Back to Journals » Drug Design, Development and Therapy » Volume 9
A thorough QT study in the context of an uptitration regimen with selexipag, a selective oral prostacyclin receptor agonist
Authors Hoch M, Darpo B, Remenova T, Stoltz R, Zhou M, Kaufmann P, Bruderer S, Dingemanse J
Received 8 October 2014
Accepted for publication 13 November 2014
Published 17 December 2014 Volume 2015:9 Pages 175—185
DOI https://doi.org/10.2147/DDDT.S75565
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Shu-Feng Zhou
Matthias Hoch,1 Borje Darpo,2,3 Tatiana Remenova,4 Randall Stoltz,5 Meijian Zhou,2 Priska Kaufmann,1 Shirin Bruderer,1 Jasper Dingemanse1
1Department of Clinical Pharmacology, Actelion Pharmaceuticals Ltd, Allschwil, Switzerland; 2iCardiac Technologies Inc, Rochester, New York, NY, USA; 3Karolinska Institute, Department of Clinical Sciences, Danderyd’s Hospital, Division of Cardiovascular Medicine, Stockholm, Sweden; 4Drug Safety Department, Actelion Pharmaceuticals Ltd, Allschwil, Switzerland; 5Covance Clinical Research Unit, Evansville, IN, USA
Abstract: The effects of selexipag and its active metabolite ACT-333679 on cardiac repolarization were assessed in a thorough QT study as per International Conference on Harmonisation E14 guidance. In this randomized, double-blind, placebo/positive-controlled, parallel-group study, healthy male and female subjects were randomized to receive escalating doses of selexipag (n=91) or placebo/moxifloxacin (n=68). Ascending multiple doses of selexipag in the range of 400-1,600 µg or placebo were administered twice daily for 21 days. Following a nested crossover design, subjects in the moxifloxacin/placebo treatment group received a single oral 400 mg dose of moxifloxacin on day 2 or 24. The primary endpoint (QT interval correction using individualized formula [QTcI]) was chosen based on a prospectively defined test applied to on-treatment data. The mean baseline-adjusted placebo-corrected ΔQTcI (ΔΔQTcI) for selexipag was small at all time points and never exceeded 1.4 msec (upper bound of 90% confidence interval [CI], 3.9 msec) on 800 µg or –0.7 msec (upper bound of 90% CI, 2.1 msec) on 1,600 µg. The mean ΔΔQTcI peak effect for moxifloxacin was 7.5 msec (lower bound of 90% CI, 4.8 msec). The exposure-response analysis did not demonstrate a relevant relationship between plasma concentrations of selexipag or ACT-333679 and ΔΔQTcI but, in contrast, a positive slope within the expected range for moxifloxacin. In conclusion, selexipag does not have an effect on cardiac repolarization.
Keywords: prostacyclin, corrected QT, TQT, selexipag, moxifloxacin
Introduction
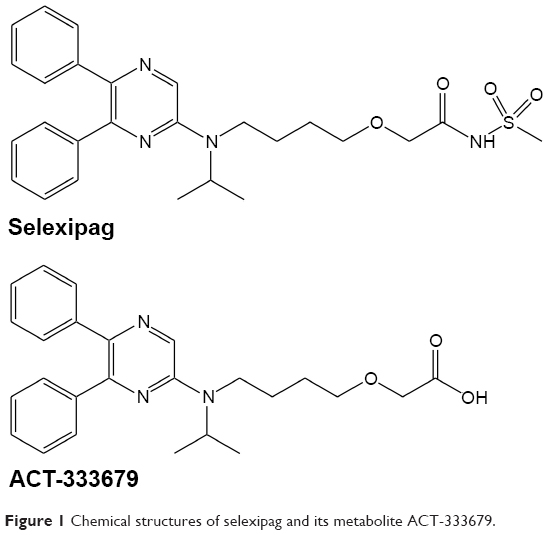
Selexipag is a potent, orally available, selective prostacyclin (IP) receptor agonist that is currently in clinical development for the treatment of pulmonary arterial hypertension. The selectivity of selexipag for the IP receptor offers the potential to overcome the limitations on tolerability (eg, nausea and vomiting) associated with prostacyclin analog therapy.1 Selexipag is rapidly absorbed and hydrolyzed in the liver by carboxylesterase 1 to yield the active metabolite ACT-333679, which has a 13-fold higher affinity than selexipag and a more than 130-fold higher affinity for the IP receptor than for other prostaglandin receptors.2,3 The chemical structures of selexipag and ACT-333679 are shown in Figure 1. The plasma protein binding is above 99% for both selexipag and ACT-333679, with no saturation of binding in a clinically relevant concentration range. High binding to human albumin and α-acid glycoprotein was demonstrated for both compounds (data on file).
|
Figure 1 Chemical structures of selexipag and its metabolite ACT-333679. |
In the multiple-dose uptitration study in healthy subjects, maximum plasma concentrations of selexipag and ACT-333679 were achieved 2 hours and 4 hours, respectively, following administration. In the same study, following a 1,800 μg dose of selexipag, a terminal half-life of 1.4 hours and 8.7 hours for selexipag and ACT-333679, respectively, was obtained.4 Thus, the pharmacokinetics of selexipag and its active metabolite allow for twice-daily dosing. A study in healthy male subjects showed that selexipag at doses of up to 1,600 μg twice daily (the highest dose tested in Phase III) can be tolerated when using a gradual uptitration regimen.4
In a Phase II proof-of-concept study in 43 patients (selexipag to placebo randomization, 3:1) with symptomatic pulmonary arterial hypertension, selexipag administered for 17 weeks on top of a single and double pulmonary arterial hypertension-specific background therapy showed a statistically significant reduction in pulmonary vascular resistance compared with placebo.5
Drug-induced QT interval (time from electrocardiogram [ECG] Q wave to the end of the T wave) prolongation is associated with an increased risk for development of torsades de pointes and sudden cardiac death. The International Conference on Harmonisation (ICH) E14 document, and the subsequent Q&A document, both recommend that all new drugs with systemic exposure be evaluated for prolonging effects on the heart rate-corrected QT (QTc) interval and provide guidance on how to perform and analyze a thorough QT study.6,7
In vitro studies in Chinese hamster ovary (CHO)-K1 cells expressing recombinant human ether-a-go-go-related gene (hERG) potassium channels showed no effects of selexipag and ACT-333679 (highest concentration tested, 30 μM) on hERG channel conductance at concentrations reached in humans. In conscious dogs, selexipag had no effect on ECG variables at doses up to 10 mg/kg.
