Back to Journals » Therapeutics and Clinical Risk Management » Volume 14
Achalasia following reflux disease: coincidence, consequence, or accommodation? An experience-based literature review
Authors Vereczkei A ![]() , Bognár L
, Bognár L ![]() , Papp A, Horváth ÖP
, Papp A, Horváth ÖP ![]()
Received 24 September 2017
Accepted for publication 24 November 2017
Published 29 December 2017 Volume 2018:14 Pages 39—45
DOI https://doi.org/10.2147/TCRM.S152429
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Garry Walsh
András Vereczkei, Laura Bognár, András Papp, Örs Péter Horváth
Department of Surgery, University of Pécs, Pécs, Hungary
Abstract: Achalasia is a motility disorder of the esophagus characterized by the defective peristaltic activity of the esophageal body and impaired relaxation of the lower esophageal sphincter due to the degeneration of the inhibitory neurons in the myenteric plexus of the esophageal wall. The histopathological and pathophysiological changes in achalasia have been well described. However, the exact etiological factors leading to the disease still remain unclear. Currently, achalasia is believed to be a multifactorial disease, involving both extrinsic and intrinsic factors. Based on our experience and the review of literature, we believe that gastroesophageal reflux disease (GERD) might be one of the triggering factors leading to the development of achalasia. However, it is also stated that the two diseases can simultaneously appear independently from each other. Considering the large number and routine treatment of patients with GERD and achalasia, the rare combination of the two may even remain unnoticed; thus, the analysis of larger patient groups with this entity is not feasible. In this context, we report four cases where long-standing reflux symptoms preceded the development of achalasia. A literature review of the available data is also given. We hypothesize that achalasia following the chronic acid exposure of the esophagus is not accidental but either a consequence of a chronic inflammation or a protective reaction of the organism in order to prevent aspiration and lessen reflux-related symptoms. This hypothesis awaits further clinical confirmation.
Keywords: achalasia, gastroesophageal reflux disease, Barrett’s esophagus, Nissen fundoplication
Introduction
Heartburn, regurgitation, and nocturnal cough accompanying dysphagia can be misleading symptoms in the diagnosis of achalasia. These symptoms can sometimes be so dominant that the patient is falsely diagnosed and treated with gastroesophageal reflux disease (GERD) and even, by mistake, may undergo antireflux surgery. That is why the exclusion of achalasia is essential before the indication of surgical treatment of GERD. On the other hand, it can be presumed that, beyond similar clinical symptoms, there may be other relations between the two diseases. Achalasia is believed to be a multifactorial disease, but the exact factors leading to the degeneration of ganglion cells in the myenteric plexus of the esophagus have so far been poorly understood.1 Regarding the pathophysiology there can be either extrinsic or intrinsic causes responsible for the loss of the inhibitory innervation of the esophagus. Extrinsic causes may include central nervous system lesions involving the dorsal motor neuron or the vagal nerve fibers, although it is also possible that the vagal nerve degeneration and the loss of dorsal motor nucleus neurons are a secondary phenomenon caused by the loss of contact with the end organ, the myenteric plexus of the esophagus.2 Intrinsic causes may lead to the primary loss of the inhibitory ganglion cells in the myenteric plexus, which results in the imbalance between the excitatory and inhibitory neurons of the myenteric plexus. In humans, nitric oxide is the primary inhibitory neurotransmitter in the myenteric plexus. In achalasia patients, a significant decrease in nitric oxide neurons in the lower esophageal sphincter (LES) has been demonstrated compared to normal controls.3 In the background of the abovementioned pathophysiological changes, several diseases can be suspected. Infection with Trypanosoma cruzi can very closely mimic the picture of primary achalasia. Also, viral infections like measles and varicella zoster were suspected to be responsible for the neuronal changes, which has not been adequately confirmed.4 Achalasia patients were also found to have higher prevalence of circulating antimyenteric autoantibodies, which may support an autoimmune etiology.5 As a familial type of achalasia is also known, it was suggested that achalasia might be an inherited disease. Currently, it is thought that genetic predisposition in such individuals probably increases their susceptibility to achalasia after exposure to the same environmental factors that may play a role in the pathogenesis.6 Our hypothesis is that one possible initial insult that leads to the development of achalasia might be GERD. Severe esophagitis can damage the ganglion cells, and, later, an autoimmune reaction may develop, maintaining a chronic inflammation in the myenteric plexus of the esophagus. The theory that GERD could be an etiological factor in the development of achalasia was first proposed by Smart et al7 in 1986, who described five patients presenting with reflux and subsequently developing achalasia over the years.
