Back to Journals » OncoTargets and Therapy » Volume 8
Ability of PITX2 methylation to predict survival in patients with prostate cancer
Authors Li J, Zhang Y, Wen B, Li M, Wang Y
Received 4 March 2015
Accepted for publication 20 July 2015
Published 24 November 2015 Volume 2015:8 Pages 3507—3512
DOI https://doi.org/10.2147/OTT.S83914
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Faris Farassati
This paper has been retracted
Jiu-zhi Li,1,2 Yu Zhang,2 Bin Wen,2 Ming Li,2 Yu-jie Wang1
1Department of Urology, First Affiliated Hospital of Xinjiang Medical University, 2Department of Urology, People’s Hospital of Xinjiang Uygur Autonomous Region, Urumqi, People’s Republic of China
Background: The aim of this study was to explore whether candidate gene methylation can effectively predict death from prostate cancer.
Methods: After reviewing the literature to identify likely candidate genes, we assembled a case-control cohort (in a 1:2 ratio) to explore the distribution of PITX2, WNT5a, SPARC, EPB41L3, and TPM4 methylation levels. The case group comprised 45 patients with a Gleason score ≤7 who had died as a result of prostate cancer, and the control group comprised 90 current prostate cancer patients or those who died of other causes. The methylation possibility of each of the candidate genes were maximized. Univariate conditional logistic was applied for data analysis and to evaluate prediction efficiency of gene methylation on prostate cancer.
Results: The results indicated that a raised level of PITX2 methylation increased the likelihood of death due to prostate cancer by 10% (odds ratio 1.56, 95% confidence interval 1.17–2.08; P=0.005). Methylation of SPARC was found to be able to distinguish between benign prostate hyperplasia and prostate cancer.
Conclusion: Methylation of PITX2 is an effective biomarker to predict death from prostate cancer, particularly in patients with a low Gleason score.
Keywords: DNA methylation, PITX2, prognostic biomarkers, prostatic cancer
Introduction
The progression of prostate cancer differs between individuals, with some patients dying of metastatic cancer within only a few months of diagnosis, whereas in others it may be years after diagnosis or not at all. At present, serum prostate-specific antigen testing is the main method used to diagnose prostate cancer in clinical practice.1 However, elevated serum prostate-specific antigen levels can also reflect pathological changes in other systems, so this test has poor specificity for prostate cancer. There is also no test presently available to predict the aggressiveness of the disease.2,3 The Gleason score is one of the most practical methods for evaluating a prostate cancer prognosis. However, errors can occur in the assessment itself or the interaction between them may also appear. Furthermore, inaccurate positioning of the needle during biopsy of the cancer tissue may result in unreliable sampling and scoring.4 Therefore, existing tools and methods available cannot specifically test for, or accurately diagnose how aggressive the disease is, and thereby identify which patients would benefit from aggressive intervention or a more conservative approach. During 12 years of comparing observations between randomized clinical studies of prostate cancer localization and conservative observation, complete resection of the prostate did not significantly reduce the related death rate of prostate cancer patients.5 New biomarkers are needed which can accurately and effectively evaluate the disease’s progression in a patient with cancer. In this study, we will explore the clinical and practical significance of these biomarkers based on their cell cycle progression score of RNA, apparent biological genetic markers, and protein expression.
