Back to Journals » Pharmacogenomics and Personalized Medicine » Volume 10
ABCG2 polymorphisms in gout: insights into disease susceptibility and treatment approaches
Authors Cleophas MC, Joosten LA ![]() , Stamp LK, Dalbeth N, Woodward OM, Merriman TR
, Stamp LK, Dalbeth N, Woodward OM, Merriman TR
Received 17 December 2016
Accepted for publication 18 February 2017
Published 20 April 2017 Volume 2017:10 Pages 129—142
DOI https://doi.org/10.2147/PGPM.S105854
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Dr Martin H Bluth
MC Cleophas,1,2 LA Joosten,1–3 LK Stamp,4 N Dalbeth,5 OM Woodward,6 Tony R Merriman7
1Department of Internal Medicine, 2Radboud Institute for Molecular Life Sciences, Radboud University Medical Center, Nijmegen, the Netherlands; 3Department of Medical Genetics, Iuliu Haţieganu University of Medicine and Pharmacy, Cluj-Napoca, Romania; 4Department of Medicine, University of Otago Christchurch, Christchurch, 5Department of Medicine, University of Auckland, Auckland, New Zealand; 6Department of Physiology, University of Maryland School of Medicine, Baltimore, MD, USA; 7Department of Biochemistry, University of Otago, Dunedin, New Zealand
Abstract: As a result of the association of a common polymorphism (rs2231142, Q141K) in the ATP-binding cassette G2 (ABCG2) transporter with serum urate concentration in a genome-wide association study, it was revealed that ABCG2 is an important uric acid transporter. This review discusses the relevance of ABCG2 polymorphisms in gout, possible etiological mechanisms, and treatment approaches. The 141K ABCG2 urate-increasing variant causes instability in the nucleotide-binding domain, leading to decreased surface expression and function. Trafficking of the protein to the cell membrane is altered, and instead, there is an increased ubiquitin-mediated proteasomal degradation of the variant protein as well as sequestration into aggresomes. In humans, this leads to decreased uric acid excretion through both the kidney and the gut with the potential for a subsequent compensatory increase in renal urinary excretion. Not only does the 141K polymorphism in ABCG2 lead to hyperuricemia through renal overload and renal underexcretion, but emerging evidence indicates that it also increases the risk of acute gout in the presence of hyperuricemia, early onset of gout, tophi formation, and a poor response to allopurinol. In addition, there is some evidence that ABCG2 dysfunction may promote renal dysfunction in chronic kidney disease patients, increase systemic inflammatory responses, and decrease cellular autophagic responses to stress. These results suggest multiple benefits in restoring ABCG2 function. It has been shown that decreased ABCG2 141K surface expression and function can be restored with colchicine and other small molecule correctors. However, caution should be exercised in any application of these approaches given the role of surface ABCG2 in drug resistance.
Keywords: ABCG2, BCRP, gout, urate, uric acid, polymorphism, allopurinol
Introduction
Gout is an inflammatory arthritis caused by an extremely painful but self-limiting innate immune response to monosodium urate (MSU) crystals deposited in synovial fluid.1 Without effective management, in some individuals, gout can become chronic with the development of tophi (organized lumps of urate and immune cells2) and permanent bony erosion and disability. Gout is also comorbid with other metabolic-based conditions such as heart and kidney disease and type 2 diabetes.3 An elevated concentration of urate (hyperuricemia) is necessary, but not sufficient, for the development of gout. Host-specific and environmental factors are required for the progression from hyperuricemia to gout. Approximately 30 genetic loci, including ABCG2, influence serum urate concentrations,4 but less is understood about the genetic control of the formation of MSU crystals and the subsequent inflammatory response.5 Urate-lowering therapy is a cornerstone of gout management; however, for various reasons, it is often not effective.1 Despite the availability of a range of cost-effective urate-lowering drugs, there is a need for the development of better therapies for acute gout.
The ATP-binding cassette G2 (ABCG2) protein first came to biomedical attention through its contribution to an ATP-dependent multidrug resistance phenotype in a breast cancer cell line.6 It is one of a superfamily of 48 human ABC transporters that, in general, transport a wide array of substrates and are dependent on ATP binding to activate transport. The ABCG2 transporter is one of three well-studied multidrug transporters (others are multidrug resistance protein 1 and P-glycoprotein encoded by the ABCC1 and ABCB1 genes, respectively). These transporters export a wide range of compounds, including in the case of ABCG2, uric acid from cells. ABCG2 is a “half transporter” ABC protein, containing one membrane-spanning domain of six transmembrane helices, a N-terminal single intracellular portion containing a nucleotide-binding domain (NBD), and a short intracellular C-terminal tail (Figure 1). “Half transporters” like ABCG2 need to homodimerize to function in vitro but more likely exist in higher-order tetrameric or greater assemblages.7,8
 | Figure 1 Schematic representation of gout- and hyperuricemia-related polymorphisms in ABCG2. The ABCG2 protein consists of 655 amino acids and can be divided into an intracellular N-terminal domain, a nucleotide binding domain, six transmembrane domains, and a C-terminal end. The nucleotide-binding domain (amino acids 44-288) contains several signature sequences, named Walker A (A), Walker B (B), and the ABC signature sequence (C). The beginning and end amino acid position numbers are given for the signature sequences and the transmembrane domains. The third extracellular loop contains two cysteine residues connected by a disulfide bridge, and a glycosylated asparagine residue, both of which influence stability of the receptor. The cysteine residue at position 603 is important for homodimer formation. The V12M polymorphism is located in the N-terminal domain. The Q126X and Q141K polymorphisms are both located in the nucleotide-binding domain between the Walker A and the ABC signature sequences. Abbreviation: ABCG2, ATP-binding cassette G2. |
ABCG2, also known as breast cancer resistance protein, is overexpressed in human and animal cell lines resistant to chemotherapeutic drugs9,10 and transports anticancer drugs such as mitoxantrone and doxorubicin6 (reviewed in reference 11). ABCG2 is important in stem cell biology.12,13 Also, the wide expression pattern in normal tissues (Figure 2) suggests that ABCG2 may fill an important physiological role as an excretor of environmental and endogenous toxins, for example, heme.14 This assertion is supported by the susceptibility of ABCG2 knockout mice to diet-induced phototoxicity and protoporphyria.15 It is expressed in the small intestine (excretion and limiting absorption), blood–brain and blood–placental barriers (mediating distribution), liver and kidney (elimination and excretion), and mammary gland (transporting into milk).16 The pattern of ABCG2 expression is consistent with its role as a uric acid efflux transporter (reviewed in reference 17).
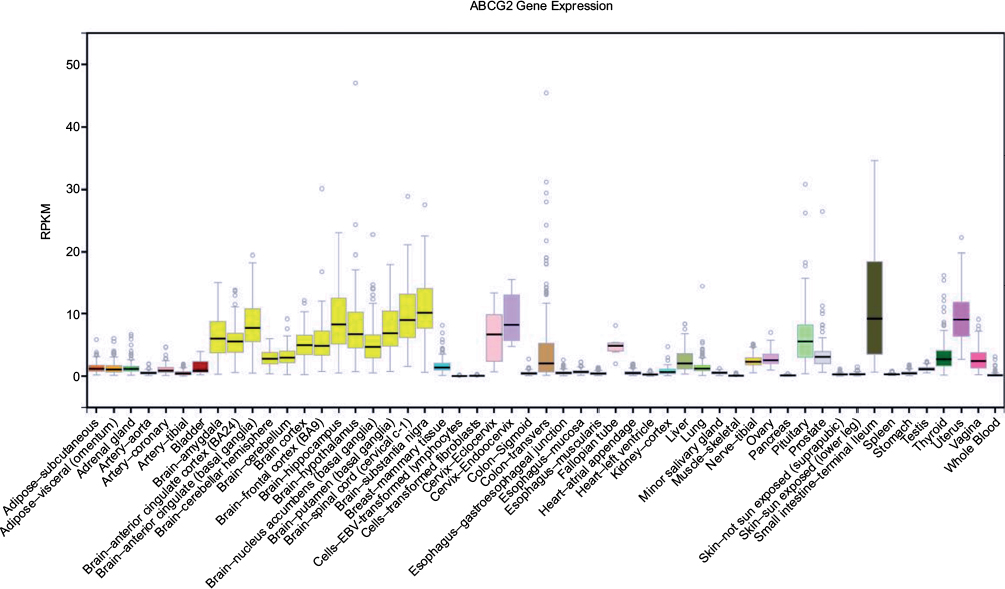 | Figure 2 Tissue expression of ABCG2 in humans. Generated from the Genotype Tissue Expression (GTeX) database, derived from deceased people (organ and tissue donors) and surgical donors. Abbreviations: ABCG2, ATP-binding cassette G2; RPKM, reads per kilobase of exon per million reads mapped. |
The ABCG2 transporter was identified as a uric acid efflux transporter only after it was associated with serum urate concentrations in a large genome-wide association study.18,19 This review explores the effects of several polymorphisms in the ABCG2 gene on susceptibility to hyperuricemia, gout, and chronic kidney disease. Furthermore, the role of ABCG2 in inflammatory responses and the possibility of ABCG2 as a treatment target are discussed.