The aim of this study was to assess whether selexipag at steady-state for two therapeutic dose levels (800 μg and 1,600 μg) has an adverse effect on QT/QTc as per ICH E14 guidance in a thorough QT/QTc study. In the Phase III study (GRIPHON, ClinicalTrials.gov ID NCT01106014), as for other prostacyclin receptor agonists, selexipag was uptitrated to the patient’s individual maximum tolerated dose. Thus, testing of a supratherapeutic dose, as recommended, was not feasible; instead, a gradual uptitration dosing schedule was used to attain the highest therapeutic dose level tested in Phase III, and exposure-response analysis was applied to the data to project QTc effects at plasma levels expected with supratherapeutic doses. Two methods for heart rate correction of the QT interval were used, ie, QT interval correction using individualized formula (QTcI) and QT interval correction with Fridericia’s formula (QTcF).
The primary endpoint was chosen based on a prospective test on on-treatment data, which evaluates each method’s ability to remove the heart rate dependence of QTc.8 In line with current guidelines, moxifloxacin 400 mg was used as a positive control to show adequate sensitivity to detect small changes in the QT interval.6,7 Due to the relatively long dosing duration (21 days), a parallel-group study design was chosen. By using a nested crossover design in a combined placebo/moxifloxacin group to assess the effect of moxifloxacin, it was possible to conduct the study more efficiently by reducing the number of treatment groups from three to two.
Materials and methods
Subjects
Eligible subjects had to be healthy (assessed by physical examination and standard laboratory tests), nonsmokers, aged 18–45 years, and with a body mass index of 18.5–32 kg/m2. Subjects with a personal or family history of long QT syndrome or hypokalemia, as well as female subjects with a QTc value >470 msec or male subjects with a value >450 msec at screening were excluded. Consumption of grapefruit or grapefruit juice and food containing poppy seeds was not permitted during the course of the study. Concomitant medications were not allowed during this study, except for hormonal contraceptives and the treatment of adverse events.
Written informed consent was obtained from all subjects. The study was conducted in accordance with good clinical practice, ICH E14 guidance, and the Declaration of Helsinki at a single center in the USA (Covance Clinical Research Unit, Evansville, IN, USA). The study was approved by an independent ethics committee (Independent Investigational Review Board Inc., Plantation, FL, USA).
Study design
This study used a double-blind, randomized, placebo/positive-controlled (moxifloxacin), double-dummy, parallel-group, multiple-dose, uptitration study design. Subjects were randomized to two treatment groups, ie, selexipag in escalating doses or placebo/moxifloxacin in a nested crossover design for moxifloxacin (ClinicalTrials.gov ID NCT02206204). Treatment randomization was done in a 4:3 fashion to selexipag or placebo/moxifloxacin. A maximum of 160 subjects was planned to be enrolled to have at least 56 evaluable subjects per treatment group completing the study.
All subjects received a single dose of placebo for selexipag on day 1. Subjects in the selexipag treatment group received selexipag on days 3 to 23 (400–1,600 μg twice daily as 200 μg tablets) and placebo for moxifloxacin on days 2 and 24. Subjects in the placebo/moxifloxacin treatment group received placebo for selexipag on days 3 to 23 and one single 400 mg dose of moxifloxacin or placebo for moxifloxacin either on day 2 or 24 (Figure 2). The dose for each subject was uptitrated after every 3 days (ie, after six dose administrations, which is thought to be sufficient to attain steady-state conditions) in steps of 200 μg up to a dose of 1,600 μg twice daily (with the exception of day 23 [only morning administration]). The study drugs were orally administered 1 hour after a meal (morning and evening) with approximately 240 mL of water. For a particular subject, the dosing interval between selexipag administrations was kept as close as possible to 12 hours. Moxifloxacin 400 mg was administered as over-encapsulated tablets (Avelox®; Bayer, Zürich, Switzerland).

|
Figure 2 Study design. |
Safety and tolerability
Safety and tolerability were evaluated by monitoring of adverse events, assessing standard blood chemistry and hematology laboratory variables, urinalysis, body weight, safety 12-lead ECG recordings, and supine and orthostatic vital signs.
Cardiodynamics/QTcI derivation
A continuous ECG (Holter) was recorded on days 1, 2, 11, 23, and 24 (12-lead, Mortara Surveyor; Mortara Instrument, Inc., Milwaukee, WI, USA). Triplicate ECGs were extracted by a central ECG laboratory (eResearch Technology, Philadelphia, PA, USA) from the continuous recordings at the following time points: prior to respective morning dose and at 0.5, 1, 1.5, 2, 3, 4, 5, 6, 7, 8, 10, and 12 hours after morning drug administration (time-matched with respect to clock time [actual time elapsed] and the individual dose schedule). Extractions were approximately 1 minute apart but did not exceed 5 minutes, so did not conflict with the pharmacokinetic sampling time. The ECGs were extracted using manual adjudication and interpreted by an experienced cardiologist. The technicians and the cardiologist were blinded with respect to time point and treatment. The same reader was assigned for all ECGs of a given subject. From at least 10 minutes before until 5 minutes after each predefined ECG extraction time point, subjects were resting in a supine position.
