Back to Archived Journals » Journal of Receptor, Ligand and Channel Research » Volume 7
Adipokines: biological functions and metabolically healthy obese profile
Authors Pereira S, Alvarez-Leite J
Received 5 November 2013
Accepted for publication 6 March 2014
Published 15 May 2014 Volume 2014:7 Pages 15—25
DOI https://doi.org/10.2147/JRLCR.S36060
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Solange Silveira Pereira,1,2 Jacqueline I Alvarez-Leite1,2
1Laboratory for Atherosclerosis and Nutritional Biochemistry, Department of Biochemistry and Immunology, Institute of Biological Sciences, 2Alfa Institute of Gastroenterology, Clinics Hospital, Medicine School, Federal University of Minas Gerais (UFMG), Belo Horizonte, MG, Brazil
Abstract: Adipose tissue is an extremely active organ, and plays a fundamental role in the genesis of comorbidities associated with obesity. Since the discovery of leptin, an important focus has been assigned to adipose tissue as a key organ in the pathogenesis of metabolic disorders. The influence on the genesis of comorbidities associated with obesity is directly related to the pattern of adipokine secretion, the bioactive molecules produced on adipose tissue. The imbalance of adipokines consequent to the expansion of adipose tissue has been implicated in the development of the low-grade chronic inflammation seen in obesity. Adipokines act in a paracrine, autocrine, and endocrine fashion, influencing cytokine and chemokine secretions and hormonal and growth factors, as well as interfering with actions of insulin and lipid and glucose metabolism. The main adipokines include leptin, adiponectin, resistin, tumor-necrosis factor, interleukin 6, chemokine (C–C motif) ligand 2, interleukin 10, and transforming growth factor-β. The imbalance between pro- and anti-inflammatory adipokines on adipose tissue results in insulin resistance and the development of metabolic syndrome, type 2 diabetes, and cardiovascular disease. However, not all obese individuals develop these comorbidities or metabolic changes. Metabolically normal obese or metabolically healthy obese individuals have been the focus of research because of their absence of comorbidities. The profile of adipokines in adipose tissue of these individuals can be protective for the development of insulin resistance and metabolic disorders. This review emphasizes the roles of adipokines, the signaling pathways involved in the pathogenesis of inflammation and insulin resistance, and the profile found in metabolically healthy obese individuals.
Keywords: adipokines, adipose tissue, obesity, metabolically healthy obese
Introduction
For a long time, adipose tissue was considered a deposit of energy. Nowadays, it is well known that the key role of adipose tissue in metabolism is as an endocrine organ responsible for the secretion of bioactive molecules termed “adipokines.”1 Adipokines have hormone function, act as growth factors that modulate insulin resistance, and act on the fat and glucose metabolism and participate in pro and anti-inflammatory responses.2,3 Deregulated adipokine expression caused by excessive adiposity and adipocyte dysfunction seen in obesity has been linked to the pathogenesis of several diseases through altered immune responses.1
Adipose tissue comprises mature adipocytes, preadipocytes, endothelial cells, fibroblasts, mast cells, and immune-system cells.4 Adipose tissue is not a uniform organ, and secretes different patterns of adipokines, depending on its location.5 Changes in specific adipokine profile lead to metabolic disturbances that play a central role in the development of insulin-resistant type 2 diabetes mellitus (T2DM) and cardiovascular diseases.6
Adipose tissue is dynamically involved in the regulation of cell function and the genesis of diseases via a complex network of signal endocrine, paracrine, and autocrine influencing the response of many tissues, including the hypothalamus, pancreas, liver, skeletal muscle, kidney, endothelium, and immune system, among others.5,7 These signals are mediated by adipokines and their mechanisms of action by binding to their receptors (Figure 1).
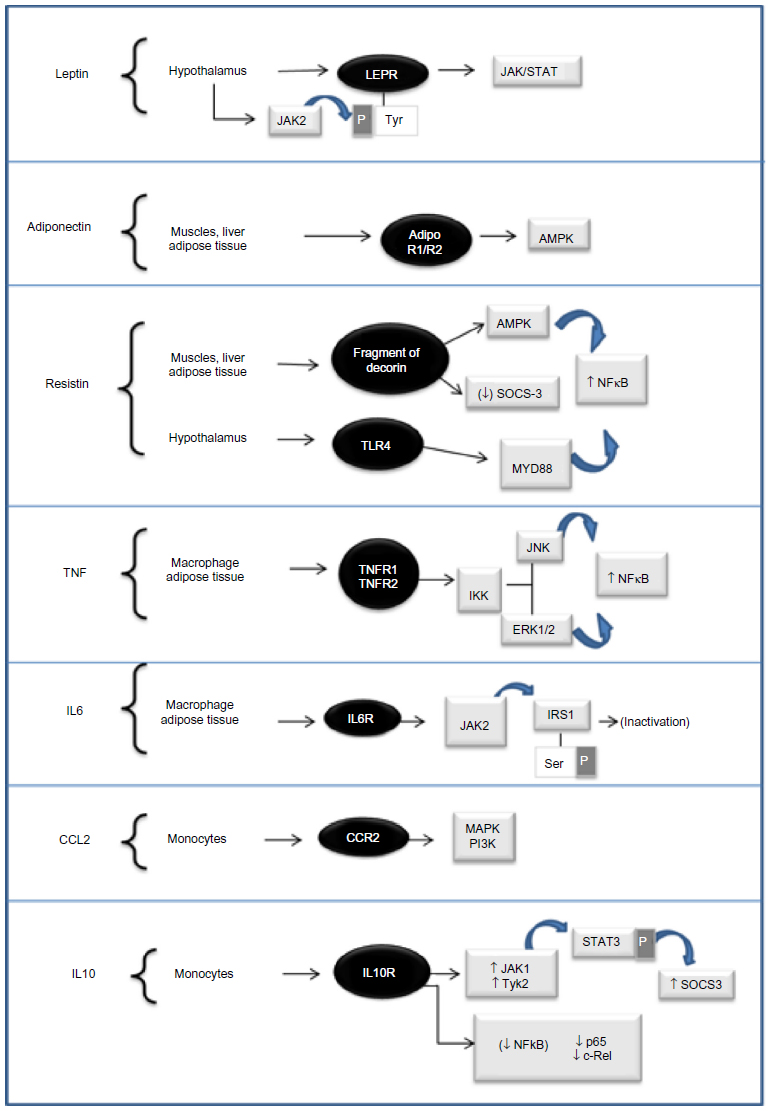 | Figure 1 Mechanisms of actions of adipokines. |
Adipokines are classified as hormones, growth factors, angiogenic factors, and cytokines. Among them, leptin, adiponectin, resistin, monocytes, and macrophage chemotactic protein 1 (chemokine [C–C motif] ligand [CCL]-2), interleukin (IL)-6, IL-1β, tumor-necrosis factor (TNF), anti-inflammatory IL-10, and transforming growth factor (TGF)-β are the most studied8 (Table 1).