Many authors deem the occurrence of gastroesophageal reflux and achalasia in the same patient as a coincidence, saying that reflux is a common disease that should, once in a while, randomly occur among achalasia patients as well. However, the fact that the standard chronological sequence of symptoms described by several authors in that long-lasting reflux is followed by dysphagia (and never vice versa) seems to suggest that achalasia might be a consequence of GERD. This theory is supported by Altorjay et al,8 who found enteric ganglionitis and smooth muscle hypertrophy in the muscle samples of the LES of reflux patients. These morphological changes, which are characteristic of achalasia, could be interpreted as consequences of long-term reflux episodes.8 At the same time, it is known that the human body has developed several aerodigestive reflexes to protect the airways from aspiration.9 Similarly, various esophageal pathologies have been presumed to be related to reflux such as hypertensive LES, hypertrophic upper esophageal sphincter (UES), Schatzki’s ring, and achalasia. All of these conditions, characterized by the narrowing of the esophageal diameter, could be regarded as defensive mechanisms or ways of accommodation to lessen acid regurgitation, aimed at relieving reflux-associated symptoms and avoiding aspiration. With our four reported cases in this paper, we would like to support a causal relation between GERD and achalasia.
Patients
Between 2000 and 2015, we operated on a total of 39 patients with achalasia, which is a rare disease in our region with more than 900,000 inhabitants, where our department is a center for esophageal surgery. After a retrospective analysis of patient data, in four patients (10%), the etiological role of reflux arose. All patients underwent endoscopy, stationary esophageal manometry, and barium swallow, with three of them also undergoing 24-hour esophageal pH monitoring.
Analysis of patient data and publication of the case details was approved by the Regional Ethics Committee of the Medical School University of Pécs. Upon signing the informed consent document, all patients approved that the data connected to their surgical care can be processed for scientific purposes. Exhaustive attempts have been made to contact the four patients or their next of kin for consent to publish the case details, but to no avail. The case details have been sufficiently anonymized not to cause harm to the patients or their families.
Case 1
A 44-year-old male had a 2-year history of the classic symptoms of GERD. These symptoms transformed into dysphagia within a year and progressed over a final 6-month period until the patient was only able to drink cold water. Over the last year, a 15 kg weight loss was observed. First, esophagogastroscopy performed at the beginning of the reflux symptoms only showed erosive esophagitis. Thus, the patient was put on acid-suppressive therapy, which resulted in the improvement of the symptoms. From that time on, dysphagia progressed rapidly, and a control endoscopy revealed a dilated esophageal body with intact mucosa. Cardia was closed, suspected of being fibrotic, but the endoscope passed through it. Barium swallow showed a dilated esophageal body with a short, smoothly tapered segment at the esophagogastric junction. Decreased peristalsis and retention of barium was thought to be consistent with achalasia. Esophageal manometry demonstrated an LES average resting pressure of 34.4 mmHg with an inappropriate LES (relaxation <90%). Twenty-four-hour pH-metry showed acid reflux with a total DeMeester score of 94.9. When (instead of the usual pH 4) pH 3 was used as a discriminant threshold for GERD, the reflux score was 62.3. The patient underwent surgery with a hypertrophic LES and a rigid cardia found during the operation. Cardiomyotomy and Nissen fundoplication were performed.
