Back to Journals » HIV/AIDS - Research and Palliative Care » Volume 9
Abnormalities in alternative splicing of angiogenesis-related genes and their role in HIV-related cancers
Authors Mthembu NN, Mbita Z, Hull R ![]() , Dlamini Z
, Dlamini Z
Received 17 October 2016
Accepted for publication 17 February 2017
Published 30 March 2017 Volume 2017:9 Pages 77—93
DOI https://doi.org/10.2147/HIV.S124911
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Bassel Sawaya
Nonkululeko N Mthembu,1 Zukile Mbita,2 Rodney Hull,1 Zodwa Dlamini1
1Research, Innovation and Engagements, Mangosuthu University of Technology, Durban, 2Department of Biochemistry, Microbiology and Biotechnology, University of Limpopo, Sovenga, South Africa
Abstract: Alternative splicing of mRNA leads to an increase in proteome biodiversity by allowing the generation of multiple mRNAs, coding for multiple protein isoforms of various structural and functional properties from a single primary pre-mRNA transcript. The protein isoforms produced are tightly regulated in normal development but are mostly deregulated in various cancers. In HIV-infected individuals with AIDS, there is an increase in aberrant alternative splicing, resulting in an increase in HIV/AIDS-related cancers, such as Kaposi’s sarcoma, non-Hodgkin’s lymphoma, and cervical cancer. This aberrant splicing leads to abnormal production of protein and is caused by mutations in cis-acting elements or trans-acting factors in angiogenesis-related genes. Restoring the normal regulation of alternative splicing of angiogenic genes would alter the expression of protein isoforms and may confer normal cell physiology in patients with these cancers. This review highlights the abnormalities in alternative splicing of angiogenesis-related genes and their implication in HIV/AIDS-related cancers. This allows us to gain an insight into the pathogenesis of HIV/AIDS-related cancer and in turn elucidate the therapeutic potential of alternatively spliced genes in HIV/AIDS-related malignancies.
Keywords: vascular endothelial growth factor, oncogenic viruses, hypoxia induced factor 1, Kaposi’s sarcoma, non-Hodgkin’s lymphoma, therapies targeting alternative splicing
Introduction
In eukaryotic multi-exonic genomes, ~95% of the genes encode transcripts that undergo alternative splicing.1,2 Alternative splicing leads to an increase in protein diversity in cells, by allowing a single gene to code for multiple protein isoforms. The resulting isoforms may be structurally and functionally distinct.3,4 The vascular endothelial growth factor (VEGF) is one of the most important regulators of angiogenesis. This growth factor functions to regulate vascularization by regulating the proliferation, survival, and permeability of vascular endothelial cells derived from arteries, veins, and lymphatics.5–8 The loss of fidelity in alternative splicing in a gene-like VEGFA is the result of mutations within cis-acting elements which results in incorrect pre-mRNA processing or alternatively from mutations that affect trans-acting components leading to changes in the regulation of splicing.1 These defects in the regulation of alternative splicing can serve as a direct causative agent of disease, including cancer. It may also increase the susceptibility of an individual to a disease or an increase in disease severity.9
Numerous RNA binding proteins play dual roles in alternative splicing regulation and RNA processing events. Defects in these RNA processing events are known to play a role in diseases such as neurodegenerative disorders and cancer.10 This highlights the importance of abnormalities in alternative splicing in diseases and emphasizes this event as a common signature of many diseases including cancers. Cancer-related genes such as those involved in apoptosis, cell proliferation, angiogenesis, adhesion, metastasis, and invasion are commonly regulated by alternative splicing.11 Cancer is a significant cause of mortality and morbidity in those infected with HIV and AIDS. Approximately 35 million HIV-infected people are prone to develop a malignancy during their life time because of increased susceptibility to opportunistic diseases as a result of the decreased functioning of the immune system.12 Angiogenesis is implicated in >20 diseases including Kaposi’s sarcoma (KS), non-Hodgkin’s lymphoma (NHL), and cervical cancer. It is also a hallmark of most malignancies, contributing to carcinogenesis.13 This review summarizes the details of the molecular machinery required for normal constitutive alternative splicing and the mechanism by which abnormalities in alternative splicing of angiogenesis-related genes contribute to the development of HIV/AIDS-related cancers. Major focus is placed on the role of abnormalities in alternative splicing of angiogenesis-related genes in KS, NHL, and cervical cancer. Lastly, this review covers therapeutic strategies targeting angiogenesis-related alternative splicing through the manipulation of some of the tumor angiogenesis-related genes, in order to restore normal cell physiology in patients with HIV/AIDS-related cancers.
Aberrant alternative splicing
Alternative splicing of RNA is a vital and precisely regulated process occurring prior to mRNA translation.2 The pre-mRNA formed by gene transcription processes is a copy of the genomic RNA that still retains the intronic regions. In order to form a mature RNA molecule, these introns must be removed during pre-mRNA processing.11 Alternative RNA splicing also occurs in several ways as depicted in Figure 1A.14 Different combinations of exons are joined together, and in some instances, introns or their segments are included in the mature mRNA, resulting in the generation of multiple protein products of different function yet encoded by a single gene.11 Alternative splicing can result from 1) exon skipping, 2) inclusion of mutually exclusive exons, 3) use of alternative 5′ or 3′ splice sites, or 4) intron retention.2
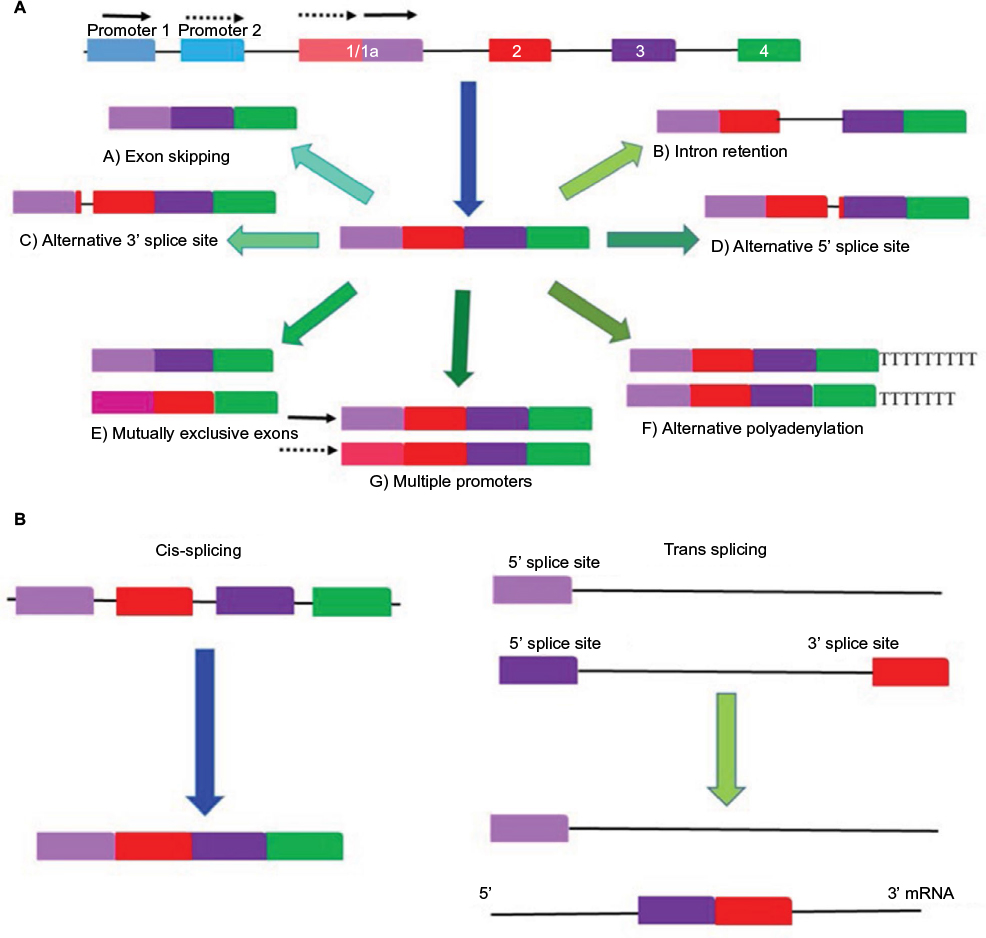 | Figure 1 Different forms of alternative splicing. Notes: (A) The process of alternative splicing differentially selects and pairs exons and splices out introns producing multiple mRNAs and uses different promoters to produce different transcripts. A gene is first transcribed by removing introns after which alternative splicing can occur in various ways. These include A) exon skipping, B) intron retention alternative, C) 3’ or 5’, D) splicing. E) Mutually exclusive exons. F) Alternative polyadenylation and finally the use of G) multiple promoters. (B) Cis splicing mRNA is assembled from the same strand RNA transcript. In trans-splicing exons from two different RNA transcripts are joined end to end. |
Alternative splicing occurs in the spliceosome, which encompasses a complex of small nuclear ribonucleoproteins, in which the two-step splicing reaction comprising intron removal and exon ligation takes place.15 Basically the spliceosome facilitates the “cut-and-paste” reaction of the intron and exon (5′ and 3′ splice site) sequences.16 Sequences that act as enhancer or repressor regulatory factors exist within the pre-mRNA. The cis-element binding proteins are known as serine-arginine-rich (SR) proteins. The trans-element binding proteins include members of the heterogeneous nuclear ribonucleoprotein (hnRNP) protein family (Figure 1B). Therefore, the RNA sequences and other cellular factors within the exons and introns play a role in positively or negatively regulating splicing.17,18 De-phosphorylation of SR proteins results in the modification of their activity and regulation. This leads to changes in alternative splicing, which results in the altered expression of isoforms involved with apoptosis and angiogenesis during cancer. This is exemplified by the expression switch from the anti-apoptotic Bcl-xL to a pro-apoptotic isoform, Bcl-xS, and from the pro-angiogenic VEGFxxx to the anti-angiogenic VEGFxxxb (Figure 2A and B).16 Changes in the activity and composition of these splice factors have been reported to modify the selection of splice sites and, therefore, interfere with normal cell development and differentiation.2,16 Point mutations during alternative splicing often occur causing aberrant alternative splicing, thereby generating variants that contribute to tumor development and perpetuate disease progression.19
