Back to Journals » Hepatic Medicine: Evidence and Research » Volume 10
Acute pediatric hyperammonemia: current diagnosis and management strategies
Authors Savy N, Brossier D, Brunel-Guitton C ![]() , Ducharme-Crevier L, Du Pont-Thibodeau G, Jouvet P
, Ducharme-Crevier L, Du Pont-Thibodeau G, Jouvet P ![]()
Received 15 February 2018
Accepted for publication 7 May 2018
Published 12 September 2018 Volume 2018:10 Pages 105—115
DOI https://doi.org/10.2147/HMER.S140711
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Dr Gerry Lake-Bakaar
Nadia Savy,1 David Brossier,2 Catherine Brunel-Guitton,1 Laurence Ducharme-Crevier,1 Geneviève Du Pont-Thibodeau,1 Philippe Jouvet1
1Department of Pediatrics, CHU Sainte-Justine, University of Montreal, Montreal, QC, Canada; 2Department of Pediatrics, Pediatric Intensive Care Unit, CHU Caen, Caen, France
Abstract: Acute hyperammonemia may induce a neurologic impairment leading to an acute life-threatening condition. Coma duration, ammonia peak level, and hyperammonemia duration are the main risk factors of hyperammonemia-related neurologic deficits and death. In children, hyperammonemia is mainly caused by severe liver failure and inborn errors of metabolism. In an acute setting, obtaining reliable plasma ammonia levels can be challenging because of the preanalytical difficulties that need to be addressed carefully. The management of hyperammonemia includes 1) identification of precipitating factors and cerebral edema presence, 2) a decrease in ammonia production by reducing protein intake and reversing catabolism, and 3) ammonia removal with pharmacologic treatment and, in the most severe cases, with extracorporeal therapies. In case of severe coma, transcranial Doppler ultrasound could be the method of choice to noninvasively monitor cerebral blood flow and titrate therapies.
Keywords: pediatrics, critical care, hyperammonemia, sodium phenylacetate, sodium benzoate, hemodialysis
Introduction
Hyperammonemia is usually defined as a plasmatic level above 80 µmol/L in infants up to 1 month of age and above 55 µmol/L in older children.1 Hyperammonemia is an acute life-threatening condition that can lead to severe neurologic impairment and cerebral edema. The most common causes of hyperammonemia in children are severe liver failure and inherited metabolic diseases. This article summarizes the pathophysiology, and the diagnostic and therapeutic management of hyperammonia in children, including medications, dialysis, and treatment novelties.
Pathophysiology
Ammonia metabolism
Ammonia, NH3, is a major source of nitrogen. Ammonia arises from the breakdown of proteins, from the amino acid metabolism and is produced by the gut bacteria. Then, ammonia is transported through the portal circulation to periportal hepatocytes where 90% of ammonia enters the urea cycle and is converted to urea. As ammonia is not excreted (not water soluble), the remaining 10% is carried on to perivenous hepatocytes where ammonia is condensed with glutamate to glutamine through the glutamine synthetase (GS), a lower capacity system. The glutamine synthase is also present in the astrocytes in the brain for the removal of ammonia, as well as in the kidney and skeletal muscle.2 Glutamine is either eliminated in urine or used for energy production by the gut.3 When there is reduced elimination, or production is increased, ammonia accumulates and is shunted to the systemic circulation resulting in brain accumulation and subsequent dysfunction.4
Different hypotheses to explain neurologic impairment (Figure 1)
 | Figure 1 Putative mechanisms underlying hyperammonemia encephalopathy and brain edema. |
At physiologic pH levels, ammonia predominantly exists as the ionic form (NH4+). NH4+ barely permeates cell membrane. Blood–brain barrier (BBB) is mostly impermeable to the ionic form but about 5 times more permeable to the gaseous form, NH3. Therefore, alkalosis leads to an increased accumulation of ammonia in the central nervous system. In the astrocyte, ammonia is transformed into glutamine through the GS. Under physiologic conditions, GS functions near the maximal capacity and, therefore, can easily be saturated. Glutamine is then released into the extracellular space through the sodium-coupled neutral amino acid transporter (SNAT5), and is captured by neurons, where the enzyme glutaminase converts glutamine to glutamate. Glutamate is released into the synaptic cleft stimulating N-methyl-d-aspartate (NMDA) receptors. Activation of NMDA receptors leads to calcium release, increased nitric oxide production, and excitotoxicity.5 Glutamine is an osmotic agent and, therefore, its accumulation will induce cellular swelling and cerebral edema. Decreased SNAT5 expression may also play a key role and lead to the glutamine trapping in astrocytes. Furthermore, the neurotransmission decrease conducts to an excess of neuroinhibition, which contributes to encephalopathy. Glutamine in the mitochondria is transformed into ammonia through the phosphate-activated glutaminase. Ammonia is known to alter mitochondrial permeability, leading to mitochondrial swelling and dysfunction.6 In addition, hyperactivation of NMDA receptors activates neurotoxic pathways conducting to axonal degeneration and cellular death.5 Another hypothesis of brain impairment is the activation of the phosphatase calcineurin, which increases the ATP consumption by 80% through the Na/K ATPase dephosphorylation. An oxidative stress with free radicals is then induced, thereby increasing cellular death.5 Finally, a high level of ammonia in the brain may affect the autoregulation and cause hyperemia.6
Untreated, hyperammonemia can induce irreversible damage to a developing brain leading to uncontrolled seizures, motor and cognitive impairment, cerebral palsy, and/or death.