Case 2
A 65-year-old woman was admitted with severe dysphagia and regular vomiting after meals. In her history, she had 7 years of typical reflux symptoms (heartburn and regurgitation) with no dysphagia experienced during that time. Her first esophagogastroscopy was performed 2 years before her admission, which showed Savary–Miller stage II esophagitis and a sliding type hiatal hernia. The patient was put on proton pump inhibitors (PPIs). A control endoscopy 1 year later showed no signs of esophagitis with a normal cardia and stomach. During the year before her admittance, her reflux symptoms disappeared, but dysphagia developed. Endoscopy found esophageal dilatation with esophagitis and narrowed cardia, while biopsies from the distal esophagus showed chronic esophagitis and Barrett’s metaplasia. Barium swallow confirmed the endoscopic findings and showed esophageal dilatation and a sliding type hiatal hernia (Figure 1). Manometry showed an LES pressure of 34.3 mmHg with 11.5% relaxation. Twenty-four-hour pH-metry showed acid reflux with multiple sharp dips, which is characteristic of typical gastroesophageal reflux, and a total DeMeester score of 94.6. After pH 3 was used as a discriminatory threshold for GERD, the reflux score was 64.2. Achalasia and concomitant GERD were diagnosed, and the patient underwent laparoscopic surgery. Hiatal hernia reconstruction (Figure 2), Heller’s myotomy, and 360° Nissen fundoplication were performed.
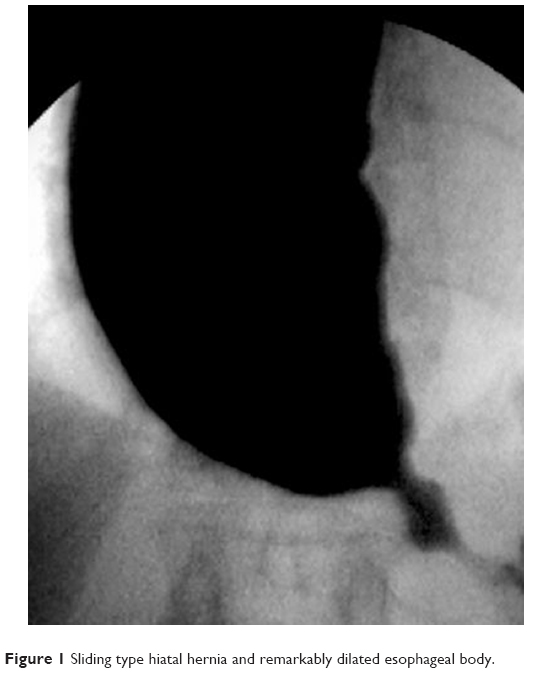 | Figure 1 Sliding type hiatal hernia and remarkably dilated esophageal body. |
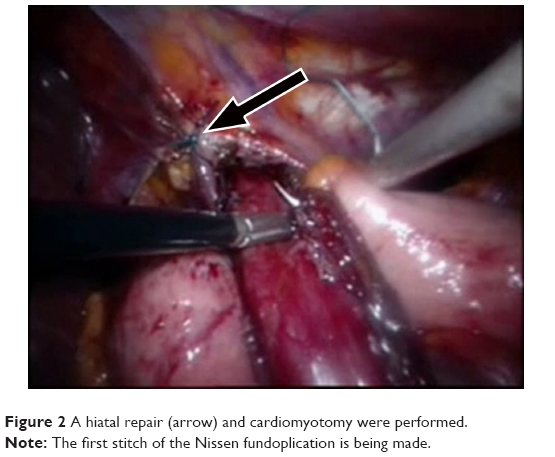 | Figure 2 A hiatal repair (arrow) and cardiomyotomy were performed. |
Case 3
A 42-year-old man came to our clinic with a leading complaint of dysphagia, which had been worsening over a 4-month period. He had typical reflux symptoms at the same time, which was then treated with PPIs. Endoscopy revealed dilatation and atony of the esophageal body plus a closed cardia through which the instrument could only be passed with some difficulty. A mild inflammation was found in the distal portion of the esophagus. Barium swallow showed a dilated esophageal body with nonpropulsive contractions and nonrelaxing sphincter, which allowed contrast medium to escape the esophagus in small quantities. The resting pressure of the LES was not increased (24.4 mmHg); however, an incomplete relaxation could be observed. A 24-hour pH-metry revealed nocturnal acid reflux. The patient underwent laparoscopic surgery, cardiomyotomy, and total fundoplication.