Major ABCG2 genetic variants
The ABCG2 transporter, expressed on erythrocytes, is the basis of a recently explained blood group system, Junior (Jr).20,21 Individuals null for ABCG2 produce an ABCG2 alloantibody upon blood transfusion that can cause transfusion reactions, in extreme cases leading to fatal hemolytic disease of the fetus and newborn. The Jr(a-) phenotype is most prevalent in Japan with the most common Jr(a-) mutations being Q126X (Japanese) and R246K (Europeans).19,20
There are only two common (≥1%) missense genetic variants in the ABCG2 gene (V12M; rs2231137 and Q141K; rs2231142) and one common nonsense variant (Q126X; rs72552713) that is specific to South East Asian populations (Figure 1). The Q141K and Q126X variants influence expression levels of ABCG2 and contribute risk to phenotypes such as serum urate levels, gout, and response to the urate-lowering drug allopurinol (Table 1). Numerous other rare (<1%) variants are identified (http://www.ensembl.org), some of which have been phenotyped.22–28 The most widely studied variant is Q141K, a variant that is clinically relevant and has significant impact on ABCG2 characteristics. It is prevalent in most major population groups with the 141K allele frequency varying between 1% in African and 29% in South East Asian populations. There is consistent evidence for the 141K variant reducing the amount of ABCG2 protein expressed both in vitro19,22–25,28,29 and, in vivo, from human liver samples.30
 | Table 1 Major genetic variation in ABCG2 and impact on protein expression and phenotype Notes: Phenotype and prevalence information is given to the minor (alternative) allele. All phenotypes are compared to wild-type. Study systems: naturally occurring humans4,30,58,67,69,70,73,82,86,87,117,130–132; HEK293 (human embryonic kidney) and derivative cells22,25,27–29,36; LLC-PKI (porcine epithelial kidney cells)25,26; Sf-9 insect cells26,27,33; PA317 mouse fibroblast cells23; Xenopus oocytes.32 The Sf-9 cells were used for ATPase activity assays. *Meta-analyses. |
Residue 141 is located in the E-helix31 of the NBD of ABCG2, an area critical for the packing of the soluble portion of the protein. The 141K variant reduces ATPase activity,26,27 uric acid transport activity, and total and surface abundance (Table 1 and Figure 3A). These defects are believed to be caused by reduced NBD stability.32 Although proteolysis and thermo-stability as assessed by melting point curves of the isolated 141K NBD showed only small differences from the wild type,33 work done on the whole protein shows evidence of increased NBD instability in mammalian expression systems. Stabilizing the 141K variant protein with either suppressor mutations or with small molecules that may bind the NBD directly34 rescues the trafficking, function, and abundance of 141K ABCG2.32 Furthermore, recent modeling of the 141K NBD suggested that a loop adjacent to the Q141K substitution seems to be shifted outward, caused by a potential clash with a histidine residue (H155) at the top of the loop. This potential packing error in the NBD seems to be corrected by a H155A substitution on the 141K background as evidenced by rescued protein abundance.35 The decreased NBD stability of the 141K variant increases the interaction of the protein with the endoplasmic reticulum-associated degradation pathway and subsequent ubiquitin-mediated proteasomal degradation.29 There is also strong evidence that the 141K variant protein may accumulate in aggresomes prior to its degradation.36 In addition, protein expression is also influenced by microRNAs (37 and references therein). In one example, the 3’ untranslated region truncation allows increased expression of ABCG2, removing an hsa-miR-519c binding site that blocks translation.38 The 141K variant enhances the hsa-miR-519c-mediated repression of translation.37 Interestingly, the ABCG2 F142 residue is in an analogous position to the ∆F508 cystic fibrosis-causing mutation in the cystic fibrosis transmembrane receptor (encoded by ABCC7).32 In vitro deletion of F142 from ABCG2 leads to major processing and folding defects with no surface expression,32,33 marking the Q141 and F142 regions as a more general ABC transporter mutational “hot spot.”
 | Figure 3 Schematic overview of the roles of ABCG2 in hyperuricemia and gout. (A) The ABCG2 transporter is highly expressed in the gut, where it regulates ATP-dependent uric acid excretion. The 141K ABCG2 variant leads to decreased surface expression, targeting of the misfolded protein to aggresomes and proteasomal degradation. In addition, the polymorphism increases miRNA-mediated degradation of mRNA. The resulting increase in circulating urate not only increases the risk of gout but can also induce a more pro-inflammatory state in monocytes due to epigenetic reprogramming.129 Colchicine and HDAC inhibitors (HDACi) can restore 141K surface expression and function. (B) Monosodium urate crystals can induce mitochondrial damage, leading to the release of mitochondrial reactive oxygen species and DNA (mtROS, mtDNA), which in turn can activate the NLRP3 inflammasome. This can be limited through autophagy-mediated clearance of damaged mitochondria. ABCG2 plays a role in this process. In defective or diminished autophagy, p62 can accumulate, bind to keap-1, and induce translocation of Nrf2 to the nucleus. Nrf2 induces transcription of heme oxygenase-1 and superoxide dismutase (HO-1, SOD), which can also induce NLRP3 inflammasome activation. Abbreviations: MSU, monosodium urate; HDACi, histone deacetylase inhibitors; TLR, Toll-like receptor; NLRP3, NBD, leucine-rich family [NLR], pyrin containing 3; ABCG2, ATP-binding cassette G2; IL, interleukin. |
Regulation of ABCG2 expression
The expression of ABCG2 is regulated by several transcription factors and hormones. A large number of cis-regulatory regions have been identified in or near the promoter region of ABCG2 (reviewed in 39). Hypoxia-inducible factor-1 (HIF-1α) stabilizes under hypoxic conditions and has been found to bind to a hypoxia-responsive element in the ABCG2 promoter and increase expression.40,41 Upstream of HIF-1α, signaling via phosphatase and tensin homolog (PTEN), phosphoinositide 3-kinase (PI3K) and protein B kinase (Akt) regulates ABCG2 expression on the cell membrane.42–45 Furthermore, nuclear factor E2-related factor 2 (Nrf2), a transcription factor involved in activating oxidative stress responses, can induce ABCG2 transcription46 in addition to inducing the expression of peroxisome proliferator-activated receptor gamma (PPARγ).47,48 Finally, the hormone estrogen has been found to up-regulate ABCG2 gene expression.49–51 To our knowledge, however, no polymorphisms in any of these regulatory genes have been associated with serum urate levels or gout. However a variant in PPARGC1B (rs45520937) has been associated with gout in Han Chinese.52 PPARGC1B encodes the PGC1β transcriptional repressor, a coactivator for PPARγ that is involved in estrogen signaling53,54 in addition to a key role in maintaining mitochondrial biogenesis.55 However the association of PPARGC1B with gout has not yet been replicated and it is unknown if rs45520937 is itself associated with ABCG2 expression. Since reduced mitochondrial DNA copy number is associated with gout56 it is possible that systematically linked changes in PGC1β and associated molecules (including perhaps ABCG2) are factors connecting mitochondrial dysfunction and gout.57
ABCG2, urate control, and risk of gout
An individual’s serum urate level, like other complex traits, is a function of a suite of inherited genetic variants and environmental exposures.5 A genome-wide association study identified the Q141K variant in the ABCG2 gene as associated with serum urate concentration in people of European ancestry, with the 141K allele associated with increased urate concentration.18 This genome-wide significant level of association has been consistently replicated in other genome-wide association studies in people of European and east Asian ancestry4,58 but not in people of African–American ancestry.59,60 Lack of association in African–Americans in GWAS is likely because of the reduced power because of lower allele frequency in African–Americans (≤3%) – there is nominal evidence for the association of the 141K variant with increased serum urate levels in African–Americans18 and the increased risk of gout.19 The Q141K variant (and other variants) has been associated with serum urate levels or hyperuricemia in other populations, for example, Tibetan61 and Korean.62 Uric acid transport assays in Xenopus oocytes showed that ABCG2 transports uric acid with the 141K variant causing ~50% reduced ability to secrete uric acid.19 The regulation of ABCG2 protein in epithelial cells is largely unknown. However, similar to other uric acid renal influx transporters, ABCG2 co-immunoprecipitates with the scaffold protein PDZK1,63 suggesting a possible common regulatory pathway.
