for further clinical studies.66 The clinical development of enzalutamide was spearheaded by Medivation, Inc. (San Francisco, CA, USA) in 2007. Enzalutamide was unique from first generation AR antagonists, since it lacked significant agonist activity, as it inhibited PSA induction as well as multiple steps in the AR signaling pathway. The first action of enzalutamide blocks binding of testosterone to the AR and blocks the conformational change induced by AR-testosterone binding (see Figure 1). Enzalutamide has approximately five to eight-fold higher binding affinity to AR than the first generation anti-androgen bicalutamide. The second action of enzalutamide reduces nuclear translocation of the AR and results in a significant fraction of the AR remaining in the cytoplasm. The remaining actions of enzalutamide include impairment of AR DNA binding and interference with co-activator recruitment, which ultimately results in decreased cell growth and apoptosis leading to decreased prostatic tumor volume.
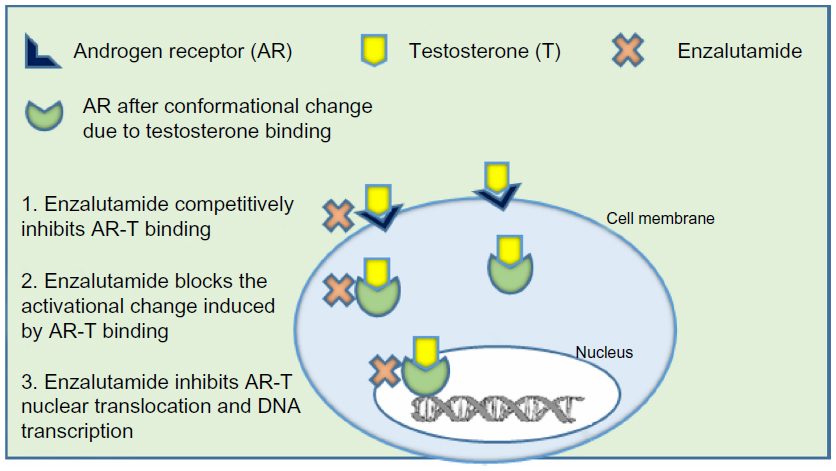 | Figure 1 Mechanism of action of enzalutamide. |
Pharmacokinetics
Enzalutamide has as its active metabolite, N-desmethyl enzalutamide. The pharmacokinetics have been described both in patients with mCRPC following an oral administration of 160 mg/day and in healthy male volunteers following a single oral administration of 160 mg with or without a fatty meal.67
Absorption and distribution
Enzalutamide was rapidly and well absorbed after oral administration, with absorption estimated to be 84% of the administered dose.69 The drug had a median time to maximum concentration (Cmax) of 1 hour (range of 30 minutes to 3 hours) and steady state plasma concentrations were achieved by day 28 of daily treatment with low daily fluctuations in plasma concentrations. After reaching the steady state, the mean plasma Cmax was 16.6 μg/mL for enzalutamide and 12.7 μg/mL for N-desmethyl enzalutamide with plasma mean pre-dose trough values of 11.4 and 13.0 μg/mL, respectively. The coefficient of variation was ≤30% for these values. The plasma enzalutamide level is not affected by the dose up to 600 mg per day.68 The mean peak to trough ratio in enzalutamide plasma concentration was 1.25, indicating a low daily fluctuation in concentration. During a 1 year period of ongoing enzalutamide therapy, the pre-dose minimum concentration of enzalutamide and its active metabolite remained constant. Similarly, the area under the concentration-time curves was notably similar in healthy volunteers after a single 160 mg oral dose either fasting or with a high fat meal. Thus, enzalutamide can be taken with or without food. The mean apparent volume of distribution after a single dose is 110 L, which indicates extensive extravascular distribution. The majority (97%) of enzalutamide binds to albumin, and 95% of N-desmethyl enzalutamide binds to plasma proteins.
Metabolism and elimination
Enzalutamide has several potential drug interactions owing to its metabolism. The drug is mainly metabolized by the liver cytochrome P450 system, specifically in vitro human CYP3A4 and CYP2C8, the latter of which is primarily responsible for the formation of the active metabolite, N-desmethyl enzalutamide. After administration of a single dose of radioactive enzalutamide, radioactivity was detected in the plasma as enzalutamide, N-desmethyl enzalutamide, and carboxylic acid (an inactive metabolite). Metabolism of enzalutamide may be modified by concomitant administration of drugs that induce CYP2C8 or CYP3A4. Enzalutamide is a strong inducer of CYP3A4 and a moderate inducer of CYP2C19 and CYP2C9, and inhibits P glycoprotein.
Enzalutamide is cleared predominantly via hepatic metabolism, being excreted 71% in the urine and 14% in the feces, primarily as an inactive metabolite. Enzalutamide’s mean total plasma clearance is 0.56 L/hour and the mean terminal half-life is 5.8 days. The mean terminal half-life for N-desmethyl enzalutamide was about 8 days.