Case 4
Our fourth patient was a 39-year-old man who had a history of gastroesophageal reflux, starting 4 years before his admittance. Endoscopy performed at the time of the onset of his reflux symptoms revealed an open cardia and esophagitis. The patient was put on PPI therapy and, consequently, his reflux symptoms stopped. However, during the year before his admission, he developed a severe dysphagia and lost 30 kg. Right before the onset of his dysphagia, the patient had a control endoscopy that showed a normal esophageal body and cardia with signs of reflux esophagitis. A year later, the endoscopy showed a dilated esophageal body with closed, spastic cardia. Esophageal manometry showed an LES pressure of 31 mmHg with incomplete relaxation (46%) and dysmotility in the tubular esophagus with “mirror wave images,” which is characteristic of achalasia. The patient underwent laparoscopic surgery and cardiomyotomy with Nissen fundoplication.
Discussion
In the title of the paper, three possibilities were raised to characterize the relationship between GERD and achalasia. Is the occurrence of the two diseases in the same patient a mere coincidence? Could achalasia be a consequence of gastroesophageal reflux? Could a sort of an accommodation develop, through which the organism tries to protect itself from excessive acid exposure?
Coincidence
Many experienced authors describe the relationship between reflux and achalasia as a pure coincidence, suggesting that GERD is a frequent condition in the population that should proportionally occur among achalasia patients as well. Fisichella et al10 found complete LES relaxation in 13% of his 145 untreated achalasia patients, having found seven patients with abnormal DeMeester scores. However, he did not think there was a direct relationship between the two conditions.10 Spechler et al11 reported lower resting LES pressures in achalasia patients who had a history of heartburn than in those without heartburn, but the authors postulated that the two diseases were coincidental. Besides, a previous study showed that antineural antibodies of very similar properties were found in the serum of both achalasia and GERD patients. Therefore, one could assume that the two diseases may not appear independently of each other but may have common autoimmune mechanisms in their pathogenesis. However, the authors concluded that these circulating antineural antibodies were unlikely to have functional consequences and did not contribute to the disease process.12
Consequence
Data published in the literature on functional esophageal diseases support the idea that GERD itself can be a causative factor for achalasia.7 In previously reported cases, where the etiological role of reflux had arisen, the typical sequence of symptoms started with a long-standing reflux disease confirmed by functional examinations and endoscopy with reflux symptoms gradually improving and dysphagia developing. In a well-documented case, Robson et al13 spectacularly demonstrated the progression of GERD to diffuse esophageal spasm and then to achalasia. In his patient with typical reflux symptoms, manometry showed an LES pressure of 10 mmHg with an abnormal reflux time of 20% (pH<4). A year later, the patient’s symptoms started to change with complaints of regurgitation of solid food, and at the same time, his heartburn began improving. Manometry showed a diffuse esophageal spasm, and his LES pressure increased to 25 mmHg with vigorous esophageal contractions. Another year later, the patient had dysphagia to solids as well as liquids, and typical clinical symptoms of achalasia developed with an LES pressure of 50 mmHg.
The 24-hour pH-metry of two of our untreated achalasia patients showed long periods of time under pH 3 in the esophagus. This abnormal pH-metry appears to be an argument for the etiology of reflux, which had very likely preceded the onset of achalasia.
A convincing argument for the chronological order and, presumably, the causal relationship between reflux and achalasia is that Barrett’s esophagus was found in untreated achalasia patients. Guo et al14 reported nine patients like this, and we also found columnar metaplasia in one of our untreated achalasia patients. The development of Barrett’s esophagus or esophageal adenocarcinoma following cardiomyotomy or pneumatic dilatation can be accounted for a posttreatment complication. However, in untreated patients, the most plausible explanation is that they had had long-standing GERD before the development of achalasia. In another patient of ours, we found sliding hernia combined with achalasia. Hiatal hernia is extremely rare in achalasia patients and, with the corollary relationship between hiatal hernia and reflux suspected, the etiological role of reflux in the development of achalasia reasonably arises.
Altorjay et al8,15 reported very interesting observations by analyzing LES muscle specimens of reflux patients and comparing them to a control group. They found hypertrophy-like changes or (more specifically) a significant increase in the concentration of structural protein S-100 and myofibrillar protein troponin I in the LES muscle. Also, they revealed the inflammatory infiltration of the myenteric plexus and the ganglion – furthermore, detecting a significantly lower Schwann cell count in the LES muscle samples of reflux patients.8,15 These morphological changes can be interpreted as consequences of a reflux disease.