 | Figure 2 VEGF splicing. Notes: (A) Alternative splice isoforms of VEGFA. All variants contain exons 1–4. Two families of VEGF isoforms arise due to differential splice-acceptor-site selection in the 3′UTR within exon 8 of the VEGF gene. The first family VEGFxxx are pro-angiogenic, whereas the members of the VEGFxxxb family are anti-angiogenic. (B) Alteration in cis- and trans-splicing regulatory elements in VEGF. A mutation in the exonic splicing enhancer (ESE) located before exon 8 prevents binding of the splicing factor 2 or alternative splicing factor 3 winner (ie, SF2/ASF) resulting in exon skipping. The modification in the activity of SR protein (phosphorylated/non-phosphorylated SR protein and trans-acting factors) induces splicing switches in the expression to either an anti-angiogenic or a pro-angiogenic isoform of VEGF.25 Abbreviation: VEGF, vascular endothelial growth factor. |
Mutations within the spliceosome machinery can also alter alternative splicing. There are three known alternatively spliced isoforms of the polypyrimidine tract binding protein, each with distinct RNA recognition activities. The presence of these isoforms directly affects alternative RNA splice site selection and contributes to disease-causing aberrant alternative splicing.20 A large-scale study of 32,000 mutations associated with inherited human diseases revealed that 14% of the point mutations that caused these disorder occurred at the intron–exon junctions, resulting in RNA splicing defects.21,22 The actual percentage is most likely much higher as defined enhancer and repressor sequences were not analyzed. For instance, 50% of the alternative RNA splicing defects that arise during the transcription of the neurofibromatosis type 1 or ataxia telangiectasia genes resulted from mutations in enhancers and repressor regions.23,24 The exonic splicing enhancer located before exon 8 prevents the binding of the splicing factor 2 or alternative splicing factor 3 winner (ie, SF2/ASF) resulting in exon skipping. The modification in the activity of SR protein (phosphorylated/non-phosphorylated SR protein and trans-acting factors) induces a splicing switch of VEGF to either an anti-antiangiogenic or pro-angiogenic isoform.25
Alternatively spliced angiogenic genes in malignancies
Angiogenesis is defined as the formation of new capillary blood vessels from the existing micro vessels. This process is initiated by the recruitment of associated supporting cells together with different segments of the vasculature.26,27 Under physiological conditions, the activity of inducers and inhibitors of various angiogenic factors are maintained in a tightly controlled and balanced manner. Angiogenesis is controlled through a series of “on” and “off” regulatory switches. The main “on” switch being the angiogenesis growth factors (cytokines), whereas the main “off” switches are endogenous angiogenesis inhibitors.28
Persistently upregulated angiogenesis is often found as the causal factor in certain pathological conditions such as cancer, atherosclerosis, and diabetic retinopathy.26 The onset of angiogenesis in most tumor cell populations is independent and expressed at different times during neoplasia; for example, the onset of angiogenesis in cervical cancer occurs before neoplasia, whereas in breast cancer, it occurs during neoplasia.13,29 The angiogenic process occurs in a series of events (Figure 3) that are controlled by receptors, cytokines, and enzymes. These include VEGF, angiopoietin, and ephrin family members.11,30
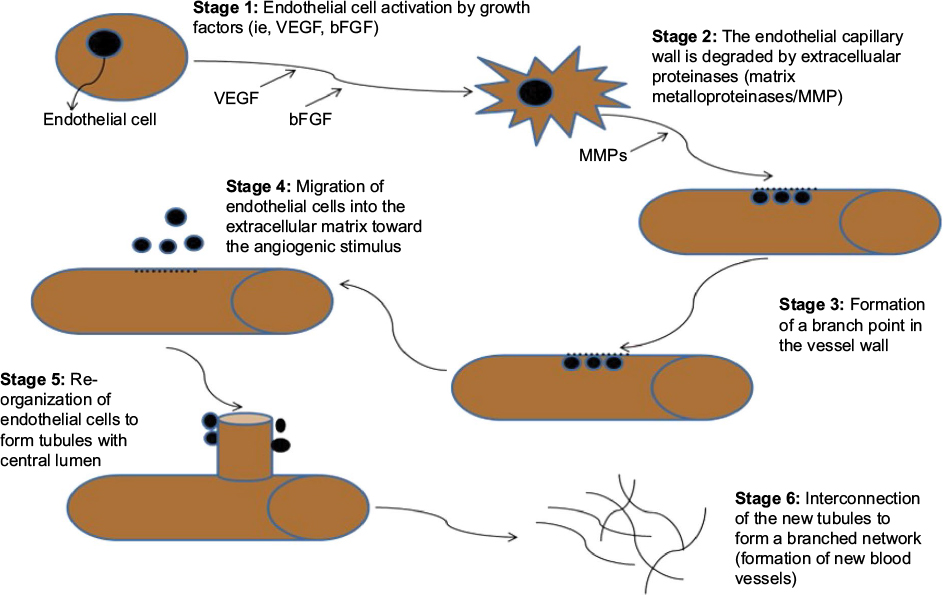 | Figure 3 The key stages of angiogenesis along with relevant markers in each stage. Notes: Stage 1: Potent stimulatory factor for endothelial cell migration and proliferation, splicing switches involved include VEGF. Stage 2: Splicing isoforms of MMPs (MMP1/2) act as extracellular matrix components. Stages 3 and 4: Integrins are expressed during this stage and allows the migrating endothelial cells to interact with specific compounds of the surrounding matrix. Stage 5: Splicing isoforms of Angiopoietin (Ang 1) are produced by surrounding cells to facilitate endothelial survival and stabilization of new capillary tubes. Stage 6: Endothelial cells of the new capillaries produce alternatively spliced isoforms of platelet-derived growth factor (PDGF) and recruit pericytes which in turn stabilize the new blood vessels.145 Abbreviation: VEGF, vascular endothelial growth factor. |
Angiogenesis stimulators such as VEGF, angionenin, proliferin, and interleukin-8 together with angiogenesis inhibitors such as interleukin-2 and -18 are tightly controlled in normal physiological conditions.31 Angiogenesis requires the endothelial cells of existing blood vessels to degrade the underlying basement membrane and invade into the stroma (which supplies blood) of the neighboring tissue in order to form new capillaries.32 Many of the various novel splicing transcripts that are associated with cancer are not expressed in normal adult tissues, but are found during embryonic development.33 The activation of varying oncogenes and an inactivation of suppressor genes (Table 1) enable cancer cells to successfully proliferate, survive, and resist apoptosis.18
 | Table 1 Alternately spliced genes involved in angiogenesis and cancer Note: The table lists some of the genes involved in angiogenesis whose alternate splicing may contribute to malignancies. |
Given the widespread function of alternatively spliced genes that play a role in angiogenesis, it is of no surprise that aberrant regulation of alternative splicing is a hallmark of tumorigenesis. Exon inclusion is controlled by splicing factors such as SF2/ASF, which determine the transition of the epithelial cell phenotype to a mesenchymal trait that is commonly observed in cancer cell invasion.34 Tumor cells are exposed to stressful conditions such as hypoxia and acidosis, leading to growth factors and cytokines, being provided by cancer or nontumor cells, activating signaling cascades that affect both the activity and/or the expression levels of splicing regulatory factors (SRFs), thereby altering target gene splicing.18 SRSFs enhance the translation of oncogenic variants involved in key aspects of cancer cell biology and are also involved in the regulation of alternative splicing of pre-mRNAs relevant to cancer progression.18 This network of alternative splicing events is susceptible to change causing various abnormalities in genes involved in cancer progression mechanisms.
Abnormalities in alternatively spliced angiogenesis-related genes
Deregulation of splicing and the expression of pro-cancer splice variants that occur in cancer can be caused by changes in the activity of trans-acting splicing regulators.4 Extensive studies on the abnormalities of alternative splicing of several candidate angiogenesis-related genes have been investigated in correlation with cancer risk or progression (Table 2). Fibroblast growth factors (FGFs), along with VEGF remain the key players in tumor angiogenesis and their inhibition represses tumor growth.30 Little is known about the involvement of alternative splicing of other angiogenesis-related genes such as angiopoietin and the ephrin gene family in comparison with VEGF in the development of HIV/AIDS-related cancers.