There were no clinically important effects of age or body weight on enzalutamide pharmacokinetics. Insufficient data in non-Caucasian populations exist to evaluate pharmacokinetic differences based on race. The apparent clearance and composite area under the curve of enzalutamide was similar in patients with pre-existing mild and moderate renal impairment (creatinine clearance ≥30 mL/minute) and mild to moderate hepatic impairment (Child-Pugh Class A and B) when compared to volunteers with normal renal and hepatic function, based on a population pharmacokinetic analysis.69 While no studies have included patients with severe hepatic or renal impairment, no adjustments for initial dosing were suggested for patients with mild to moderate renal or hepatic impairment,69 making the use of enzalutamide attractive for patients with such dysfunction.
Efficacy, safety, and tolerability studies, including comparative studies
Given the inevitable resistance or progression to first-generation AR inhibitors that limits effective prostate cancer therapy, efforts to investigate resistance patterns and mechanisms to these inhibitors have evolved. The partial agonist activity seen with the older-generation AR inhibitors and the greater binding affinity of the more potent androgen DHT lend insights into the design of second-generation anti-androgens.70,71 While earlier AR inhibitors such as bicalutamide or flutamide have demonstrated PSA responses, improvement in OS was uniformly lacking.72–74
Early preclinical trials with enzalutamide showed greater affinity with lower nanomolar concentrations required for binding, compared to earlier generations of anti-androgens such as bicalutamide. Given enzalutamide’s greater affinity relative to bicalutamide as well as its function as a pure AR antagonist,75 though there has been emerging small reports of possible enzalutamide withdrawal responses,76 initiation of a phase I/II trial in humans was undertaken.
The initial phase I/II trial enrolled 140 patients with mCRPC across five centers in the US, 78% of whom had metastatic disease, and they received enzalutamide orally at varying daily doses (range: 30–600 mg).67 The trial sought to establish the safety, tolerability, and anti-tumor effects with positron emission tomography (PET) scan and circulating tumor cell (CTC) correlation.
The patients were relatively heavily pre-treated, with nearly half of the patients having had prior chemotherapy, and over three-quarters of patients having received at least two lines of hormonal therapy, with 45% having prior ketoconazole exposure. Antitumor activity was observed at all tested dosages, including stabilized bone disease in 61 (56%) of 109 patients, >50% or more declines in PSA in 78 (56%) patients, soft tissue responses in 13 (22%) of 59 patients and conversion from unfavorable to favorable CTC counts in 25 (49%) of the 51 patients, signifying a favorable effect in this adverse prognostic group of patients. PSA responses were similar between those who received prior chemotherapy versus not. The maximum tolerated dose for sustained treatment (>28 days) was 240 mg and no additional benefit was obtained from higher dosages, and the tested dose later used for the phase III trial was 160 mg. There was a beneficial effect on objective radiological regression with an observed 47 weeks of median time to radiological progression for all patients, although it was notably more prolonged in the chemotherapy-naïve group (60 weeks compared to the chemotherapy pre-treated group at 29 weeks). There were no safety concerns in this early study but of note, three patients who developed seizures were receiving 360 mg, 480 mg, and 600 mg, respectively. Efforts to determine any risk factors unveiled two of these patients were on medications that lowered the seizure threshold. Hence later exclusion in the phase III AFFIRM (A Study Evaluating the Efficacy and Safety of the Investigational Drug MDV3100) trial of patients with any risk factors that may lower such seizure threshold, such as a history of prior stroke, aneurysms, concomitant medications that lower seizure thresholds. At longer follow-up and at the time of updated analysis, 18 of the enrolled patients remained in the study, with a median time on therapy of 131 weeks.77
Phase III study: the AFFIRM trial
The phase 3 AFFIRM trial was a randomized, double-blind, placebo-controlled, international trial evaluating enzalutamide at 160 mg/day versus placebo in 1,199 men with advanced prostate cancer who were previously treated with docetaxel-based chemotherapy.78 Distinct from the COU-AA-301 or COU-AA-302 trials, steroids were not mandated but allowed at study entry and were found to be used in a third of patients, in both arms. The trial had OS as its primary end point with secondary end points of radiographic PFS, time to PSA progression, quality of life (QoL), and time to the first SRE. Given analysis showing improved OS favoring enzalutamide, the Data and Safety Monitoring Committee recommended halting the trial and patients on placebo were offered to cross over to receive enzalutamide, although only about 19 patients (5%) still remained on placebo at the time of unblinding. At a median follow-up of 14 months, the median OS was still significantly improved in the enzalutamide arm versus the placebo arm (18.4 months versus 13.6 months, respectively; HR, 0.63; 95% confidence interval [CI], 0.53–0.75; P<0.001). This translated to a 37% reduction in the risk of death of any cause in the enzalutamide arm. The efficacy of enzalutamide translated across all other end points, such as time to PSA progression (8.3 versus 3 months), median rPFS (8.3 versus 2.9 months), and risk-categories. Overall response rates (ORRs) via Response Evaluation Criteria In Solid Tumors (RECIST) was also significantly different at 29% in the enzalutamide arm compared to 4% in the placebo arm (P<0.001). The effects on pain and SREs showed improvement in the time to first SRE, as 16.7 months versus 13.3 months; HR, 0.62; P<0.001. Given the use of steroids in about a third of patients, a highlighted difference from the COU-AA-301 and COU-AA-302 trials wherein all patients were on steroids, further post hoc analysis was examined and found that patients who were on steroids upon study entry had worse survival compared to those who were not on steroids,79 suggesting the potential detrimental role of glucocorticoid receptor expression as a means of resistance seen in enzalutamide therapy,80 though potentially confounded by a patient population that could have been more symptomatic, hence the need for steroid use in the first place. The positive findings seen from the AFFIRM trial became instrumental in the subsequent FDA approval of enzalutamide for men with mCRPC who have failed prior chemotherapy.