The QTcI correction factor was derived from linear mixed-effects modeling on the log scale log(QT) = log(a) + bi·log(R wave to R wave [RR] interval) with sex-specific slope and intercept included as fixed effects, and subject included as a random effect for both slope and intercept. This was done for all subjects on baseline data (ie, nominal time points) derived from day 1. The QTcI correction factor (ie, log[RR] coefficient) for each subject, bi, was then used to calculate the individually corrected QT for each subject as follows: QTcI = QT/RRbi. QTcF was derived using Fridericia’s formula: QTcF = QT/(RR)1/3. Categorical QTcI outliers (defined as QTcI values >450, >480, or 500 msec, respectively) were analyzed descriptively. Cardiodynamic analysis of the raw Holter data was performed by iCardiac Technologies Inc., (Rochester, NY, USA).
Pharmacokinetics
Blood samples (approximately 3 mL) for measurement of selexipag and its active metabolite ACT-333679 were collected in lithium heparin tubes at the following time points: within 30 minutes prior to dosing and then at 0.5, 1, 1.5, 2, 3, 4, 5, 6, 7, 8, 10, and 12 hours post-dose on days 11 and 23. In addition, trough (predose) samples for the measurement of selexipag and its active metabolite ACT-333679 were taken on days 5, 8, 10, 11, 14, 17, 20, and 22. To prevent degradation of selexipag and of a metabolite of ACT-333679, the plasma samples were acidified with 10% of plasma volume of 1.0 M HCl. Exposure of plasma samples to light was minimized and opaque tubes were used. Moxifloxacin blood samples (approximately 3 mL) were collected on days 2 and 24 in ethylenediaminetetraacetic acid tubes at the same time points as for selexipag and ACT-333679 on days 11 and 23. Plasma samples were stored at −80°C±10°C pending analysis. Plasma concentrations were determined using validated liquid chromatography coupled to tandem mass spectrometry assays. The lower limit of quantification for selexipag and ACT-333679 was 0.01 ng/mL. For moxifloxacin the lower limit of quantification was 5 ng/mL. The inter-assay accuracy for selexipag was 98.6%–100.7% and the inter-assay precision was 5.7%–10.6%. The inter-assay accuracy for ACT-333679 was 100.7%–103.3% and the interassay precision was 4.4%–13.3%. The inter-assay accuracy for moxifloxacin was 99.6%–102.7% and the inter-assay precision was 4.0%–6.8%. The pharmacokinetic parameters were calculated by noncompartmental analysis from the plasma concentration–time data using Phoenix WinNonlin software version 6.1 (Pharsight Corp., Mountain View, CA, USA).
Statistical analysis
Sample size
The sample size was calculated for a noninferiority margin of 10 msec. A standard deviation of 8 msec was assumed for ΔQTc, and a ΔΔQTc difference between selexipag 1,600 μg and placebo of ≤5 msec at five time points (1, 2, 3, 4, and 5 hours post-dose). Fifty-six evaluable subjects per treatment arm at study end were calculated to be sufficient to ensure with a probability of at least 85% (power) that the upper bound of the 95% one-sided confidence interval (CI) for the difference between ΔQTc for the highest dose group of selexipag (1,600 μg) and the placebo group would be <10 msec at all five time points.
Evaluation of optimal QT-RR correction method
To determine which QT correction (either QTcF or QTcI) corrected best for the effect of heart rate, graphical assessment and statistical analysis for regression between QTc values and RR data were used. As a next step, the relationship between QTc (QTcF and QTcI) and RR interval was investigated using on-treatment data (selexipag 800 μg and 1,600 μg, moxifloxacin, and placebo) by linear regression modeling: QTc = a + bi × RR. The RR coefficient for each subject, bi, was then used to calculate the average sum of squared slopes for each of the different QT-RR correction methods, as proposed by Tornoe et al.8 The correction method that resulted in the average on-treatment slope closest to zero (on placebo, selexipag 800 μg, and selexipag 1,600 μg) was deemed the most appropriate heart rate correction method and was therefore used as the primary endpoint.
Primary analysis
The primary analysis for each of the two doses of selexipag was based on a linear mixed effects model with the change from time-matched baseline QTc as a dependent variable, treatment, time (categorical), and treatment-by-time as factors. “Subject” was included in the model as a random effect for the intercept. A two-sided 90% CI was calculated for the contrast “treatment (selexipag) – placebo”. This model was fitted across all time points on day 11 (800 μg) and on day 23 (1,600 μg) separately. The study was judged negative if the upper one-sided 95% CI (ie, the upper bound of the two-sided 90% CI) was below 10 msec for the largest time-matched mean effect, ie, time-matched mean difference from placebo, ΔΔQTc. For heart rate, PR interval, and QRS interval, descriptive statistics including mean, standard error, and 90% two-sided CI from the Student’s t-test are given.
Concentration-QTc analysis
The relationship between plasma concentrations (of selexipag, its metabolite ACT-333679, and moxifloxacin) and ΔΔQTcI (primary endpoint) was quantified using a linear mixed effects modeling approach. Three different models were to be considered, ie, a linear model with an intercept, a linear model with the mean intercept fixed to 0, and a linear model with no intercept. The time-matched concentration was included in the model as a covariate and subject as a random effect for both intercept and slope, whenever applicable. The model that fitted the data best (ie, had the smallest Akaike information criterion and predicted CIs similar to the observed ones) was used for predicting population average ΔΔQTcI and the corresponding 90% two-sided CI intervals at the geometric mean peak plasma concentration (Cmax) of selexipag, its metabolite ACT-333679, and moxifloxacin.