Development of prostate cancer is associated with methylation of DNA, and there is increasing evidence to suggest that methylation has the potential to improve sensitivity in the diagnosis of prostate cancer, and that DNA methylation can play a complementary role, or even replace the cell cycle progression score and immunohistochemical analysis, in this regard.6,7 The advantages of methylation testing include its robustness as a test for stable DNA and its low cost. However, as a clinical method for detecting the aggressiveness of the prostate cancer and its potential risk of death, its prognostic value is limited to primary stages instead of terminal points, in detecting its postsurgical biochemical recurrence. In one observational cohort study, HSPB1 methylation was examined as a means to predict the death rate of patients with prostate cancer.8 In our study, which focused on patients with localized prostate cancer and a Gleason score ≤7, in both the case group and control group, we searched for genes similar to HSPB1 in order to explore whether methylation in any of the identified genes could be used as an index of survival prognosis in patients with prostate cancer. The study included two phases, ie, an evaluation of methylation levels of WNT5a, CTNNB1, SPARC, EPB41L3, PITX2, TOP2a, CDC20, LINE1, miR-23b, and TPM4 in samples of frozen prostate cancer and benign prostatic hyperplasia. The aforementioned genes, in particular PITX2I, were identified through a literature review as being abnormally expressed or methylated in prostate cancer, and a number of studies indicated their ability to predict biochemical recurrence in patients with prostate cancer. We investigated the expression of PTIX2 by real-time polymerase chain reaction (PCR), and undertook further testing to explore whether the expression of this gene differed between those found in prostate cancer or in benign prostatic hyperplasia. Our hypothesis was that there would be significant differences in methylation levels between the case group and control group and that untreated patients with prostate cancer with a low Gleason score may still have a high risk of mortality.
Materials and methods
Specimens and patient information
Prostate tissue specimens were taken from patients with either prostate cancer or benign prostatic hyperplasia. DNA from 20 cases were extracted from frozen tissue (ten cases with prostatic cancer, and ten cases with benign prostatic hyperplasia), and applied in the first stage of this study. In the second stage of the study, 367 DNA samples was extracted from prostate tissue specimens of prostate cancer patients by formalin-fixed transurethral resection. These patients had presented typical clinical symptoms and did not receive any surgery or radiation therapy for half a year. The primary endpoint in the study was death from prostatic cancer of which the relevant information was obtained from hospital medical records. Of the 367 samples, 275 of these patients had a Gleason grade of ≤7 points, with 229 of these cases being still alive at the time or had died of other systematic diseases, leaving 46 cases with death specially due to prostatic cancer, which were used as our case group. Ninety of the 229 cases who were either still alive or had died from other systematic diseases were used as the control group. To make up the 90 patients in the control group, all patients who were still alive from the 367 cases were included, and the patients who died of other systematic diseases with the longest preliminary end point, decided by survival status, were chosen to make up the rest of the group. Survival was longer in the control group that in the case group. This study was approved by the ethics committee of Xinjiang Medical University, Xinjiang, People’s Republic of China.
Methylation analysis
We used an EpiTect bisulfite reagent kit (Qiagen NV, Venlo, the Netherlands) to transform 120–200 ng of DNA, and performed a PCR analysis of transformational DNA using a PyroMark PCR reagent kit (Qiagen) following the manufacturer’s instructions. The transformational DNA was equal to 1,000 formalin-fixed, paraffin-embedded tissues or DNA from 400 frozen tissue cells. PyroMark Assay Design v 2.0.1.15 (Qiagen) was used to design the primers and avoid the overlapping of CpG positions on the basis of previous reports. The same 3–6 CpG positions for the targets of PITX2, LINE1, TPM4, and SPARC were chosen. In order to internally control the total bisulfite conversion, non-CpG cytosine was added when analyzed, with each PCR reaction containing 12.5 μL PCR main mixture, 2.5 μL Coral red, 2 μL DNA, 5 pmol primer, and double distilled water. The total volume was adjusted to 25 μL with a further 2.5 mM MgCl2 added to each reaction for the determination of LINE1. The mixture was circulated for 10 minutes at 95°C followed by 45 circulations at 94°C (30 seconds), and then underwent annealing for a further 10 minutes at the temperature of 72°C. The accuracy of amplification was confirmed by QIAxcel (Qiagen) and by pyrosequencing. A standard curve was drawn for the positive control (0.50% and 100% human gene methylation), and for the non-template control. Lastly, 200 ng of non-methylation and supermethylation DNA was used to obtain different ratios of DNA methylation and to evaluate the hydrogen sulfate conversion.
Statistical analysis
The Mann–Whitney U-test was used to compare mean gene methylation levels between cancer tissues and benign prostate hyperplasia tissues. In this condition, univariate conditional logistic regression model at maximum conditional likelihood was used to analyze the methylation of each gene and its primary end point. Each kind of gene was analyzed separately, and P<0.05 indicated a statistically significant difference. In order to reduce errors, we used the Benjamini–Hochberg method to keep the false positive rate at 5%.