The magnitude of association of Q141K with serum urate levels is the second strongest genome-wide behind SLC2A9, which is consistently the strongest in Europeans and East Asians. Each copy of the 141K allele associates with a 0.22 mg/dL increase in serum urate in Europeans4 and an 0.14 mg/dL increase in east Asians.58 The magnitude is stronger in men than women and is modulated by body mass index (BMI), with the magnitude being stronger in lean people than those who are overweight and obese. The sex difference is most marked in lean people.64 The sex difference in association with serum urate levels is an interesting observation given the regulation of ABCG2 expression by estrogen.49–51 Compared to other typical genetic variants controlling complex phenotypes, Q141K has a very large magnitude of association, explaining ~1% of variance in serum urate levels. This compares with ~0.3% for the strongest genetic association with BMI at the FTO/IRX3 locus.65
When associations of other SNPs within the ABCG2 locus with serum urate levels are tested conditionally on Q141K genotype, there is evidence for a second independent association at the ABCG2 locus, marked by intronic SNP rs2622629.66 This second association is of clinical relevance as the rs2622629-correlated SNPs (ie those SNPs in linkage disequilibrium) include rs10011796 (r2=0.84 in Europeans). Rs10011796 is also implicated in gout and in allopurinol response (Table 1).67 The molecular mechanism whereby rs2622629 (or tightly correlated variants) influence serum urate levels is not known. However, it maps within a DNaseI hypersensitivity cluster of ~150 bp (genome.ucsc.edu) identified by the Encode project (www.encodeproject.org) consistent with the effect of the association being mediated through the control of gene expression and/or mRNA editing. Interestingly, Q141K was not associated with gout in a Han Chinese study of 143 patients,68 whereas other variants in ABCG2 were, further supporting the existence of multiple gout-associated variants in ABCG2.
Aside from Q141K, the only other common (>1%) missense variant in ABCG2 is V12M situated in a short N-terminal section of ABCG2 prior to the structurally well-defined NBD (Figure 3). This variant is genetically independent of both Q141K (r2=0.002 in Europeans) and rs2622629 (r2=0.009 in Europeans). However, the impact on ABCG2 characteristics are, compared to Q141K, very moderate and inconsistent (Table 1). There are some data on association with serum urate and gout, suggesting an independent role for V12M in urate control and risk of gout (given the genetic independence from Q141K and rs2622629). Hence, V12M can be considered a third independent effect on gout in ABCG2, after Q141K and rs10011796/rs2622629. The largest genome-wide association study in serum urate in Europeans reported an increase in serum urate of 0.077 mg/dL4 and in East Asians an increase of 0.108 mg/dL58 per copy of the 12V allele. In gout, the common 12V allele consistently confers risk in five separate samples drawn from four ancestral groups: Europeans within the UK Biobank (odds ratio [OR] =1.24 [T Merriman, unpublished data, October 2016]), Taiwanese Aborigines (OR =1.36),69 Han Chinese in Taiwan (OR =1.33),69 Han Chinese in Shanghai (OR =1.82),70 and New Zealand Māori and Pacific (Polynesian) (OR =1.31 [T Merriman, unpublished data, October 2016]). Any impact of this variant on the uric acid transport activity of ABCG2 is currently unknown. A combined meta-analysis is shown in Figure 4. Based on the impact of the V12M variant on ABCG2 characteristics (Table 1), it is not obvious how the 12V allele increases serum urate levels and risk of gout. It is possible that the V12M variant is itself not causal and in genetic correlation (linkage disequilibrium) with a separate causal variant (which is not Q141K or rs10011796/rs2622629).
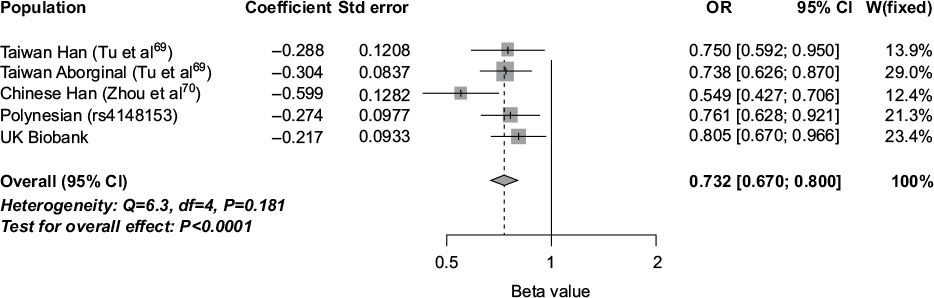 | Figure 4 Meta-analysis of association of ABCG2 V12M (rs2231137) with gout: Han Chinese (China),70 Han Chinese (Taiwan),69 Taiwanese Aboriginal,69 New Zealand Polynesian (982 cases and 941 controls), and the UK Biobank (2,422 cases and 152,249 controls). Effect is shown to the minor allele (12M). Variant rs4148153 is a surrogate marker for rs2231137 in people of Polynesian ancestry. Abbreviations: ABCG2, ATP-binding cassette G2; OR, odds ratio; CI, confidence interval; Std, standard. |
The kidney is the major regulator of serum urate levels, which are determined by the balance of secretion and reabsorption. This is controlled by reuptake transporters such as GLUT9 (SLC2A9) and URAT1 (SLC22A12) and secretory transporters ABCG271 and NPT1/NPT4 (SLC17A1, SLC17A3),72 all of which are genetically associated with serum urate concentration.4 Expression of most of these transporters is relatively high in the kidney or, for URAT1, restricted to the kidney. However, expression of ABCG2 is also relatively high in the gut (Figure 2).71 Recent work in a Japanese gout cohort has demonstrated the role of ABCG2 in uric acid excretion in both the gut and kidney. In this well-defined Japanese population sample, it was possible to create grades of ABCG2 dysfunction based on Q141K and Q126X genotype combinations. Individuals positive for the dysfunctional variants 126X and 141K had the highest serum urate levels and highest risk for gout, and those homozygous for 141Q and 126Q had the lowest serum urate levels and lowest risk for gout.73 ABCG2 dysfunction has been shown to influence extra-renal excretion by correlating with an increase in total urinary uric acid excretion. This results in a renal overload (ROL) type of hyperuricemia.74 The presence of 141K (and 126X) alleles reduces excretion through the gut and adds to the circulating urate, overloading the kidney excretion system, resulting in increased urinary uric acid levels. Further study also revealed that ABCG2 dysfunction resulted in a renal under-excretion (RUE) type of hyperuricemia consistent with a critical role in ABCG2-mediated uric acid secretion in the kidney.75 Other studies support a primary role for ABCG2 in excretion of uric acid by the gut. A Spanish study demonstrated considerably stronger association of the 141K allele with gout in patients classified as normo-excretors than in those classified as under-excretors,76 and two New Zealand studies demonstrated 141K-positive individuals not to be renal under-excretors of uric acid.77,78
Predictably, therefore, ABCG2 Q141K associates with gout in most, but not all, ancestral groups analyzed – European, Han Chinese, Japanese, Korean, African American, Taiwanese Aborigine, and Pacific Islanders but not New Zealand Māori.69,79–84 The 126X variant, specific to East Asian populations, confers a higher risk to gout than 141K in Han Chinese and Japanese sample sets.73 In the context that the magnitude of association of ABCG2 on serum urate concentration is less than that of SLC2A9,4 it is interesting that the magnitude of association of ABCG2 Q141K on the risk of gout is consistently larger than that of SLC2A9.4,5,85 This does not equate with the hypothesis that ABCG2 influences gout solely through contributing to hyperuricemia. Emerging evidence (described in the ‘ABCG2: a regulator of gouty inflammation?’ section) indicates that ABCG2 has pleiotropic effects in gout, contributing to the etiology of gout in the presence of hyperuricemia.
ABCG2: a regulator of acute gouty inflammation?
The key initial checkpoint in gout is hyperuricemia.1 Further checkpoints are formation of MSU crystals, the acute inflammatory response to these crystals (gout flare), and tophus formation. A gout flare typically presents as an acutely inflamed and extremely painful lower limb joint. The symptoms peak over the first 24 h and then gradually resolve over the next 10–14 days. Flares are usually recurrent, and the patient is well in between flares. In some patients with prolonged hyperuricemia, tophi can develop both in the joint and non-articular tissues; these are collections of MSU crystals surrounded by a chronic inflammatory granulomatous response.2
As described earlier, it is well established from genome-wide association studies that ABCG2 variants are associated with elevated serum urate concentrations. However, as pointed out above, these studies show that the influence of ABCG2 variants on serum urate is lower than SLC2A9 variants. In contrast, ABCG2 variants are more strongly associated with risk of gout than SLC2A9; Köttgen et al4 reported ORs for gout of 1.73, P=1.7×10−39 for ABCG2 and 1.56, P=1.9×10−31, for SLC2A9. In a more recent analysis,86 ABCG2 was associated with an increased risk of gout; compared with asymptomatic hyperuricemia controls, the OR for gout was 1.83, P=2.6×10−14, in European people, and was 2.35, P=3.9×10−5, in Polynesian people. In contrast, SLC2A9 variants were not associated with increased risk of gout when compared with asymptomatic hyperuricemia controls.86 These are intriguing findings that suggest a role for ABCG2 in developing gout once hyperuricemia is established. They do, however, require replication in other sample sets.