Accommodation
The anatomic contiguity between the gastroesophageal and pharyngolaryngeal pathways can predispose the airways to the risk of aspiration during the retrograde transit of gastric juices. To avoid this, the airways are protected by several aerodigestive reflexes such as the esophago-UES contractile reflex, the esophagoglottal closure reflex, and the reflexive pharyngeal swallow.9 These reflexes provide transient, momentary protection to prevent the potential spillage of refluxate into the airways. Beyond these particular, neuron-mediated quick responses, we suggest that chronic acid exposure may slowly lead to various structural and functional changes in the esophagus. All of these secondary pathologies could be considered to prevent the refluxate getting into the esophagus or beyond – to the oral cavity, larynx, or lungs – by narrowing the lumen of the esophagus. Frequent muscle contraction ending up in hypertrophy (achalasia) could be one of these protective reactions of the esophagus, aimed at avoiding uncomfortable reflux symptoms or even life-threatening aspiration. A few reports can be found in the literature where gastroesophageal reflux has been well documented to occur in patients prior to the development of achalasia.7,16 In our clinic (in the past 15 years), we have operated on 39 patients with achalasia, and only in 4 (10%) did the etiological role of reflux arise. This rate equals the frequency of reflux detected as a complication after Heller’s myotomy was completed with only a partial fundoplication.17 One can assume that patients who developed achalasia as a consequence of long-standing reflux disease will be more susceptible to postmyotomy or postdilatation reflux. The development of achalasia in patients with GERD can be considered as a protective reaction of the esophagus against reflux. Considering reflux as a potential etiological factor in the development of achalasia may have consequences to the therapy as well. If, based on the anamnesis and functional examinations, the etiological role of gastroesophageal reflux emerges, dilatation therapy is not recommended at all, and surgical intervention should always include an antireflux procedure – most preferably a 360° fundoplication – to minimize the risk of postoperative reflux.
A search through the literature resulted in finding further defensive mechanisms of the esophagus, which may all be triggered by reflux disease.
To date, probably the most proven protective mechanism of the esophagus against reflux and its complications is the development of a hypertensive LES. In these patients, LES pressure may increase threefold (as high as 45–60 mmHg). Interestingly, nearly half of the patients present with abnormal esophageal acid exposure on 24-hour pH monitoring. The main evidence for hypertensive LES being secondary to acid reflux is the fact that following a Nissen fundoplication sphincter pressure, and pH-metry, values get in the normal range again and reflux symptoms disappear.18,19 A further protective mechanism to reduce excessive acid exposure of the esophagus is the development of a lower esophageal or Schatzki’s ring. GERD has been reported to be present in 31%–60% of the cases, with hiatal hernia reported to be a cofinding in all patients with Schatzki’s ring. In a prospective study, Sgouros et al20 found that the high relapse rate after a successful esophageal dilatation due to the reformation of the ring could be prevented by a postdilatational, long-term, acid-suppressive therapy with PPIs. This finding indirectly supports the idea that acid reflux may play an etiological role in the development of the ring. The cause–effect relationship between GERD and hypertrophic UES is also presumed in the literature, supposing that acidic fluid regurgitating to the hypopharynx may result in the hypertrophy of the cricopharyngeal muscle and (what is more), as a consequence, these patients usually develop Zenker’s diverticulum too.21 A rare type of gastroesophageal reflux-triggered accommodation is the subglottic tracheal stenosis. The presence of pepsin, shown in the larynx of patients with subglottic tracheal stenosis, is indicative of excessive esophageal reflux, which, if chronic, results in fibrosis of the subglottic area.22 Also, there are reports that appear to suggest that there is an association between GERD and proximal esophageal stenosis.23
Besides these pathologies, Barrett’s esophagus could also be considered as a special form of accommodation of the esophagus against reflux. In typical cases, severe reflux symptoms lasting for decades gradually cease, which is not due to a successful treatment but to the transformation of esophageal mucosa to acid-resistant metaplastic epithelium.24
It is to be noted that the line between what we call consequence and accommodation cannot unambiguously be drawn.