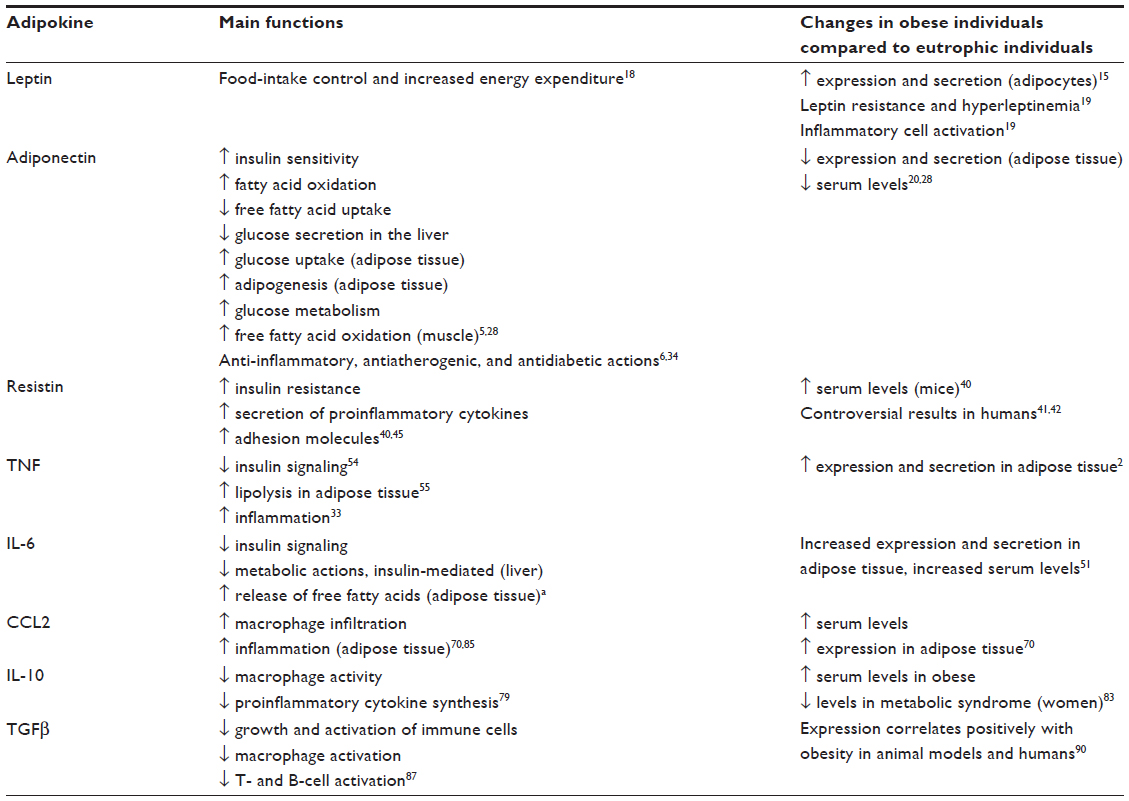 | Table 1 Main adipokines and their functions |
The influence of adipose tissue in the development of metabolic disorders related to abdominal obesity has been well described.9 However, the presence of obesity-related metabolic disorders varies widely among obese individuals.10 A group of obese individuals with a favorable metabolic profile has been described.10–12 Individuals classified as metabolically normal obese or metabolically healthy obese (MHO) have normal cardiovascular risk, high insulin sensitivity, absence of dyslipidemia, and a favorable inflammatory profile.10,13 The mechanisms that may explain the favorable metabolic profile in these individuals are still unknown. Characteristics of adipose tissue, such as proinflammatory profile and expression of adipokines, may be involved.10 The interest in understanding the metabolically normal obese is increasing, due to the potential to elucidate the mechanisms of chronic diseases and translate them into treatment options.
This review aims to describe the main biological functions of some adipokines and their mechanisms of action by binding to their receptors, and describes the profile of adipokines found in the adipose tissue of the MHO.
Adipokines, biologic functions, mechanisms of action, and profile in the MHO
Leptin
Leptin is a 16 kDa nonglycosylated peptide hormone, consisting of 167 amino acids.14 Leptin is the product of the LEP gene, which is located on chromosome 7 and mainly expressed and produced by differentiated mature adipocytes of white adipose tissue. Subcutaneous adipose tissue is the main source of secretion.6 The production of leptin is higher in women than in men, and positively related to the expansion of adipose tissue and total body fat.15 It has been shown that low levels of leptin inhibit insulin release, and higher levels have a stimulating effect on this. This could explain some conflicting results regarding the influence of leptin in T2DM.16 Additionally, leptin modulates lipid metabolism, hematopoiesis, thermogenesis, and ovarian and β-cell function.3 The human gene of the leptin receptor (LEPR) is located on chromosome 1p31, and the interaction of genetic alterations of LEPR and obesity has been investigated.17 Leptin binds to its receptors, and is highly expressed in the hypothalamus (long-isoform LEPRb), that induces such actions as the control of food intake and increase of energy expenditure.18 LEPR binding activates the Janus kinase (JAK)/signal transducer and activator of transcription (STAT) signaling pathway. The JAK2 kinase, associated with LEPRb, phosphorylates tyrosine residues on LEPRb, which subsequently recruits and phosphorylates members of the STAT family, which translocate to the nucleus, where they regulate gene transcription.16
Obesity is associated with leptin resistance, which manifests as hyperleptinemia with possible consequences in immune-cell activation.19 It is unclear whether leptin resistance is the main contribution to obesity or whether it is secondary to increased production in obese individuals.20 Hyperleptinemia followed by leptin resistance may be an important cause of adipocyte dysfunction and ectopic deposition of lipids in peripheral tissues, propitiating insulin resistance.21 Leptin resistance may be explained by various mechanisms, including the defective transport of leptin across the blood–brain barrier and impaired signaling in neurons and other target cells. It is also believed that leptin resistance may be due to a defect in the leptin-receptor gene that results in the absence of portions of its cytoplasmic region. Therefore, individuals with this mutation tend to have excessive food intake and weight gain.22 Increased expression of proteins that block leptin signaling, such as protein tyrosine phosphatase (PTP)-1B and suppressor of cytokine signaling (SOCS)-3, shows an additional mechanism of neuronal resistance.6 Leptin induces the expression of proinflammatory cytokines in macrophages and T cells, and activates inflammation pathways used by proinflammatory cytokine receptors, including mitogen-activated protein kinases (MAPKs), JAK-STAT3, and phosphatidylinositol-4,5-bisphosphate 3-kinase. The induction of these signaling pathways and their expression pattern suggests that leptin also mediates inflammatory responses.1,20
When compared with obese individuals at cardiovascular risk of becoming MHO, no differences were found in serum leptin levels; however, these were increased when both obese groups were compared to normal-weight individuals.23 A similar result was found when morbidly obese insulin-sensitive individuals were compared to insulin-resistant ones. Individuals from both groups had similar leptin levels, both men and women.24 However the leptin:adiponectin serum ratio was higher in young morbidly obese patients at risk compared to the MHO. This ratio was associated with cardiovascular risk in these individuals, along with serum triglycerides and male sex.25 Previously, it was shown that the leptin:adiponectin ratio contributed to the development of metabolic syndrome in the severe obese and negatively correlated with insulin sensitivity in people without obesity or diabetes.26 There are few published data on leptin expression in adipose tissue and leptin levels in the MHO and its contribution to the inflammatory profile found in these individuals, so more studies are needed.