ABCG2 variants have also been associated with tophaceous disease in people with gout. In a Taiwanese study of people with gout, 141K was associated with tophi in both Han and Aboriginal Taiwanese populations (OR 1.51 and 1.50, respectively, for the 141K allele, pooled OR 1.55, P=7.8×10−5).69 Recently, a New Zealand study of people with gout has reported that risk alleles for two ABCG2 SNPs, Q141K and rs10011796, were associated with tophi (compared with gout patients without tophi), with effects observed in participants of Western Polynesian (Samoan, Tongan, Niuean, and Tokelaun) ancestry (OR for rs2231142 1.71, P=0.017 and for rs10011796, 3.76, P=0.002) but not in people of other ancestries (European and Eastern Polynesian [NZ Māori, Cook Island Māori]).87 The ABCG2 associations persisted in Western Polynesian people even after accounting for serum urate and disease duration in the regression models. Given that only two studies have thus far associated ABCG2 with tophus in gout, further studies are required to substantiate this relationship.
These genetic data suggest a role of ABCG2 in the presentation of gout that is, at least in part, independent of hyperuricemia. Supporting this, cyclosporine inhibits ABCG2 in vitro,88 hyperuricemia and gout can be induced by cyclosporine,89 and there is an interaction between cyclosporine and colchicine90 (colchicine is commonly used to treat gout attacks and is able to correct the dysfunctional internalization of ABCG2 141K36). Collectively, these observations suggest that ABCG2 may be influenced by off-target effects of cyclosporine. It is possible that ABCG2 influences other checkpoints, such as MSU crystal formation or the inflammatory response to these crystals. Although the mechanisms of MSU crystal formation are incompletely understood, it is conceivable that ABCG2 variants lead to preferential transport of urate into the joint (or reduced efflux out of the joint), resulting in higher saturating urate concentrations and MSU crystal formation.
Mechanistic aspects of ABCG2 in inflammation
A schematic diagram of the role of ABCG2 in inflammation is shown in Figure 3B. NLRP3 (NBD, leucine-rich family [NLR], pyrin containing 3) inflammasome activation leading to mature interleukin (IL)-1β release through caspase-1 is a key step in the initiation of the acute gout flare.91 An initial signal (signal 1) is required to stimulate the expression of pro-IL-1β and inflammasome components.92 Signal 1 includes Toll-like receptor-4 (TLR4) and TLR2 ligands, as well as unsaturated long-chain free fatty acids,93,94 and can be inhibited by omega-3 fatty acids and HDL-cholesterol.95,96 MSU crystals act as signal 2, inducing NLRP3 inflammasome complex oligomerization and activation of caspase-1.91 Genes in this system have been associated with gout. A variant in TLR4, that likely influences TLR4 expression, has been associated with gout in two studies.97,98 Two SNPs in NLRP3-related genes have been associated with gout.99–101 The first one is a nonsense variation in the caspase recruitment domain-containing protein 8 (CARD8, rs2043211), a negative regulator of the NLRP3 inflammasome. One of the studies also found a multiplicative interaction between this SNP and an IL-1β polymorphism (rs1143623)101 consistent with a pathogenic model whereby greater inflammasome activity from reduced CARD8 expression, combined with higher levels of pre-IL-1β expression, leads to increased production of mature IL-1β and an amplified immune response. Furthermore, the PPARGC1B SNP associated with gout (rs45529037) upregulates the expression of NLRP3 and IL-1β.52
The role of ABCG2 in regulating inflammation has been described in nonarticular cells. In HEK293 cells cultured with H2O2, ABCG2 inhibited reactive oxygen species generation, enhanced anti-oxidant capacity of the cells, and protected cells from reactive oxygen species-induced toxicity.102 This may be mediated by the role of ABCG2 in exporting urate, an established pro-oxidant in the intracellular setting103 by mechanisms that include promotion of mitochondrial dysfunction.104 In animals, ABCG2 also inhibited NF-κB and the expression of the inflammatory gene IL-8 induced by reactive oxygen species.102 In rheumatoid arthritis, a chronic autoimmune inflammatory arthritis, ABCG2 is highly expressed in the intimal lining layer and on macrophages and endothelial cells in the synovial sub-lining layer.105 The synovium is also a very important tissue in gouty arthritis. In several cell culture systems, the expression and function of ABCG2 is reduced when stimulated with pro-inflammatory cytokines.106–108 Collectively, these results suggest both a suppressive effect of ABCG2 on pro-inflammatory signaling pathways and that ABCG2 is itself a target of pro-inflammatory cytokines, where its expression and activity is suppressed. To date, the influence of ABCG2 variants on the inflammatory response to MSU crystals has not been reported, although genetic and other data suggest a role for the 141K variant in promoting gout once hyperuricemia is established (see the ‘ABCG2: a regulator of gouty inflammation?’ section).
ABCG2 and autophagy
In addition to its effects through increased serum urate concentration and the inflammatory responses described earlier, ABCG2 has been reported to play a role in autophagy. In a study by Ding et al,109 ABCG2 overexpressing cell lines were more resistant to stressors such as nutrient deprivation due to increased autophagy. The authors observed more degradation of autophagy receptor p62 and increased accumulation of LC3-II, a protein essential for autophagosome formation and function. Knockdown of ABCG2 in these cells subsequently abolished these effects.109 It was shown previously that effective autophagy is essential in acute gouty arthritis to clear mitochondria that have been damaged by MSU crystals. This prevents the release of mitochondrial DNA and the production of reactive oxygen species, which can activate the NLRP3 inflammasome.110,111 When autophagy is not effective, p62 accumulates, causing nuclear factor E2-related factor 2 (Nrf2), a transcription factor involved in oxidative stress responses, to be released from its repressor, kelch-like ECH-associated protein 1 (keap-1), and translocate to the nucleus. Previously, Nrf2 has been shown to induce transcription of heme oxygenase-1 and superoxide dismutase, which can in turn also activate the NLRP3 inflammasome.112 Finally, ABCG2 expression has been shown to be upregulated by Nrf2, which promotes cancer stem cell survival.46 Taken together, ABCG2 plays a role in autophagy and its expression is induced through Nrf2. Decreased or a lack of ABCG2 function could therefore induce or prolong NLRP3 inflammasome activation in acute gout through defective autophagy of damaged mitochondria in response to MSU crystals.
ABCG2 as a drug target
Restoring the expression and function of ABCG2 molecules that harbor detrimental genetic polymorphisms may be an important next step to limit urate levels and inflammatory responses. Recently, several papers have shown that histone deacetylase (HDAC) inhibitors can restore the function of the 141K ABCG2 variant. Woodward et al demonstrated that the HDAC inhibitor 4-phenylbutyrate restores trafficking and dimer expression of 141K ABCG2.32 Basseville et al showed that several different HDAC inhibitors, as well as colchicine, inhibit targeting of 141K ABCG2 to the aggresome and promote relocalization on the cell surface.36 Colchicine is an anti-inflammatory agent that acts by inhibiting microtubule polymerization through binding to tubulin. This suggests that HDAC inhibitors, as does colchicine, inhibit trafficking of ABCG2 to the aggresome along microtubules. Finally, in addition to restoring surface localization of 141K ABCG2, the HDAC inhibitor romidepsin was shown to increase gene transcription of ABCG2 by activating the aryl hydrocarbon pathway.113 Recently, small molecules that are effective at rescuing the trafficking, abundance, and function of the 141K variant protein have been found. The compounds VRT-325 and 4a, both from the corrector class of molecules found in efforts to rescue mutant CFTR protein, are effective rescue agents for mutant ABCG2 as well.32 These results should encourage further research into the use of less toxic small molecules than HDAC inhibitors to restore defective ABCG2 function in patients with the 141K polymorphism.