Diagnosis and etiologies of hyperammonemia
Diagnosis of hyperammonemia
Acute hyperammonemia is usually triggered by protein catabolism caused by prolonged fasting, fever, infections, gastrointestinal bleeding, dehydration, high protein intake, anesthesia, and surgery. Clinical symptoms are numerous including digestive symptoms (nausea, vomiting, anorexia, abdominal pain, and failure to thrive) and neuropsychiatric symptoms (headaches, ataxia, dysarthria, behavioral modifications, neurodevelopmental delay, and hypotonia) and, in severe cases, seizures, alteration of consciousness, and central hyperventilation. Sometimes clinical signs of liver failure (jaundice and bleeding) and/or multiorgan failure (hypotension and oliguria) are present. In neonates, the clinical signs are nonspecific, sepsis-like.
In any undiagnosed neurologic signs in children, a plasma ammonia level is needed. In practice, having a reliable plasma ammonia level remains a challenge. Majority of laboratories use a method based on the amino reduction of α-ketoglutarate, which requires a reduced nicotinamide adenine dinucleotide phosphate (NADPH). This reduction is proportionally correlated to the plasmatic level of ammonia, and NADPH variations of concentration can be measured by spectrometry. The disadvantage of this method is that ammonia blood samples cannot be collected using a tourniquet and must be put on ice immediately and rapidly routed to the laboratory, ammonia being stable for <15 min at 4°C. Smaller laboratories use another method faster and cheaper but less accurate. Ammonemia is measured by reflectance meters on a dry slide chemistry strip soaked with whole blood. In this method, ammonia reacts with bromocresol green, and the intensity of color is measured accurately. The main disadvantage is the cutoff range of 286 µmol/L.7
False positives are commonly due to preanalytical conditions because of hemolysis and delay in transport to laboratory. According to a consensus statement from the Urea Cycle Disorders Conference Group, if the assay cannot be run immediately, plasma should be stored at −70°C and capillary sample should not be used.8 Because of these technical difficulties, it is recommended to repeat the blood sample in case of hyperammonemia (positive predictive value about 60%).7,9
Neuroimaging techniques may help in the diagnostic workup of hyperammonemic encephalopathy. Cerebral computed tomography can eliminate another life-threatening diagnosis such as an intracranial bleeding, or an expanding process. Magnetic resonance imaging (MRI) typically shows either a diffuse pattern involving cerebral cortex, basal ganglia, thalami, and brain stem or a central pattern involving basal ganglia, perirolandic region, and internal capsule.10 According to Gunz et al,10 more extensive cerebral involvement on MRI was associated with severe developmental outcomes in 11 neonates with urea cycle defects (UCDs). However, diffuse patterns were also seen in patients with mild and moderate outcomes. Central pattern on MRI did not correlate with outcome. Diffusion tensor imaging may be particularly helpful as this sequence highlights cytotoxic edema on apparent diffusion coefficient maps.11
Magnetic resonance spectroscopy (MRS) permits to noninvasively measure cerebral concentrations of specific biochemical compounds, especially glutamine. MRS can also be used successfully to assess the response to treatment by following the evolution of glutamine concentration.11,12
Transcranial Doppler ultrasound (TCD) is a novel tool of high value in hyperammonemic encephalopathy. TCD can assess dynamic cerebral blood flow, can point toward cerebral edema, and allows the evaluation of cerebral. Of note, cerebral blood flow could increase before symptomatic intracranial hypertension.13 Careful TCD monitoring could, therefore, permit an early intracranial hypertension diagnosis. From our center’s experience, TCD is the method of choice to routinely and noninvasively monitor cerebral blood flow and guide medical therapy.
Electroencephalography has shown similar characteristics in 3 newborns with hyperammonemia: multifocal spikes or repetitive paroxysmal activity during crisis13 but is not specific. Its interest lay mostly in detecting seizures in a comatose or sedated patient.
Investigations of the causes of hyperammonemia (Figure 2)
 | Figure 2 Diagnostic algorithm facing hyperammonemia Note: Adapted from Haberle J, Boddaert N, Burlina A, et al. Suggested guidelines for the diagnosis and management of urea cycle disorders. Orphanet J Rare Dis. 2012;7:32.27 Abbreviations: AR1D, arginase 1 deficiency; ASLD, arginosuccinate lyase deficiency; ASSD, arginosuccinate synthetase deficiency; CPS1-D, carbamoylphosphate synthetase 1 deficiency; NAGS-D, N-acetylglutamate synthetase deficiency; OTCD, ornithine transcarbamylase deficiency. |
Multiples diseases can lead to acute hyperammonemia in children admitted in pediatric intensive care: liver failure (64% of cases), UCDs (23%), and others (13%) such as exposure to toxics and medications.1 The initial workup of hyperammonemia is guided by a clinical examination and can include the following laboratory investigations: blood gas, electrolytes and anion gap measurement, ketonuria, glycemia, lactic acid, liver enzymes and function test (aspartate transaminase , alanine transaminase , bilirubin, gamma-glutamyl transferase, factor V, prothrombin ratio, international normalized ratio), renal function tests (blood urea nitrogen and creatinine), plasma and urine amino acids and urine organic acids chromatographies, plasma acylcarnitine profile on dried blood, dosage of plasma total, esterified and free carnitine, and urinary orotic acid.