 | Table 2 Aberrantly spliced genes in malignancies Note: A list of genes whose aberrant splicing contribute to abnormal angiogenesis in tumor cells. |
VEGF
Aberrant splicing of the pre-mRNA of genes such as VEGFA leads to isoforms with altered functions in angiogenesis because the isoforms produced have different C-termini and different affinities for receptors.2 The alternative splicing of VEGFA transcripts promotes the formation of new blood vessels. For instance, VEGF4 results from alternative splicing of the 3’ splice site and is implicated in the formation of new blood vessels.35 In cancer, hypoxic cells with VEGF encoded by VEGFA interacts with hypoxia-inducible factor (HIF-1α) and diffuses through the tumor mass to the normal neighboring blood vessels. There, they bind to VEGFR2, stimulating proliferation and migration of endothelial cells in order to form new capillaries.18
There are at least four structurally related human VEGF isoforms. These include VEGF121, VEGF165, VEGF189, and VEGF20636–39 (Figure 2A). VEGF 165 is one of the most abundant isoforms and has the highest activity. The pro-angiogenic VEGF165 and anti-angiogenic VEGF165b result from the mutually exclusive inclusion of alternate terminal exons. The pro-angiogenic isoform (VEGFxxx/VEGF165) is generated from a proximal 5’ splice site (PPS) in exon 8a, whereas the anti-angiogenic isoform (VEGFxxxb/VEGF165b) is generated from a distal 5’ splice site (DSS) in exon 8b. These isoforms bind to a receptor (VEGFR2) to either induce or inhibit angiogenesis, respectively (Figure 2 A and B).18,40 The expression and secretion of VEGF165 is regulated by numerous growth factors and cytokines such as interleukin 1β, platelet-derived growth factor (PDGF), and transforming growth factor β.38 In several studies, it was discovered that VEGFxxxb/VEGF165b is expressed in normal tissues such as kidney, pancreas, and lung but is downregulated in many cancers such as renal cell carcinoma and malignant prostate cancer where angiogenesis plays a crucial role in the survival of the tumour.19,40,41 These studies indicate that VEGF is required for tumor survival and progression and its inhibition suppresses tumor growth in a wide variety of tumor cell lines in murine models.2,42
Formation of branched vascular vessels depends on VEGF-A to regulate endothelial cell division, migration, and survival in a dose-dependent manner. VEGF-A acts through the tyrosine kinases, FLK-1 (VEGFR2) and FLT-1 (VEGFR1) (Figure 4).43 FLT-1 comprises a ligand-binding domain splice variant, sFLT-1 (soluble VEGR receptor-1), which results from alternative splicing of FLT-1 pre-mRNA and functions to inhibit VEGF.44 It accomplishes this by binding to VEGF-A with high affinity and blocking VEGF/FLT-1 signaling.43
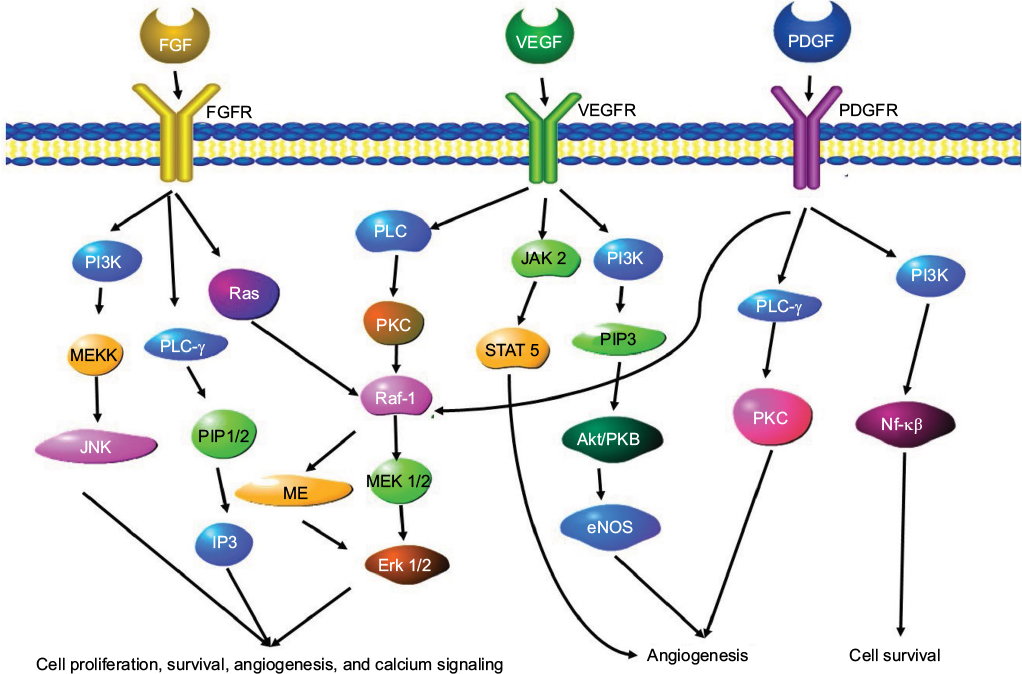 | Figure 4 A schematic overview of the angiogenesis gene signaling pathways. Notes: Three signaling pathways involved in angiogenesis are the FGF signaling pathway, the VEGF signaling pathway and the Ang-1/Ang-2 signaling pathway. Abbreviations: FGF, fibroblast growth factor; FGFR, FGF receptor; PDGF, platelet derived growth factor; PDGFR, platelet derived growth factor receptor; VEGF, vascular endothelial growth factor; VEGFR, VEGF receptor. |
Matrix metalloproteinases (MMPs)
MMPs act as both pro-angiogenic and anti-angiogenic proteolytic enzymes. Their function depends on the type and stage of a particular cancer.30 The activity of MMPs is controlled through their tissue inhibitors (tissue inhibitor of metalloproteinases). MMPS and tissue inhibitor of metalloproteinases function to regulate the breakdown of extracellular matrix (ECM), which is an essential step in the formation of new blood vessels. They achieve this by degrading the structural components of the ECM. This leads to changes in cellular proliferation, cell–cell adhesion, angiogenesis, invasion, and metastases.45 In humans, there are >50 MMPs. These are very similar but can be divided further into separate families. These include the MMPs (23 in humans), the A disintegrin and metalloproteinases (ADAMs) (13 in humans) and ADAMs with thrombospondin motifs (19 in humans). The MMP family can be further divided into collagenases (MMP-1, MMP-8, and MMP-13), stromelysins (MMP-3, MMP-10, and MMP-11), and gelatinases (gelatinase A or MMP-2; gelatinase B or MMP-9).46
MMPs that play a more vital role than others in angiogenesis include MMP-2 (gelatinase A) and MMP-9 (gelatinase B), which cleave the major components of the blood vessel basement membrane, including collagen type IV, fibronectin, and laminin.47 Many studies suggest that MMP-2 and MMP-9 as well as aberrant splice variants of MMP-9 and MMP-7 are required for the release of pro-angiogenic isoforms of VEGF from the tumor matrix leading to the initiation of tumor angiogenesis.48,49 At the same time, MMP-9 seems to be important for angiogenesis as it is expressed in blood vessels at proliferative margins.50 Other angiogenic factors function to upregulate the expression of the gelatinases (MMP-2 and MMP-9) and MMP-2, respectively.48 Excessive breakdown of the basement membrane can have negative effects on angiogenesis as the MMPs begin to damage essential support for endothelial cells, as well as releasing anti-angiogenic factors such as endostatin.51
Angiopoietin
The angiopoietin family of proteins comprise three structurally related proteins, angiopoietin-1 (Ang-1), Ang-2, and Ang-3/4.52 Ang-1 phosphorylates and activates endothelial cell-specific receptor tyrosine kinase (Tie-2) receptor. Ang-2 can inhibit the activation of Tie-2 by Ang-1.53 Ang-1 interacts with Tie2 to enhance the production and stabilization of new blood vessels through the Akt signaling pathways.54,55 Ang-1 acts as an anti-angiogenic factor by preventing cell migration by affecting junction molecules,56 tightening the attachment of mesenchymal cells to endothelial tube. This leads to the maturation of mesenchymal cells into pericytes, Ang-1 then binds to Tie-2 and promotes the attachment of these pericytes to form mature, “nonleaky” blood vessels.57 Ang-2 acts as a pro-angiogenic factor by blocking the vessel stabilizing action of angiopoietin-1 promoting endothelial cell migration. This leads to the establishment of new vasculature. These activities of Ang-2 are known to occur in the presence of VEGF or other pro-angiogenic factors. However, it is may also act alone.58,59
Alternatively spliced isoforms that have both pro- and anti-tumorigenic effects have been identified for both Ang-1 and Ang-2.60 The Ang-1 coding region of 1.5 kb gives rise to three isoforms of Ang-1 that are 0.7, 0.9, and 1.3 kb in size.61 One of the alternately spliced exons is exon 7, and this exon is responsible for receptor binding.62 The shorter versions that lack this domain may act as dominant negative form of the protein and regulate the binding and activity of longer forms. An isoform of Ang-2 encoding a protein of 443 amino acids was identified and named Ang2443. This isoform lacks exon 2 and codes for a glycosylated dimer that binds Tie-2 but not Tie-1. It can act as a negative regulator of both Ang-1 and wild-type Ang-2 by competing for receptor binding.63 Another Ang-2 isoform has an alternate version of exon 1, exon 1B, which results from an alternate transcription start site.64
Fibroblast growth factor
Basic fibroblast growth factor (bFGF) is a pro-angiogenic growth factor that encourages capillary endothelial cell growth and division in vitro and in vivo.65 This molecule initiates ECM remodeling and is stored in the ECM in anticipation of this step in angiogenesis.66 When the growth factor binds to the growth factor receptor, the receptors dimerize, which initiates the intracellular signaling cascade. Multiple isoforms of the bFGF receptor type 1 (FGFR1) are generated by alternative splicing of mRNA.67 To date, 23 FGF isoforms and five FGFRs have been identified. Three receptors have a very similar structure consisting of an extracellular immunoglobulin (Ig)-like domain and an intracellular tyrosine kinase domain. The fourth isoform lacks the trans membrane and intracytoplasmic domains.67 The fifth isoform (FGFR-5) only has two Ig-like domains.68 The presence of these isoforms may alter the ability of ligands to bind, leading to changes in the signaling cascade, contributing to cancer progression and agiogenesis.69 In cancers such as ovarian cancer, FGF is suspected to play a direct role in tumor cell proliferation and angiogenesis by interacting with pro-angiogenic factors such as VEGF. The FGF signaling pathway also involves downstream proteins such as MAPK proteins and proteins of the PI3K/Akt cascade. There is also “crosstalk” between the FGF pathway and the Notch and IP3 cascades.69 A schematic overview of some elements of the FGF-related pathways are shown in Figure 4.
Oncogenic viruses, angiogenesis, and AIDS-related cancers
The cancers that affect HIV-positive people can be described as those cancers that are AIDS-defining and opportunistic cancers. The AIDS-defining cancers are those that can be described as cancers that constitute an AIDS diagnosis in patients with HIV. These include KS, NHL, and invasive cervical cancer.70 Oncoviruses contribute to carcinogenesis by affecting cell signaling pathways and their downstream targets that play a role in the development of cancer. One of the major pathways affected by oncoviruses is that of the HIF family of transcription factors. The HIF proteins play a major role in the response of normal cells to hypoxic conditions.71 Oxygen limitation in normal cells is resolved by suppressing the degradation of HIF-1α. HIF-1α is degraded by the action of von Hippel–Lindau tumor suppressor protein and the 26S proteasome. When oxygen supply is sufficient, HIF-1α is ubiquinylated and degraded, preventing its dimerization with HIF-1β. The HIF dimer associates with the hypoxia response element (RE) of the promoter target gene. These target genes play a role in processes such as angiogenesis, glucose metabolism, cell proliferation, apoptosis, and invasion/metastasis.71,72 Stabilization and increased transcription and expression of HIF-1 is commonly affected by human oncogenic viruses. This is achieved by disrupting the degradation of HIF-1α or through increased HIF-1α mRNA translation via the PI3K/Akt/mTOR or MAPK signaling pathways.71 Table 3 gives a list of oncogenic viruses, the cancers they cause, and alternatively spliced genes involved in their oncogenic activity.