ABCG2 and allopurinol response
Allopurinol is the most commonly used urate-lowering therapy for people with gout. It is rapidly metabolized to oxypurinol which is responsible for the majority of the urate-lowering effect through the inhibition of xanthine oxidase. Despite widespread use of allopurinol, many people fail to achieve the recommended target serum urate of <0.36 mmol/L. For example, in the Febuxostat versus Allopurinol Controlled Trial, only 21% of those receiving allopurinol 300 mg daily achieved the primary endpoint of the last three serum urates being ≤0.36 mmol/L.114 Although poor adherence and restricted dosing based on kidney function are frequently associated with failure to reach target serum urate, genes encoding the enzymes involved in the metabolism, mechanism of action, and excretion of allopurinol may also have a role.115
Most studies related to the pharmacogenetics of allopurinol have focused on adverse events rather than predicting efficacy.116 However, two recent studies have highlighted a potential role for ABCG2 in allopurinol response. A significant association between ABCG2 141K and reduced allopurinol response was initially identified in a genome-wide association study.67 However, the definition of response used in this study was allopurinol-related change in serum urate, and there was only assessment of adherence with allopurinol using administrative data. A subsequent study, in which poor response was defined as serum urate ≥0.36 mmol/L despite allopurinol >300 mg daily, and with adherence confirmed by plasma oxypurinol concentrations, replicated the finding with 141K conferring a significantly increased risk of poor response to allopurinol (OR 2.71 (1.70–4.48), P=6.0×10−5).117 This effect remained significant after adjustment for various variables that influence serum urate and allopurinol response including age, sex, BMI, ethnicity, eGFR, diuretic use, and serum urate off urate-lowering therapy. The association between ABCG2 Q141K and allopurinol response is a very interesting observation that should be further replicated in further studies.
The mechanism by which ABCG2 modulates allopurinol response is not clear. Wen et al67 reported ABCG2 to be an efficient allopurinol and oxypurinol efflux pump. HEK 293 cells transfected with 141K had significantly impaired ABCG2 function leading to intracellular accumulation of allopurinol and oxypurinol.67 These authors suggested that the ABCG2 141K variant might impair allopurinol response by increasing allopurinol and oxypurinol concentrations in renal tubular cells and decreasing concentration in the tubule fluid, thereby reducing the inhibition of uric acid influx pumps within the kidney. However, this theory is unproven, and modeling the complexity of the relative contribution of reabsorbed allopurinol118 versus nonsecreted allopurinol is extremely difficult; therefore, in vivo experiments are required to further explore this possible mechanism.
ABCG2 and chronic kidney disease
It is clinically established that chronic kidney disease is a cause of hyperuricemia and gout. However, the physiological response to disrupted renal uric acid handling in kidney disease is a question of clinical interest. Bhatnagar et al119 addressed this question by evaluating the strength of genetic association with urate of variants in a suite of genes encoding uric acid transporters (including SCL2A9, ABCG2, SLC17A1-A3, and SLC22A11-A12) in 3,598 individuals with chronic kidney disease. In contrast to the situation in healthy individuals where SLC2A9 has the strongest magnitude of association on serum urate levels (0.37 mg/dL increase in urate per risk allele compared with 0.22 mg/dL for ABCG2 in sex-adjusted analysis),4 each ABCG2 risk allele increased serum urate by 0.68 mg/dL in people of European ancestry, compared with 0.30 mg/dL for SLC2A9 in sex-adjusted analysis.119 Although this finding requires replication, it does suggest that the extra-renal (gut) urate handling of ABCG2 necessarily compensates in renal injury.119 This is consistent with the observation, in nephrectomized rats, of increased gut expression of ABCG2 and maintenance of normal serum urate concentration.120 Bhatnagar et al119 evoke the “remote sensing and signaling” hypothesis whereby the action of multi-specific SLC and ABC transporters is critical in disrupted homeostasis resulting from organ injury.121 The hypothesis posits that the transporters cooperate in sensing and controlling fluctuations in important substances, with the ABC (and SLC) transporters facilitating interorgan communication through small molecules,121 including those important in oxidant status such as urate.122 One specific example of this is the ability of ABCG2 to transport and transfer heme, through an extracellular loop, to albumin.123
In a study of fructose-induced hyperuricemia in humans, the urate-increasing 141K allele was associated with a lower increase in serum urate but greater increase in fractional excretion of uric acid when compared with the 141Q allele,78 consistent with the previously reported ROL type of hyperuricemia. Supporting ABCG2 as a “remote sensor and signaller,” the 141K allele was associated with a lower glucose response to the fructose load.78 Whether or not the urate and glucose effects are related is unclear; however, the remote sensing hypothesis could relate to differential ATP utilization by the 141Q and 141K alleles given that the 141K variant has significantly reduced ATPase activity.32
Hyperuricemia predicts reduced renal function and chronic kidney disease, independent of measured confounders, in observational studies.124 However, whether or not urate is causal of reduced renal function is unresolved. Mendelian randomization studies, in which urate-associated genetic variants inherited at conception are used as an un-confounded proxy for exposure to urate,125 suggest that urate is not causal of reduced renal function in healthy people126 but could promote progression of declining renal function in chronic kidney disease.127 Based on the observation in mice that uric acid may compete with toxins as an efflux substrate of ABCG2, Dankers et al postulate that, in the kidney, hyperuricemia promotes reduced renal ABCG2-mediated secretion of toxic molecules.128
Conclusion
The ABCG2 transporter is an important molecule in urate excretion. Decreased ABCG2 expression and function due to genetic polymorphisms leads to both ROL hyperuricemia and RUE hyperuricemia. The most extensively studied genetic variant is Q141K. Besides significantly increasing serum urate concentration, the 141K ABCG2 variant has also been associated with acute gout, tophaceous gout, and poor allopurinol response. In addition, 141K-induced hyperuricemia may lead to excessive inflammatory responses and decreased ABCG2 function may cause defective autophagy. All of these effects warrant further research to the restoration of 141K ABCG2 function and surface expression, for example, by small molecules.
Acknowledgments
The authors thank the anonymous reviewers for their insightful and constructive comments. MC is supported by a grant of the Dutch Arthritis Foundation (nr. 12-02-303). LJ is supported by a Competitiveness Operational Programme grant of the Romanian Ministry of European Funds (P_37_762). TRM is supported by the Health Research Council of New Zealand.
Disclosure
The authors report no conflicts of interest in this work.
References
Dalbeth N, Merriman TR, Stamp LK. Gout. Lancet. 2016;388(10055):2039–2052. | ||
Dalbeth N, Pool B, Gamble GD, et al. Cellular characterization of the gouty tophus: a quantitative analysis. Arthritis Rheum. 2010;62(5):1549–1556. | ||
Choi HK, Atkinson K, Karlson EW, Curhan G. Obesity, weight change, hypertension, diuretic use, and risk of gout in men: the health professionals follow-up study. Arch Internal Med. 2005;165(7):742–748. | ||
Köttgen A, Albrecht E, Teumer A, et al. Genome-wide association analyses identify 18 new loci associated with serum urate concentrations. Nat Genet. 2013;45(2):145–154. | ||