Acute liver failure (ALF)
ALF can lead to hyperammonemic hepatic encephalopathy (HE)14 (Table 1). Primary causes of liver failure are numerous and can all lead to hyperammonemia if severe. Drug-induced liver injury (e.g., acetaminophen) accounts for 50% of ALF cases in the USA. Eighteen percent to 47% remain of unknown etiology.15 Inborn errors of metabolism are reported to be the cause of ALF in 10%–34% of cases.15 Galactosemia most often manifests itself during the neonatal period after milk introduction and usually presents with Gram-negative septicemia. Diagnosis is confirmed by the dosage of a metabolite, galactose-1-phosphate, or the enzyme, galactose-1-phosphate uridyl transferase, in erythrocytes or by molecular genetic testing. Tyrosinemia usually presents with ALF during the second month of life with a typical nodular liver on the ultrasound. Hereditary fructose intolerance presents after the fructose-containing food introduction, with liver and digestive symptoms and hypoglycemia. Other inborn errors of metabolism causing liver failure include mitochondrial hepatopathies, fatty acid oxidation disorders, congenital disorders of glycosylation, among others. Viral infections notably due to enteroviruses and herpes simplex virus account for 6%–22% of cases of ALF. Other etiologies include hemophagocytic lymphohistiocytosis, fetal alloimmune hepatitis (previously called neonatal hemochromatosis), autoimmune (giant cells hepatitis with autoimmune haemolytic anemia), or vascular disease (veno-occlusive disease, Budd–Chiari syndrome, and so on). In pediatric intensive care unit (ICU), liver failure can also be secondary to a multiorgan failure syndrome.
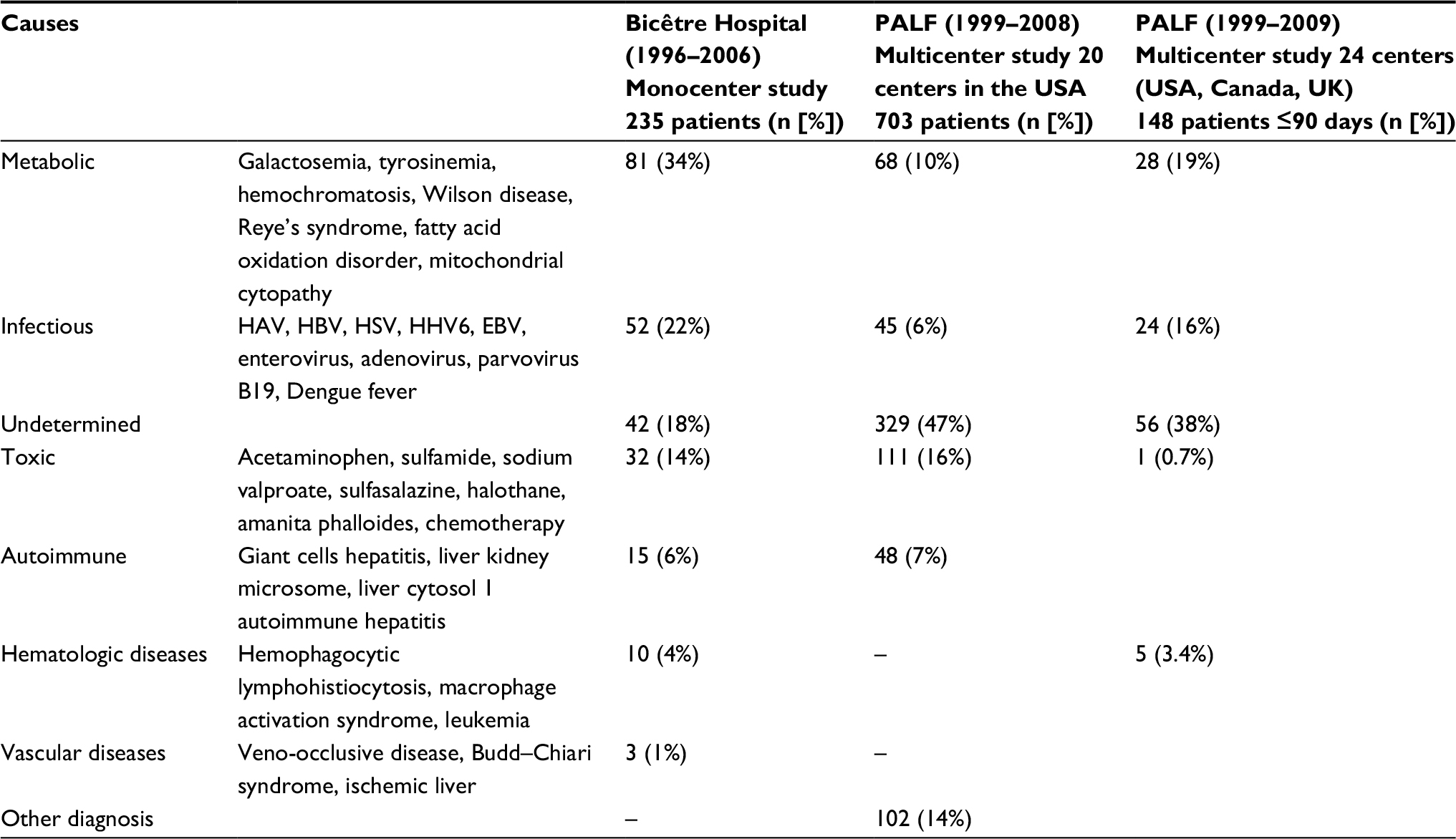 | Table 1 Comparison of 3 large studies on etiologies of acute liver failure in children Note: Reproduced from Devictor D, Tissieres P, Afanetti M, Debray D. Acute liver failure in children. Clin Res Hepatol Gastroenterol. 2011;35(6–7):430–437. Copyright ©2011, published by Elsevier Masson SAS. All rights reserved.15 Abbreviations: EBV, Epstein–Barr virus; HAV, hepatitis A virus; HBV, hepatitis B virus; HHV6, herpes virus-6; HSV, herpes simplex virus; PALF, Pediatric Acute Liver Failure Study Group. |
Urea cycle defects
UCDs are responsible for 23% of acute hyperammonemia in critically ill children.1 Prevalence and incidence of these diseases seem to differ between continents (Figure 3). All these diseases can result in acute and/or chronic hyperammonemia, according to the severity of enzyme deficiency. Most of the UCDs are mostly diagnosed on amino acid plasma chromatography and can be detected on newborn screening programs.