Adiponectin
Adiponectin is a 30 kDa plasmatic protein encoded by the ADIPOQ gene, which is located on chromosome 3q27. Although the ADIPOQ gene is expressed primarily in adipocytes, studies have shown that adiponectin expression can be induced in other cells.27 It is structurally related to the C1q factor of the complement system, and also has similarity with collagens VIII and X. Various multimeric forms, such as low-molecular-weight trimers and high-molecular-weight dodecamers, are found in abundance in blood. It has been suggested that the high-molecular-weight form is the main and most active contributor to the peripheral metabolic effects of adiponectin.27
Different from other adipokines, adiponectin is mostly expressed in subcutaneous adipose tissue, and its expression and blood concentration decreases with the increase of adiposity.20,28 However, a different profile in the MHO has been found. Obese African Americans with hyperadiponectinemia had a healthier metabolic profile, including higher high-density lipid cholesterol, lower insulin levels, smaller waist circumference, and insulin levels, compared to those without hyperadiponectinemia. Furthermore, a significant association was found between hyperadiponectinemia and a metabolically healthy obese phenotype in these individuals.29 It was also shown that adipose tissue of MHO individuals had a favorable inflammatory profile in association with high levels of adiponectin.30 Bik et al31 found higher serum concentrations of total adiponectin in metabolically healthy patients compared to those diagnosed with metabolic syndrome. Similar results were found by Klöting et al,24 who showed that obese individuals had higher insulin-sensitive adiponectin levels than obese insulin-resistant subjects. In contrast, in other studies, no differences between adiponectin levels among MHO individuals or the unhealthy obese were found.23,32
Adiponectin has a wide spectrum of metabolic and anti-inflammatory effects, acting mainly via two receptors: AdipoR1, found in the skeletal muscle, liver, and adipose tissue, and AdipoR2, found in the liver and adipose tissue.33,34 The AdipoR1 receptor mediates the metabolic action of adiponectin in the liver and muscle, primarily through an increase in adenosine monophosphate kinase (AMPK) activity,1,35–37 while AdipoR2 is involved in the activation of peroxisome proliferator-activated receptor (PPAR)-α in the liver, leading to increased insulin sensitivity.36 The action of adiponectin occurs through binding of its receptors AdipoR1 and -2 to APPL1 (adaptor protein, phosphotyrosine interaction, PH domain, and leucine zipper-containing protein 1),37,38 an adaptor protein identified as a facilitator of adiponectin signaling. In adipose tissue, the main metabolic effects of adiponectin are an improvement in insulin sensitivity, fatty acid oxidation, reduced secretion of glucose from liver, increased glucose uptake, and adipogenesis. In muscle, adiponectin stimulates glucose metabolism and accelerates the oxidation of free fatty acids.5,28 Adiponectin also suppresses the migration of monocytes/macrophages and their transformation into foam cells in the vascular wall. These actions can be responsible for the anti-inflammatory and antiatherogenic effects of adiponectin.34 In vitro studies have shown that adiponectin may reduce the inflammatory response of endothelial cells through inhibition of TNF-α induced nuclear factor-kappaB (NF-κB) activation.6 Interestingly it has been shown that adiponectin receptors have different capacities to regulate the expression of genes involved in inflammation and lipid uptake by macrophages and foam cells. In response to adiponectin, AdipoR2 but not AdipoR1 suppresses lipid uptake via scavenger receptor AI, and induces the IL-1 receptor antagonist (RA), which exerts an anti-inflammatory effect. AdipoR1, in turn, preferentially suppresses proinflammatory molecules, such as TNF and CCL2. The axis adiponectin–ADIPOR1/2–APPL1 in human macrophages is believed to play an important role in lipid metabolism, inflammation, and foam-cell formation.38 Altogether, these studies suggest that adiponectin is the only hormone adipocyte derived that has antidiabetic, anti-inflammatory, and antiatherogenic characteristics.
Resistin
Resistin was initially discovered in mice in 2001, and was named for its capacity to interfere with the action of insulin, leading to insulin resistance.39 It is a 12.5 kDa polypeptide secreted by adipocytes on rodents and by macrophages on humans.40 Human resistin is expressed by the RETN gene, located at position 19 of chromosome 19p 13.3. The relationship between serum resistin and insulin resistance, T2DM, and obesity on humans is still controversial. Some studies have found no changes in circulating resistin levels in obesity, insulin resistance, or T2DM, while others reported a significant increase between circulating levels of resistin and these conditions.41,42 The mechanisms by which resistin exerts biological effects are not fully elucidated. These effects may be mediated by paracrine and endocrine fashion, and probably via binding of resistin to a surface receptor on target cells.43 The probable receptor for murine resistin is a fragment of decorin without the glycosaminoglycan-binding site, and thus without an associated carbohydrate chain. Decorin is a connective tissue extracellular matrix protein, a member of a family of leucine-rich proteoglycans that bind to type I collagen and influence matrix modeling.41 Benomar et al40 showed that the binding resistin to Toll-like receptor 4 in the hypothalamus activates proinflammatory pathways in that organ, contributing to the comprehension of molecular mechanisms involved in the inflammation and insulin resistance induced by resistin.
Downstream resistin targets have highlighted its indirect role in intracellular pathways involving the inhibition of insulin signaling, as well as the inflammatory response. In rodents, resistin inhibited AMPK in liver and muscle.43 In addition to activating cytokines, such as IL-6, IL-12, and TNF via NFκB pathway, resistin also activates SOCS-3, recognized by reducing insulin signaling in adipose tissue and other tissues.43 In agreement with this finding, Palanivel et al44 demonstrated improvement in systemic insulin sensitivity by inhibiting the production of SOC3 in the adipose tissue of obese mice.