Merriman TR. An update on the genetic architecture of hyperuricemia and gout. Arthritis Res Ther. 2015;17:98. | ||
Doyle LA, Yang W, Abruzzo LV, et al. A multidrug resistance transporter from human MCF-7 breast cancer cells. Proc Natl Acad Sci U S A. 1998;95(26):15665–15670. | ||
Wong K, Briddon SJ, Holliday ND, Kerr ID. Plasma membrane dynamics and tetrameric organisation of ABCG2 transporters in mammalian cells revealed by single particle imaging techniques. Biochim Biophys Acta. 2016;1863(1):19–29. | ||
Xu J, Liu Y, Yang Y, Bates S, Zhang JT. Characterization of oligomeric huma half-ABC transporter ATP-binding cassette G2. J Biol Chem. 2004;279:19781–19789. | ||
Allen JD, Brinkhuis RF, Wijnholds J, Schinkel AH. The mouse Bcrp1/Mxr/Abcp gene amplification and overexpression in cell lines selected for resistance to topotecan, mitoxantrone, or doxorubicin. Cancer Res. 1999;59(17):4237–4241. | ||
Ross DD, Yang W, Abruzzo LV, et al. Atypical multidrug resistance: breast cancer resistance protein messenger RNA expression in mitoxantrone-selected cell lines. J Natl Cancer Inst. 1999;91(5):429–433. | ||
Leslie EM, Deeley RG, Cole SP. Multidrug resistance proteins: role of P-glycoprotein, MRP1, MRP2, and BCRP (ABCG2) in tissue defense. Toxicol Applied Pharmacol. 2005;204(3):216–237. | ||
Padmanabhan R, Chen KG, Gillet JP, et al. Regulation and expression of the ATP-binding cassette transporter ABCG2 in human embryonic stem cells. Stem Cells. 2012;30(10):2175–2187. | ||
Szepesi Á, Matula Z, Szigeti A, et al. ABCG2 is a selectable marker for enhanced multilineage differentiation potential in periodontal ligament stem cells. Stem Cells Dev. 2014;24(2):244–252. | ||
Wagener FA, Dankers AC, van Summeren F, et al. Heme oxygenase-1 and breast cancer resistance protein protect against hemeinduced toxicity. Curr Pharmaceutical Design. 2013;19(15):2698–2707. | ||
Jonker JW, Buitelaar M, Wagenaar E, et al. The breast cancer resistance protein protects against a major chlorophyll-derived dietary phototoxin and protoporphyria. Proc Natl Acad Sci U S A. 2002;99(24):15649–15654. | ||
Horsey AJ, Cox MH, Sarwat S, Kerr ID. The multidrug transporter ABCG2: still more questions than answers. Biochemical Soc Trans. 2016;44(3):824–830. | ||
Woodward OM, Köttgen A, Köttgen M. ABCG transporters and disease. FEBS J. 2011;278(18):3215–3225. | ||
Dehghan A, Köttgen A, Yang Q, et al. Association of three genetic loci with uric acid concentration and risk of gout: a genome-wide association study. Lancet. 2008;372(9654):1953–1961. | ||
Woodward OM, Kottgen A, Coresh J, Boerwinkle E, Guggino WB, Kottgen M. Identification of a urate transporter, ABCG2, with a common functional polymorphism causing gout. Proc Natl Acad Sci U S A. 2009;106(25):10338–10342. | ||
Saison C, Helias V, Ballif BA, et al. Null alleles of ABCG2 encoding the breast cancer resistance protein define the new blood group system Junior. Nat Genet. 2012;44(2):174–177. | ||
Zelinski T, Coghlan G, Liu X-Q, Reid ME. ABCG2 null alleles define the Jr (a-) blood group phenotype. Nat Genet. 2012;44(2):131–132. | ||
Deppe S, Ripperger A, Weiss J, Ergün S, Benndorf RA. Impact of genetic variability in the ABCG2 gene on ABCG2 expression, function, and interaction with AT1 receptor antagonist telmisartan. Biochem Biophys Res Comm. 2014;443(4):1211–1217. | ||
Imai Y, Nakane M, Kage K, et al. C421A polymorphism in the human breast cancer resistance protein gene is associated with low expression of Q141K protein and low-level drug resistance. Mol Cancer Ther. 2002;1(8):611–616. | ||
Kobayashi D, Ieiri I, Hirota T, et al. Functional assessment of ABCG2 (BCRP) gene polymorphisms to protein expression in human placenta. Drug Metab Dispos. 2005;33(1):94–101. | ||
Kondo C, Suzuki H, Itoda M, et al. Functional analysis of SNPs variants of BCRP/ABCG2. Pharm Res. 2004;21(10):1895–1903. | ||
Mizuarai S, Aozasa N, Kotani H. Single nucleotide polymorphisms result in impaired membrane localization and reduced atpase activity in multidrug transporter ABCG2. Int J Cancer. 2004;109(2):238–246. | ||
Morisaki K, Robey RW, Özvegy-Laczka C, et al. Single nucleotide polymorphisms modify the transporter activity of ABCG2. Cancer Chemother Pharmacol. 2005;56(2):161–172. | ||
Tamura A, Wakabayashi K, Onishi Y, et al. Re-evaluation and functional classification of non-synonymous single nucleotide polymorphisms of the human ATP-binding cassette transporter ABCG2. Cancer Sci. 2007;98(2):231–239. | ||
Furukawa T, Wakabayashi K, Tamura A, et al. Major SNP (Q141K) variant of human ABC transporter ABCG2 undergoes lysosomal and proteasomal degradations. Pharm Res. 2009;26(2):469–479. | ||
Prasad B, Lai Y, Lin Y, Unadkat JD. Interindividual variability in the hepatic expression of the human breast cancer resistance protein (BCRP/ABCG2): effect of age, sex, and genotype. J Pharm Sci. 2013;102(3):787–793. | ||
Lee JY, Kinch LN, Borek DM, et al. Crystal structure of the human sterol transporter ABCG5/ABCG8. Nature. 2016;533:561–564. | ||
Woodward OM, Tukaye DN, Cui J, et al. Gout-causing Q141K mutation in ABCG2 leads to instability of the nucleotide-binding domain and can be corrected with small molecules. Proc Natl Acad Sci U S A. 2013;110:5223–5228. | ||
Sarankó H, Tordai H, Telbisz A, et al. Effects of the gout-causing Q141K polymorphism and a CFTR ∆508 mimicking mutation on processing and stability of the ABCG2 protein. Biochem Biophys Res Commun. 2103;437:140–145. | ||
Yu W, Kim Chiaw P, Bear CE. Probing conformational rescue induced by a chemical corrector of F508del-cystic fibrosis transmembrane conductance regulator (CFTR) mutant. J Biol Chem. 2011;286:24714–24725. | ||
Woodward OM. ABCG2: the molecular mechanisms of urate secretion and gout. Am J Physiol Renal Physiol. 2015;309:F485–F488. | ||
Basseville A, Tamaki A, Ierano C, et al. Histone deacetylase inhibitors influence chemotherapy transport by modulating expression and trafficking of a common polymorphic variant of the ABCG2 efflux transporter. Cancer Res. 2012;72(14):3642–3651. | ||
Ripperger A, Benndorf RA. The C421A (Q141K) polymorphism enhances the 3`-untranslated region (3`-UTR)-dependent regulation of ATP-binding cassette transporter ABCG2. Biochem Pharmacol. 2016;104:139–147. | ||
To KK, Robey RW, Knutsen T, Zhan Z, Ried T, Bates SE. Escape from hsa-miR-519c enables drug-resistant cells to maintain high expression of ABCG2. Mol Cancer Ther. 2009;8(10):2959–2968. | ||
Nakanishi T, Ross DD. Breast cancer resistance protein (BCRP/ABCG2): its role in multidrug resistance and regulation of its gene expression. Chin J Cancer. 2012;31:73–99. | ||
He X, Wang J, Wei W, et al. Hypoxia regulates ABCG2 activity through the activation of ERK1/2/HIF-1α and contributes to chemoresistance in pancreatic cancer cells. Cancer Biol Ther. 2016;17:188–198. | ||
Krishnamurthy P, Ross DD, Nakanishi T, et al. The stem cell marjer Bcrp/ABCG2 enhances hypoxic cell survival through interactions with heme. J Biol Chem. 2004;279:24128–24225. | ||
Mogi M, Yang J, Lambert CF, et al. Akt signaling regulates side population cell phenotype via Bcrp1 translocation. J Biol Chem. 2003;278:39068–39075. | ||
Bleau AM, Hambardzumyan D, Ozawa T, et al. PTEN/PI3K/Akt pathway regulates the side population phenotype and ABCG2 activity in glioma tumor stem-like cells. Cell Stem Cell. 2009;6:226–235. | ||
Hu CF, Huang YY, Wang YJ, Gao FG. Upregulation for ABCG2 via the PI3K-Akt pathway contributes to acidic microenvironment-induced cisplatin resistance in A549 and LTEP-a-2 ling cancer cells. Oncol Rep. 2016;36:455–461. | ||
Huang FF, Zhang L, Wu DS, et al. PTEN regulates BCRP/ABCG2 and the side population through the PI3K/Akt pathway in chronic myeloid leukemia. PLoS One. 2014;9:e88298. | ||
Jia Y, Chen J, Zhu H, Jia ZH, Cui MH. Aberrantly elevated redox sensing factor Nrf2 promotes cancer stem cell survival via enhanced transcriptional regulation of ABCG2 and Bcl-2/Bmi-1 genes. Oncol Rep. 2015;34(5):2296–2304. | ||