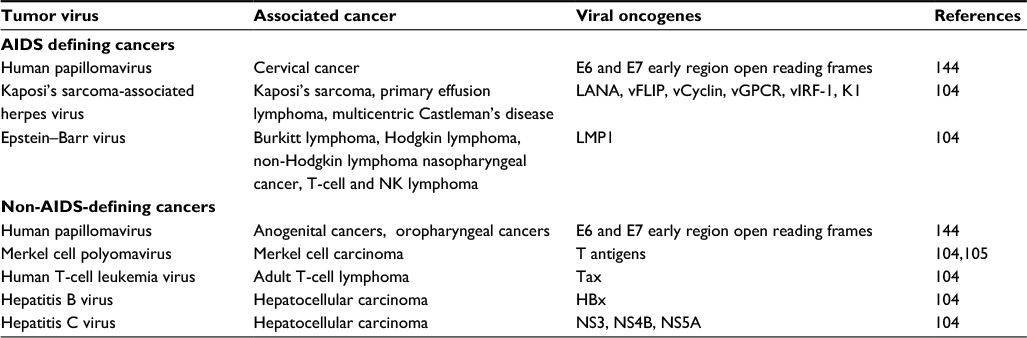 | Table 3 Oncogenic viruses, the cancers they cause, and alternatively spliced genes involved in their oncogenic activity |
Kaposi’s sarcoma-associated herpes virus (KHSV)
KS is a type of skin cancer, commonly found in HIV/AIDS patients, and is related to an infection with the human herpes virus 8 (HHV-8). KS results in skin, lymph node, liver, spleen, lungs, and digestive tract lesion. HHV-8 is a DNA virus whose genome consists of a large 165 kb double-stranded DNA molecule and is enveloped by an icosahedral capsid. The virus was originally isolated from KS lesions from an AIDS patient and has been linked to other malignancies besides KS, including primary effusion lymphoma, body cavity-based lymphoma, multicentric Castleman’s disease, germinotropic lymphoproliferative disease, multiple myeloma, angiosarcomas, malignant skin tumors, and squamous cell carcinomas.73 In KS lesions, most of the cells infected by the virus are in the latent phase and do not proliferate or express angiogenic cytokines. A small percentage remain active and express angiogenic cytokines such as cytokines such as VEGF, IL-6, CXCL8, and bFGF.73 Multiple cell types can be infected by KHSV, but the infection of endothelial cells plays a major role in viral proliferation and angiogenesis. These endothelial cells have increased levels of proliferation and display chemotactic migration and invasion characteristics.74 KSHV can promote angiogenesis through the action of proteins encoded by the virus itself. During the lytic phase of the life cycle of the virus, all the viral proteins are expressed. Many of these are able to induce angiogenesis.73
KSHV viral interleukin-6 (vIL-6) shares 49.7% sequence similarity with human IL-6 (hIL-6)73 and is capable of upregulating VEGF expression and angiogenesis in experimental models.75 The pro-angiogenic activities of vIL-6 are enhanced through interaction with HIV-1-encoded Nef protein.76 vIL-6 is capable of initiating multiple downstream pathways transcription factors and REs, including JAK/STAT, MAPK, PI3K/AKT, STAT1/3, STAT5, IL-6 RE, C/EBP, and c-jun.73 The KSHV homolog of the angiogenic IL-8 receptor, viral G-protein-coupled receptor (vGPCR) controls the secretion of various angiogenic factors. These include VEGF, bFGF, IL-1α, IL-2, IL-4, IL-6, and IL-8, and TNF α.73 vGPCR modulates the nuclear factor of activated T-cells (NFAT) pathway by elevating cytosolic calcium through sarcoplasmic reticulum calcium ATPase.77 The ability of vGPCR to control multiple pathways including the PLC, PKC, MAPK, PI3K/AKT/mTOR, NF-кB, AP1, and NFAT networks allows this protein to cause multifocal and aberrantly vascularized tumors.73 vGPCR also upregulate HIF expression leading to the increased expression of VEGF, PDGF, TGFa, TGFb, ANGPT-2, and ANGPTL-4.73 HIF also increases the expression of pyruvate kinase M2 (PKM2), which leads to an increase in angiogenesis in KS.78 KSHV-encoded viral interferon regulatory factor (vIRF-3) is a cellular interferon regulatory factor homolog and is also known as latency-associated nuclear antigen-2 (LANA2). LANA inhibits p53, modulates G1–S transition, and interacts with c-Myc, angiogenin (ANG), and Annexin A2, leading to an increase in the cells ability to resist apoptosis.73 It promotes angiogenesis by inducing the expression of VEGF-A by stabilizing HIF-1α.79 LANA also upregulates EMPIRIN80,81 and suppresses immune responses by inhibiting antigen presentation MHC-I peptide presentation and suppressing MHC-II gene expression.73
The trans-membrane glycoprotein K1 promotes angiogenesis in cell culture82 and promotes VEGF expression in primary human endothelial cells.83 The activation of VEGF signaling pathways is expected to be one of the primary means, whereby KSHV infection leads to increased angiogenesis.73,84 It is important to note that K1 can interact with HIV proteins to promote angiogenesis. HIV Tat promotes K1-induced angiogenesis through the activation of the NF-кB signaling pathway. This is due to HIV Tat and K1 promoting the transcription of the microRNA miR-891a-5p which inhibits IкB expression.85 HIV-1 Nef and K1 also act together to promote angiogenesis by initiating the transcription of the microRNA miR-718 in chicken and mice models. This miRNA promotes the degradation of PTEN, resulting in the activation of the AKT/mTOR pathway.86 K1 also induces angiogenesis by enhancing the expression of MMP-9. KSHV also upregulates MMPs through the increased levels of EMMPRIN,80 and KS tumors express higher levels of MMP-1, MMP-2, MMP-3, MMP-9, and MMP-19. For this reason, MMP inhibitors have been used to treat AIDS-related KS.87 The three proteins K4, K4.1, and K6 are homologs of cellular chemokines that also promote angiogenesis by inducing VEGF expression.88
KSHV also influences angiogenesis through the transcription of its own non-coding microRNAs. KSHV encodes 12 microRNAs k12-1 through to k12-12. Thrombospondin 1 is an anti-angiogenic factor and is a target of the KSHV-encoded miRNAs miR-K12-1, miR-K12-3, miR-K12-6, and miR-K12-11.89 The transcription factor MAF is also repressed by miR-K12-6 and miR-K12-1, resulting in blood vessel endothelial cells being reprogrammed.90 K5 is also known as a modulator of immune recognition (MIR-2), and is an E3 ligase that blocks immune function and promotes angiogenesis by degrading cadherin, promoting the re-modeling of endothelial adherens junctions.91 K5 achieves this by modulating AKT and extracellular signal-regulated kinase 1 and 2 (Erk 1/2) activity.92 The KHSV proteins Kaposin B and K12 interact with MK2 kinase and enhance its activity, leading to the stabilization of pro-inflammatory and angiogenic mRNAs.93 K15 activates the Ras/MAPK, JNK/SAPK, and NF-кB pathways, leading to the induction of angiogenesis94 and increased expression of IL-6, IL-8, CCL20, CCL2, CXCL3, IL-1a/b, and COX-2. K15 can also induce angiogenesis through the RCAN1/DSCR1 pathway by upregulating calcineurin and NFAT1.73
NHL (Epstein–Barr virus)
HIV/AIDS-related NHL is the second most common AIDS-related cancer. NHLs are heterogeneous lymphoproliferative malignancies and can be divided based on the type of lymphocyte that becomes malignant. The most common lymphomas are B-cell lymphomas and represent 88% of all lymphomas. In HIV/AIDS patients, 80% of all lymphomas are high-grade B-cell lymphoma. Lymphomas of the T and natural killer (NK) cells make up 12% of all NHLs. In HIV/AIDS patients, the most common types of NHL are primary central nervous system lymphomas which affect the brain and spinal fluid.95 Lymphoma cells are able to induce or promote angiogenesis in numerous ways; first, through increased release of pro-angiogenic factors such as MMP2 and MMP996 and second, by increasing their ability to bind and interact with the substratum by interacting with the ECM components vitronectin and fibronectin. This leads to increased proliferation and protease secretion.97
The true effect that lymphomas have on angiogenesis can be observed by comparing the effect that cancerous and non-cancerous tissue has on the development of blood vessels. Implantation of normal lymph nodes in the chick embryo CAM model results in the development of a small number of growing vessels and increased levels of microvascular density (MVD).98 Benign lymphadenopathies show increased levels of MVD in reactive parts of affected lymph nodes. B-NHL displays alterations in laminin and type IV collagen expression that corresponds to the expected expression patterns that would occur during endothelial sprouting in outgrowing capillaries.95
The expression of tenascin is increased in the stroma of B-NHL, leading to increased levels of angiogenesis and endothelial cell migration.99 Intermediate and high-grade NHLs contain immature capillaries that lack the basement membrane,100 whereas low grade b-NHLs contain more developed capillaries that is surrounded by a continuous basement membrane.101 VEGF-A is overexpressed in tumor and endothelial cells in angioimmunoblastic T-cell lymphoma.102 The levels of VEGF expression in NHL can be used as an accurate predictor of tumor stage as well as a prognostic indicator of patient survival.103 Other angiogenic growth factors whose expression is increased in NHL include FGF-2 and PDGF-β.95
Epstein–Barr virus is a human c-herpes virus that has been associated with NHL. The major viral oncogene expressed in EBV-associated lymphoma is latent membrane protein-1 (LMP1). This is a 62-kDa integral membrane protein that immortalizes and transforms human B cells, promoting cell proliferation. LMP1 activation leads to overexpression of anti-apoptotic genes and inhibits p53-induced apoptosis. LMP1 is also able to influence angiogenesis through the overexpression of HIF-1α by increasing the level of reactive oxygen species such as H2O2. The increase in oxidative stress activates downstream signaling, leading to increased HIF activity.71 LMP1 is transcribed from two different promoters producing two LMP1 isoforms, with one isoform being a shorter version lacking the N terminus. This shorter version is non-coding and acts as a negative regulator of the larger oncogenic isoform.104 Other oncogenes expressed by EBV include the EBV nuclear antigens (EBNAs). EBNA-1 enhances the transcriptional activity of activator protein 1, which is responsible for the transcription of HIF-1α.