Results
DNA methylation in frozen tissues
Figure 1 shows the methylation levels of the genes of interest in tissue from ten cases of prostate cancer and ten cases of benign prostatic hyperplasia. The methylation levels of EPB41L3, SPARC, PITX2, WNT5a, miR-23b, TOP2a, and CDC20 genes in the cancer tissue were significantly higher than those in the benign prostatic hyperplasia; the most obvious difference between the two types of tissue was in SPARC (P=0.0002, area under the curve 1.0). Methylation levels for CDC20, TOP2A, and CTNNB1 were relatively lower, <5%, and this low level may be due to the reduced accuracy of pyrosequencing that is caused by measurement error. At the other end of the spectrum, methylation levels of miR-23b and LINE1 were higher in all cancer detected samples. The maximum variation between cancer tissue and benign prostatic hyperplastic tissue can be as high as 10% to 20%.
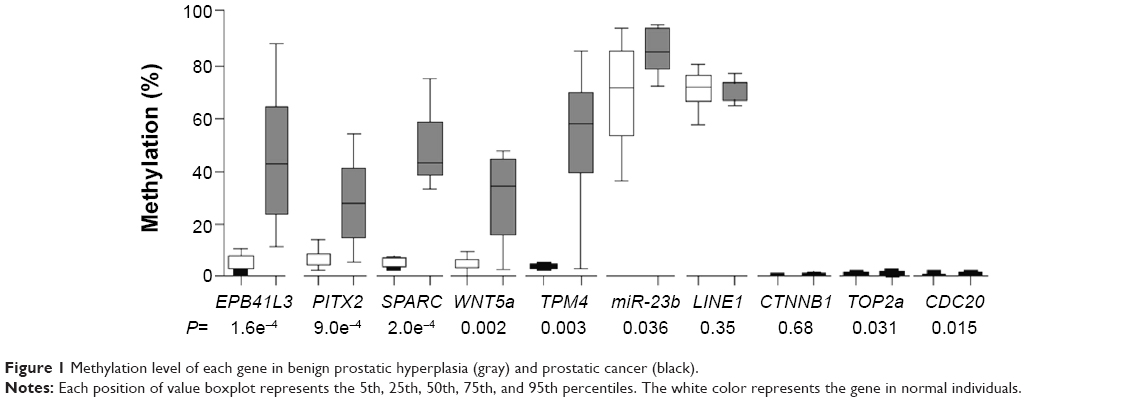 | Figure 1 Methylation level of each gene in benign prostatic hyperplasia (gray) and prostatic cancer (black). |
DNA methylation in the case-control cohort
Gene methylation levels for EPB41L3, SPARC, PITX2, WNT5a, and TPM4 were tested in 135 patients (Figure 2), and the number of genes detected in each patient differed. Table 1 shows the case number and the number of patients included in the analysis of each gene. Table 1 shows that each gene methylation with a kind of layered distribution in case group and control group. The mean duration of follow-up was 7.8 years (median was 6.1 years) in the case group, and 15.3 years (interquartile range 6.8) in the control group. The methylation of each gene, univariate conditional logistic regression analysis of death from prostate cancer, and odds ratio (OD) are illustrated in Table 1. Before multiple testing adjustment, the methylation level of PITX2 and WNT5a was evaluated to determine which of the two genes may be related to a patient’s death due to prostate cancer (OD 1.56, 95% confidence interval 1.17–2.08 vs OD 1.28, 95% confidence interval 1.02–1.60). After adjusting for a 5% false positive rate, the methylation level of only PITX2 remained significant for predicting the risk of death from prostate cancer (P=0.005). Since the methylation level of PITX2 increased by 10%, the prostate cancer-related death events will increase by at least 1.56 times.