Wang J, Zhu XX, Liu L, et al. SIRT1 prevents hyperuricemia via the PGC-1α/PPARγ-ABCG2 pathway. Endocrine. 2016;53:443–452. | ||
Szatmari I, Vamosi G, Brazda P, et al. Peroxisome proliferator-activated receptor gamma-regulated ABCG2 expression confers cytoprotection to human dendritic cells. J Biol Chem. 2006;281:23812–23823. | ||
Ee PL, Kamalakaran S, Tonetti D, et al. Identification of a novel estrogen responsive element in the breast cancer resistance protein gene. Cancer Res. 2004;15:1247–1251. | ||
Evseenko DA, Paxton JW, Keelan JA. Independent regulation of apical and basolateral drug transporter expression and function in placental trophoblasts by cytokines, steroids, and growth factors. Drug Metab Disppos. 2007;35:595–601. | ||
Chang FW, Fan HC, Liu JM, et al. Estrogen enhances the expression of the multidrug transporter gene ABCG2 – increasing drug resistance of breast cancer cells through estrogen receptors. Int J Mol Sic. 2017;18:E163. | ||
Chang W-C, Wu Y-J J, Chung W-H, et al. Genetic variants of PPAR-gamma coactivator 1B augment NLRP3-mediated inflammation in gouty arthritis. Rheumatology. 2017;56(3):457–466. | ||
Galmés-Pascual BM, Nadal-Casellas A, Bauza-Thorbrügge M, et al. 17β-estradiol imrpoves hepatic mitochondrial biogenesis and function through PGC1B. J Endcrinol. 2017;232:297–308. | ||
Hentschke M, Süsens U, Borgmeyer U. PGC-1 and PERC, coactivators of the estrogen receptor-related receptor gamma. Biochem Biophys Res Commun. 2002;299:872–879. | ||
Scarpulla RC. Transcriptional paradigms in mammalian mitochondrial biogenesis and function. Physiol Rev. 2008;88:611–638. | ||
Merriman TR, Boocock J, Dalbeth N, et al., Mitochondrial genetic variation and susceptibility to gout in Polynesians. Ann Rheum Dis. 2016;75(S2):115. | ||
Merriman T, Terkeltaub R. PPARGC1B: insight into the expression of the gouty inflammation phenotype: PPARGC1B and gouty inflammation. Rheumatology. 2017;56(3):323–325. | ||
Okada Y, Sim X, Go MJ, et al. Meta-analysis identifies multiple loci associated with kidney function-related traits in east Asian populations. Nat Genet. 2012;44(8):904–909. | ||
Charles BA, Shriner D, Doumatey A, et al. A genome-wide association study of serum uric acid in African Americans. BMC Med Genomics. 2011;4:17. | ||
Tin A, Woodward OM, Kao WHL, et al. Genome-wide association study for serum urate concentrations and gout among African Americans identifies genomic risk loci and a novel URAT1 loss-of-function allele. Hum Mol Genet. 2011;20(20):4056–1068. | ||
Ren YC, Jin TB, Sun XD et al. PKD2 and ABCG2 genes polymorphisms are correlated with blood glucose levels and uric acid in Tibetan gout patients. Genet Mol Res. 2016;15. | ||
Sull JW, Yang SJ, Kim S, Jee SH. The ABCG2 polymorphism rs2725220 is associated with hyperuricemia in the Korean population. Genom Inform. 2014;12:231–235. | ||
Shimizu T, Sugiura T, Wakayama T, et al. PDZK1 regulates breast cancer resistance protein in small intestine. Drug Metab Dispos. 2011;39(11):2148–2154. | ||
Huffman JE, Albrecht E, Teumer A, et al. Modulation of genetic associations with serum urate levels by body-mass-index in humans. PLoS One. 2015;10(3):e0119752. | ||
Speliotes EK, Willer CJ, Berndt SI, et al. Association analyses of 249,796 individuals reveal 18 new loci associated with body mass index. Nat Genet. 2010;42(11):937–948. | ||
Stahl E, Choi H, Cadzow M, Flynn T, Topless R, Merriman TR. Conditional analysis of 30 serum urate loci identifies 25 additional independent effects. Arthritis Rheum. 2014;66:S1294. | ||
Wen C, Yee S, Liang X, et al. Genome-wide association study identifies ABCG2 (BCRP) as an allopurinol transporter and a determinant of drug response. Clin Pharm Therap. 2015;97(5):518–525. | ||
Jiri M, Zhang L, Lan B, et al. Genetic variation in the ABCG2 gene is associated with gout risk in the Han Chinese population. Clin Rheumatol. 2016;35:159–163. | ||
Tu H-P, Ko AM-S, Chiang S-L, et al. Joint effects of alcohol consumption and ABCG2 Q141K on chronic tophaceous gout risk. J Rheumatol. 2014;41(4):749–758. | ||
Zhou D, Liu Y, Zhang X, et al. Functional polymorphisms of the ABCG2 gene are associated with gout disease in the Chinese Han male population. Int J Mol Sci. 2014;15(5):9149–9159. | ||
Huls M, Brown CDA, Windass AS, et al. The breast cancer resistance protein transporter ABCG2 is expressed in the human kidney proximal tubule apical membrane. Kidney Int. 2008;73:220–225. | ||
Mandal AK, Mount DB. The molecular physiology of uric acid homeostasis. Ann Rev Physiol. 2015;77:323–345. | ||
Matsuo H, Takada T, Ichida K, et al. Common defects of ABCG2, a high-capacity urate exporter, cause gout: a function-based genetic analysis in a Japanese population. Sci Trans Med. 2009;1(5):5ra11. | ||
Ichida K, Matsuo H, Takada T, et al. Decreased extra-renal urate excretion is a common cause of hyperuricemia. Nat Commun. 2012;3:764. | ||
Matsuo H, Nakayama A, Sakiyama M, et al. ABCG2 dysfunction causes hyperuricemia due to both renal urate underexcretion and renal urate overload. Sci Rep. 2014;4:3755. | ||
Torres RJ, de Miguel E, Bailén R, et al. Tubular urate transporter gene polymorphisms differentiate patients with gout who have normal and decreased urinary acid excretion. J Rheumatol. 2014;41:1863–870. | ||
Kannangara DR, Phipps-Green AJ, Dalbeth N, et al. Hyperuricemia: contributions of urate transporter ABCG2 and the fractional renal clearance of urate. Ann Rheum Dis. 2016;75:1363–1366. | ||
Dalbeth N, House M, Gamble GD, et al. Influence of the ABCG2 gout risk 141 K allele on urate metabolism during a fructose challenge. Arthritis Res Ther. 2014;30:R34. | ||
Kim YS, Kim Y, Park G, et al. Genetic analysis of ABCG2 and SLC2A9 gene polymorphisms in gouty arthritis in a Korean population. Korean J Intern Med. 2015;30(6):913. | ||
Li C, Li Z, Liu S, et al. Genome-wide association analysis identifies three new risk loci for gout arthritis in Han Chinese. Nat Commun. 2015;6:7041. | ||
Matsuo H, Yamamoto K, Nakaoka H, et al. Genome-wide association study of clinically defined gout identifies multiple risk loci and its association with clinical subtypes. Ann Rheum Dis. 2016;75(4):652–659. | ||
Phipps-Green AJ, Hollis-Moffatt J, Dalbeth N, et al. A strong role for the ABCG2 gene in susceptibility to gout in New Zealand Pacific Island and Caucasian, but not Maori, case and control sample sets. Hum Mol Genet. 2010;19:4813–4819. | ||
Wan W, Xu X, Zhao DB, Pang YF, Wang YX. Polymorphisms of uric acid transporter proteins in the pathogenesis of gout in a Chinese Han population. Genet Mol Res. 2015;14:2546–1550. | ||
Stark K, Reinhard W, Grassl M, et al. Common polymorphisms influencing serum uric acid levels contribute to susceptibility to gout, but not to coronary heart disease. PLoS One. 2009;4:e7729. | ||
Phipps-Green A, Merriman M, Topless R, et al. Twenty-eight loci that influence serum urate levels: analysis of association with gout. Ann Rheum Dis. 2016;75:124–130. | ||
Merriman TR, Phipps-Green A, Boocock J, et al. Pleiotropic effect of ABCG2 in gout. In: 2016 ACR/ARHP Annual Meeting; November 11–16, 2016; Washington, DC; Abstract Number 2276. | ||
He W, Phipps-Green A, Stamp LK, Merriman TR, Dalbeth N. Population-specific association between ABCG2 variants and tophaceous disease in people with gout. Arthritis Res Ther. 2017;19:43 | ||
Xia CQ, Liu N, Miwa GT, Gan LS. Interactions of cyclosporin a with breast cancer resistance protein. Drug Metab Dispos. 2007;35:576–582. | ||
Lin HY, Rocher LL, McQuillan MD, et al. Cyclosporine-induced hyperuricemia and gout. N Engl J Med. 1990;322:334–336. | ||
Watson S, Digiacinto JL, Davis MW. Effect of cyclosporine on the pharmacokinetics of colchicine in healthy subjects. Postgrad Med. 2012;124:189–196. | ||
Martinon F, Petrilli V, Mayor A, Tardivel A, Tschopp J. Gout-associated uric acid crystals activate the NALP3 inflammasome. Nature. 2006;440(7081):237–241. | ||