Non-AIDS-defining cancers
Merkel cell polyomavirus causes an aggressive non-melanoma skin tumor known as Merkel cell carcinoma. The levels of Merkel cell polyomavirus infection is higher in HIV-infected individuals.105 Alternative splicing of the early pre-mRNAs of the virus produce four isoforms (LT, sT1, sT2, and 57kT).104 The T antigens have different functions, with the large T- and small t-antigen exerting their oncogenic effects by inhibiting tumor suppressors, activating tumor promoters, preventing apoptosis, inducing angiogenesis, stimulating metastasis, translation, and inhibiting E3 ubiquitin ligase activity.106
Hepatitis C virus is an RNA virus with a 9.6-kb genome encoding for a single polyprotein that is cleaved into 10 protein products. HIV and hepatitis C virus have shared transmission routes, and this results in patients commonly being infected with both the viruses. HIV infection increases the severity and worsens the outcomes for patients with HCV by increasing the risk of hepatocellular carcinoma. Furthermore, the effect of HIV on the immune system combined with cross reactivity between proteins expressed by both viruses leads to increased viral proliferation.107 HIF-1α is overexpressed in hepatocellular carcinoma, leading to increased VEGF expression and cell survival. This is partly due to viral infection inducing oxidative stress as these cells are in hypoxic conditions in the damaged liver where there is a reduction in vascularization. Non-structural hepatitis C viral proteins are able to induce HIF pathways as can the HCV core protein.71,108 Alternative splicing does not occur in CV mRNA, but the virus requires the activity of the host cells RNA helicase DDX3. DDX3 plays a role in splicing and the innate immune response to viruses.109
HTLV-1 is a delta retrovirus capable of causing adult T-cell lymphomas or leukaemia’s.71 The viral oncogene of HTLV-1, Tax, is a 40-kDa nuclear phosphoprotein that promotes viral transcription and leads infected T cells to becoming immortalized.109 Tax activates the PI3K/Akt pathway, leading to an increase in the levels of HIF-1α protein. The single HTLV-1 pre-mRNA undergoes extensive splicing to give rise to nine types of spliced and unspliced mRNAs.104
Therapies targeting alternative splicing
The widespread and vital role played by angiogenesis in the occurrence of all cancer types and its relatively limited role in normal physiology has resulted in the suggestion that the inhibition of angiogenesis may be effective in the treatment of cancer.11 This therapeutic inhibition of angiogenesis may be achieved through the modulation of alternative splicing. Currently, multiple strategies are under investigation as a means of correcting aberrant alternative splicing in cancer. However, one of the most promising strategies is the use of small molecules that can target alternative splicing that alter angiogenesis in cancer. Many of these compounds are already used as chemotherapy drugs and have been shown to alter alternative splicing.110 Some of these compounds and their modes of action are listed in Table 4.
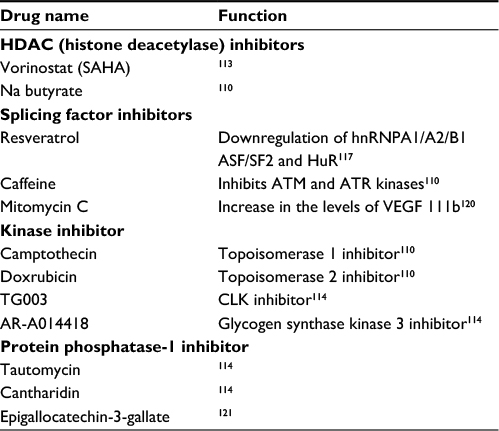 | Table 4 Small molecules that affect alternative splicing Note: A list of examples of small compounds that have effects on alternative splicing, their targets ,and modes of action. Abbreviations: HDAC, histone deacetylase; VEGF, vascular endothelial growth factor; CLK, CDC-like kinase. |
Specific RNAs to interfere with splicing
Alternative splicing can be targeted at multiple stages including at the transcription stage by targeting the splicing machinery, by preventing RNA binding proteins from recognizing their RNA targets, by preventing RNA binding proteins from performing their actions on the splicing machinery, and finally, through controlling agents that alter the ability of RNA binding proteins to carry out their splicing actions. One strategy is to prevent splicing factors from gaining access to specific RNA sequences. This can be achieved through the use of specifically designed oligonucleotides that act to block the recruitment of splicing factors to specific pre-mRNA as in antisense oligonucleotides.110,111 Alternatively, RNA sequences can be designed to be targets for specific splicing factors. These RNA sequences can then be used to position splicing factors at a specific splice point or sequester these splicing factors.112
Histone deacetylase (HDAC) inhibitors
The reversible acetylation of histones plays an important role in the regulation of transcription and HDACs repress transcription by condensing chromatin. Aberrant HDAC recruitment is associated with cell growth and tumor cell proliferation, most likely due to inappropriate transcriptional repression mediated by HDAC.113,114 HDAC inhibitors cause cells to undergo growth arrest, terminal differentiation, and/or apoptosis. By inhibiting the activity of HDACs, these small molecules change the expression pattern of splicing factors leading to a change in splicing. One of these molecules sodium butyrate changes SR protein expression in mice.114 However, the same compound was able to alter the splicing of the prohibitin protein, leading to an increase in the levels of the anti-cancer long isoform in a splicing factor independent manner.115 These compounds my also lead to increased levels of protein acetylation and activity.114 Another compound named vorinostat-SAHA (suberoylanilide hydroxamic acid) induces growth arrest or apoptosis in cancer cells, and antitumor activity has been observed in patients. Vorinostat leads to the accumulation of acetylated proteins such as BCL6, p53, and Hsp90.113
Splicing factors
These small molecules have the advantage of being able to target multiple splicing factors and affecting the splicing of multiple gene products. This increases their effectiveness, but the decrease in specificity might limit their usefulness. Treatment of cells with DNA damaging agents such as UV and a series of chemotherapeutic drugs including camptothecin, doxorubicin, and cisplatinum leads to the expression of the VEGF isoform VEGF111. It is a pro-angiogenic factor and is resistant to degradation by plasmin and MMP. This makes it a very attractive target for the development of anti-cancer drugs.116 There are many small compounds that inhibit the upregulation of VEGF111 caffeine, epigallocatechin gallate, and resveratrol. Resveratrol may accomplish this by through the downregulation of several RNA processing factors including hnRNPA1/A2/B1 ASF/SF2 and HuR.117 Caffeine inhibits ATM and ATR kinases leading to the disruption of multiple DNA damage-responsive cell cycle checkpoints and inhibit the repair of DNA lesions. This may lead to a downregulation in the expression of the VEGF111 isoform.110,118 The SR splicing factor, SC35, was upregulated by caffeine treatment. This caffeine-induced upregulation of SC35 led to an increase in the expression of the alternatively spliced tumor suppressor gene Kruppel-like factor 6 transcript110 as well as the p53β isoform that promotes senescence.119 Caffeine induces alternative splicing in multiple targets and, therefore, lacks specificity and is not suitable for use in clinical practice.110 Treatment of ovarian cancer cells with the chemotherapy agent mitomycin C leads to an increase in the levels of the anti-apoptotic isoform of VEGF111, VEGF 111b. This leads to a decrease in cell growth.120 Another strategy is to interfere with the structural proteins that make up the spliceosome or the proteins that regulate RNA binding. An example of this would be the compound borrelidin. This antifungal compound binds to the splicing protein FBP21. This results in the use of an alternative splice site in VEGF pre-mRNA, resulting in the expression of a protein with an alternate C terminus and that prevents angiogenesis.112
Kinase inhibitors
In order to regulate pre-mRNA splicing, SR proteins are phosphorylated to allow their nuclear import and dephosphorylated in order to allow for the assembly of the spliceosome. As a result of this both hypo- and hyperphosphorylation result in the inhibition of SR protein function. DNA damaging agents such as camptothecin exert their activity by inhibiting the topoisomerase 1 SR protein phosphorylating factor, whereas doxrubicin inhibits topoisomerase 2. These drugs are able to alter splice site selection in numerous pre-mRNA targets including VEGFA.110,114 Other drugs that have been developed to target proteins that phosphorylate SR proteins include TG003 that inhibits members of the CDC-like kinases, AR-A014418 and LiCl both of which inhibit glycogen synthase kinase 3.114
Phosphatase and protease inhibitors
Protein phosphatase-1 (PP1) is able to affect splice site selection by directly binding to a RNA-recognition motif of different splicing regulatory proteins. Tautomycin and cantharidin inhibit PP1 and PP2A, leading to changes in alternative splicing patterns.114 Epigallocatechin-3-gallate (EGCG) and ibuprofen synergistically act to suppress proliferation and enhance apoptosis of prostate cancer cell lines by increasing the level of pro-apoptotic variants of Bcl-X and Mcl-1 while lowering the levels of the anti-apoptotic splice variants. This is accomplished through the increased activity of PP1.121 Yet another strategy is the use of protease inhibitors. Inhibitors of metalloproteinase activity, such as the synthetic inhibitor AG3340, are being tested for their ability to treat brain tumors. This molecule inhibits glioma growth, invasion, and angiogenesis.122 The use of HIV protease inhibitor (HIV-PI)-based therapies was discovered to be closely associated with the regression of KS or NHL, in HIV-infected people.123 This is due to various HIV PIs down regulating the expression of pro-angiogenic MMP. This was shown in animal models and in humans, where HIV-PI blocks MMP-2 proteolytic activation, thus inhibiting tumour angiogenesis in vitro.124
Conclusion
The immunosuppression resulting from infection with HIV increases the risk for development of several cancers. The spread and progression of cancer relies upon the establishment of new blood vessels. Angiogenesis involves multiple cellular signaling pathways and results from the altered expression of pro-inflammatory and pro-angiogenic chemokine factors. AIDS-related cancers such as KS and NHL show high degrees of vascularization with the increased expression of multiple angiogenic factors such as bFGF and VEGF-A (VEGFHIV proteins also contribute to angiogenesis. Therefore, anti-angiogenic therapy may represent an important tool for the treatment of AIDS-related cancers. Since angiogenesis is not common in normal adult tissues, the process may be targeted with little adverse effects. With the multitude of genes whose regulation of alternative splicing goes awry in cancer development, there is no shortage of potential targets. The varied pathways that many alternatively spliced oncogenic gene products contribute to angiogenesis in cancers resulting from viral infections all converge on the activation of HIF-1. This makes the upregulation of HIF-1 an attractive target for the development of new therapies. Nevertheless, the chance of adverse effects on normal physiology must always be assessed.