The chronic central infusion of resistin on normal mice markedly affects the hypothalamus and peripheral responses to insulin. Resistin treatment inhibited insulin signaling pathways, such as the expression and phosphorylation of insulin-receptor substrate (IRS) and Akt, increased phosphorylation of the serine residue in IRS1, and increased expression of PTP1B and SOCS3, both negative regulators of insulin signaling.40 Resistin also activates endothelial cells by increasing the expression of endothelin 1, the intercellular adhesion molecule, and the vascular cell-adhesion molecule, which can contribute to atherosclerosis in humans.45
In addition to the effects of resistin on insulin resistance, diabetes and atherosclerosis, it has also been implicated in a variety of diseases such as autoimmune diseases, nonalcoholic fatty liver disease, malignancies, asthma, inflammatory bowel disease and chronic kidney disease.39
Lower levels of resistin were found in metabolically healthy patients compared to those diagnosed with metabolic syndrome.31 In contrast, no differences were found in resistin levels between the obese at cardiovascular risk and the MHO, even when compared with normal-weight individuals.23 It is important to consider that the role of resistin in human obesity is still unclear, and there are few studies on the profile of resistin in the MHO.
Tumor-necrosis factor
TNF is synthesized as a 26 kDa transmembrane monomer (TNFm), which after proteolytic cleavage by the TNF-converting enzyme results in soluble 17 kDa TNF (TNFs) molecules. Both TNFm and TNFs have biological effects and metabolic responses, suggesting that TNFm mediates autocrine and paracrine actions, while TNFs mediates endocrine responses.46 TNF is able to bind to two structurally related transmembrane receptors: TNFR1 (p60) and TNFR2 (p80).47,48 The TNF–TNFR complex suffers a proteolytic cleavage, releasing its soluble forms sTNFR1 and sTNFR2 in the circulation.47 These soluble forms are related to the clearance and excretion of TNF, and thus can modulate TNF activity. These soluble receptors may also be used as more accessible biomarkers for TNF activity.46
TNF has been described as the first factor derived from adipose tissue to represent an association between obesity, inflammation, and diabetes. TNF expression is increased in obesity, and has since been implicated in the pathogenesis of insulin resistance.49 Although adipocytes are capable of producing TNF-α, M1 macrophage from the stromal vascular fraction are the primary source of adipose tissue50,51 derived TNF-α in obese individuals.5,52
High levels of TNF were observed in individuals with metabolic syndrome.52 In contrast, the MHO have lower levels of TNF and a reduced proinflammatory profile.30 Despite equivalent levels of fat mass, MHO women had significantly lower circulating TNF levels compared to women with metabolic syndrome.53 However, in another study, it was demonstrated that the messenger ribonucleic acid (mRNA) expression of TNF in peripheral blood mononuclear cells was similar in individuals of normal weight, obese at cardiovascular risk, and the MHO.23
The relationship between elevated levels of TNF and metabolic syndrome is related to TNF’s ability to mediate insulin resistance. TNF induces the phosphorylation of a serine residue of IRS1 by action of the c-Jun NH2-terminal kinase, inhibiting the normal phosphorylation of the tyrosine residue of IRS1 and consequently disturbing insulin signaling.54 TNF can also inhibit lipoprotein lipase in adipocytes, favoring the lipolysis and release of free fatty acids to systemic circulation, causing insulin resistance in such peripheral tissues as the liver and muscle of obese patients.55,56 Although the exact mechanisms by which TNF and other inflammatory cytokines contribute to insulin resistance are not yet fully elucidated, several downstream mediators seem to cross-talk among inflammatory and metabolic diseases. Among these are such protein kinases as c-Jun N-terminal kinase, IκB kinase, and MAPK, including extracellular signal-regulated kinase 1/2, play an important role.57–59 Additionally, Song et al60 showed more influence of TNF in the pathogenesis of complications of obesity. It has been shown that TNF reduces the differentiation of 3T3-L1 preadipocytes in vitro, probably due to the increase of PTP1B expression. In addition, TNF is also involved in the synthesis of proinflammatory cytokines, such as IL-6, CCL2, and TNF itself, through NFκB activation.14,34 This NF is a well-known factor involved in diet-induced obesity and insulin resistance, as well as in the low grade inflammation related to adipose-tissue expansion.61
Interleukin 6
IL-6 is a pluripotent cytokine secreted by several tissues and cells, such as immune cells, endothelial cells, myocytes, and adipocytes. It is involved in multiple physiological processes, including inflammatory responses, cell growth and differentiation, and response to tissue injury and host defense.34,62,63 Besides its role in inflammation and host defense, IL-6 is involved in the regulation of insulin signaling, and lipid metabolism in peripheral tissues.64 To exert these biological functions, IL-6 first binds to the specific membrane receptor IL-6R. The IL-6–IL-6R complex then associates with the signal transducer receptor gp130.65
Approximately a third of circulating IL-6 is synthesized by adipose tissue. High blood levels of IL-6 are positively correlated with obesity, glucose intolerance, and insulin resistance, and are predictive for the development of T2DM, metabolic syndrome, and cardiovascular disease.51 The production of IL-6 is three times higher in visceral compared to subcutaneous adipose tissue, and provides a link between visceral fat and insulin resistance, as well as visceral fat and inflammation.51 Both adipocyte hypertrophy and inflammatory stimuli, such as TNF, favor the increase of IL-6. It has been described that weight loss reduces both circulating levels and expression by adipose tissue of IL-6.1,56
Reduced levels of IL-6 in insulin-sensitive obese individuals compared to insulin-resistant obese have been demonstrated. Similar results were found for levels of C-reactive protein, which is known to be related to IL-6.24 In accordance with these data, similar IL-6 and C-reactive protein serum levels in lean and MHO individuals were found, but were lower than those in the unhealthy obese.32 Additionally, lower levels of IL-6 were found, as well as TNF, in some murine models of the MHO.66
The mechanisms associated with insulin resistance induced by IL6 are similar to those described for TNF: deregulation on insulin signaling via phosphorylation of a serine residue in IRS1 and inhibition of lipoprotein lipase, with a consequent increase in the release of free fatty acids from adipose tissue. IL-6 suppresses insulin actions in hepatocytes by a mechanism mediated by the expression of SOCS3.1 With regard to cardiovascular diseases, IL-6 induces hypertriglyceridemia associated with obesity by stimulating hepatic secretion of very low-density lipoprotein, and also increases C-reactive protein, an important risk factor.6 The IL-6 signaling pathways differ markedly between myocytes and macrophages. The intramuscular expression of IL-6 is regulated by a cascade of signaling that includes Ca2+/NF of activated T cells and glycogen/p38 MAPK pathways while in macrophages; IL-6 signaling is dependent on the activation of NFκB. Therefore, IL-6 in monocytes/macrophages induces a proinflammatory response, while in muscle it induces a TNF- and NFκB-independent response.67,68
Chemokine ligand 2
CCL2/monocyte chemotactic protein 1 is a chemokine with the C–C motif that participates in inflammation by recruiting monocytes toward the inflammatory site.69 It is produced principally by macrophages and endothelial cells with increased expression in atherosclerotic lesion areas.70,71 The chemotactic activity of CCL2 is regulated by binding to chemokine receptor 2 (CCR2),72 which induces monocyte chemoattraction, activation, and transmigration.73 CCR2 is coupled to a seven-transmembrane protein expressed on monocytes.74 The absence of CCL2 or CCR2 in low-density lipoprotein-receptor-/- and apolipoprotein E-/- mice protected these animals from atherosclerosis, a condition in which the recruitment of macrophages plays a crucial role.69 Elevated plasma levels and adipose-tissue overexpression of CCL2 have been found in insulin-resistant obese individuals.75 CCL2 is positively regulated by leptin, and has increased expression in the adipose tissue of genetically obese diabetic (db/db) mice and those with diet-induced obesity.70 It is expressed more in cells of vascular stroma compared to adipocytes, and in visceral compared to subcutaneous adipose tissue.51 Kanda et al70 demonstrated that increased CCL2 expression is positively associated with macrophage infiltration in adipose tissue, insulin resistance, and increased hepatic triglyceride content in mice. Furthermore, the deletion of the CCL2 gene reduced the extent of macrophage accumulation in adipose tissue, insulin resistance, and hepatic obesity-associated steatosis.