 | Figure 3 Urea cycle and therapies for hyperammonemia. Notes: Each number corresponds to a urea cycle deficiency disease: 1 = N-acetylglutamate synthase; 2 = carbamoylphosphate synthetase 1; 3 = ornithine transcarbamylase; 4 = arginosuccinate synthetase; 5 = arginosuccinate lyase; 6 = arginase deficiency. Abbreviation: CoA, coenzyme A. |
Ornithine transcarbamylase (OTC) deficiency
OTC deficiency is the most common inherited defect of ureagenesis (>50% of all UCDs), with a prevalence of 1 for 14,000 to 1 for 62,000–77,000 live births.16 OTC deficiency is an X-linked disorder. In males, OTC is usually lethal in the neonatal period, although milder variants are also described. In females, there is a wide variability of symptoms due to the X-inactivation pattern in females. Fifteen percent to 20% will present more severe symptoms. In OTC deficiency, citrulline is low or normal and urinary orotic acid is increased.3,16
N-Acetylglutamate synthetase (NAGS) deficiency
NAGS deficiency is an autosomal-recessive disorder. The activity of carbamoylphosphate synthetase 1 (CPS1) is decreased, thus leading to hyperammonemia.17,18 Citrulline is normal or decreased on plasma amino acid chromatography and urinary orotic acid is not increased. Carglumic acid is the analog of NAGS and the treatment of choice in this disease. In the context of acute hyperammonemia and given that carglumic acid is an oral drug, standard hyperammonemia management should be initiated in parallel (see below).
CPS1 deficiency
CPS1 deficiency is an autosomal-recessive disorder. The biochemical phenotype is indistinguishable from NAGS deficiency.19
Arginosuccinate synthetase deficiency
Arginosuccinate synthetase deficiency or citrullinemia type I is an autosomal-recessive disorder. Citrulline and urinary orotic acids are increased.
Arginosuccinate Lyase deficiency
Arginosuccinate lyase deficiency is an autosomal-recessive disorder. Citrulline and arginosuccinic acid are increased on plasma amino acid chromatography.20 This is the only UCD where there is a high rate of neurologic complications even without hyperammonemia and a progressive chronic liver disease and hepatic fibrosis21.
Arginase 1 deficiency
Arginase 1 deficiency is an autosomal-recessive disorder. Its deficiency leads to an accumulation of arginine on plasma amino acid chromatography and increases urinary orotic acid. Acute hyperammonemia is less frequent, and patients mostly present with progressive spasticity, intellectual disability, and sometimes seizures after the second year of age.3
Organic acidurias (OA) and hyperammonemia
OA are all autosomal-recessive inherited disorders mainly affecting branched-chain amino acid degradation. Hyperammonemia is due to acetyl coenzyme A (CoA) decrease and inhibition of NAGS and CPS1 activities22 by toxic metabolites. In these diseases, hyperammonemia is accompanied by a severe metabolic acidosis with high anion gap and ketonuria. The diagnosis is made by urinary organic acid chromatography and plasma acylcarnitine profile.
Others causes of hyperammonemia
Other inherited metabolic diseases such as fatty acid oxidation defects lead to hyperammonemia probably because of a generalized mitochondrial dysfunction7 and the lack of acetyl-CoA resulting from acylcarnitine degradation blockade.22 These diseases will manifest with acute liver disease, hypoglycemia, high creatine kinase, and cardiomyopathy, associated to hyperammonemia. HMG-CoA lyase deficiency associates the feature of organic acidemia and fatty acid oxidation defects. Pyruvate carboxylase deficiency and other mitochondrial defects might associate liver failure with hyperammonemia. Presence of lactic acidosis should prompt careful use of intravenous (IV) glucose and insulin. Hyperammonemia is also observed in lysinuric protein intolerance. Citrin deficiency is a specific inherited metabolic disease where high-carbohydrate diet can result in hyperammonemia and require a specific management different from the standard approach described below.23
Khalessi et al report perinatal asphyxia as a cause of hyperammonemia, because of the hypoxic stress, which can induce an increasing catabolism with a decrease in hepatic urea synthesis, leading to mild hyperammonemia. In their results among 100 patients with perinatal asphyxia, 20% patients had hyperammonemia above 90 µmol/L, with a mean plasma level of 117±41 µmol/L in asphyxia stages 2 and 3.24
Transient hyperammonemia of the newborn is a condition of potentially severe hyperammonemia due to transient platelet activation and portosystemic shunting through a large ductus venosus. The neurologic outcomes of these patients can range from normal development to profound mental retardation and seizures.3,24 Survival with favorable neurologic outcome can be achieved with suitable treatment.7
Drug-related causes of hyperammonemia are as follows: valproic acid that interferes with glutamine synthesis, carbamazepine, salicylates (Reye’s syndrome25), topiramate, tranexamic acid, and chemotherapy.