Acknowledgment
The authors would like to thank the MRC for providing funding.
Disclosure
The authors report no conflicts of interest in this work.
References
Ward AJ, Cooper TA. The pathobiology of splicing. J Pathol. 2010;220(2):152–163. | ||
Kim Y-J, Kim H-S. Alternative splicing and its impact as a cancer diagnostic marker. Genomics Inform. 2012;10(2):74–80. | ||
Caceres JF, Kornblihtt AR. Alternative splicing: multiple control mechanisms and involvement in human disease. Trends Genet. 2002;18(4):186–193. | ||
Ghigna C, Riva S, Biamonti G. Alternative splicing of tumor suppressors and oncogenes. Cancer Treat Res. 2013;158:95–117. | ||
Conn G, Bayne ML, Soderman DD, et al. Amino acid and cDNA sequences of a vascular endothelial cell mitogen that is homologous to platelet-derived growth factor. Proc Natl Acad Sci U S A. 1990;87(7):2628–2632. | ||
Ferrara N. Vascular endothelial growth factor and the regulation of angiogenesis. Recent Prog Horm Res. 2000;55:15–35; discussion 35–16. | ||
Ferrara N, Henzel WJ. Pituitary follicular cells secrete a novel heparin-binding growth factor specific for vascular endothelial cells. Biochem Biophys Res Commun. 1989;161(2):851–858. | ||
Plouet J, Schilling J, Gospodarowicz D. Isolation and characterization of a newly identified endothelial cell mitogen produced by AtT-20 cells. EMBO J. 1989;8(12):3801–3806. | ||
Faustino NA, Cooper TA. Pre-mRNA splicing and human disease. Genes Dev. 2003;17(4):419–437. | ||
Tazi J, Bakkour N, Stamm S. Alternative splicing and disease. Biochim Biophys Acta. 2009;1792(1):14–26. | ||
Ladomery MR, Harper SJ, Bates DO. Alternative splicing in angiogenesis: the vascular endothelial growth factor paradigm. Cancer Lett. 2007;249(2):133–142. | ||
Spano JP, Atlan D, Breau JL, Farge D. AIDS and non-AIDS-related malignancies: a new vexing challenge in HIV-positive patients. Part II. Cervical and anal squamous epithelial lesions, lung cancer, testicular germ cell cancers, and skin cancers. Eur J Intern Med. 2002;13(4):227–232. | ||
Verheul HM, Voest EE, Schlingemann RO. Are tumours angiogenesis-dependent? J Pathol. 2004;202(1):5–13. | ||
Lopez C, Ayllon MA, Navas-Castillo J, Guerri J, Moreno P, Flores R. Molecular variability of the 5’- and 3’-terminal regions of citrus tristeza virus RNA. Phytopathology. 1998;88(7):685–691. | ||
Garcia-Blanco MA, Baraniak AP, Lasda EL. Alternative splicing in disease and therapy. Nat Biotechnol. 2004;22(5):535–546. | ||
Pajares MJ, Ezponda T, Catena R, Calvo A, Pio R, Montuenga LM. Alternative splicing: an emerging topic in molecular and clinical oncology. Lancet Oncol. 2007;8(4):349–357. | ||
Fairbrother WG, Yeh RF, Sharp PA, Burge CB. Predictive identification of exonic splicing enhancers in human genes. Science. 2002;297(5583):1007–1013. | ||
Bonomi S, Gallo S, Catillo M, Pignataro D, Biamonti G, Ghigna C. Oncogenic alternative splicing switches: role in cancer progression and prospects for therapy. Int J Cell Biol. 2013;2013:962038. | ||
Venables JP. Aberrant and alternative splicing in cancer. Cancer Res. 2004;64(21):7647–7654. | ||
Wollerton MC, Gooding C, Robinson F, Brown EC, Jackson RJ, Smith CW. Differential alternative splicing activity of isoforms of polypyrimidine tract binding protein (PTB). RNA. 2001;7(6):819–832. | ||
Krawczak M, Ball EV, Fenton I, et al. Human gene mutation database-a biomedical information and research resource. Hum Mutat. 2000;15(1):45–51. | ||
Cartegni L, Chew SL, Krainer AR. Listening to silence and understanding nonsense: exonic mutations that affect splicing. Nat Rev Genet. 2002;3(4):285–298. | ||
Ars E, Serra E, Garcia J, et al. Mutations affecting mRNA splicing are the most common molecular defects in patients with neurofibromatosis type 1. Hum Mol Genet. 2000;9(2):237–247. | ||
Teraoka SN, Telatar M, Becker-Catania S, et al. Splicing defects in the ataxia-telangiectasia gene, ATM: underlying mutations and consequences. Am J Hum Genet. 1999;64(6):1617–1631. | ||
Kaida D. [Extra-splicing functions of spliceosome]. Seikagaku. 2012;84(7):562–565. Japanese. | ||
Folkman J. Tumor angiogenesis in women with node-positive breast cancer. Cancer J Sci Am. 1995;1(2):106–108. | ||
Nakagawa T, Kosugi T, Haneda M, Rivard CJ, Long DA. Abnormal angiogenesis in diabetic nephropathy. Diabetes. 2009;58(7):1471–1478. | ||
Sullivan LA, Brekken RA. The VEGF family in cancer and antibody-based strategies for their inhibition. mAbs. 2010;2(2):165–175. | ||
Folkman J. What is the evidence that tumors are angiogenesis dependent? J Natl Cancer Inst. 1990;82(1):4–6. | ||
Amankwah EK, Sellers TA, Park JY. Gene variants in the angiogenesis pathway and prostate cancer. Carcinogenesis. 2012;33(7):1259–1269. | ||
Malecki M, Kolsut P, Proczka R. Angiogenic and antiangiogenic gene therapy. Gene Ther. 2005;12(Suppl 1):S159–S169. | ||
Liekens S, De Clercq E, Neyts J. Angiogenesis: regulators and clinical applications. Biochem Pharmacol. 2001;61(3):253–270. | ||
Zhou YQ, He C, Chen YQ, Wang D, Wang MH. Altered expression of the RON receptor tyrosine kinase in primary human colorectal adenocarcinomas: generation of different splicing RON variants and their oncogenic potential. Oncogene. 2003;22(2):186–197. | ||
Kelemen O, Convertini P, Zhang Z, et al. Function of alternative splicing. Gene. 2013;514(1):1–30. | ||
David CJ, Manley JL. Alternative pre-mRNA splicing regulation in cancer: pathways and programs unhinged. Genes Dev. 2010;24(21):2343–2364. | ||
Alon T, Hemo I, Itin A, Pe’er J, Stone J, Keshet E. Vascular endothelial growth factor acts as a survival factor for newly formed retinal vessels and has implications for retinopathy of prematurity. Nat Med. 1995;1(10):1024–1028. | ||
Benjamin LE, Golijanin D, Itin A, Pode D, Keshet E. Selective ablation of immature blood vessels in established human tumors follows vascular endothelial growth factor withdrawal. J Clin Invest. 1999;103(2):159–165. | ||
Gerber HP, McMurtrey A, Kowalski J, et al. Vascular endothelial growth factor regulates endothelial cell survival through the phosphatidylinositol 3’-kinase/Akt signal transduction pathway. Requirement for Flk-1/KDR activation. J Biol Chem. 1998;273(46):30336–30343. | ||
Yuan F, Chen Y, Dellian M, Safabakhsh N, Ferrara N, Jain RK. Time-dependent vascular regression and permeability changes in established human tumor xenografts induced by an anti-vascular endothelial growth factor/vascular permeability factor antibody. Proc Natl Acad Sci U S A. 1996;93(25):14765–14770. | ||
Bates DO, Cui TG, Doughty JM, et al. VEGF165b, an inhibitory splice variant of vascular endothelial growth factor, is down-regulated in renal cell carcinoma. Cancer Res. 2002;62(14):4123–4131. | ||
Biselli-Chicote PM, Oliveira AR, Pavarino EC, Goloni-Bertollo EM. VEGF gene alternative splicing: pro- and anti-angiogenic isoforms in cancer. J Cancer Res Clin Oncol. 2012;138(3):363–370. | ||
Niu G, Chen X. Vascular endothelial growth factor as an anti-angiogenic target for cancer therapy. Curr Drug Targets. 2010;11(8):1000–1017. | ||
Kearney JB, Kappas NC, Ellerstrom C, DiPaola FW, Bautch VL. The VEGF receptor flt-1 (VEGFR-1) is a positive modulator of vascular sprout formation and branching morphogenesis. Blood. 2004;103(12):4527–4535. | ||
He Y, Smith SK, Day KA, Clark DE, Licence DR, Charnock-Jones DS. Alternative splicing of vascular endothelial growth factor (VEGF)-R1 (FLT-1) pre-mRNA is important for the regulation of VEGF activity. Mol Endocrinol. 1999;13(4):537–545. | ||
Egeblad M, Werb Z. New functions for the matrix metalloproteinases in cancer progression. Nat Rev Cancer. 2002;2(3):161–174. | ||
Pepper MS. Extracellular proteolysis and angiogenesis. Thromb Haemost. 2001;86(1):346–355. | ||
Burbridge MF, Coge F, Galizzi JP, Boutin JA, West DC, Tucker GC. The role of the matrix metalloproteinases during in vitro vessel formation. Angiogenesis. 2002;5(3):215–226. | ||
Burrell K, Zadeh G. Molecular mechanisms of tumor angiogenesis. In: Ran S, editor. Tumor Angiogenesis. Rijeka, Croatia: InTech; 2012:275–296. | ||
Bergers G, Brekken R, McMahon G, et al. Matrix metalloproteinase-9 triggers the angiogenic switch during carcinogenesis. Nat Cell Biol. 2000;2(10):737–744. | ||
Nakada M, Kita D, Futami K, et al. Roles of membrane type 1 matrix metalloproteinase and tissue inhibitor of metalloproteinases 2 in invasion and dissemination of human malignant glioma. J Neurosurg. 2001;94(3):464–473. | ||