Improvement in obesity and associated metabolic disorders, such as insulin resistance and hepatic steatosis, was seen after pharmacological inhibition of CCR2 in db/db mice.70,76 A similar effect was seen after CCR2-antagonist treatment that reduced injury in target organs on T2DM mice, by improving metabolic disorders as well as inhibiting proinflammatory and prothrombotic processes.74 However, the association of CCL2 serum levels and obesity or insulin resistance was not confirmed in obese postmenopausal women.73
Several groups have demonstrated the action of CCL2 and/or CCR2 in increasing inflammatory infiltrate on adipose tissue, and thus insulin resistance and other metabolic disorders in obese humans and animals.70,74,77 MHO individuals display a reduced inflammatory profile, with lower numbers of infiltrating adipose-tissue macrophages and crown-like structures in adipose tissue and reduced serum levels of CCL2.24,30
These results suggest that the CCL2–CCR2 axis plays an important role in the development of insulin resistance by inducing inflammatory response on adipose tissue, and also plays an important role in the pathogenesis of metabolic syndrome and obesity-associated T2DM.76,77
Interleukin 10
IL-10 is a Th2 cytokine produced by various types of immune cells, mainly M2 macrophages and Th2 lymphocytes.20,78 It is also expressed by adipocytes, and creates an anti-inflammatory environment in adipose tissue under physiological conditions.54 IL-10 plays a central role in the regulation of immune responses by limiting inflammation over a variety of mechanisms, including inhibition of macrophage activity and the synthesis of such proinflammatory cytokines as TNF and IL-12 by suppression of p65 and c-Rel subunits of NFκB. IL-10 is also an important negative regulator of the release of reactive oxygen species and nitrogen intermediates, and also suppresses cytotoxic T-cell responses and antigen presentation.79
The human IL-10 receptor belongs to the cytokine-receptor class II family, and is composed of two α-molecules (IL-10Rα) and two β-molecules (IL-10Rβ). IL-10Rα mediates high-affinity binding to the ligand and subsequent signal transduction. IL-10Rβ is believed to contribute only to the signaling process.80 The binding of IL-10 to the receptor results in activation of the tyrosine kinases JAK1 and Tyk2, associated with the receptor, resulting in STAT3 phosphorylation and induction of STAT3-dependent genes, including SOCS3.81,82
High levels of IL-10 transcripts block the production of inflammatory mediators, such as TNF, IL-6, IL-1β, and chemokines, and increase such anti-inflammatory cytokines as IL-1Rα.54,69 Elevated serum levels of IL-10 have been observed in obesity,54,83 while low levels are associated with metabolic syndrome in women with or without obesity. Low serum levels of IL-10 were found in obese children and were negatively correlated with IL-1β, suggesting that the decline in IL-10 concentration is related to the inflammatory environment seen in these individuals.79 So far, the profile of IL-10 in the MHO is unknown, but considering its anti-inflammatory properties, it is possible to suggest that its levels are positively correlated with this phenotype.
Treatment with IL-10 reduced inflammation of the liver and adipose tissue, and improved lipid and glucose hepatic metabolism in obese mice.84 Mice with diet-induced obesity that overexpressed IL-10 had greater insulin sensitivity and insulin signaling in muscle, due to attenuation of the inflammatory response.82 Forkhead box (FOX)-P3+ regulatory T cells of obese mice have higher IL-10 secretion compared to cells from control mice.85 Coadministration of IL-10 and TNF in culture adipocytes inhibits the expression of all proinflammatory genes induced by TNF, as well as reverts TNF-induced glucose transporter type 4 (GLUT4) and IRS1 downregulation, thus restoring appropriate insulin signaling.85
Transforming growth factor-β
TGFβ is a member of the growth-factor family, and comprises at least 30 members in mammals.86 Almost all cells in rodents and humans can produce and respond to TGFβ.87 At the cellular level, members of the TGF superfamily regulate fundamental processes, such as proliferation, differentiation, death, cytoskeletal organization, cell adhesion, and migration.86 First discovered as a critical factor for the growth of nonimmune cells, TGFβ has gradually been recognized as a critical cytokine in the regulation of immune responses.87 TGFβ is a recognized potent neutralizer of macrophage activation, and exerts an inhibitory effect on growth and activation of immune cells. In addition, TGFβ has recently been involved in the induction of FOXP3 expression in CD4+CD25+FOXP3– cells, converting those cells in FOXP3+ regulatory T cells in both mice and humans.88
TGFβ signaling occurs through type I and type II serine/threonine kinase transmembrane receptors, which recruit and phosphorylate receptor-activated Smads, including Smad2 and Smad3, and TGFβ canonical intracellular mediators.75 However, a number of Smad-independent signaling cascades have also been elucidated. However, a number of Smad-independent signaling cascades have also been elucidated. Among these are the GTPase, MAPK and phosphatidylinositol 3-kinase (PI3K).86
The TGFβ level on adipose tissue is strongly associated with class III obesity.75,89 The exact role of TGFβ in adipogenesis and obesity remains unclear, and needs further study. Although TGFβ positively correlates with obesity in animal models and humans, it inhibits adipogenesis in 3T3-F442A cell culture. Alternatively, TGFβ promotes adipogenesis in pluripotent progenitor cells during the initial phase of adipose-tissue expansion, but inhibits it in populations of committed preadipocytes.90
MHO TGFβ data are still scarce. The mRNA expression of TGFβ in peripheral blood mononuclear cells of MHO individuals was similar to cardiovascular risk in obese and lean individuals.23
Other adipokines
Several other adipokines have been identified and implicated in insulin resistance associated with obesity and inflammation. Among them is the proinflammatory cytokine IL-1β, produced by macrophages infiltrating the adipose tissue of obese patients. IL-1β, like TNF, impairs insulin signaling and increases lipolysis.57 Interestingly, IL-1β mRNA expression in peripheral blood mononuclear cells was lower in MHO compared to lean individuals, however, higher compared to obese at cardiovascular risk.23
IL-1RA is an IL-1β antagonist that binds to IL-1β receptors without inducing cellular response, thus antagonizing the inflammatory action of IL1β. In humans, the secretion of IL-RA in adipose tissue is induced by interferon and IL-1β through an autocrine and paracrine regulatory response. In obese patients with hyperleptinemia, circulating IL-RA levels are seven times higher than in nonobese patients.91
Retinol-binding protein (RBP)-4 is expressed in the liver, adipocytes, and macrophages, and is increased in obese diabetic rodents and humans. RBP4 inhibits the insulin-induced phosphorylation of IRS1, and its expression is inversely correlated with glucose transporter type 4 in adipocytes.92 The serum RBP4 concentration is twofold higher in insulin-resistant obese subjects when compared with the insulin-sensitive obese.24
Omentin, vaspin, and secreted frizzled-related protein 5 are anti-inflammatory adipokines that improve insulin resistance. Lipocalin 2 and visfatin are proinflammatory adipokines that induce TNF expression.92 A range of adipokines have recently been identified; however, more studies are needed to elucidate their action on insulin resistance and their comorbidity influence associated with obesity and even the MHO profile.