Assay sensitivity
The assay sensitivity hypothesis was that the study design enabled detection of QT prolongation of the active control (moxifloxacin) compared with placebo. Estimates of time-matched, baseline-adjusted mean differences from placebo and their 90% CIs for the primary endpoint (ΔΔQTcI) were derived at each time point of 2, 3, and 4 hours post-dose. A linear mixed model was fitted with sequence, period, time (categorical), treatment, and time-by-treatment interaction as fixed effects, and subject as a random effect for the intercept. For the time points of 2, 3, and 4 hours, the contrast in treatment ΔΔQTcI = “moxifloxacin – placebo” was tested against the one-sided null hypothesis ΔΔQTcI ≤5 msec at the 5% level. Multiplicity was controlled using a Hochberg procedure.9 If the lower bound of the 90% CI of the placebo-corrected ΔQTcI exceeded 5 msec at any of the predefined time points, assay sensitivity was considered to be shown.
Results
Demographics and baseline characteristics
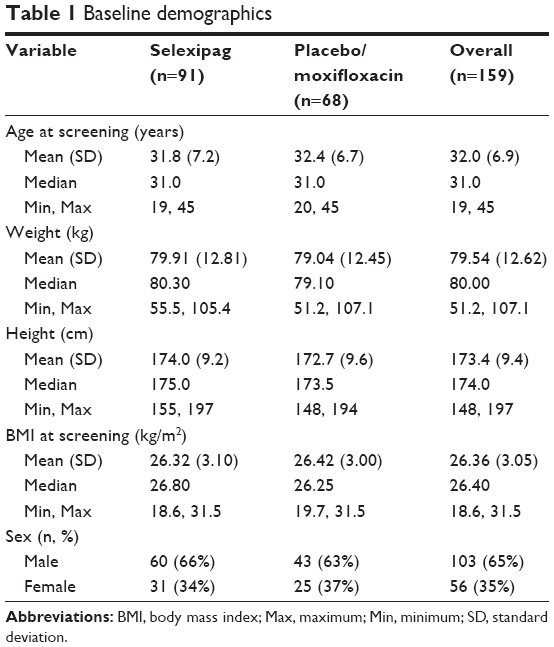
A total of 159 subjects were enrolled in this study; of these, 91 subjects (60 males, 31 females) were randomized to the selexipag treatment group and 68 subjects (43 males, 25 females) to the placebo/moxifloxacin treatment group. In total, 122 subjects (77%) completed the study, with 56 subjects (62%; 40 males, 16 females) completing in the selexipag treatment group and 66 subjects (97%; 43 males, 23 females) completing in the placebo/moxifloxacin treatment group.
The two treatment groups were balanced with regard to age, weight, height, body mass index, and sex (Table 1). In the selexipag group, 47.3% of subjects were Black/African American, 40.7% were White/Caucasian, 2.2% were American Indian or Alaska Native, 3.3% were Asian, one subject (1.1%) was Native Hawaiian or other Pacific Islander, and 5.5% were of other races. In the placebo/moxifloxacin group, 60.3% of subjects were White/Caucasian, 35.3% were Black/African American, one subject (1.5%) was American Indian or Alaska Native, and 2.9% were of other races.
|
Table 1 Baseline demographics |
Safety and tolerability
The most frequently reported adverse events following multiple dosing with selexipag (days 3 to 23), which occurred at a higher frequency than following placebo/moxifloxacin were headache, dizziness, myalgia, jaw pain, nausea, diarrhea, and vomiting. In total, 26 subjects (14 males, 12 females) discontinued due to adverse events following multiple doses of selexipag (days 3 to 23) and two following multiple doses of placebo/moxifloxacin. The most frequent adverse event leading to premature discontinuation in the selexipag treatment group was headache (16 subjects), followed by myalgia (eight subjects), nausea (eight subjects), and jaw pain (six subjects). Study discontinuations due to adverse events occurred at all selexipag dose levels. None of the adverse events leading to discontinuation were related to ECG abnormalities or could be attributed to QT-prolonging effects.
There was one serious adverse event reported by a female subject in the selexipag group (symptomatic hypotension following administration of selexipag 1,200 μg). Three other female subjects discontinued from the study due to hypotensive events following multiple doses of selexipag (at 800–1,000 μg). These events were moderate in intensity and considered to be related to the study treatment by the investigator. No clinically relevant changes in clinical laboratory variables or safety ECGs were observed.
Cardiodynamics
QTcI was shown to remove the heart rate dependence somewhat better than QTcF, with a consistently smaller mean sum of squared individual slopes than for QTcF, and was therefore chosen for the primary endpoint analysis (Table 2). The mean slope of the correction factor for QTcI was 0.3076 (90% CI 0.3027–0.3124), as compared with the fixed slope of 0.33 for QTcF.