Luttun A, Dewerchin M, Collen D, Carmeliet P. The role of proteinases in angiogenesis, heart development, restenosis, atherosclerosis, myocardial ischemia, and stroke: insights from genetic studies. Curr Atheroscler Rep. 2000;2(5):407–416. | ||
Reiss Y. Angiopoietins. Recent Results Cancer Res. 2010;180:3–13. | ||
Maisonpierre PC, Suri C, Jones PF, et al. Angiopoietin-2, a natural antagonist for Tie2 that disrupts in vivo angiogenesis. Science. 1997;277(5322):55–60. | ||
Papapetropoulos A, Fulton D, Mahboubi K, et al. Angiopoietin-1 inhibits endothelial cell apoptosis via the Akt/survivin pathway. J Biol Chem. 2000;275(13):9102–9105. | ||
Falcon BL, Hashizume H, Koumoutsakos P, et al. Contrasting actions of selective inhibitors of angiopoietin-1 and angiopoietin-2 on the normalization of tumor blood vessels. Am J Pathol. 2009;175(5):2159–2170. | ||
Duncan WC, Nio-Kobayashi J. Targeting angiogenesis in the pathological ovary. Reprod Fertil Dev. 2013;25(2):362–371. | ||
Frumovitz M, Sood AK. Vascular endothelial growth factor (VEGF) pathway as a therapeutic target in gynecologic malignancies. Gynecol Oncol. 2007;104(3):768–778. | ||
Masood R, Cai J, Zheng T, Smith DL, Hinton DR, Gill PS. Vascular endothelial growth factor (VEGF) is an autocrine growth factor for VEGF receptor-positive human tumors. Blood. 2001;98(6):1904–1913. | ||
Petrillo M, Scambia G, Ferrandina G. Novel targets for VEGF-independent anti-angiogenic drugs. Expert Opin Investig Drugs. 2012;21(4):451–472. | ||
Tait CR, Jones PF. Angiopoietins in tumours: the angiogenic switch. J Pathol. 2004;204(1):1–10. | ||
Huang YQ, Li JJ, Karpatkin S. Identification of a family of alternatively spliced mRNA species of angiopoietin-1. Blood. 2000;95(6):1993–1999. | ||
Procopio WN, Pelavin PI, Lee WM, Yeilding NM. Angiopoietin-1 and -2 coiled coil domains mediate distinct homo-oligomerization patterns, but fibrinogen-like domains mediate ligand activity. J Biol Chem. 1999;274(42):30196–30201. | ||
Kim I, Kim JH, Ryu YS, Jung SH, Nah JJ, Koh GY. Characterization and expression of a novel alternatively spliced human angiopoietin-2. J Biol Chem. 2000;275(24):18550–18556. | ||
Mezquita J, Mezquita P, Montserrat P, et al. Genomic structure and alternative splicing of chicken angiopoietin-2. Biochem Biophys Res Commun. 2000;275(2):643–651. | ||
Folkman J, Klagsbrun M, Sasse J, Wadzinski M, Ingber D, Vlodavsky I. A heparin-binding angiogenic protein – basic fibroblast growth factor – is stored within basement membrane. Am J Pathol. 1988;130(2):393–400. | ||
Gospodarowicz D. Growth factors and their action in vivo and in vitro. J Pathol. 1983;141(3):201–233. | ||
Johnson DE, Lu J, Chen H, Werner S, Williams LT. The human fibroblast growth factor receptor genes: a common structural arrangement underlies the mechanisms for generating receptor forms that differ in their third immunoglobulin domain. Mol Cell Biol. 1991;11(9):4627–4634. | ||
Lee PL, Johnson DE, Cousens LS, Fried VA, Williams LT. Purification and complementary DNA cloning of a receptor for basic fibroblast growth factor. Science. 1989;245(4913):57–60. | ||
Gavalas NG, Liontos M, Trachana SP, et al. Angiogenesis-related pathways in the pathogenesis of ovarian cancer. Int J Mol Sci. 2013;14(8):15885–15909. | ||
Frisch M, Biggar RJ, Goedert JJ. Human papillomavirus-associated cancers in patients with human immunodeficiency virus infection and acquired immunodeficiency syndrome. J Natl Cancer Inst. 2000;92(18):1500–1510. | ||
Cuninghame S, Jackson R, Zehbe I. Hypoxia-inducible factor 1 and its role in viral carcinogenesis. Virology. 2014;456–457:370–383. | ||
Semenza GL. Targeting HIF-1 for cancer therapy. Nat Rev Cancer. 2003;3(10):721–732. | ||
Purushothaman P, Uppal T, Sarkar R, Verma SC. KSHV-mediated angiogenesis in tumor progression. Viruses. 2016;8(7):pii:E198. | ||
Qian LW, Xie J, Ye F, Gao SJ. Kaposi’s sarcoma-associated herpesvirus infection promotes invasion of primary human umbilical vein endothelial cells by inducing matrix metalloproteinases. J Virol. 2007;81(13):7001–7010. | ||
Aoki Y, Jaffe ES, Chang Y, et al. Angiogenesis and hematopoiesis induced by Kaposi’s sarcoma-associated herpesvirus-encoded interleukin-6. Blood. 1999;93(12):4034–4043. | ||
Zhu X, Guo Y, Yao S, et al. Synergy between Kaposi’s sarcoma-associated herpesvirus (KSHV) vIL-6 and HIV-1 Nef protein in promotion of angiogenesis and oncogenesis: role of the AKT signaling pathway. Oncogene. 2014;33(15):1986–1996. | ||
Zhang J, He S, Wang Y, et al. Herpesviral G protein-coupled receptors activate NFAT to induce tumor formation via inhibiting the SERCA calcium ATPase. PLoS Pathog. 2015;11(3):e1004768. | ||
Ma T, Patel H, Babapoor-Farrokhran S, et al. KSHV induces aerobic glycolysis and angiogenesis through HIF-1-dependent upregulation of pyruvate kinase 2 in Kaposi’s sarcoma. Angiogenesis. 2015;18(4):477–488. | ||
Shin YC, Joo CH, Gack MU, Lee HR, Jung JU. Kaposi’s sarcoma-associated herpesvirus viral IFN regulatory factor 3 stabilizes hypoxia-inducible factor-1 alpha to induce vascular endothelial growth factor expression. Cancer Res. 2008;68(6):1751–1759. | ||
Qin Z, Dai L, Slomiany MG, Toole BP, Parsons C. Direct activation of emmprin and associated pathogenesis by an oncogenic herpesvirus. Cancer Res. 2010;70(10):3884–3889. | ||
Dai L, Bratoeva M, Toole BP, Qin Z, Parsons C. KSHV activation of VEGF secretion and invasion for endothelial cells is mediated through viral upregulation of emmprin-induced signal transduction. Int J Cancer. 2012;131(4):834–843. | ||
Wang L, Damania B. Kaposi’s sarcoma-associated herpesvirus confers a survival advantage to endothelial cells. Cancer Res. 2008;68(12):4640–4648. | ||
Bowser BS, DeWire SM, Damania B. Transcriptional regulation of the K1 gene product of Kaposi’s sarcoma-associated herpesvirus. J Virol. 2002;76(24):12574–12583. | ||
Wang L, Wakisaka N, Tomlinson CC, et al. The Kaposi’s sarcoma-associated herpesvirus (KSHV/HHV-8) K1 protein induces expression of angiogenic and invasion factors. Cancer Res. 2004;64(8):2774–2781. | ||
Yao S, Hu M, Hao T, et al. MiRNA-891a-5p mediates HIV-1 Tat and KSHV Orf-K1 synergistic induction of angiogenesis by activating NF-kappaB signaling. Nucl Acids Res. 2015;43(19):9362–9378. | ||
Xue M, Yao S, Hu M, et al. HIV-1 Nef and KSHV oncogene K1 synergistically promote angiogenesis by inducing cellular miR-718 to regulate the PTEN/AKT/mTOR signaling pathway. Nucl Acids Res. 2014;42(15):9862–9879. | ||
Cianfrocca M, Cooley TP, Lee JY, et al. Matrix metalloproteinase inhibitor COL-3 in the treatment of AIDS-related Kaposi’s sarcoma: a phase I AIDS malignancy consortium study. J Clin Oncol. 2002;20(1):153–159. | ||
Stine JT, Wood C, Hill M, et al. KSHV-encoded CC chemokine vMIP-III is a CCR4 agonist, stimulates angiogenesis, and selectively chemoattracts TH2 cells. Blood. 2000;95(4):1151–1157. | ||
Samols MA, Skalsky RL, Maldonado AM, et al. Identification of cellular genes targeted by KSHV-encoded microRNAs. PLoS Pathog. 2007;3(5):e65. | ||
Hansen A, Henderson S, Lagos D, et al. KSHV-encoded miRNAs target MAF to induce endothelial cell reprogramming. Genes Dev. 2010;24(2):195–205. | ||
Mansouri M, Viswanathan K, Douglas JL, et al. Molecular mechanism of BST2/tetherin downregulation by K5/MIR2 of Kaposi’s sarcoma-associated herpesvirus. J Virol. 2009;83(19):9672–9681. | ||
Gantt S, Casper C. Human herpesvirus 8-associated neoplasms: the roles of viral replication and antiviral treatment. Curr Opin Infect Dis. 2011;24(4):295–301. | ||
McCormick C, Ganem D. The kaposin B protein of KSHV activates the p38/MK2 pathway and stabilizes cytokine mRNAs. Science. 2005;307(5710):739–741. | ||
Tsai YH, Wu MF, Wu YH, et al. The M type K15 protein of Kaposi’s sarcoma-associated herpesvirus regulates microRNA expression via its SH2-binding motif to induce cell migration and invasion. J Virol. 2009;83(2):622–632. | ||
Ribatti D, Nico B, Ranieri G, Specchia G, Vacca A. The role of angiogenesis in human non-Hodgkin lymphomas. Neoplasia. 2013;15(3):231–238. | ||
Vacca A, Ribatti D, Ria R, et al. Proteolytic activity of human lymphoid tumor cells. Correlation with tumor progression. Dev Immunol. 2000;7(2–4):77–88. | ||