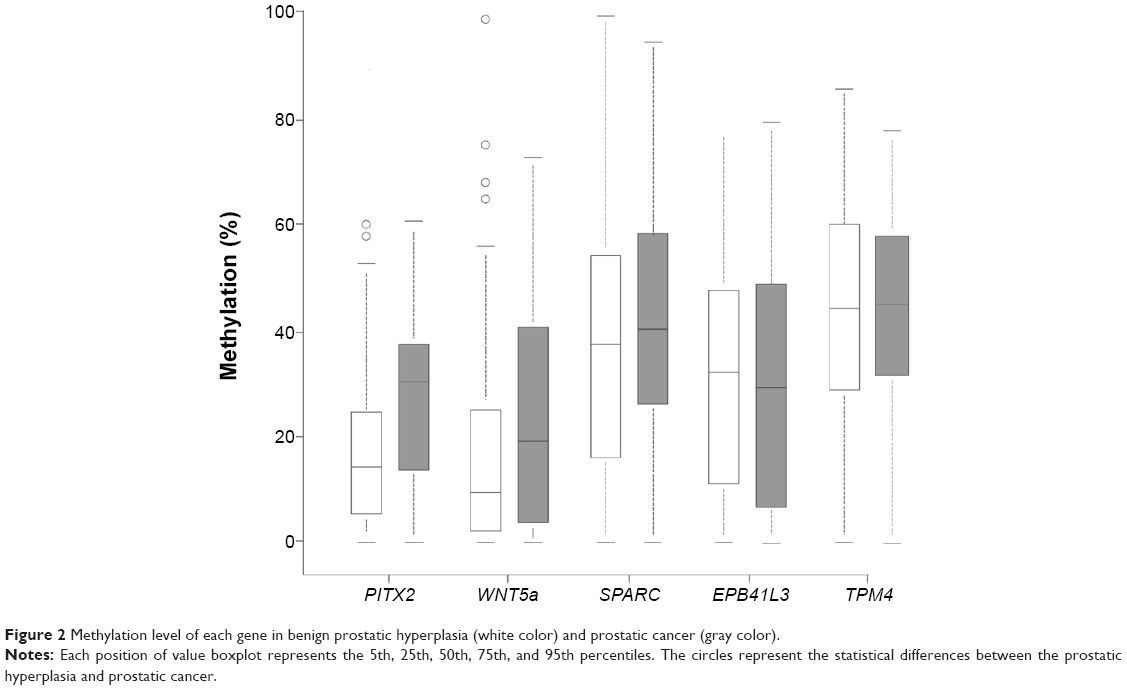 | Figure 2 Methylation level of each gene in benign prostatic hyperplasia (white color) and prostatic cancer (gray color). |
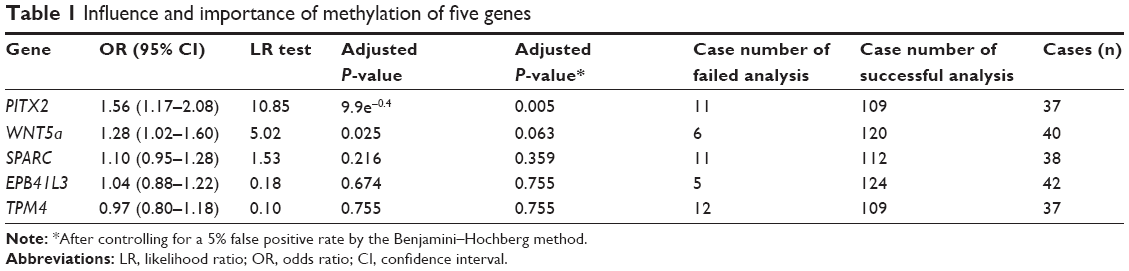 | Table 1 Influence and importance of methylation of five genes |
Discussion
PITX2 and methylation of WNT5a can be regarded as biological markers in the prognosis of prostate cancer, which evaluate Gleason score and the risk of death of patients with prostate cancer. Both PITX2 and methylation of WNT5a react in the Wnt signaling pathway. PITX2 is thought to be a transcription factor that is related to the beta-chain protein-dependent or independent pathways. WNT5a can be regarded as a typical ligand that can activate the beta-chain protein-dependent pathway.9,10 In this study, we did not deliberately choose the Wnt signaling pathway biological factors, but obtained them through a literature review at the initial stage of this study.