|
Table 2 Average sum of squared slopes for different QT-RR correction methods across treatments |
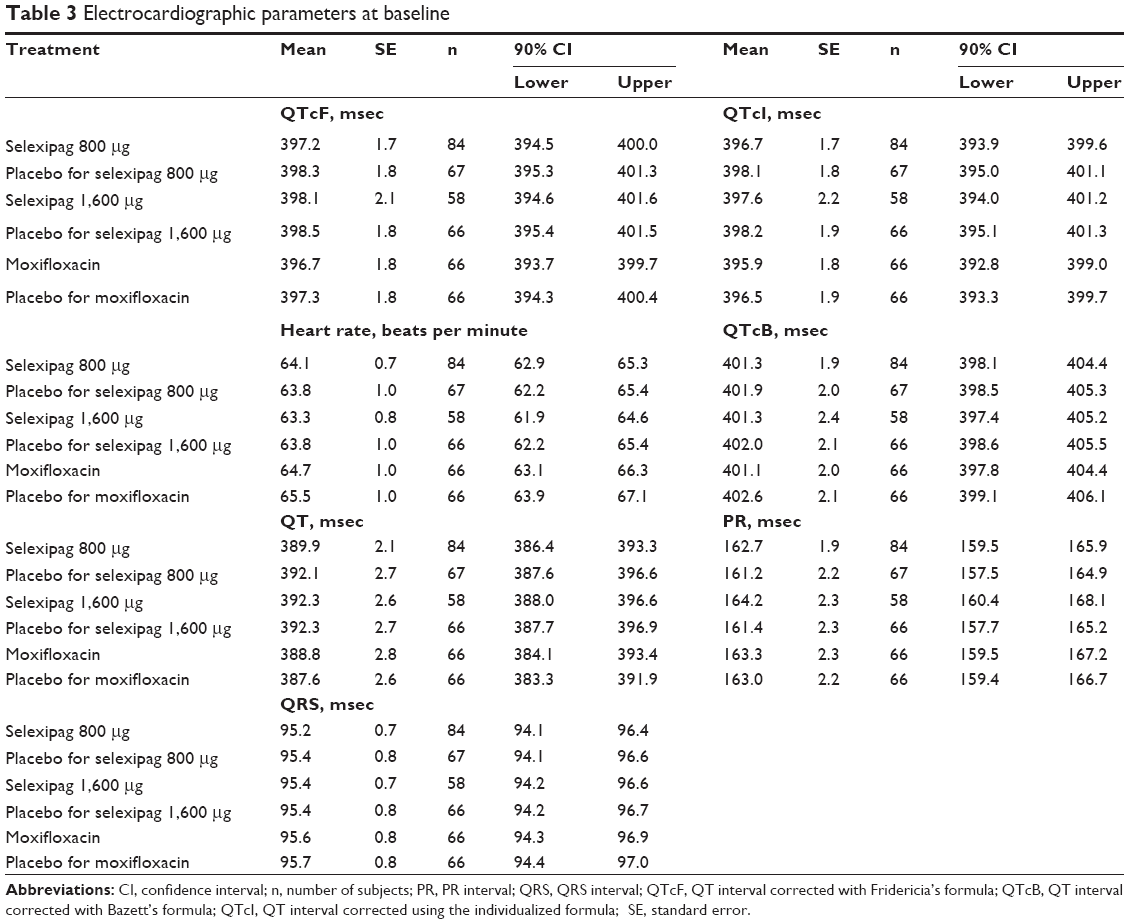
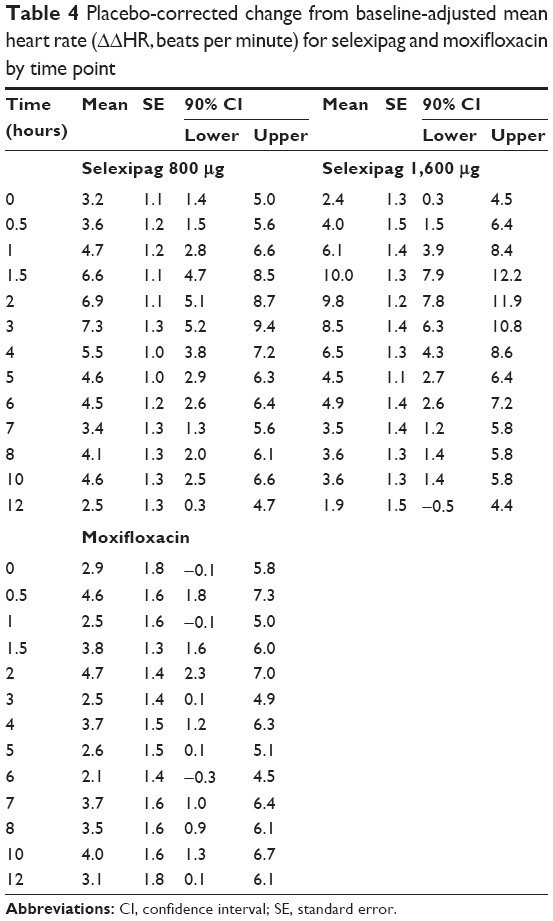
Baseline ECG parameters were well balanced across treatment groups, with only minor differences (Table 3). The largest difference in mean heart rate and QTcI was 2.2 beats per minute and 2.3 msec, respectively. The results of the placebo-corrected change from baseline-adjusted mean ECG parameters for selexipag and moxifloxacin by time point are summarized in Tables 4–7.
|
Table 3 Electrocardiographic parameters at baseline |
|
Table 4 Placebo-corrected change from baseline-adjusted mean heart rate (ΔΔHR, beats per minute) for selexipag and moxifloxacin by time point |

|
Table 5 Placebo-corrected change from baseline-adjusted mean QTcI (ΔΔQTcI, msec) for selexipag and moxifloxacin by time point |

|
Table 6 Placebo-corrected change from baseline-adjusted mean PR (ΔΔPR, msec) for selexipag and moxifloxacin by time point |

|
Table 7 Placebo-corrected change from baseline-adjusted mean QRS (ΔΔQRS, msec) for selexipag and moxifloxacin by time point |
Selexipag caused a mild increase in heart rate. The placebo-corrected heart rate reached 6–7 beats per minute 1.5–3 hours after dosing with selexipag 800 μg (day 11) and 9–10 beats per minute at the same time points on selexipag 1,600 μg (day 23). This effect on heart rate was slightly greater in females as compared with males after both selexipag doses (7–9 beats per minute in males and 12–16 beats per minute in females at the 1,600 μg dose).