Vacca A, Ria R, Presta M, et al. Alpha(v)beta(3) integrin engagement modulates cell adhesion, proliferation, and protease secretion in human lymphoid tumor cells. Exp Hematol. 2001;29(8):993–1003. | ||
Passalidou E, Stewart M, Trivella M, et al. Vascular patterns in reactive lymphoid tissue and in non-Hodgkin’s lymphoma. Br J Cancer. 2003;88(4):553–559. | ||
Vacca A, Ribatti D, Fanelli M, et al. Expression of tenascin is related to histologic malignancy and angiogenesis in b-cell non-Hodgkin’s lymphomas. Leuk Lymphoma. 1996;22(5–6):473–481. | ||
Ribatti D, Vacca A, Nico B, Fanelli M, Roncali L, Dammacco F. Angiogenesis spectrum in the stroma of B-cell non-Hodgkin’s lymphomas. An immunohistochemical and ultrastructural study. Eur J Haematol. 1996;56(1–2):45–53. | ||
Crivellato E, Nico B, Vacca A, Ribatti D. B-cell non-Hodgkin’s lymphomas express heterogeneous patterns of neovascularization. Haematologica. 2003;88(6):671–678. | ||
Zhao WL, Mourah S, Mounier N, et al. Vascular endothelial growth factor-A is expressed both on lymphoma cells and endothelial cells in angioimmunoblastic T-cell lymphoma and related to lymphoma progression. Lab Invest. 2004;84(11):1512–1519. | ||
Salven P, Orpana A, Teerenhovi L, Joensuu H. Simultaneous elevation in the serum concentrations of the angiogenic growth factors VEGF and bFGF is an independent predictor of poor prognosis in non-Hodgkin lymphoma: a single-institution study of 200 patients. Blood. 2000;96(12):3712–3718. | ||
Ajiro M, Zheng ZM. Oncogenes and RNA splicing of human tumor viruses. Emerg Microb Infect. 2014;3(9):e63. | ||
Wieland U, Kreuter A. Merkel cell polyomavirus infection and Merkel cell carcinoma in HIV-positive individuals. Curr Opin Oncol. 2011;23(5):488–493. | ||
Moens U, Rasheed K, Abdulsalam I, Sveinbjornsson B. The role of Merkel cell polyomavirus and other human polyomaviruses in emerging hallmarks of cancer. Viruses. 2015;7(4):1871–1901. | ||
Liberto MC, Zicca E, Pavia G, et al. Virological mechanisms in the coinfection between HIV and HCV. Mediators Inflamm. 2015;2015:320532. | ||
Lin D, Wu J. Hypoxia inducible factor in hepatocellular carcinoma: a therapeutic target. World J Gastroenterol. 2015;21(42):12171–12178. | ||
Hernandez-Lopez HR, Graham SV. Alternative splicing in human tumour viruses: a therapeutic target? Biochem J. 2012;445(2):145–156. | ||
Munaut C, Colige AC, Lambert CA. Alternative splicing: a promising target for pharmaceutical inhibition of pathological angiogenesis? Curr Pharm Des. 2010;16(35):3864–3876. | ||
Baralle D, Lucassen A, Buratti E. Missed threads. The impact of pre-mRNA splicing defects on clinical practice. EMBO Rep. 2009;10(8):810–816. | ||
Bates DO, Morris JC, Oltean S, Donaldson LF. Pharmacology of modulators of alternative splicing. Pharmacol Rev. 2017;69(1):63–79. | ||
Richon VM. Targeting histone deacetylases: development of vorinostat for the treatment of cancer. Epigenomics. 2010;2(3):457–465. | ||
Sumanasekera C, Watt DS, Stamm S. Substances that can change alternative splice-site selection. Biochem Soc Trans. 2008;36(Pt 3):483–490. | ||
Puppin C, Passon N, Franzoni A, Russo D, Damante G. Histone deacetylase inhibitors control the transcription and alternative splicing of prohibitin in thyroid tumor cells. Oncol Rep. 2011;25(2):393–397. | ||
Mineur P, Colige AC, Deroanne CF, et al. Newly identified biologically active and proteolysis-resistant VEGF-A isoform VEGF111 is induced by genotoxic agents. J Cell Biol. 2007;179(6):1261–1273. | ||
Markus MA, Marques FZ, Morris BJ. Resveratrol, by modulating RNA processing factor levels, can influence the alternative splicing of pre-mRNAs. PLoS One. 2011;6(12):e28926. | ||
Sabisz M, Skladanowski A. Modulation of cellular response to anticancer treatment by caffeine: inhibition of cell cycle checkpoints, DNA repair and more. Curr Pharm Biotechnol. 2008;9(4):325–336. | ||
Lu GY, Huang SM, Liu ST, Liu PY, Chou WY, Lin WS. Caffeine induces tumor cytotoxicity via the regulation of alternative splicing in subsets of cancer-associated genes. Int J Biochem Cell Biol. 2014;47:83–92. | ||
Li X, Gu F, Niu C, et al. VEGF111b, a C-terminal splice variant of VEGF-A and induced by mitomycin C, inhibits ovarian cancer growth. J Transl Med. 2015;13:164. | ||
Kim MH. Protein phosphatase 1 activation and alternative splicing of Bcl-X and Mcl-1 by EGCG + ibuprofen. J Cell Biochem. 2008;104(4):1491–1499. | ||
Schlomann U, Rathke-Hartlieb S, Yamamoto S, Jockusch H, Bartsch JW. Tumor necrosis factor alpha induces a metalloprotease-disintegrin, ADAM8 (CD 156): implications for neuron-glia interactions during neurodegeneration. J Neurosci. 2000;20(21):7964–7971. | ||
Monini P, Sgadari C, Barillari G, Ensoli B. HIV protease inhibitors: antiretroviral agents with anti-inflammatory, anti-angiogenic and anti-tumour activity. J Antimicrob Chemother. 2003;51(2):207–211. | ||
Barillari G, Sgadari C, Toschi E, Monini P, Ensoli B. HIV protease inhibitors as new treatment options for Kaposi’s sarcoma. Drug Resist Updat. 2003;6(4):173–181. | ||
Gacche RN, Meshram RJ. Angiogenic factors as potential drug target: efficacy and limitations of anti-angiogenic therapy. Biochim Biophys Acta. 2014;1846(1):161–179. | ||
Yu Y, Flint A, Dvorin EL, Bischoff J. AC133-2, a novel isoform of human AC133 stem cell antigen. J Biol Chem. 2002;277(23):20711–20716. | ||
Aoka Y, Johnson FL, Penta K, et al. The embryonic angiogenic factor Del1 accelerates tumor growth by enhancing vascular formation. Microvasc Res. 2002;64(1):148–161. | ||
Cheng N, Brantley DM, Chen J. The ephrins and Eph receptors in angiogenesis. Cytokine Growth Factor Rev. 2002;13(1):75–85. | ||
Sheen-Chen SM, Liu YW, Eng HL, Chou FF. Serum levels of hepatocyte growth factor in patients with breast cancer. Cancer Epidemiol Biomarkers Prev. 2005;14(3):715–717. | ||
Shanmugalingam T, Bosco C, Ridley AJ, Van Hemelrijck M. Is there a role for IGF-1 in the development of second primary cancers? Cancer Med. 2016;5(11):3353–3367. | ||
Craig MJ, Loberg RD. CCL2 (monocyte chemoattractant protein-1) in cancer bone metastases. Cancer Metastasis Rev. 2006;25(4):611–619. | ||
Kato M, Maeta H, Kato S, Shinozawa T, Terada T. Immunohistochemical and in situ hybridization analyses of midkine expression in thyroid papillary carcinoma. Mod Pathol. 2000;13(10):1060–1065. | ||
Mook OR, Frederiks WM, Van Noorden CJ. The role of gelatinases in colorectal cancer progression and metastasis. Biochim Biophys Acta. 2004;1705(2):69–89. | ||
Karjalainen K, Jaalouk DE, Bueso-Ramos CE, et al. Targeting neuropilin-1 in human leukemia and lymphoma. Blood. 2011;117(3):920–927. | ||
Yang GY, Taboada S, Liao J. Induced nitric oxide synthase as a major player in the oncogenic transformation of inflamed tissue. Methods Mol Biol. 2009;512:119–156. | ||
Okumura N, Yoshida H, Kitagishi Y, Nishimura Y, Matsuda S. Alternative splicings on p53, BRCA1 and PTEN genes involved in breast cancer. Biochem Biophys Res Commun. 2011;413(3):395–399. | ||
Tanimoto R, Lu KG, Xu SQ, et al. Mechanisms of progranulin action and regulation in genitourinary cancers. Front Endocrinol. 2016;7:100. | ||
Raimondi C, Falasca M. Targeting PDK1 in cancer. Curr Med Chem. 2011;18(18):2763–2769. | ||
Su SC, Lin CW, Yang WE, Fan WL, Yang SF. The urokinase-type plasminogen activator (uPA) system as a biomarker and therapeutic target in human malignancies. Expert Opin Ther Targets. 2016;20(5):551–566. | ||
Moody TW, Nuche-Berenguer B, Jensen RT. Vasoactive intestinal peptide/pituitary adenylate cyclase activating polypeptide, and their receptors and cancer. Curr Opin Endocrinol Diabetes Obes. 2016;23(1):38–47. | ||
Breier G, Grosser M, Rezaei M. Endothelial cadherins in cancer. Cell Tissue Res. 2014;355(3):523–527. | ||
Tiscornia AC, Cayota A, Landoni AI, et al. Post-transcriptional regulation of inducible nitric oxide synthase in chronic lymphocytic leukemia B cells in pro- and antiapoptotic culture conditions. Leukemia. 2004;18(1):48–56. | ||
Keith B, Johnson RS, Simon MC. HIF1α and HIF2α: sibling rivalry in hypoxic tumor growth and progression. Nat Rev Cancer. 2011;12(1):9–22. | ||
Wang CJ, Sparano J, Palefsky JM. Human immunodeficiency virus/AIDS, human papillomavirus, and anal cancer. Surg Oncol Clin North Am. 2017;26(1):17–31. | ||
Karamysheva AF. Mechanisms of angiogenesis. Biochemistry. 2008;73(7):751–762. |
 © 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.