Acute management
The therapeutic measures to treat hyperammonemia are specifically targeted to decrease ammonia plasma level and to protect the brain from hyperammonemia toxicity (Figure 4). Rapid diagnosis and management are primordial to reduce the risk of irreversible brain damage. Suspected patients should rapidly be transferred to centers with a pediatric ICU, where first-line medications and consensus-based written protocols should be 24 h/7d easily accessible. Indeed coma duration and level of ammonia blood concentration are the main factors for determining neurologic outcome.22 Concentration of plasmatic ammonia above 300 µmol/L is usually accepted to be a sign of severity, but a threshold at 200 µmol/L within 48 h after admission in pediatric intensive care has been demonstrated to be of better prediction of mortality at 28 days (odds ratio 5.07; 95% CI [1.22–21.02]).4 The hyperammonemia duration of >3 days is the major criterion of brain edema according to Kumar et al.26 According to these data, there is no defined threshold for prognosis, but aggressive correction and control of persistently high NH3 levels should be the aim of every management protocol.
 | Figure 4 Suggested algorithm for management of hyperammonemia symptomatic patients according to Guideline Development Group – Grade of recommendation C–D. Source: Adapted from Haberle J, Boddaert N, Burlina A, et al. Suggested guidelines for the diagnosis and management of urea cycle disorders. Orphanet J Rare Dis. 2012;7:32.27 Abbreviations: CPS1D, carbamoylphosphate synthetase 1 deficiency; IV, intravenous; NAGSD, N-acetylglutamate synthetase deficiency; OTCD, ornithine transcarbamylase deficiency; TCD, transcranial Doppler; UCD, urea cycle defect. |
Initial management of hyperammonemia
The first-line treatment for hyperammonemia consists of stopping all protein intake for 24–48 h, while ensuring adequate hydration with high glucose content (dextrose 10%) at 1–1.5 times maintenance to provide sufficient calories to promote anabolism and reverse catabolism. The continuous glucose infusion could be 8–10 mg/kg/min in neonates. Insulin, which has anabolic properties, can be added if needed to maintain glycemia levels within the normal range. However, presence of lactic acidosis should prompt careful use of IV glucose and insulin. IV lipids, at 1–2 g/kg/day, can also be introduced to reach 130–150 kcal/kg/day in neonates or 1,500 kcal/m2/day caloric intake. Lipids can be given, once a fatty acid oxidation defect has been ruled out. As soon as possible, enteral feeding should be started to maximize caloric intake with a formula containing only carbohydrates and lipids. When blood ammonia drops below 100 µmol/L, proteins can be reintroduced slowly. According to age, it can be reintroduced initially at 0.4–0.5 g/kg/day and slowly increase with clinical and ammonia monitoring, under the genetic team and nutritionist supervision.
Nitrogen scavengers’ drugs are key drugs in hyperammonemia caused by inborn errors of metabolism and can help to reduce the ammonia plasma level in ALF (Figure 3). Sodium benzoate binds with glycine to form hippurate that is excreted in urine. After a bolus dose of 250 mg/kg in 90–120 min, the maintenance dose is 250–500 mg/kg/day given in continuous infusion.27 Sodium phenylacetate or sodium phenylbutyrate binds with glutamine to produce phenylacetylglutamine which is also eliminated in urine. The dosage is the same as sodium benzoate.27 Ammonul® (Ucyclyd Pharma Inc, Scottsdale, AZ, USA) is an IV drug that combines sodium benzoate and sodium phenylacetate and, as for all NH3 IV scavengers, contains a large amount of sodium.28
Other treatments
Lactulose and different antibiotics are used in HE to decrease ammonia level. Lactulose promotes NH4+ form to decrease its passage through BBB and acidifies the colonic environment, rendering it more hostile to urease-producing bacteria. However, its efficacy is still not clearly documented.29 Lactulose is not used in inherited metabolic diseases. Rifaximin is an oral antibiotic with a broad-spectrum activity against aerobic and anaerobic Gram-positive and Gram-negative bacteria. It may inhibit ammonia-producing enteral bacteria.30 Neomycin inhibits the activity of glutaminase in intestinal mucosa, decreasing ammonia production. Their side effects include ototoxicity, nephrotoxicity, and enterocolitis.31 Oral metronidazole and oral vancomycin have also an activity against anaerobic gut flora. Reducing the amount of these bacteria decreases ammonia production. Out of rifaximin, the other antibiotics should be combined to lactulose.31 Probiotics are used to enhance the growth of non-urease-producing bacteria in cirrhotic patients and seem to decrease the ammonia blood level in long-term therapy in these patients. However, probiotics effectiveness on clinical outcomes is not proven.29
IV L-arginine and IV L-citrulline will help to promote the nitrogen excretion through the urea cycle as arginine becomes an essential amino acid in UCD. Arginine should not be given in arginase deficiency. The recommended dosage of L-arginine is a bolus of 250–500 mg/kg in 90–120 min followed by a maintenance dosage of 250 mg/kg/day.27
L-Carnitine is an essential element in fatty acid beta-oxidation by transferring long-chain fatty acids from the cytoplasm to the inner mitochondrial membrane, which facilitates mitochondrial energy metabolism, and consequently acetyl-CoA production. L-Carnitine is necessary in the treatment of OA and could be given in undiagnosed hyperammonemia IV at 200 mg/kg/day. In rats, 1H- and 13C-MRS have shown to improve the mitochondrial energy metabolism in the brain and in muscles with L-Carnitine.32
Neuroprotection measures
Comatous children must be considered as having cerebral edema and treated accordingly: head raised at 30°, neck in the neutral position, sedation and analgesia to avoid agitation, and mechanical ventilation according to coma severity. Sedation agents need to be adjusted because of the risk of accumulation in case of altered liver function and/or renal function. Short-acting anesthetic agents should be preferred such as propofol or fentanyl. Alpha-2-agonist agents (clonidine, for example) have been studied for their suggested ability to reduce the release of glutamate, which would decrease NMDA receptor activation and thus hyperexcitability. Calligaris et al have demonstrated IV clonidine efficacy in a 7-month-old infant with UCDs.33
Cerebral perfusion monitoring with TCD is helpful and can detect a decrease in cerebral blood flow because of intracranial hypertension. The use of intracranial pressure (ICP) monitoring is still debated in children despite its interest to monitor cerebral edema given the hemorrhagic risks especially in children with ALF.