Phase III study: PREVAIL
At the 2014 American Society of Clinical Oncology Genitourinary Cancers Symposium, data from the much anticipated phase III PREVAIL study (A Safety and Efficacy Study of Oral MDV3100 in Chemotherapy-Naive Patients With Progressive Metastatic Prostate Cancer) was presented,81 and is now published.82 PREVAIL was a double-blinded, international, placebo-controlled trial that enrolled 1,717 chemotherapy-naïve mCRPC patients who were stratified by site and given enzalutamide 160 mg/day or placebo in a 1:1 fashion. The co-primary end points included OS and radiographic PFS. The findings were strikingly positive with statistical improvement in the OS of 32.4 months in the enzalutamide arm versus 30.2 months in the placebo arm. The co-primary end point of median rPFS was similarly superior and was not reached (95% CI: 13.8 – upper limit not reached) in the enzalutamide arm versus only 3.9 months (95% CI: 3.7–5.4) in those who received placebo, upon reporting of the trial. There was a corresponding 29% reduction in risk of death for the enzalutamide arm compared to placebo (OS: HR 0.706; 95% CI: 0.59–0.83; P<0.0001) and an 81% reduction in risk of radiographic progression or death (rPFS: HR 0.19; 95% CI: 0.15–0.23; P<0.0001). Given these findings, the Independent Data Monitoring Committee recommended halting the study and study patients were offered a cross over from placebo to enzalutamide. It is also interesting to note that the use of enzalutamide delayed the median time to the institution of chemotherapy by about 17 months, with a time to chemotherapy at 28 months in the enzalutamide arm versus 10.8 months in the placebo arm (HR 0.35; 95% CI:0.3–0.4; P<0.0001).
Certain differences in trial characteristics between the Cougar abiraterone studies23,25 compared to the enzalutamide trials can be noted. While the abiraterone COU-AA-302 trial excluded men with visceral disease, the PREVAIL trial included about 11.2% of patients with visceral metastases. The median time to delay in chemotherapy was about 17 months in the PREVAIL trial versus 8.4 months in the COU-AA-302 trial although the latter used prednisone as the control arm, rather than just placebo.
Patterns of resistance
While the results from both AFFIRM and PREVAIL studies are both unprecedented, questions remain as to the proper sequencing of these agents.51 It is increasingly recognized that responses of enzalutamide are modest post-abiraterone use and the issue of cross-resistance is increasingly being recognized.83 In one retrospective multicenter trial that included 183 men who had received prior abiraterone, about 30% of men who had no significant prior response to abiraterone still achieved response to enzalutamide.84 However, overall responses are still modest. There is increasing recognition that emergence of splice variants in the AR, AR mutations especially in the ligand binding domain, confers resistance to enzalutamide.85 This has become an active area of research with determination of splice variants such as AR-V7 that may help discern the patients who may or may not respond to enzalutamide, as well as development of novel therapeutics to inhibit such variants.86
Safety and tolerability
Enzalutamide appears to be generally well tolerated at the administered 160 mg/day dose employed in the phase III trials. There were a few toxicities noted in the AFFIRM study that were more common in the enzalutamide arm,78 including fatigue (all grades, 34% versus 29.1%), diarrhea, musculoskeletal pain, headache, hypertension, and hot flashes. The patients on the enzalutamide arm had a lower incidence of grade 3–4 adverse events; 45.3%, versus 53.1% in the placebo group. One adverse event of note was the occurrence of seizures which was reported in five patients in the enzalutamide arm (versus none in the placebo arm) during the phase III AFFIRM trial with another two seizure events reported in the follow-up data. This occurrence was noted despite exclusion of patients in the AFFIRM trial with known predisposition to seizures, such as those with recent history of cerebrovascular accidents, febrile seizures, transient ischemic attacks within the past 12 months, and even use of certain drugs that could lower seizure threshold or prolong QT interval such as insulin, tricyclic anti-depressants, antiarrhythmics, among others. Certain risk factors for decreasing the seizure threshold have been identified in these studies as a potential explanation for occurrence of seizures. Of the five patients who experienced seizures, one received lidocaine, one patient had brain atrophy due to alcohol, and two had brain metastases. Including the two patients who experienced seizures in the longer follow-up of the AFFIRM study, the overall combined seizure risk was about 1%. The PREVAIL trial showed reported seizure events in only two patients.81
Patient focused perspectives: quality of life, patient satisfaction/acceptability and adherence
Health related QoL is increasingly incorporated in clinical trials as one of multiple end points being assessed. This is particularly true in diseases with a poor prognosis like mCRPC where QoL, and not just efficacy alone, may be of significant importance and relevance to the patient and family, as well as the treating physicians. Prostate cancer significantly affects QoL in many patients especially given pain-related symptoms due to bone metastases and treatment-related issues like urinary, sexual, bowel, and hormonal functions, which when affected, greatly influence satisfaction of the patients and their families with treatment outcome.87
Although assessing QoL is not as objective as assessing OS or radiographic tumor response, many tools are being used to assess QoL in patients with various tumors and many of these tools are validated and used in a wide array of studies. One such tool is the use of the Functional Assessment of Cancer Therapy-Prostate (FACT-P). FACT-P is a QoL instrument used with prostate cancer patients which assesses patients in the physical, social/family, emotional, and functional well-being domains. Combining all four modalities scores represents a global QoL score with higher scores representing better QoL response.