A small dose-independent PR interval shortening of approximately 5 msec was observed on selexipag treatment, which is consistent with the observed effect of the drug on heart rate. No relevant effect was observed on the QRS interval, with the mean ΔΔQRS ranging between −1.1 msec and 0.0 msec and with the highest upper bound of the 90% CI being 1.1 msec on selexipag 1,600 μg.
Change-from-baseline QTcI (ΔQTcI) was small at all time points and the mean placebo-corrected change-from-baseline QTcI (ΔΔQTcI) did not exceed 1.4 msec (upper bound of 90% CI, 3.9 msec) on selexipag 800 μg and −0.7 msec (upper bound of 90% CI, 2.1 msec) on selexipag 1,600 μg (Figure 3). Following moxifloxacin 400 mg, a peak effect on ΔΔQTcI of 7.5 msec at 3 hours post-dose, with a lower bound of the 90% CI of 4.8 msec was observed. Effects on QTcF were consistent with the QTcI results, with a largest mean ΔΔQTcF of 2.3 msec (upper bound of 90% CI, 4.8 msec) at 0.5 hours on selexipag 800 μg and 0.6 msec (upper bound of 90% CI, 3.5 msec) at 2 hours on selexipag 1,600 μg. There were no subjects with a QTcI exceeding 480 msec (Table 8) or change from baseline QTcI >30 msec on selexipag. The number of time points at which T-wave morphology changes were observed was small and similar across all treatment groups.
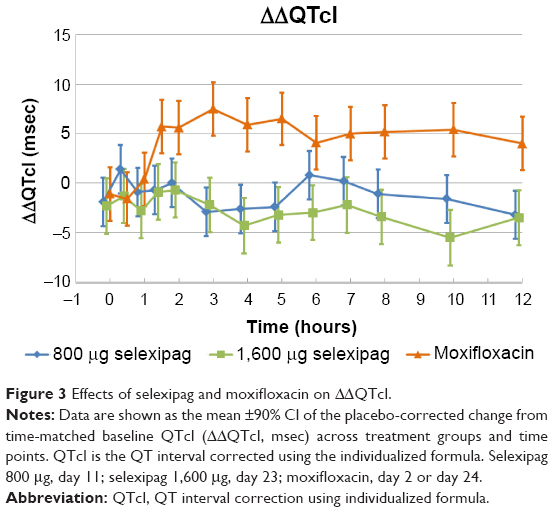
|
Figure 3 Effects of selexipag and moxifloxacin on ΔΔQTcI. |

|
Table 8 QT interval corrected using the individualized formula QTcI per absolute categories (>450 msec, >480 msec, and >500 msec) |
For the concentration-QTc analysis, a linear model with an intercept was identified to best fit the data obtained. No concentration-dependent effect of selexipag and ACT-333679 on QTcI was identified. The estimated population intercept for selexipag was −1.72 msec with a slope of 0.0520 msec per ng/mL (90% CI −0.0357, 0.1396; P=0.3259). The estimated population intercept for ACT-333679 was −1.22 msec with a slope of −0.0374 msec per ng/mL (90% CI −0.102, 0.0276; P=0.3410). A strong concentration-dependent effect of moxifloxacin on QTcI was identified with an estimated slope of 0.005 msec per ng/mL (90% CI 0.0037–0.0063; P<0.0001).
Pharmacokinetics
The mean concentration-time profiles of selexipag following the third day (fifth dose) of 800 μg on day 11 and following the third day (fifth dose) of 1,600 μg on day 23 are shown in Figure 4. The predose trough (Ctrough) concentrations of selexipag and ACT-333679 were low, indicating very little accumulation. Visual inspection of the mean Ctrough concentrations for selexipag and ACT-333679 showed that approximate steady-state conditions were reached on day 11 and on day 23, ie, following 2 days of 800 μg twice daily and 1,600 μg twice daily dosing, respectively (data on file).
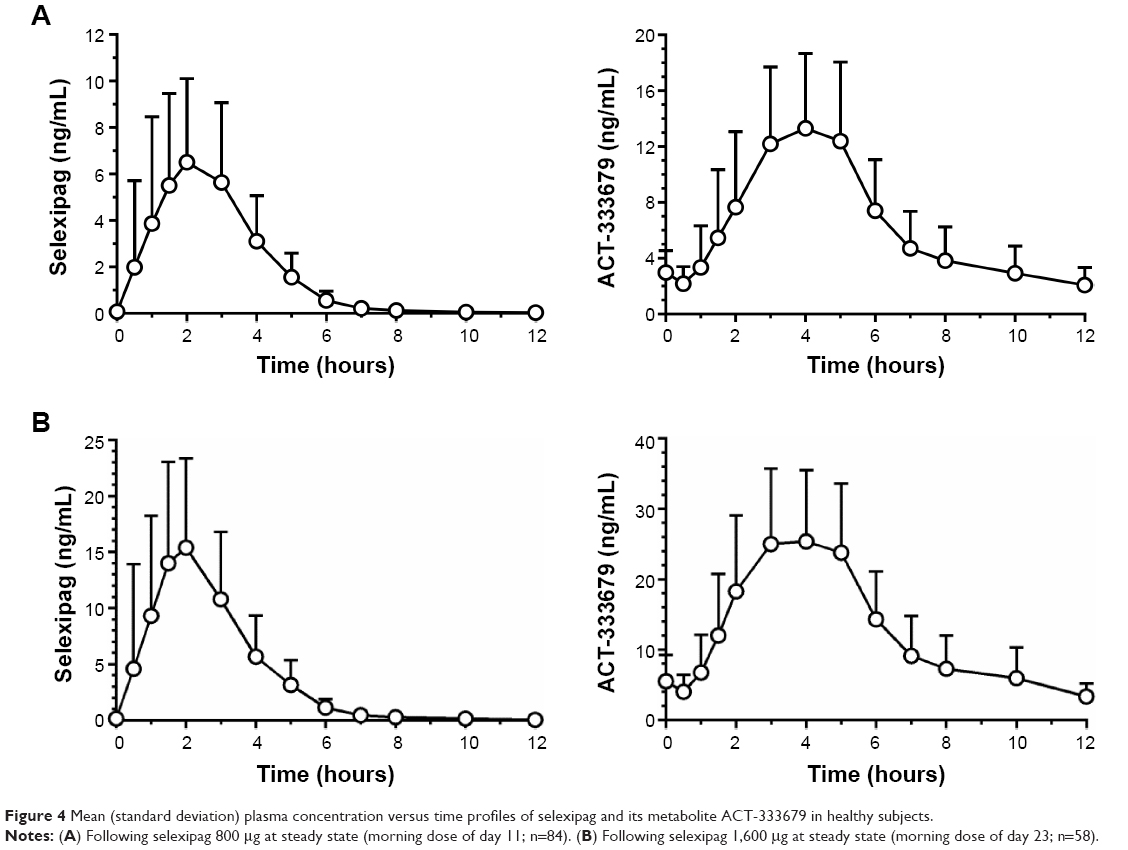
|
Figure 4 Mean (standard deviation) plasma concentration versus time profiles of selexipag and its metabolite ACT-333679 in healthy subjects. |