Conclusion
Adipokines are bioactive molecules that have a decisive role in the genesis of inflammation and insulin resistance associated with obesity. Different pathways are activated as a result of connecting different adipokines with their respective receptors. There is still much to elucidate about this interesting environment in the adipose tissue of obese individuals. Unraveling the mechanisms involved in the genesis of inflammation and metabolic disorders is of fundamental importance in the search for therapeutic targets against the growing epidemic of obesity and associated comorbidities. Additionally, the profile of adipokines in MHO can be an important tool to elucidate protective factors for the development of metabolic imbalance and comorbidities associated with obesity.
Disclosure
The authors report no conflicts of interest in this work.
References
Ouchi N, Parker JL, Lugus JJ, Walsh K. Adipokines in inflammation and metabolic disease. Nat Rev Immunol. 2011;11(2):85–97. | |
Ouchi N, Ohashi K, Shibata R, Murohara T. Adipocytokines and obesity-linked disorders. Nagoya J Med Sci. 2011;74(1–2):19–30. | |
Ntaios G, Gatselis NK, Makaritsis K, Dalekos GN. Adipokines as mediators of endothelial function and atherosclerosis. Atherosclerosis. 2013;227(2):216–221. | |
Lee MJ, Wu Y, Fried SK. Adipose tissue remodeling in pathophysiology of obesity. Curr Opin Clin Nutr Metab Care. 2010;13(4):371–376. | |
Coelho M, Oliveira T, Fernandes R. Biochemistry of adipose tissue: an endocrine organ. Arch Med Sci. April 20, 2013;9(2):191–200. | |
Leal VO, Mafra D. Adipokines in obesity. Clin Chim Acta. 2013;419: 87–94. | |
Li ZY, Wang P, Miao CY. Adipokines in inflammation, insulin resistance and cardiovascular disease. Clin Exp Pharmacol Physiol. 2011;38(12):888–896. | |
Lemoine AY, Ledoux S, Larger E. Adipose tissue angiogenesis in obesity. Thromb Haemost. 2013;110(4):661–668. | |
Bigornia SJ, Farb MG, Mott MM, et al. Relation of depot-specific adipose inflammation to insulin resistance in human obesity. Nutr Diabetes. 2012;2:e30. | |
Primeau V, Coderre L, Karelis AD, et al. Characterizing the profile of obese patients who are metabolically healthy. Int J Obes (Lond). 2011;35(7):971–981. | |
Wildman RP, Muntner P, Reynolds K, et al. The obese without cardiometabolic risk factor clustering and the normal weight with cardiometabolic risk factor clustering: prevalence and correlates of 2 phenotypes among the US population (NHANES 1999–2004). Arch Intern Med. 2008;168(15):1617–1624. | |
Karelis AD, Brochu M, Rabasa-Lhoret R, Garrel D, Poehlman ET. Clinical markers for the identification of metabolically healthy but obese individuals. Diabetes Obes Metab. 2004;6(6):456–457. | |
Karelis AD, Faraj M, Bastard JP, et al. The metabolically healthy but obese individual presents a favorable inflammation profile. J Clin Endocrinol Metab. 2005;90(7):4145–4150. | |
Gualillo O, González-Juanatey JR, Lago F. The emerging role of adipokines as mediators of cardiovascular function: physiologic and clinical perspectives. Trends Cardiovasc Med. 2007;17(8):275–283. | |
Pujanek M, Bronisz A, Malecki P, Junik R. Pathomechanisms of the development of obesity in some endocrinopathies – an overview. Endokrynol Pol. 2013;64(2):150–155. | |
Dunmore SJ, Brown JE. The role of adipokines in β-cell failure of type 2 diabetes. J Endocrinol. 2013;216(1):T37–T45. | |
Becer E, Mehmetçik G, Bareke H, Serakinci N. Association of leptin receptor gene Q223R polymorphism on lipid profiles in comparison study between obese and non-obese subjects. Gene. 2013;529(1):16–20. | |
Jung CH, Kim MS. Molecular mechanisms of central leptin resistance in obesity. Arch Pharm Res. 2013;36(2):201–207. | |
de Heredia FP, Gómez-Martínez S, Marcos A. Obesity, inflammation and the immune system. Proc Nutr Soc. 2012;71(2):332–338. | |
Ye J, McGuinness OP. Inflammation during obesity is not all bad: evidence from animal and human studies. Am J Physiol Endocrinol Metab. 2013;304(5):E466–E477. | |
Vázquez-Vela ME, Torres N, Tovar AR. White adipose tissue as endocrine organ and its role in obesity. Arch Med Res. 2008;39(8):715–728. | |
Etemad A, Ramachandran V, Pishva SR, et al. Analysis of Gln223Agr polymorphism of leptin receptor gene in type II diabetic mellitus subjects among Malaysians. Int J Mol Sci. 2013;14(9):19230–19244. | |
Telle-Hansen VH, Halvorsen B, Dalen KT, et al. Altered expression of genes involved in lipid metabolism in obese subjects with unfavourable phenotype. Genes Nutr. 2013;8(4):425–434. | |
Klöting N, Fasshauer M, Dietrich A, et al. Insulin-sensitive obesity. Am J Physiol Endocrinol Metab. 2010;299(3):E506–E515. | |
Labruna G, Pasanisi F, Nardelli C, et al. High leptin/adiponectin ratio and serum triglycerides are associated with an “at-risk” phenotype in young severely obese patients. Obesity (Silver Spring). 2011;19(7):1492–1496. | |
Finucane FM, Luan J, Wareham NJ, et al. Correlation of the leptin:adiponectin ratio with measures of insulin resistance in non-diabetic individuals. Diabetologia. 2009;52(11):2345–2349. | |
Yadav A, Kataria MA, Saini V. Role of leptin and adiponectin in insulin resistance. Clin Chim Acta. 2013;417:80–84. | |
Lenz A, Diamond FB Jr. Obesity: the hormonal milieu. Curr Opin Endocrinol Diabetes Obes. 2008;15(1):9–20. | |
Doumatey AP, Bentley AR, Zhou J, Huang H, Adeyemo A, Rotimi CN. Paradoxical hyperadiponectinemia is associated with the metabolically healthy obese (MHO) phenotype in African Americans. J Endocrinol Metab. 2012;2(2):51–65. | |