However, all of the pathologies mentioned in this paper are originally meant to serve a good cause, that is, protection against aspiration. These functional and structural changes can, at the same time, lead to unwanted and harmful consequences as well. The narrowing of the esophagus at different levels may cause obstructive symptoms with all of its complications (prestenotic dilatation, formation of Zenker’s diverticulum, etc), while the transformation of the esophageal mucosa to acid-resistant metaplastic epithelium insensitive to reflux may result in a delay in the diagnosis in patients with Barrett’s esophagus.
The limitations of this case series review lie in its retrospective nature. Furthermore, the supposed connection between achalasia and GERD is a rare phenomenon, with achalasia having a low incidence itself. Thus, special attention during the preoperative assessment must be paid to identify these cases. This may have not happened in some cases, as patients with complete preoperative assessment are frequently referred to our tertiary care department from many hospitals.
Conclusion
In summary, the potential relationship between gastroesophageal reflux and achalasia is not clearly defined yet, but we have reason to believe that it can very well be. Based on a rational interpretation of our own findings and literature data (Table 1), it can be assumed that chronic acid exposure may trigger diverse structural and functional changes in the esophagus. We consider achalasia as one of these changes – a sort of defensive mechanism – which the esophagus has developed in response to long-lasting GERD. We believe that reflux may play an etiological role in about 10% of all achalasia patients. Our recommendation is that the history of GERD in achalasia patients should be explored and involved in therapeutic decision making. Dilatation therapy in such cases should be avoided, and surgical intervention as the primary option should always include an antireflux procedure as well – preferably a 360° fundoplication – to minimize the risk of postprocedure reflux. This must also be kept in mind when the decision about such an intervention is made or when the possibility of a competent antireflux procedure is not given such as with peroral endoscopic myotomy.
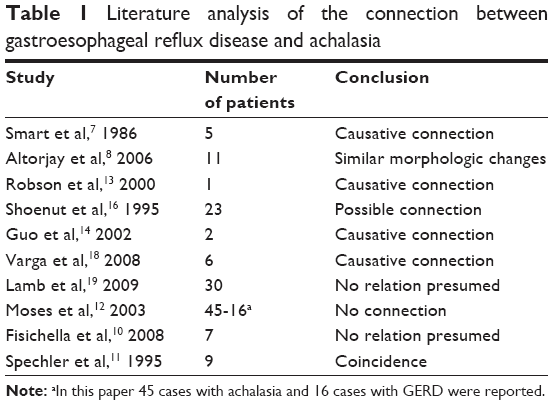 | Table 1 Literature analysis of the connection between gastroesophageal reflux disease and achalasia |
Acknowledgments
The present scientific contribution is dedicated to the 650th anniversary of the foundation of the University of Pécs, Hungary.
Author contributions
A Vereczkei and L Bognár performed the research and wrote the paper, A Papp and ÖP Horváth made critical revisions. All authors contributed toward data analysis, drafting and critically revising the paper and agree to be accountable for all aspects of the work.
Disclosure
The authors report no conflicts of interest in this work.