Nearly all prostate cancer patients with metastatic disease will become castration-resistant at some point during the course of their disease. Therefore, research has focused on finding drugs that will serve to increase not only survival but QoL. Of the medications that are proven to increase survival in mCRPC, as well as QoL parameters (docetaxel, cabazitaxel, sipuleucel-T, radium 223, abiraterone acetate, and enzalutamide), abiraterone and enzalutamide are the only orally-available medications and have favorable safety profiles. Hence, these agents are being increasingly and favorably used by physicians. Therefore, assessing QoL measures in these two medications is of significant importance.
The two studies that showed improvement in OS and studied QoL measures with abiraterone are the COU AA-301 (post-docetaxel) and COU AA-302 (pre-docetaxel). In the COU AA-301 trial,23 the rate of pain palliation was significantly better with abiraterone/prednisone versus prednisone alone (44% versus 27%, P=0.002) and the median time to FACT-P total scale deterioration was also significantly better with abiraterone (14 months versus 8.4 months, HR 0.607, P<0.0001).88 In the COU AA-302, median time to increased pain was significantly better with abiraterone versus prednisone (26.7 months versus 18.4 months, HR 0.82, P=0.049) and median time to opiate use was significantly delayed with abiraterone. As for the median time to FACT-P score deterioration, it was 12.7 months versus 8.3 months (HR 0.78, P=0.003) in favor of abiraterone.25
Enzalutamide was also studied in the pre-docetaxel (PREVAIL) and post-docetaxel (AFFIRM) chemotherapy trials with positive results with respect to OS and QoL measures when compared to placebo. In the AFFIRM trial, FACT-P QoL response was significantly better with enzalutamide compared to placebo (43% versus 18%, P<0.001).78 The median time to pain progression on the FACT-P scale was not reached for enzalutamide versus 13.8 months for placebo (HR 0.56, P=0.0004).89 Pain palliation, defined as more than 30% reduction in median pain score after 12 weeks of treatment compared with pretreatment pain score without more than 30% increase in use of opiates, was achieved in 45% of patients on enzalutamide versus 7% of patients on placebo. Results of the PREVAIL study reported that the median time to FACT-P global score decline was 11.3 months versus 5.6 months in favor of enzalutamide versus placebo with HR 0.625 and P<0.0001.81
Conclusion
Substantial evidence exists for improving not only the rigorous end points of OS, rPFS but also pain and QoL data with the use of these newer agents for mCRPC. However, much work remains to be done with trying to overcome resistance and choice of sequencing of these agents. Decision about which therapy comes first and the designation of terms such as “pre-docetaxel” or “post-docetaxel” are arbitrary since the timing of the FDA approval of drugs currently dictates practice pattern of which agents are being used first. Once resistance occurs, it is unclear whether subsequent regimens would exert the same benefit, since randomized head-to-head comparisons do not exist for these currently available drugs. Much is therefore left to the clinicians to determine factors such as patients’ comorbidities, pace of disease progression, presence of predominantly bone or visceral metastases, symptoms, toxicity profile, patient preference, cost, and accessibility, to enable making appropriate decisions about choice of therapy.
Disclosure
Jeanny B Aragon-Ching has served on the Advisory Board of Amgen and Algeta/Bayer, and the Speakers’ Bureau of Astellas/Medivation and Janssen/Ortho-Biotech. All other authors have no conflicts of interest to disclose.