O’Connell J, Lynch L, Cawood TJ, et al. The relationship of omental and subcutaneous adipocyte size to metabolic disease in severe obesity. PLoS One. 2011;5(4):e9997. | |
Bik W, Ostrowski J, Baranowska-Bik A, et al. Adipokines and genetic factors in overweight or obese but metabolically healthy Polish women. Neuro Endocrinol Lett. 2010;31(4):497–506. | |
Perreault M, Zulyniak MA, Badoud F, et al. A distinct fatty acid profile underlies the reduced inflammatory state of metabolically healthy obese individuals. PLoS One. 2014;9(2):e88539. | |
Lago F, Gómez R, Gómez-Reino JJ, Dieguez C, Gualillo O. Adipokines as novel modulators of lipid metabolism. Trends Biochem Sci. 2009;34(10):500–510. | |
Bray GA, Clearfield MB, Fintel DJ, Nelinson DS. Overweight and obesity: the pathogenesis of cardiometabolic risk. Clin Cornerstone. 2009;9(4):30–40; discussion 41–42. | |
Yamauchi T, Kadowaki T. Physiological and pathophysiological roles of adiponectin and adiponectin receptors in the integrated regulation of metabolic and cardiovascular diseases. Int J Obes (Lond). 2008;32 Suppl 7:S13–S18. | |
Yamauchi T, Kadowaki T. Adiponectin receptor as a key player in healthy longevity and obesity-related diseases. Cell Metab. 2013;17(2):185–196. | |
Luo N, Chung BH, Wang X, et al. Enhanced adiponectin actions by overexpression of adiponectin receptor 1 in macrophages. Atherosclerosis. 2013;228(1):124–135. | |
Tian L, Luo N, Zhu X, Chung BH, Garvey WT, Fu Y. Adiponectin-AdipoR1/2-APPL1 signaling axis suppresses human foam cell formation: differential ability of AdipoR1 and AdipoR2 to regulate inflammatory cytokine responses. Atherosclerosis. 2012;221(1):66–75. | |
Jamaluddin MS, Weakley SM, Yao Q, Chen C. Resistin: functional roles and therapeutic considerations for cardiovascular disease. Br J Pharmacol. 2012;165(3):622–632. | |
Benomar Y, Gertler A, De Lacy P, et al. Central resistin overexposure induces insulin resistance through Toll-like receptor 4. Diabetes. 2013;62(1):102–114. | |
Onuma H, Tabara Y, Kawamura R, et al. Plasma resistin is associated with single nucleotide polymorphisms of a possible resistin receptor, the decorin gene, in the general Japanese population. Diabetes. 2013;62(2):649–652. | |
Singh AK, Tiwari S, Gupta A, Natu SM, Mittal B, Pant AB. Association of resistin with metabolic syndrome in Indian subjects. Metab Syndr Relat Disord. 2012;10(4):286–291. | |
Schwartz DR, Lazar MA. Human resistin: found in translation from mouse to man. Trends Endocrinol Metab. 2011;22(7):259–265. | |
Palanivel R, Fullerton MD, Galic S, et al. Reduced Socs3 expression in adipose tissue protects female mice against obesity-induced insulin resistance. Diabetologia. 2012;55(11):3083–3093. | |
Tsiotra PC, Boutati E, Dimitriadis G, Raptis SA. High insulin and leptin increase resistin and inflammatory cytokine production from human mononuclear cells. Biomed Res Int. 2013;2013:487081. | |
Cawthorn WP, Sethi JK. TNF-alpha and adipocyte biology. FEBS Lett. 2008;582(1):117–131. | |
Cartier A, Cote M, Bergeron J, et al. Plasma soluble tumour necrosis factor-alpha receptor 2 is elevated in obesity: specific contribution of visceral adiposity. Clin Endocrinol (Oxf). 2010;72(3):349–357. | |
Lecube A, Sampol G, Muñoz X, Ferrer R, Hernández C, Simó R. TNF-α system and lung function impairment in obesity. Cytokine. 2011;54(2):121–124. | |
Hotamisligil GS, Shargill NS, Spiegelman BM. Adipose expression of tumor necrosis factor-alpha: direct role in obesity-linked insulin resistance. Science. 1993;259(5091):87–91. | |
Zou C, Shao J. Role of adipocytokines in obesity-associated insulin resistance. J Nutr Biochem. 2008;19(5):277–286. | |
Maury E, Brichard SM. Adipokine dysregulation, adipose tissue inflammation and metabolic syndrome. Mol Cell Endocrinol. 2010;314(1):1–16. | |
Galic S, Oakhill JS, Steinberg GR. Adipose tissue as an endocrine organ. Mol Cell Endocrinol. 2010;316(2):129–139. | |
Poelkens F, Eijsvogels TM, Brussee P, Verheggen RJ, Tack CJ, Hopman MT. Physical fitness can partly explain the metabolically healthy obese phenotype in women. Exp Clin Endocrinol Diabetes. 2014;122(2):87–91. | |
Balistreri CR, Caruso C, Candore G. The role of adipose tissue and adipokines in obesity-related inflammatory diseases. Mediators Inflamm. 2010;2010:802078. | |
Guilherme A, Virbasius JV, Puri V, Czech MP. Adipocyte dysfunctions linking obesity to insulin resistance and type 2 diabetes. Nat Rev Mol Cell Biol. 2008;9(5):367–377. | |
Hermsdorff HH, Angeles Zulet M, Bressan J, Alfredo Martínez J. Effect of diet on the low-grade and chronic inflammation associated with obesity and metabolic syndrome. Endocrinol Nutr. 2008;55(9):409–419. | |
Jager J, Gremeaux T, Gonzalez T, et al. Tpl2 kinase is upregulated in adipose tissue in obesity and may mediate interleukin-1beta and tumor necrosis factor-{alpha} effects on extracellular signal-regulated kinase activation and lipolysis. Diabetes. 2010;59(1):61–70. | |
Hotamisligil GS. Inflammatory pathways and insulin action. Int J Obes Relat Metab Disord. 2003;27 Suppl 3:S53–S55. | |
Plomgaard P, Keller P, Keller C, Pedersen BK. TNF-alpha, but not IL-6, stimulates plasminogen activator inhibitor-1 expression in human subcutaneous adipose tissue. J Appl Physiol (1985). 2005;98(6):2019–2023. | |
Song DD, Chen Y, Li ZY, Guan YF, Zou DJ, Miao CY. Protein tyrosine phosphatase 1B inhibits adipocyte differentiation and mediates TNFα action in obesity. Biochim Biophys Acta. 2013;1831(8):1368–1376. | |