A careful monitoring of the osmolarity is also required and osmotic agents should be considered to decrease cerebral edema.
Dialysis if ammonia continues to rise after 4 h
Dialysis is considered if ammonia blood level is >500 µmol/L or if it does not decrease despite a 4-h well-conducted medical management.27,34,35 Patients treated with conventional hemodialysis (HD) had a significantly faster ammonium reduction rate at 4 h (p<0.05) when compared to continuous venovenous hemodiafiltration (CVVHD).34 Thus, HD is the more efficient dialysis method to rapidly decrease the ammonia level and should be the method of choice in case of threatening high level of ammonemia. An HD session is then followed by CVVHD to control ammonemia the following hours.27,35 There is a risk of osmolarity shift with HD, especially in infants (personal experience). To avoid the risk of cerebral herniation due to osmolarity shift during HD, osmolarity should be monitored carefully (hourly) during HD session, and dialysate formula adapted to avoid any rapid decrease in osmolarity.36 Peritoneal dialysis is of limited efficacy to remove NH3 in children.35
Novel therapies
Hypothermia to control ICP in ALF with hyperammonemia has been demonstrated to be beneficial in rats with extension of the survival time, preventing brain edema and decreasing cerebral spinal fluid ammonia. Mild hypothermia may be to limit the transfer of ammonia through BBB. Jalan et al have studied mild hypothermia (32°C–35°C) in adult populations of patients with ALF. Mean ICP was at 36.5±2.7 mmHg before cooling and decreased to 17±0.9 mmHg at 4 h and remained <20 mmHg at 24 h (p<0.001). Arterial ammonium concentration was reduced by 30% and by 66% in the brain.37,38 In a small cohort of 14 children with UCDs, Lichter-Konecki et al reported a faster reduction of ammonia blood level in patients treated with CVVHD and maintained hypothermic when compared to patients treated with CVVHD and maintained normothermic. No unanticipated adverse event was observed.39
Liver transplantation stays one of the options in ALF with threatening hyperammonemia when detoxification measures are insufficient or can be elective in some inborn errors of metabolism.
Carbamylglutamate is a costly synthetic structural analog of N-acetylglutamate (NCG). In case of its deficiency, carbamylglutamate may reduce ammonemia by substituting this essential cofactor of CPS1 to produce carbamylphosphate, the first by-product of ammonia in urea cycle. NAGS deficiency can be due to a primary genetic defect as described previously or can be secondary in OA. If CPS1 is partially deficient, NCG may stimulate the remaining CPS1.40,41 Initially advocated to NAGS deficiency, its safety, its quick action, and its oral administration incite some authors to propose its use in neonates as soon as they are diagnosed with severe hyperammonemia even before diagnosis confirmation.27,28,40,42,43 The initial dosage is 200 mg/kg (100–300 mg/kg) in bolus per nasogastric tube and then 200 mg/kg divided q3h to 6 h.
L-Ornithine-L-aspartate enhances the urea cycle in residual hepatocytes resulting in ammonia detoxification. Moreover, L-Ornithine can be transformed into glutamate semialdehyde by ornithine aminotransferase, and then to glutamate, which removes 1 ammonia molecule. Its effect on clinical outcome is still debated, and despite encouraging findings, this treatment is not currently considered part of the usual care of these patients.29 L-Ornithine phenylacetate activates the urea cycle and glutamine synthase, thanks to L-Ornithine, and conjugates glutamine and glycine to eliminate them, thanks to phenylacetate. In animal studies, this medication decreased ammonia blood level, extracellular cerebral ammonia, and ICP. In a phase I study in cirrhotic patients, the adverse effects were mild and blood ammonia concentration decreased by 50% in 36 h under continuous infusion.29
Flumazenil has been posed as an alternative in HE because of its anti-GABAergic activity, but a meta-analysis recently published does conclude neither in mortality decrease nor in clinical improvement in patients with HE.44 Flumazenil may be considered for selected patients with HE and chronic liver disease.31
Activated carbon microparticles have been shown to decrease intestinal ammonia absorption and thus lower ammonia blood concentration in animals and humans.29
Gene therapy is the future of genetic disease therapy. Various studies on animal models are currently ongoing. Cunningham et al have worked on the delivery of OTC through an adeno-associated virus 2/8 vector in OCTD mouse model (spfash mouse models). This model restores the enzymatic activity and controls the ammonia blood level. Hepatocyte proliferation, few weeks after the neonatal virus administration, decreases the viral copy number and thus the efficacy of the model, which, therefore, keeps a sufficient minimal activity to achieve a therapeutic effect.45–47 There is an ongoing open-label clinical trial in adults with late-onset OTCD (ClinicalTrials.gov Identifier: NCT02991144).