References
Siegel R, Ma J, Zou Z, et al. Cancer statistics, 2014. CA Cancer J Clin. 2014;64(1):9–29. | |
Mohler JL, Kantoff PW, Armstrong AJ, et al. Prostate cancer, version 1. 2014. J Natl Compr Canc Netw. 2013;11(12):1471–1479. | |
Cookson MS, Roth BJ, Dahm P, et al. Castration-resistant prostate cancer: AUA Guideline. J Urol. 2013;190(2):429–438. | |
Mezynski J, Pezaro C, Bianchini D, et al. Antitumour activity of docetaxel following treatment with the CYP17A1 inhibitor abiraterone: clinical evidence for cross-resistance? Ann Oncol. 2012;23(11):2943–2947. | |
Noonan KL, North S, Bitting RL, et al. Clinical activity of abiraterone acetate in patients with metastatic castration-resistant prostate cancer progressing after enzalutamide. Ann Oncol. 2013;24(7):1802–1807. | |
Pezaro CJ, Omlin AG, Altavilla A, et al. Activity of Cabazitaxel in Castration-resistant Prostate Cancer Progressing After Docetaxel and Next-generation Endocrine Agents. Eur Urol. In press 2013. | |
Mohler JL, Kantoff PW, Armstrong AJ, et al. Prostate cancer, version 2.2014. J Natl Compr Canc Netw. 2014;12(5):686–718. | |
El-Amm J, Aragon-Ching JB. The changing landscape in the treatment of metastatic castration-resistant prostate cancer. Ther Adv Med Oncol. 2013;5(1):25–40. | |
Huggins C, Hodges CV. Studies on prostatic cancer. I. The effect of castration, of estrogen and androgen injection on serum phosphatases in metastatic carcinoma of the prostate. CA Cancer J Clin. 1972;22(4):232–240. | |
McLeod DG. Hormonal therapy: historical perspective to future directions. Urology. 2003;61(2 Suppl 1):3–7. | |
Hellerstedt BA, Pienta KJ. The current state of hormonal therapy for prostate cancer. CA Cancer J Clin. 2002;52(3):154–179. | |
Sharifi N, Gulley JL, Dahut WL. Androgen deprivation therapy for prostate cancer. JAMA. 2005;294(2):238–244. | |
Montgomery RB, Mostaghel EA, Vessella R, et al. Maintenance of intratumoral androgens in metastatic prostate cancer: a mechanism for castration-resistant tumor growth. Cancer Res. 2008;68(11):4447–4454. | |
Mohler JL, Titus MA, Bai S, et al. Activation of the androgen receptor by intratumoral bioconversion of androstanediol to dihydrotestosterone in prostate cancer. Cancer Res. 2011;71(4):1486–1496. | |
Auchus RJ. Overview of dehydroepiandrosterone biosynthesis. Semin Reprod Med. 2004;22(4):281–288. | |
Goyal J, Antonarakis ES. Clinical Evaluation of Abiraterone in the Treatment of Metastatic Prostate Cancer. Clin Med Insights Urol. 2013;2013(7):1–14. | |
Attard G, Reid AH, Olmos D, et al. Antitumor activity with CYP17 blockade indicates that castration-resistant prostate cancer frequently remains hormone driven. Cancer Res. 2009;69(12):4937–4940. | |
Attard G, Reid AH, A’Hern R, et al. Selective inhibition of CYP17 with abiraterone acetate is highly active in the treatment of castration-resistant prostate cancer. J Clin Oncol. 2009;27(23):3742–3748. | |
Pia A, Vignani F, Attard G, et al. Strategies for managing ACTH dependent mineralocorticoid excess induced by abiraterone. Cancer Treat Rev. 2013;39(8):966–973. | |
Attard G, Reid AH, Yap TA, et al. Phase I clinical trial of a selective inhibitor of CYP17, abiraterone acetate, confirms that castration-resistant prostate cancer commonly remains hormone driven. J Clin Oncol. 2008;26(28):4563–4571. | |
Danila DC, Morris MJ, de Bono JS, et al. Phase II multicenter study of abiraterone acetate plus prednisone therapy in patients with docetaxel-treated castration-resistant prostate cancer. J Clin Oncol. 2010;28(9):1496–1501. | |
Reid AH, Attard G, Danila DC, et al. Significant and sustained antitumor activity in post-docetaxel, castration-resistant prostate cancer with the CYP17 inhibitor abiraterone acetate. J Clin Oncol. 2010;28(9):1489–1495. | |
de Bono JS, Logothetis CJ, Molina A, et al. Abiraterone and increased survival in metastatic prostate cancer. N Engl J Med. 2011;364(21):1995–2005. | |
Fizazi K, Scher HI, Molina A, et al. Abiraterone acetate for treatment of metastatic castration-resistant prostate cancer: final overall survival analysis of the COU-AA-301 randomised, double-blind, placebo-controlled phase 3 study. Lancet Oncol. 2012;13(10):983–992. | |
Ryan CJ, Smith MR, de Bono JS, et al. Abiraterone in metastatic prostate cancer without previous chemotherapy. N Engl J Med. 2013;368(2):138–148. | |