Supermethylation of PITX2 has been confirmed to be a prognostic marker in prostate cancer, and can be used to assess for biochemical recurrence. It has also been found to be to the reduction in mRNA transcription.11–14 Current studies related to PITX2 and cancer biochemical recurrence rates use microarray and real-time PCR technology as the main methods to evaluate its methylation.15 To our knowledge, this is the first study to confirm a direct correlation between methylation of PITX2 and a Gleason score <7 and the risk of death in patients with prostate cancer. As a prognostic biomarker, its dominance ratio is 1.56 (as an independent predictor, its dominance ratio is more than 1.5).
In this study, we use consecutive methylation data to define the value as either positive or negative. Further study is required to find the critical value of each gene and define them as positive or negative markers. Predictable biomarkers can be used in multivariate classifications, and their correlation to other genes can affect the clinical values. The critical values of multivariate classification are always the sum of the whole genome value, rather than the methylation level based at a singular gene level. In the present study, we also found that methylation of SPARC can be used to distinguish between benign prostate hyperplasia and prostatic cancer (Figure 1), and there have been several reports suggesting that methylation of SPARC can also be a potential biomarker in diagnosis.16–18 Further research with a larger sample size of prostate biopsy results are needed to confirm this.
The main limitation of this study is that we used formalin-fixed tissue, but we found that it is more suitable to use patients with prostate cancer who had never received any treatment in the last 20 years, although the preliminary conclusion is still needed to be confirmed by biopsy specimens evaluations. Cell cycle progression as the first validated scoring, and then confirmed by biopsy specimens, show that formalin-fixed tissue samples are suitable for the study of prostate cancer as prognostic biomarkers.19,20 A second limitation of this study is that the sample size was small, with only 45 cases of patients who had died as an outcome of prostate cancer and 90 cases as a control for comparison. However, the aim of this study was basically achieved, since we found at least one significant odds ratio of biomarker. The two obtained values as possible genetic biomarkers need to be further tested by a larger sample evaluation. By using molecular detection of body fluid such as a blood sample, compared with other invasive procedures such as biopsy, biomarkers are the ideal detection method. In some studies, aberrant gene methylation can be detected in blood and urine,21,22 so methylation of PICX2 should also be able to be detected in body fluids.
In conclusion, patients with a low Gleason score are generally considered to be a low-risk population suitable for conservative treatment.23–25 However, it is still very important to identify biological factors that can predict the aggressiveness of a tumor. In this study, we found high PITX2 levels in patients who were given a low Gleason score but still went on to die from prostate cancer. Therefore, methylation of PITX2 can be used as a potential biomarker to predict the risk of death in prostate cancer patients with lower Gleason scores. However, this needs to be further confirmed by a biopsy study from a larger sample size.
Disclosure
The authors report no conflicts of interest in this work.