The pharmacokinetic parameters for selexipag and ACT-333679 are summarized in Table 9. Maximum plasma concentrations were reached at 2 and 4 hours post-dose (median) for selexipag and ACT-333679, respectively, on both day 11 and day 23. Selexipag and ACT-333679 mean Cmax and area under the plasma concentration-time curve during a dose interval (AUCτ) (0–12 hour) values increased roughly two-fold between days 11 and 23, which corresponded to a two-fold increase in selexipag dose. Relative to selexipag on days 11 and 23, ACT-333679 exposure was approximately 3.1-fold to 3.5-fold higher. Maximum plasma concentrations of moxifloxacin were reached at 2 hours post-dose. Mean Cmax and AUC (0–12 hour) values for moxifloxacin were 1,987 ng/mL and 13,340 ng·hour/mL, respectively. There was a trend for higher exposures to selexipag and ACT-333679 in females compared with males (selexipag AUCτ: +10% on day 11, +21% on day 23; ACT-333679 AUCτ: +18% on day 11, +32% on day 23).

|
Table 9 Pharmacokinetic parameters of selexipag and ACT-333679 |
Discussion
Repeat administration of selexipag, at oral doses uptitrated from a starting dose of 400 μg (in 200 μg steps) to a maximum dose of 1,600 μg twice daily was tolerated by most healthy male and female subjects. Frequently reported adverse events were in accordance with the known adverse event profile of selexipag.4 In the selexipag treatment group, a higher percentage of females (12 of 31 [38.7%]) than males (14 of 60 [23.3%]) prematurely discontinued the study due to adverse events. Study discontinuations due to adverse events occurred at all selexipag dose levels. No adverse experiences that could be attributed to QT-prolonging effects, such as torsades de pointes, ventricular tachycardia, ventricular fibrillation or flutter, seizures, or sudden death, were reported.
The pharmacokinetic results of selexipag and ACT-333679 were comparable with previous data obtained in Phase I studies in male subjects.4
Selexipag was associated with a mild but not clinically relevant increase in heart rate, but did not show an effect on cardiac repolarization. ΔQTcI was small at all time points, and the placebo-corrected ΔQTcI (ΔΔQTcI) did not exceed 1.4 msec (upper bound of 90% CI, 3.9 msec) on selexipag 800 μg or −0.7 msec (upper bound of 90% CI, 2.1 msec) on selexipag 1,600 μg. Effects on QTcF were entirely consistent with these results. There were no subjects with QTcI exceeding 480 msec or ΔQTcI >30 msec on selexipag. The study was thereby negative, as defined by the ICH E14 guidance (ie, the upper bound of the two-sided 90% CI of ΔΔQTcI on both tested doses of selexipag was below the threshold of concern, ie, 10 msec). Concentration-effect modeling did not demonstrate a relevant relationship between plasma concentrations of selexipag or the main metabolite, ACT-333679, and the effect on the QTc interval. It is very unlikely that any patient on selexipag 1,600 μg will reach drug exposures potentially leading to a relevant QTc effect. This exposure-response analysis provided further evidence to support the conclusion that selexipag does not have a clinically meaningful effect on cardiac repolarization.