References
Ghoshal UC, Daschakraborty SB, Singh R. Pathogenesis of achalasia cardia. World J Gastroenterol. 2012;18(24):3050–3057. | ||
Atkinson M, Ogilvie AL, Robertson CS, Smart HL. Vagal function in achalasia of the cardia. Q J Med. 1987;63(240):297–303. | ||
De Giorgio R, Di Simone MP, Stanghellini V, et al. Esophageal and gastric nitric oxide synthesizing innervation in primary achalasia. Am J Gastroenterol. 1999;94(9):2357–2362. | ||
Robertson CS, Martin BA, Atkinson M. Varicella-zoster virus DNA in the oesophageal myenteric plexus in achalasia. Gut. 1993;34(3):299–302. | ||
Ruiz-de-León A, Mendoza J, Sevilla-Mantilla C, et al. Myenteric antiplexus antibodies and class II HLA in achalasia. Dig Dis Sci. 2002;47(1):15–19. | ||
Annese V, Napolitano G, Minervini MM, et al. Family occurrence of achalasia. J Clin Gastroenterol. 1995;20(4):329–330. | ||
Smart HL, Mayberry JF, Atkinson M. Achalasia following gastro-oesophageal reflux. J R Soc Med. 1986;79(2):71–73. | ||
Altorjay A, Szilagyi A, Arato G, et al. Morphological changes in the lower esophageal sphincter influencing the result of antireflux surgical interventions in chronic gastroesophageal reflux disease. Hepatogastroenterology. 2006;53:342–347. | ||
Dua K, Surapaneni SN, Kuribayashi S, Hafeezullah M, Shaker R. Protective role of aerodigestive reflexes against aspiration: study on subjects with impaired and preserved reflexes. Gastroenterology. 2011;140(7):1927–1933. | ||
Fisichella PM, Raz D, Palazzo F, Niponmick I, Patti MG. Clinical, radiological, and manometric profile in 145 patients with untreated achalasia. World J Surg. 2008;32(9):1974–1979. | ||
Spechler SJ, Souza RF, Rosenberg SJ, Ruben RA, Goyal RK. Heartburn in patients with achalasia. Gut. 1995;37:305–308. | ||
Moses PL, Ellis LM, Anees MR, et al. Antineuronal antibodies in idiopathic achalasia and gastro-oesophageal reflux disease. Gut. 2003;52:629–636. | ||
Robson K, Rosenberg S, Lembo T. GERD progressing to diffuse esophageal spasm and then to achalasia. Dig Dis Sci. 2000;45(1):110–113. | ||
Guo JP, Gilman PB, Thomas RM, Fisher RS, Parkman HP. Barrett’s esophagus and achalasia. J Clin Gastroenterol. 2002;34(4):439–443. | ||
Altorjay A, Juhasz A, Kellner V, Sohar G, Fekete M, Sohar I. Metabolic changes in the lower esophageal sphincter influencing the result of antireflux surgical interventions in chronic gastroesophageal reflux disease. World J Gastroenterol. 2005;11(11):1623–1628. | ||
Shoenut JP, Micflikier AB, Yaffe CS, Den Boer B, Teskey JM. Reflux in untreated achalasia patients. J Clin Gastroenterol. 1995;20(1):6–11. | ||
Campos GM, Vittinghoff E, Rabl C, et al. Endoscopic and surgical treatments for achalasia: a systematic review and meta-analysis. Ann Surg. 2009;249(1):45–57. | ||
Varga G, Kiraly A, Cseke L, Kalmar K, Horvath OP. Effect of laparoscopic fundoplication on hypertensive lower esophageal sphincter associated with gastroesophageal reflux. J Gastrointest Surg. 2008;12(2):304–307. | ||
Lamb PJ, Myers JC, Thompson SK, Jamieson GG. Laparoscopic fundoplication in patients with a hypertensive lower esophageal sphincter. J Gastrointest Surg. 2009;13(1):61–65. | ||
Sgouros SN, Vlachogiannakos J, Karamanolis G, et al. Long-term acid suppressive therapy may prevent the relapse of lower esophageal (Schatzki’s) rings: a prospective, randomized placebo-controlled study. Am J Gastroenterol. 2005;100(9):1929–1934. | ||
Morales-Divo C, Jecker P, Lippert B, Mann WJ. Extraesophageal reflux in patients suffering from Zenker’s diverticulum. HNO. 2007;55(7):546–550. German. | ||
Blumin JH, Johnston N. Evidence of extraesophageal reflux in idiopathic subglottic stenosis. Laryngoscope. 2011;121(6):1266–1273. | ||
Jaspersen D, Diehl KL, Geyer P, Martens E, Arps H. Die benigne proximale Ösophagusstenose – zumeist Komplikation der gastroösophagealen Refluxkrankheit [Benign proximal esophageal stenosis – mostly a complication of gastroesophageal reflux disease]. Dtsch Med Wochenschr. 1999;124(8):205–208. German. | ||
Jovov B, Van Itallie CM, Shaheen NJ, et al. Claudin-18: a dominant tight junction protein in Barrett’s esophagus and likely contributor to its acid resistance. Am J Physiol Gastrointest Liver Physiol. 2007;293(6):G1106–G1113. |
 © 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.