Rathkopf DE, Smith MR, de Bono JS, et al. Updated interim efficacy analysis and long-term safety of abiraterone acetate in metastatic castration-resistant prostate cancer patients without prior chemotherapy (COU-AA-302). Eur Urol. In press 2014. | |
Bostwick DG, Pacelli A, Blute M, et al. Prostate specific membrane antigen expression in prostatic intraepithelial neoplasia and adenocarcinoma: a study of 184 cases. Cancer. 1998;82(11):2256–2261. | |
Goldfarb DA, Stein BS, Shamszadeh M, Petersen OR. Age-related changes in tissue levels of prostatic acid phosphatase and prostate specific antigen. J Urol. 1986;136(6):1266–1269. | |
Patel PH, Kockler DR. Sipuleucel-T: a vaccine for metastatic, asymptomatic, androgen-independent prostate cancer. Ann Pharmacother. 2008;42(1):91–98. | |
Sheikh NA, Petrylak D, Kantoff PW, et al. Sipuleucel-T immune parameters correlate with survival: an analysis of the randomized phase 3 clinical trials in men with castration-resistant prostate cancer. Cancer Immunol Immunother. 2013;62(1):137–147. | |
Small EJ, Fratesi P, Reese DM, et al. Immunotherapy of hormone-refractory prostate cancer with antigen-loaded dendritic cells. J Clin Oncol. 2000;18(23):3894–3903. | |
Small EJ, Schellhammer PF, Higano CS, et al. Placebo-controlled phase III trial of immunologic therapy with sipuleucel-T (APC8015) in patients with metastatic, asymptomatic hormone refractory prostate cancer. J Clin Oncol. 2006;24(19):3089–3094. | |
Higano CS, Schellhammer PF, Small EJ, et al. Integrated data from 2 randomized, double-blind, placebo-controlled, phase 3 trials of active cellular immunotherapy with sipuleucel-T in advanced prostate cancer. Cancer. 2009;115(16):3670–3679. | |
Kantoff PW, Higano CS, Shore ND, et al. Sipuleucel-T immunotherapy for castration-resistant prostate cancer. N Engl J Med. 2010;363(5):411–422. | |
Mita AC, Denis LJ, Rowinsky EK, et al. Phase I and pharmacokinetic study of XRP6258 (RPR 116258A), a novel taxane, administered as a 1-hour infusion every 3 weeks in patients with advanced solid tumors. Clin Cancer Res. 2009;15(2):723–730. | |
de Bono JS, Oudard S, Ozguroglu M, et al. Prednisone plus cabazitaxel or mitoxantrone for metastatic castration-resistant prostate cancer progressing after docetaxel treatment: a randomised open-label trial. Lancet. 2010;376(9747):1147–1154. | |
Pal SK, Twardowski P, Sartor O. Critical appraisal of cabazitaxel in the management of advanced prostate cancer. Clin Interv Aging. 2010;5:395–402. | |
Bahl A, Oudard S, Tombal B, et al. Impact of cabazitaxel on 2-year survival and palliation of tumour-related pain in men with metastatic castration-resistant prostate cancer treated in the TROPIC trial. Ann Oncol. 2013;24(9):2402–2408. | |
Lewington VJ, McEwan AJ, Ackery DM, et al. A prospective, randomised double-blind crossover study to examine the efficacy of strontium-89 in pain palliation in patients with advanced prostate cancer metastatic to bone. Eur J Cancer. 1991;27(8):954–958. | |
Laing AH, Ackery DM, Bayly RJ, et al. Strontium-89 chloride for pain palliation in prostatic skeletal malignancy. Br J Radiol. 1991;64(765):816–822. | |
Serafini AN, Houston SJ, Resche I, et al. Palliation of pain associated with metastatic bone cancer using samarium-153 lexidronam: a double-blind placebo-controlled clinical trial. J Clin Oncol. 1998;16(4):1574–1581. | |
Sartor O, Reid RH, Hoskin PJ, et al. Samarium-153-Lexidronam complex for treatment of painful bone metastases in hormone-refractory prostate cancer. Urology. 2004;63(5):940–945. | |
Finlay IG, Mason MD, Shelley M. Radioisotopes for the palliation of metastatic bone cancer: a systematic review. Lancet Oncol. 2005;6(6):392–400. | |
Goyal J, Antonarakis ES. Bone-targeting radiopharmaceuticals for the treatment of prostate cancer with bone metastases. Cancer Lett. 2012;323(2):135–146. | |
El-Amm J, Freeman A, Patel N, Aragon-Ching JB. Bone-targeted therapies in metastatic castration-resistant prostate cancer: evolving paradigms. Prostate Cancer. 2013;2013:210686. | |
Parker C, Nilsson S, Heinrich D, et al. Alpha emitter radium-223 and survival in metastatic prostate cancer. N Engl J Med. 2013;369(3):213–223. | |
Bruland OS, Nilsson S, Fisher DR, et al. High-linear energy transfer irradiation targeted to skeletal metastases by the alpha-emitter 223Ra: adjuvant or alternative to conventional modalities? Clin Cancer Res. 2006;12(20 Pt 2):6250s–6257s. | |