Currently, studies on cell therapy are conducted with the aim to populate the liver of UCD patients with functional hepatocytes.29
Finally, bioartificial livers could be the next generation of acute hyperammonemia treatment.48
Conclusion
Hyperammonemia must be rapidly suspected in case of neurologic symptoms in children, and an adequate management should start rapidly as coma duration and ammonia peak level are both the main risk factors of death. Children with hyperammonemia require a multidisciplinary management with geneticists/metabolicians, nephrologists, intensivists, neurologists, pharmacists, and nutritionists. Even in case of ALF, nitrogen scavengers can be helpful to decrease ammonia blood levels combined or not to dialysis according to ammonia levels’ evolution with time.
Acknowledgments
Dr. Jouvet has presented at conferences supported by Medunik Inc and Air Liquide Santé. Philips Medical lent medical devices to Dr. Jouvet.
Disclosure
P. Jouvet has received financial support from the Fonds de Recherche Santé Québec and from the Quebec Ministry of Health as well as from Sainte-Justine University Hospital. The authors report no other conflicts of interest in this work.
References
Ozanne B, Nelson J, Cousineau J, et al. Threshold for toxicity from hyperammonemia in critically ill children. J Hepatol. 2012;56(1):123–128. | ||
Hakvoort TB, He Y, Kulik W, et al. Pivotal role of glutamine synthetase in ammonia detoxification. Hepatology. 2017;65(1):281–293. | ||
Auron A, Brophy PD. Hyperammonemia in review: pathophysiology, diagnosis, and treatment. Pediatr Nephrol. 2012;27(2):207–222. | ||
Wijdicks EF. Hepatic encephalopathy. N Engl J Med. 2016;375(17):1660–1670. | ||
Paprocka J, Jamroz E. Hyperammonemia in children: on the crossroad of different disorders. Neurologist. 2012;18(5):261–265. | ||
Ducharme-Crevier L, Dupont-Thibodeau G, Lortie A, Maranda B, Tasker R, Jouvet P. Metabolic encephalopathies in children. In: Nichols D, ed. Rogers Textbook of Pediatric Intensive Care. Philadelphia, PA: Lippincott, Williams & Wilkins;2016:1053–1065. | ||
Broomfield A, Grunewald S. How to use serum ammonia. Arch Dis Child Educ Pract Ed. 2012;97(2):72–77. | ||
Urea Cycle Disorders Conference group. Consensus statement from a conference for the management of patients with urea cycle disorders. J Pediatr. 2001;138(1 Suppl):S1–S5. | ||
Maranda B, Cousineau J, Allard P, Lambert M. False positives in plasma ammonia measurement and their clinical impact in a pediatric population. Clin Biochem. 2007;40(8):531–535. | ||
Gunz AC, Choong K, Potter M, Miller E. Magnetic resonance imaging findings and neurodevelopmental outcomes in neonates with urea-cycle defects. Int Med Case Rep. J. 2013;6:41–48. | ||
Zhang XD, Zhang LJ, Wu SY, Lu GM. Multimodality magnetic resonance imaging in hepatic encephalopathy: an update. World J Gastroenterol. 2014;20(32):11262–11272. | ||
O’Donnell-Luria AH, Lin AP, Merugumala SK, et al. Brain MRS glutamine as a biomarker to guide therapy of hyperammonemic coma. Mol Genet Metab. 2017;121(1):9–15. | ||
Su PH, Chen JY, Chen YJ, Niu DM, Hsu JH, Lee IC. Electroencephalography and transcranial Doppler ultrasonography in neonatal citrullinemia. J Formos Med Assoc. 2014;113(11):857–861. | ||
Bernal W, Wendon J. Acute liver failure. N Engl J Med. 2013;369(26):2525–2534. | ||
Sundaram SS, Alonso EM, Narkewicz MR, Zhang S, Squires RH; Pediatric Acute Liver Failure Study Group. Characterization and outcomes of young infants with acute liver failure. J Pediatr. 2011;159(5):813–818. e1. | ||
Caldovic L, Abdikarim I, Narain S, Tuchman M, Morizono H. Genotype-phenotype correlations in ornithine transcarbamylase deficiency: a mutation update. J Genet Genomics. 2015;42(5):181–194. | ||
Spodenkiewicz M, Diez-Fernandez C, Rufenacht V, Gemperle-Britschgi C, Haberle J. Minireview on glutamine synthetase deficiency, an ultra-rare inborn error of amino acid biosynthesis. Biology (Basel). 2016;5(4). | ||
Ah Mew N, Caldovic L. N-Acetylglutamate synthase deficiency: an insight into the genetics, epidemiology, pathophysiology, and treatment. Appl Clin Genet. 2011;4:127–35. | ||
Ali EZ, Khalid MK, Yunus ZM, et al. Carbamoylphosphate synthetase 1 (CPS1) deficiency: clinical, biochemical, and molecular characterization in Malaysian patients. Eur J Pediatr. 2016;175(3):339–346. | ||
Nagamani SC, Erez A, Lee B. Argininosuccinate lyase deficiency. Genet Med. 2012;14(5):501–507. | ||
Baruteau J, Jameson E, Morris AA, et al. Expanding the phenotype in argininosuccinic aciduria: need for new therapies. J Inherit Metab Dis. 2017;40(3):357–368. | ||
Haberle J. Clinical and biochemical aspects of primary and secondary hyperammonemic disorders. Arch Biochem Biophys. 2013;536(2):101–108. | ||