A thorough QT study with iloprost, a stable prostacyclin analog approved for the treatment of pulmonary hypertension, scleroderma, Raynaud’s phenomenon, and ischemia, showed results similar to selexipag.10 Iloprost given as an inhalation at the maximum tolerated dose (20 μg) was shown to cause a small but clinically nonsignificant increase in QTc of 3 msec. An exposure-QTc response relationship could not be established.10
Moxifloxacin was included as a positive control to validate the sensitivity of the assay in detecting small increases from baseline in QTc. In thorough QT studies, moxifloxacin 400 mg has been demonstrated to increase ΔΔQTc by 10–15 msec, generally between 1 and 6 hours post-dose.11,12 In this study, the QTc effect of moxifloxacin 400 mg was lower than expected (mean peak ΔΔQTcI 7.5 msec with a lower bound of the 90% CI of 4.8 msec, just below the approximate threshold of 5 msec indicated by the ICH E14 guidance). Due to these rather small QTc effects observed with moxifloxacin, it was decided to analyze the moxifloxacin concentrations in plasma. The moxifloxacin geometric mean Cmax (1,987 ng/mL) was lower than expected from similar studies in healthy subjects.12 This may be explained by the over-encapsulation of moxifloxacin tablets (done for blinding reasons), which may lead to altered absorption and lower drug exposure. Food is also known to lower the exposure to moxifloxacin.12 In this study, for tolerability reasons, all study drugs were administered 1 hour after a meal. These factors might explain the low plasma concentrations and consequently the relatively small QTc effect of moxifloxacin by time point observed in this study. Also of note, there was a slightly higher variability in QTc measurement. The sample size calculation assumed a standard deviation of between-subject change-from-baseline QTc of 8 msec, whereas the employed semi-automated methodology resulted in a mean standard deviation of 9.7 msec for moxifloxacin and placebo for moxifloxacin. However, the concentration-QTc analysis for moxifloxacin showed that the slope of the concentration/ΔΔQTcI relationship was significantly positive (0.005 msec per ng/mL) and was within, or even exceeding, reported values.12,13 Using the concentration-QTc model from the moxifloxacin data in this study, ΔΔQTcI at Cmax values normally seen with moxifloxacin 400 mg (around 2,900 ng/mL)12 could be estimated to 13.1 msec with a lower bound 90% CI of 9.3 msec. This complementary analysis demonstrated that the QTc effect observed after administration of moxifloxacin was within expectations when related to the actual plasma levels. Since the concentration-QTc analysis was clearly negative for selexipag and its metabolite, it appears highly unlikely that the study was not sufficiently sensitive to detect a small QTc effect of selexipag, in case there had been an effect.
Conclusion
Selexipag does not prolong the QTc interval and does not have any adverse effect on cardiac repolarization.
Acknowledgments
The authors thank Eric Ertel from Actelion Pharmaceuticals Ltd for the electrophysiological analyses, Olaf Mackenroth from Swiss BioQuant AG, Reinach, Switzerland, for the bioanalytical analyses, Pierre-Eric Juif, Actelion Pharmaceuticals Ltd, for preparing the pharmacokinetic graphs, and eRT Inc., Philadelphia, PA, USA, for handling and analysis of the raw Holter ECG data.
Disclosure
This study was sponsored by Actelion Pharmaceuticals Ltd. MH, TR, PK, SB, and JD are employees of Actelion Pharmaceuticals Ltd. Covance Clinical Research Unit received funding from Actelion Pharmaceuticals Ltd. RS is an employee of Covance Clinical Research Unit. iCardiac Technologies received funding from Actelion Pharmaceuticals Ltd. BD is the Global Medical Director of iCardiac Technologies, and holds shares in the company. MZ is an employee of iCardiac Technologies.
References
Skoro-Sajer N, Lang IM. Selexipag for the treatment of pulmonary arterial hypertension. Expert Opin Pharmacother. 2014;15(3):429–436. | ||
Mubarak KK. A review of prostaglandin analogs in the management of patients with pulmonary arterial hypertension. Respir Med. 2010; 104(1):9–21. | ||
Kuwano K, Hashino A, Asaki T, et al. 2-[4-[(5,6-diphenylpyrazin-2-yl)(isopropyl)amino]butoxy]-N-(methylsulfonyl)acetamide (NS-304), an orally available and long-acting prostacyclin receptor agonist prodrug. J Pharmacol Exp Ther. 2007;322(3):1181–1188. | ||
Bruderer S, Hurst N, Kaufmann P, Dingemanse J. Multiple-dose up-titration study to evaluate safety, tolerability, pharmacokinetics, and pharmacodynamics of selexipag, an orally available non-prostanoid prostacyclin receptor agonist, in healthy subjects. Pharmacology. 2014;94:148–156. | ||
Simonneau G, Torbicki A, Hoeper MM, et al. Selexipag: an oral, selective prostacyclin receptor agonist for the treatment of pulmonary arterial hypertension. Eur Respir J. 2012;40(4):874–880. | ||
International Conference on Harmonisation. Harmonized Tripartite Guideline E14 (2005). The clinical evaluation of QT/QTc interval prolongation and proarrhythmic potential for non-antiarrhythmic drugs. Available from: http://www.ich.org/products/guidelines/efficacy/article/efficacy-guidelines.html. Accessed November 16, 2014. | ||
International Conference on Harmonisation. E14 Q&A (R1) (2012). E14 Implementation working group questions and answers. Available from: http://www.ich.org/products/guidelines/efficacy/article/efficacy-guidelines.html. Accessed November 16, 2014. | ||
Tornoe CW, Garnett CE, Wang Y, Florian J, Li M, Gobburu JV. Creation of a knowledge management system for QT analyses. J Clin Pharmacol. 2011;51(7):1035–1042. | ||
Hochberg Y. A sharper Bonferroni procedure for multiple tests of significance. Biometrika. 1988;75:800–802. | ||
Center for Drug Evaluation and Research. Approval Package for Iloprost (Ventavis). 2004. Available from: http://www.accessdata.fda.gov/drugsatfda_docs/nda/2004/21-779_Ventavis_medr.pdf. Accessed November 16, 2014. | ||
Yan LK, Zhang J, Ng MJ, Dang Q. Statistical characteristics of moxifloxacin-induced QTc effect. J Biopharm Stat. 2010;20(3):497–507. | ||
Florian JA, Tornoe CW, Brundage R, Parekh A, Garnett CE. Population pharmacokinetic and concentration-QTc models for moxifloxacin: pooled analysis of 20 thorough QT studies. J Clin Pharmacol. 2011; 51(8):1152–1162. | ||
Bloomfield DM, Kost JT, Ghosh K, et al. The effect of moxifloxacin on QTc and implications for the design of thorough QT studies. Clin Pharmacol Ther. 2008;84(4):475–480. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.