Liepe K. Alpharadin, a 223Ra-based alpha-particle-emitting pharmaceutical for the treatment of bone metastases in patients with cancer. Curr Opin Investig Drugs. 2009;10(12):1346–1358. | |
Kerr C. (223)Ra targets skeletal metastases and spares normal tissue. Lancet Oncol. 2002;3(8):453. | |
Nilsson S, Larsen RH, Fossa SD, et al. First clinical experience with alpha-emitting radium-223 in the treatment of skeletal metastases. Clin Cancer Res. 2005;11(12):4451–4459. | |
Sartor O, Gillessen S. Treatment sequencing in metastatic castrate-resistant prostate cancer. Asian J Androl. 2014;16(3):426–431. | |
Pisters LL, Troncoso P, Zhau HE, et al. c-met proto-oncogene expression in benign and malignant human prostate tissues. J Urol. 1995;154(1):293–298. | |
Borre M, Offersen BV, Nerstrom B, Overgaard J. Microvessel density predicts survival in prostate cancer patients subjected to watchful waiting. Br J Cancer. 1998;78(7):940–944. | |
Smith DC, Smith MR, Sweeney C, et al. Cabozantinib in patients with advanced prostate cancer: results of a phase II randomized discontinuation trial. J Clin Oncol. 2013;31(4):412–419. | |
Lee RJ, Saylor PJ, Michaelson MD, et al. A dose-ranging study of cabozantinib in men with castration-resistant prostate cancer and bone metastases. Clin Cancer Res. 2013;19(11):3088–3094. | |
Isaacs JT, Pili R, Qian DZ, et al. Identification of ABR-215050 as lead second generation quinoline-3-carboxamide anti-angiogenic agent for the treatment of prostate cancer. Prostate. 2006;66(16):1768–1778. | |
Isaacs JT. The long and winding road for the development of tasquinimod as an oral second-generation quinoline-3-carboxamide antiangiogenic drug for the treatment of prostate cancer. Expert Opin Investig Drugs. 2010;19(10):1235–1243. | |
Bratt O, Haggman M, Ahlgren G, et al. Open-label, clinical phase I studies of tasquinimod in patients with castration-resistant prostate cancer. Br J Cancer. 2009;101(8):1233–1240. | |
Pili R, Haggman M, Stadler WM, et al. Phase II randomized, double-blind, placebo-controlled study of tasquinimod in men with minimally symptomatic metastatic castrate-resistant prostate cancer. J Clin Oncol. 2011;29(30):4022–4028. | |
Weber J. Review: anti-CTLA-4 antibody ipilimumab: case studies of clinical response and immune-related adverse events. Oncologist. 2007;12(7):864–872. | |
Drake C, Kwon E, Fizazi K, et al. Results of subset analyses on overall survival (OS) from study CA184-043: Ipilimumab (Ipi) versus placebo (Pbo) in post-docetaxel metastatic castration-resistant prostate cancer (mCRPC). J Clin Oncol. 2014;32(Suppl 4):Abstr 2. | |
Kwon ED, Drake CG, Scher HI, et al. Ipilimumab versus placebo after radiotherapy in patients with metastatic castration-resistant prostate cancer that had progressed after docetaxel chemotherapy (CA184-043): a multicentre, randomised, double-blind, phase 3 trial. Lancet Oncol. 2014;15(7):700–712. | |
Ning YM, Pierce W, Maher VE, et al. Enzalutamide for treatment of patients with metastatic castration-resistant prostate cancer who have previously received docetaxel: US Food and Drug Administration drug approval summary. Clin Cancer Res. 2013;19(22):6067–6073. | |
Lamb AD, Massie CE, Neal DE. The transcriptional programme of the androgen receptor (AR) in prostate cancer. BJU Int. 2014;113(3):358–366. | |
El-Amm J, Patel N, Freeman A, Aragon-Ching JB. Metastatic castration-resistant prostate cancer: critical review of enzalutamide. Clin Med Insights Oncol. 2013;7:235–245. | |
Jung ME, Ouk S, Yoo D, et al. Structure-activity relationship for thiohydantoin androgen receptor antagonists for castration-resistant prostate cancer (CRPC). J Med Chem. 2010;53(7):2779–2796. | |
Scher HI, Beer TM, Higano CS, et al. Antitumour activity of MDV3100 in castration-resistant prostate cancer: a phase 1–2 study. Lancet. 2010;375(9724):1437–1446. | |
Bennett LL, Ingason A. Enzalutamide (Xtandi) for patients with metastatic, resistant prostate cancer. Ann Pharmacother. 2014;48(4):530–537. | |
Enzalutamide (Xtandi) [package insert]. Northbrook, IL: Astellas Pharma US, 2014. | |
Chen CD, Welsbie DS, Tran C, et al. Molecular determinants of resistance to antiandrogen therapy. Nat Med. 2004;10(1):33–39. | |
Kolvenbag GJ, Furr BJ, Blackledge GR. Receptor affinity and potency of non-steroidal antiandrogens: translation of preclinical findings into clinical activity. Prostate Cancer Prostatic Dis. 1998;1(6):307–314. | |