Skeldon AM, Faraj M, Saleh M. Caspases and inflammasomes in metabolic inflammation. Immunol Cell Biol. 2014;92(4):304–313. | ||
Giamarellos-Bourboulis EJ, Mouktaroudi M, Bodar E, et al. Crystals of monosodium urate monohydrate enhance lipopolysaccharide-induced release of interleukin 1β by mononuclear cells through a caspase 1-mediated process. Ann Rheum Dis. 2009;68(2):273–278. | ||
Joosten LA, Netea MG, Mylona E, et al. Engagement of fatty acids with toll-like receptor 2 drives interleukin-1β production via the ASC/caspase 1 pathway in monosodium urate monohydrate crystal–induced gouty arthritis. Arthritis Rheum. 2010;62(11):3237–3248. | ||
Thacker SG, Zarzour A, Chen Y, et al. High-density lipoprotein reduces inflammation from cholesterol crystals by inhibiting inflammasome activation. Immunology. 2016;149(3):306–319. | ||
Williams-Bey Y, Boularan C, Vural A, et al. Omega-3 free fatty acids suppress macrophage inflammasome activation by inhibiting NF-κB activation and enhancing autophagy. PLoS One. 2014;9(6):e97957. | ||
Qing YF, Zhou JG, Zhang QB, et al. Association of TLR4 gene rs2149356 polymorphism with primary gouty arthritis in a case-control study. PLoS One. 2013;8:e64845. | ||
Rasheed H, McKinney C, Stamp LK, et al. The toll-like receptor 4 (TLR4) variant rs2149356 and risk of gout in European and Polynesian sample sets. PLoS One. 2016;11:e1047939. | ||
Chen Y, Ren X, Li C, et al. CARD8 rs2034211 polymorphism is associated with gout in a Chinese male population. Cell Physiol Biochem. 2015;35:1394–1400. | ||
Lee SW, Lee SS, Oh DH, et al. Genetic association for P2X7R rs3751142 and CARD8 rs2043211 polymorphisms for susceptibility of gout in Korean men: multi-center study. J Korean Med Sci. 2016;31:1566–1570. | ||
McKinney C, Stamp LK, Dalbeth N, et al. Multiplicative interaction of functional inflammasome genetic variants in determining the risk of gout. Arthritis Res Ther. 2015;17:288. | ||
Shen S, Callaghan D, Juzwik C, Xiong H, Huang P, Zhang W. ABCG2 reduces ROS-mediated toxicity and inflammation: a potential role in Alzheimer’s disease. J Neurochem. 2010;114(6):1590–1604. | ||
Sautin YY, Nakagawa T, Zharikov S, Johnson RJ. Adverse effects of the classic antioxidant uric acid in adipocytes: NADPH oxidase-mediated oxidative/nitrosative stress. Am J Physiol. 2007;293(2):C584–C596. | ||
Lanaspa MA, Sanchez-Lozada LG, Choi Y-J, et al. Uric acid induces hepatic steatosis by generation of mitochondrial oxidative stress potential role in fructose-dependent and-independent fatty liver. J Biol Chem. 2012;287(48):40732–40744. | ||
van der Heijden JW, Oerlemans R, Tak PP, et al. Involvement of breast cancer resistance protein expression on rheumatoid arthritis synovial tissue macrophages in resistance to methotrexate and leflunomide. Arthritis Rheum. 2009;60(3):669–677. | ||
von Wedel-Parlow M, Wolte P, Galla HJ. Regulation of major efflux transporters under inflammatory conditions at the blood-brain barrier in vitro. J Neurochem. 2009;111(1):111–118. | ||
Poller B, Drewe J, Krahenbuhl S, Huwyler J, Gutmann H. Regulation of BCRP (ABCG2) and P-glycoprotein (ABCB1) by cytokines in a model of the human blood-brain barrier. Cell Mol Neurobiol. 2010;30(1):63–70. | ||
Le Vee M, Lecureur V, Stieger B, Fardel O. Regulation of drug transporter expression in human hepatocytes exposed to the proinflammatory cytokines tumor necrosis factor-alpha or interleukin-6. Drug Metab Dispos. 2009;37(3):685–693. | ||
Ding R, Jin S, Pabon K, Scotto KW. A role for ABCG2 beyond drug transport: Regulation of autophagy. Autophagy. 2016;12(5):737–751. | ||
Zhong Z, Umemura A, Sanchez-Lopez E, et al. NF-κB restricts inflammasome activation via elimination of damaged mitochondria. Cell. 2016;164(5):896–910. | ||
Zhou R, Yazdi AS, Menu P, Tschopp J. A role for mitochondria in NLRP3 inflammasome activation. Nature. 2011;469(7329):221–225. | ||
Jhang JJ, Cheng YT, Ho CY, Yen GC. Monosodium urate crystals trigger Nrf2- and heme oxygenase-1-dependent inflammation in THP-1 cells. Cell Mol Immunol. 2015;12(4):424–434. | ||
To KK, Robey R, Zhan Z, Bangiolo L, Bates SE. Upregulation of ABCG2 by romidepsin via the aryl hydrocarbon receptor pathway. Mol Cancer Res. 2011;9(4):516–527. | ||
Becker M, Schumacher HR, Wortmann R, et al. Febuxostat compared with allopurinol in patients with hyperuricaemia and gout. N Engl J Med. 2005;353:2450–2461. | ||
Stamp L, Merriman T, Barclay M, et al. Inadequate response to allopurinol in the treatment of gout – defining the population and potential causes. Semin Arthritis Rheum. 2014;44(2):170–174. | ||
Saito Y, Stamp L, Caudle K, et al. Clinical Pharmacogenetics Implementation Consortium (CPIC) Guidelines for Human Leukocyte Antigen B (HLA-B) Genotype and Allopurinol Dosing: 2015 update. Clin Pharm Ther. 2016;99(1):36–37. | ||
Roberts R, Wallace M, Phipps-Green A, et al. ABCG2 loss-of-function polymorphism predicts poor response to allopurinol in patients with gout. Pharmacogenomics J. Epub 2016 Jan 26. | ||
Turnheim K, Krivanek P, Oberbauer R. Pharmacokinetics and pharmacodynamics of allopurinol in elderly and young subjects. Br J Clin Pharmacol. 1999;48(4):501–509. | ||
Bhatnagar V, Richard EL, Wu W, et al. Analysis of ABCG2 and other urate transporters in uric acid homeostasis in chronic kidney disease: potential role of remote sensing and signaling. Clin Kidney J. 2016;9(3):444–453. | ||
Yano H, Tamura Y, Kobayashi K, Tanemoto M, Uchida S. Uric acid transporter ABCG2 is increased in the intestine of the 5/6 nephrectomy rat model of chronic kidney disease. Clin Exp Nephrol. 2014;18(1):50–55. | ||
Wu W, Dnyanmote AV, Nigam SK. Remote communication through solute carriers and ATP binding cassette drug transporter pathways: an update on the remote sensing and signaling hypothesis. Mol Pharmacol. 2011;79(5):795–805. | ||
Ames BN, Cathcart R, Schwiers E, Hochstein P. Uric acid provides an antioxidant defense in humans against oxidant-and radical-caused aging and cancer: a hypothesis. Proc Natl Acad Sci U S A. 1981;78(11):6858–6862. | ||
Desuzinges-Mandon E, Arnaud O, Martinez L, et al. ABCG2 transports and transfers heme to albumin through its large extracellular loop. J Biol Chem. 2010;285(43):33123–33133. | ||
Bellomo G, Venanzi S, Verdura C, et al. Association of uric acid with change in kidney function in healthy normotensive individuals. Am J Kidney Dis. 2010;56(2):264–272. | ||
Robinson PC, Choi HK, Do R, Merriman TR. Insight into rheumatological cause and effect through the use of Mendelian randomization. Nat Rev Rheumatol. 2016;12(8):486–496. | ||
Hughes K, Flynn T, de Zoysa J, Dalbeth N, Merriman TR. Mendelian randomization analysis associates increased serum urate, due to genetic variation in uric acid transporters, with improved renal function. Kidney Int. 2013;85(2):344–351. | ||
Testa A, Mallamaci F, Spoto B, et al. Association of a polymorphism in a gene encoding a urate transporter with CKD progression. Clin J Am Soc Nephrol. 2014;9(6):1059–1065. | ||
Dankers AC, Mutsaers HA, Dijkman HB, et al. Hyperuricemia influences tryptophan metabolism via inhibition of multidrug resistance protein 4 (MRP4) and breast cancer resistance protein (BCRP). Biochim Biophys Acta. 2013;1832(10):1715–1722. | ||
Crișan TO, Cleophas MC, Oosting M, et al. Soluble uric acid primes TLR-induced proinflammatory cytokine production by human primary cells via inhibition of IL-1Ra. Ann Rheum Dis. 2016;75(4):755–762. | ||
Kasza I, Várady G, Andrikovics H, et al. Expression levels of the ABCG2 multidrug transporter in human erythrocytes correspond to pharmacologically relevant genetic variations. PLoS One. 2012;7:e48423. | ||
Li R, Miao L, Xiang Y, et al. A meta-analysis of the associations between the Q141K and Q126X ABCG2 gene variants and gout risk. Int J Clin Exp Pathol. 2015;8:9812–9823. | ||
Dong Z, Guo S, Yang Y, et al. Association between ABCG2 Q141K polymorphism and gout risk affected by ethnicity and gender: a systematic review and meta-analysis. Int J Rheum Dis. 2015;18:382–391. |
 © 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.