Imamura Y, Kobayashi K, Shibatou T, et al. Effectiveness of carbohydrate-restricted diet and arginine granules therapy for adult-onset type II citrullinemia: a case report of siblings showing homozygous SLC25A13 mutation with and without the disease. Hepatol Res. 2003;26(1):68–72. | ||
Khalessi N, Khosravi N, Mirjafari M, Afsharkhas L. Plasma ammonia levels in newborns with asphyxia. Iran J Child Neurol. 2016; 10(1):42–46. | ||
Glasgow JF, Middleton B. Reye syndrome – insights on causation and prognosis. Arch Dis Child. 2001;85(5):351–353. | ||
Kumar R, Shalimar, Sharma H, et al. Persistent hyperammonemia is associated with complications and poor outcomes in patients with acute liver failure. Clin Gastroenterol Hepatol. 2012;10(8):925–931. | ||
Haberle J, Boddaert N, Burlina A, et al. Suggested guidelines for the diagnosis and management of urea cycle disorders. Orphanet J Rare Dis. 2012;7:32. | ||
Haberle J. Clinical practice: the management of hyperammonemia. Eur J Pediatr. 2011;170(1):21–34. | ||
Matoori S, Leroux JC. Recent advances in the treatment of hyperammonemia. Adv Drug Deliv Rev. 2015;90:55–68. | ||
Kaji K, Takaya H, Saikawa S, et al. Rifaximin ameliorates hepatic encephalopathy and endotoxemia without affecting the gut microbiome diversity. World J Gastroenterol. 2017;23(47):8355–8366. | ||
Jawaro T, Yang A, Dixit D, Bridgeman MB. Management of hepatic encephalopathy: a primer. Ann Pharmacother. 2016;50(7):569–577. | ||
Missler J, Zwingmann C. L-Carnitine in hyperammonemia and hepatic encephalopathy. In: Tanasescu R, ed. Miscellanea on Encephalopathies – A Second Look. Shanghai: In Tech; 2012:365–390. | ||
Calligaris L, Vidoni A, Bruno I, Vidoni M, Barbi E. Efficacy of clonidine in hyperammonemia induced hyperexcitability syndrome. Paediatr Anaesth. 2013;23(2):202–204. | ||
Picca S, Dionisi-Vici C, Bartuli A, et al. Short-term survival of hyperammonemic neonates treated with dialysis. Pediatr Nephrol. 2015;30(5):839–847. | ||
Jouvet P, Schaefer F. Dialytic therapy of inborn errors of metabolism. In: Warady B, Schaefer F, Alexander S, eds. Pediatric Dialysis. 2nd ed. Boston: Springer Verlag; 2012:765–774. | ||
Liotta EM, Romanova AL, Lizza BD, et al. Osmotic shifts, cerebral edema, and neurologic deterioration in severe hepatic encephalopathy. Crit Care Med. 2018;46(2):280–289. | ||
Jalan R, Olde Damink SW, Deutz NE, Hayes PC, Lee A. Moderate hypothermia in patients with acute liver failure and uncontrolled intracranial hypertension. Gastroenterology. 2004;127(5):1338–1346. | ||
Jalan R, Rose C. Hypothermia in acute liver failure. Metab Brain Dis. 2004;19(3–4):215–221. | ||
Lichter-Konecki U, Nadkarni V, Moudgil A, et al. Feasibility of adjunct therapeutic hypothermia treatment for hyperammonemia and encephalopathy due to urea cycle disorders and organic acidemias. Mol Genet Metab. 2013;109(4):354–359. | ||
Daniotti M, la Marca G, Fiorini P, Filippi L. New developments in the treatment of hyperammonemia: emerging use of carglumic acid. Int J Gen Med. 2011;4:21–28. | ||
Kuchler G, Rabier D, Poggi-Travert F, et al. Therapeutic use of carbamylglutamate in the case of carbamoyl-phosphate synthetase deficiency. J Inherit Metab Dis. 1996;19(2):220–222. | ||
Valayannopoulos V, Baruteau J, Delgado MB, et al. Carglumic acid enhances rapid ammonia detoxification in classical organic acidurias with a favourable risk-benefit profile: a retrospective observational study. Orphanet J Rare Dis. 2016;11:32. | ||
Kasapkara CS, Ezgu FS, Okur I, Tumer L, Biberoglu G, Hasanoglu A. N-Carbamylglutamate treatment for acute neonatal hyperammonemia in isovaleric acidemia. Eur J Pediatr. 2011;170(6):799–801. | ||
Reyes D, Barrera F. Is flumazenil an alternative for the treatment of hepatic encephalopathy? Medwave. 2017;17(9):e7113. | ||
Cunningham SC, Kok CY, Spinoulas A, Carpenter KH, Alexander IE. AAV-encoded OTC activity persisting to adulthood following delivery to newborn spf(ash) mice is insufficient to prevent shRNA-induced hyperammonaemia. Gene Ther. 2013;20(12):1184–1187. | ||
Cunningham SC, Kok CY, Dane AP, et al. Induction and prevention of severe hyperammonemia in the spfash mouse model of ornithine transcarbamylase deficiency using shRNA and rAAV-mediated gene delivery. Mol Ther. 2011;19(5):854–859. | ||
Cunningham SC, Siew SM, Hallwirth CV, et al. Modeling correction of severe urea cycle defects in the growing murine liver using a hybrid recombinant adeno-associated virus/piggyBac transposase gene delivery system. Hepatology. 2015;62(2):417–428. | ||
van Wenum M, Adam AA, Hakvoort TB, et al. Selecting cells for bioartificial liver devices and the importance of a 3D culture environment: a functional comparison between the HepaRG and C3A cell lines. Int J Biol Sci. 2016;12(8):964–978. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.