Fossa SD, Slee PH, Brausi M, et al. Flutamide versus prednisone in patients with prostate cancer symptomatically progressing after androgen-ablative therapy: a phase III study of the European organization for research and treatment of cancer genitourinary group. J Clin Oncol. 2001;19(1):62–71. | |
Akaza H, Hinotsu S, Usami M, et al. Combined androgen blockade with bicalutamide for advanced prostate cancer: long-term follow-up of a phase 3, double-blind, randomized study for survival. Cancer. 2009;115(15):3437–3445. | |
Crawford ED, Eisenberger MA, McLeod DG, et al. A controlled trial of leuprolide with and without flutamide in prostatic carcinoma. N Engl J Med. 1989;321(7):419–424. | |
Tran C, Ouk S, Clegg NJ, et al. Development of a second-generation antiandrogen for treatment of advanced prostate cancer. Science. 2009;324(5928):787–790. | |
Rodriguez-Vida A, Bianchini D, Van Hemelrijck M, et al. Is there an anti-androgen withdrawal syndrome with enzalutamide? BJU Int. In press 2014. | |
Higano CS, Beer TM, Taplin ME. Antitumor activity of MDV3100 in pre- and post-docetaxel advanced prostate cancer: long-term follow-up of a phase I/II study. J Clin Oncol. 2011;29(Supp 7): Abstr 134. | |
Scher HI, Fizazi K, Saad F, et al. Increased survival with enzalutamide in prostate cancer after chemotherapy. N Engl J Med. 2012;367(13):1187–1197. | |
Scher H, Fizazi K, Saad F, et al. Association of baseline corticosteroid with outcomes in a multivariate analysis of the phase 3 AFFIRM study of Enzalutamide (ENZA), an androgen receptor signaling inhibitor (ARSI). Annals of Oncology. 2012;23(Suppl 9): Abstr 899PD. | |
Arora VK, Schenkein E, Murali R, et al. Glucocorticoid receptor confers resistance to antiandrogens by bypassing androgen receptor blockade. Cell. 2013;155(6):1309–1322. | |
Beer TM, Armstrong AJ, Sternberg CN, et al. Enzalutamide in men with chemotherapy-naive metastatic prostate cancer (mCRPC): Results of phase III PREVAIL study. J Clin Oncol. 2014; 32(Suppl 4):LBA1. | |
Beer TM, Armstrong AJ, Rathkopf DE, et al. Enzalutamide in Metastatic Prostate Cancer before Chemotherapy. N Engl J Med. Epub June 1, 2014. | |
van Soest RJ, van Royen ME, de Morree ES, et al. Cross-resistance between taxanes and new hormonal agents abiraterone and enzalutamide may affect drug sequence choices in metastatic castration-resistant prostate cancer. Eur J Cancer. 2013;49(18):3821–3830. | |
Cheng H, Nadal R, Gulati R, et al. The effect of prior abiraterone (Abi) use on the activity of enzalutamide (Enza) in men with mCRPC. J Clin Oncol. 2014;32(Suppl 4):Abstr 8. | |
Chism DD, De Silva D, Whang YE. Mechanisms of acquired resistance to androgen receptor targeting drugs in castration-resistant prostate cancer. Expert Rev Anticancer Ther. 2014;13:1–10. | |
Liu C, Lou W, Zhu Y, et al. Niclosamide inhibits androgen receptor variants expression and overcomes enzalutamide resistance in castration-resistant prostate cancer. Clin Cancer Res. 2014;20(12):3198–3210. | |
Sanda MG, Dunn RL, Michalski J, et al. Quality of life and satisfaction with outcome among prostate-cancer survivors. N Engl J Med. 2008;358(12):1250–1261. | |
Harland S, Staffurth J, Molina A, et al. Effect of abiraterone acetate treatment on the quality of life of patients with metastatic castration-resistant prostate cancer after failure of docetaxel chemotherapy. Eur J Cancer. 2013;49(17):3648–3657. | |
Miller K, Scher HI, Fizazi K, et al. Effect of enzalutamide on health-related quality of life (HRQoL) in men with metastatic castration-resistant prostate cancer (mCRPC) following docetaxel-based therapy: Results from the AFFIRM study. ASCO Meeting Abstracts. 2013;31(6_Suppl):17. | |
Dreicer R, Jones R, Oudard S, et al. Results from a phase 3, randomized, double-blind, multicenter, placebo-controlled trial of orteronel (TAK-700) plus prednisone in patients with metastatic castration-resistant prostate cancer (mCRPC) that has progressed during or following docetaxel-based therapy (ELM-PC 5 trial). J Clin Oncol. 2014;32 (4_Suppl):Abstr 7. | |
Tannock IF, de Wit R, Berry WR, et al. Docetaxel plus prednisone or mitoxantrone plus prednisone for advanced prostate cancer. N Engl J Med. 2004;351(15):1502–1512. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.