References
Sha J, Bo J, Pan J, et al. Ductal adenocarcinoma of the prostate: immunohistochemical findings and clinical significance. Onco Targets Ther. 2013;6:1501–1506. | ||
Cuzick J, Fisher G, Kattan MW, et al. Long-term outcome among men with conservatively treated localised prostate cancer. Br J Cancer. 2006;95(9):1186–1194. | ||
Egevad L, Ahmad AS, Algaba F, et al. Standardization of Gleason grading among 337 European pathologists. Histopathology. 2013;62(2):247–256. | ||
Wilt TJ, MacDonald R, Rutks I, Shamliyan TA, Taylor BC, Kane RL. Systematic review: comparative effectiveness and harms of treatments for clinically localized prostate cancer. Ann Intern Med. 2008; 148(6):435–448. | ||
Chou R, Croswell JM, Dana T, et al. Screening for prostate cancer: a review of the evidence for the US Preventive Services Task Force. Ann Intern Med. 2011;155(11):762–771. | ||
Wilt TJ, Brawer MK, Jones KM, et al. Radical prostatectomy versus observation for localized prostate cancer. N Engl J Med. 2012;367(3):203–213. | ||
Cuzick J, Swanson GP, Fisher G, et al. Prognostic value of an RNA expression signature derived from cell cycle proliferation genes in patients with prostate cancer: a retrospective study. Lancet Oncol. 2011;12(3):245–255. | ||
Hessels D, Verhaegh GW, Schalken JA, Witjes JA. Applicability of biomarkers in the early diagnosis of prostate cancer. Expert Rev Mol Diagn. 2004;4(4):513–526. | ||
Berney DM, Gopalan A, Kudahetti S, et al. Ki-67 and outcome in clinically localised prostate cancer: analysis of conservatively treated prostate cancer patients from the Trans-Atlantic Prostate Group study. Br J Cancer. 2009;100(6):888–893. | ||
Yang M, Park JY. DNA methylation in promoter region as biomarkers in prostate cancer. Methods Mol Biol. 2012;863:67–109. | ||
Rodriguez-Paredes M, Esteller M. Cancer epigenetics reaches mainstream oncology. Nat Med. 2011;17:330–339. | ||
Li LC. Epigenetics of prostate cancer. Front Biosci. 2007;12:3377–3397. | ||
Vasiljević N, Ahmad AS, Beesley C, et al. Association between DNA methylation of HSPB1 and death in low Gleason score prostate cancer. Prostate Cancer Prostatic Dis. 2013;16(1):35–40. | ||
Richiardi L, Fiano V, Vizzini L, et al. Promoter methylation in APC, RUNX3, and GSTP1 and mortality in prostate cancer patients. J Clin Oncol. 2009;27(19):3161–3168. | ||
Hernández HG, Tse MY, Pang SC, Arboleda H, Forero DA. Optimizing methodologies for PCR-based DNA methylation analysis. Biotechniques. 2013;55(4):181–197. | ||
Chao C, Chi M, Preciado M, Black MH. Methylation markers for prostate cancer prognosis: a systematic review. Cancer Causes Control. 2013;24(9):1615–1641. | ||
Wong SY, Haack H, Kissil JL, et al. Protein 4.1B suppresses prostate cancer progression and metastasis. Proc Natl Acad Sci U S A. 2007;104(31):12784–12789. | ||
Yamamoto H, Oue N, Sato A, et al. Wnt5a signaling is involved in the aggressiveness of prostate cancer and expression of metalloproteinase. Oncogene. 2010;29(14):2036–2046. | ||
Derosa CA, Furusato B, Shaheduzzaman S, Srikantan V. Elevated osteonectin/SPARC expression in primary prostate cancer predicts metastatic progression. Prostate Cancer Prostatic Dis. 2012;15:150–156. | ||
Majid S, Dar AA, Saini S, et al. miR-23b represses proto-oncogene Src kinase and functions as methylation-silenced tumor suppressor with diagnostic and prognostic significance in prostate cancer. Cancer Res. 2012;72(24):6435–6446. | ||
Weiss G, Cottrell S, Distler J, et al. DNA methylation of the PITX2 gene promoter region is a strong independent prognostic marker of biochemical recurrence in patients with prostate cancer after radical prostatectomy. J Urol. 2009;181(4):1678–1685. | ||
Yegnasubramanian S, Haffner MC, Zhang Y, et al. DNA hypomethylation arises later in prostate cancer progression than CpG island hypermethylation and contributes to metastatic tumor heterogeneity. Cancer Res. 2008;68(21):8954–8967. | ||
Lin PC, Giannopoulou EG, Park K, et al. Epigenomic alterations in localized and advanced prostate cancer. Neoplasia. 2013;15(4): 373–383. | ||
Bañez LL, Sun L, van Leenders GJ, et al. Multicenter clinical validation of PITX2 methylation as a prostate specific antigen recurrence predictor in patients with post-radical prostatectomy prostate cancer. J Urol. 2010;184(1):149–156. | ||
Hou CP, Lee WC, Lin YH, et al. Neoadjuvant hormone therapy following treatment with robotic-assisted radical prostatectomy achieved favorable in high-risk prostate cancer. Onco Targets Ther. 2014;8:15–19. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.