Lee J. Adipose tissue macrophages in the development of obesity-induced inflammation, insulin resistance and type 2 diabetes. Arch Pharm Res. 2013;36(2):208–222. | |
Franckhauser S, Elias I, Rotter Sopasakis V, et al. Overexpression of Il6 leads to hyperinsulinaemia, liver inflammation and reduced body weight in mice. Diabetologia. 2008;51(7):1306–1316. | |
Madeleine MM, Johnson LG, Malkki M, et al. Genetic variation in proinflammatory cytokines IL6, IL6R, TNF-region, and TNFRSF1A and risk of breast cancer. Breast Cancer Res Treat. 2011;129(3):887–899. | |
Mitrou P, Boutati E, Lambadiari V, et al. Insulin resistance in hyperthyroidism: the role of IL6 and TNF alpha. Eur J Endocrinol. 2010;162(1):121–126. | |
Chalaris A, Rabe B, Paliga K, et al. Apoptosis is a natural stimulus of IL6R shedding and contributes to the proinflammatory trans-signaling function of neutrophils. Blood. 2007;110(6):1748–1755. | |
Denis GV, Obin MS. ‘Metabolically healthy obesity’: origins and implications. Mol Aspects Med. 2013;34(1):59–70. | |
Brandt C, Pedersen BK. The role of exercise-induced myokines in muscle homeostasis and the defense against chronic diseases. J Biomed Biotechnol. 2010;2010:520258. | |
Hoene M, Weigert C. The role of interleukin-6 in insulin resistance, body fat distribution and energy balance. Obes Rev. 2008;9(1):20–29. | |
Rull A, Camps J, Alonso-Villaverde C, Joven J. Insulin resistance, inflammation, and obesity: role of monocyte chemoattractant protein-1 (or CCL2) in the regulation of metabolism. Mediators Inflamm. 2010;2010. | |
Kanda H, Tateya S, Tamori Y, et al. MCP-1 contributes to macrophage infiltration into adipose tissue, insulin resistance, and hepatic steatosis in obesity. J Clin Invest. 2006;116(6):1494–1505. | |
Shoelson SE, Herrero L, Naaz A. Obesity, inflammation, and insulin resistance. Gastroenterology. 2007;132(6):2169–2180. | |
Gutierrez DA, Kennedy A, Orr JS, et al. Aberrant accumulation of undifferentiated myeloid cells in the adipose tissue of CCR2-deficient mice delays improvements in insulin sensitivity. Diabetes. 2011;60(11):2820–2829. | |
Pandzic Jaksic V, Gizdic B, Miletic Z, Trutin-Ostovic K, Jaksic O. Association of monocyte CCR2 expression with obesity and insulin resistance in postmenopausal women. Clin Invest Med. 2013;36(1):E24–E31. | |
Kang YS, Lee MH, Song HK, et al. CCR2 antagonism improves insulin resistance, lipid metabolism, and diabetic nephropathy in type 2 diabetic mice. Kidney Int. 2010;78(9):883–894. | |
Tan CK, Chong HC, Tan EH, Tan NS. Getting ‘Smad’ about obesity and diabetes. Nutr Diabetes. 2012;2:e29. | |
Tamura Y, Sugimoto M, Murayama T, et al. Inhibition of CCR2 ameliorates insulin resistance and hepatic steatosis in db/db mice. Arterioscler Thromb Vasc Biol. 2008;28(12):2195–2201. | |
Bourlier V, Bouloumie A. Role of macrophage tissue infiltration in obesity and insulin resistance. Diabetes Metab. 2009;35(4):251–260. | |
Itoh M, Suganami T, Hachiya R, Ogawa Y. Adipose tissue remodeling as homeostatic inflammation. Int J Inflam. 2011;2011:720926. | |
Chang JS, Chang CC, Chien EY, et al. Association between interleukin 1β and interleukin 10 concentrations: a cross-sectional study in young adolescents in Taiwan. BMC Pediatr. 2013;13(1):123. | |
Koukouikila-Koussounda F, Ntoumi F, Ndounga M, Tong HV, Abena AA, Velavan TP. Genetic evidence of regulatory gene variants of the STAT6, IL10R and FOXP3 locus as a susceptibility factor in uncomplicated malaria and parasitaemia in Congolese children. Malar J. 2013;12:9. | |
Glocker EO, Kotlarz D, Boztug K, et al. Inflammatory bowel disease and mutations affecting the interleukin-10 receptor. N Engl J Med. 2009;361(21):2033–2045. | |
Hong EG, Ko HJ, Cho YR, et al. Interleukin-10 prevents diet-induced insulin resistance by attenuating macrophage and cytokine response in skeletal muscle. Diabetes. 2009;58(11):2525–2535. | |
Esposito K, Pontillo A, Giugliano F, et al. Association of low interleukin-10 levels with the metabolic syndrome in obese women. J Clin Endocrinol Metab. 2003;88(3):1055–1058. | |
Gotoh K, Inoue M, Masaki T, et al. A novel anti-inflammatory role for spleen-derived interleukin-10 in obesity-induced inflammation in white adipose tissue and liver. Diabetes. 2012;61(8):1994–2003. | |
Feuerer M, Herrero L, Cipolletta D, et al. Lean, but not obese, fat is enriched for a unique population of regulatory T cells that affect metabolic parameters. Nat Med. 2009;15(8):930–939. | |
Weiss A, Attisano L. The TGFbeta superfamily signaling pathway. Wiley Interdiscip Rev Dev Biol. 2013;2(1):47–63. | |
Chen W, Konkel JE. TGF-beta and ‘adaptive’ Foxp3(+) regulatory T cells. J Mol Cell Biol. 2010;2(1):30–36. | |
Chen X, Oppenheim JJ. Resolving the identity myth: key markers of functional CD4+FoxP3+ regulatory T cells. Int Immunopharmacol. 2011;11(10):1489–1496. | |
Yadav H, Quijano C, Kamaraju AK, et al. Protection from obesity and diabetes by blockade of TGF-beta/Smad3 signaling. Cell Metab. 2011;14(1):67–79. | |
Cristancho AG, Lazar MA. Forming functional fat: a growing understanding of adipocyte differentiation. Nat Rev Mol Cell Biol. 2011;12(11):722–734. | |
Perrier S, Darakhshan F, Hajduch E. IL-1 receptor antagonist in metabolic diseases: Dr Jekyll or Mr Hyde? FEBS Lett. 2006;580(27):6289–6294. | |
Kwon H, Pessin JE. Adipokines mediate inflammation and insulin resistance. Front Endocrinol (Lausanne). 2013;4:71. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.