Back to Journals » International Journal of Nanomedicine » Volume 17
A Synergistic and Efficient Thrombolytic Nanoplatform: A Mechanical Method of Blasting Combined with Thrombolytic Drugs
Authors Hu L, Xu J, Zhang W, Wang J, Fang N, Luo Y, Xu L, Liu J, Zhang Y, Ran H, Guo D, Zhou J
Received 21 July 2022
Accepted for publication 7 October 2022
Published 9 November 2022 Volume 2022:17 Pages 5229—5246
DOI https://doi.org/10.2147/IJN.S382964
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Dongwoo Khang
Liu Hu1,2 *, Jie Xu1 *, Wenli Zhang,1,2 Junrui Wang,1,2 Ni Fang,1,2 Ying Luo,1,2 Lian Xu,1,2 Jia Liu,1 Yu Zhang,1 Haitao Ran,3 Dajing Guo,1 Jun Zhou1
1Department of Radiology, The Second Affiliated Hospital of Chongqing Medical University, Chongqing, People’s Republic of China; 2Chongqing Key Laboratory of Ultrasound Molecular Imaging & Department of Ultrasound, The Second Affiliated Hospital of Chongqing Medical University, Chongqing, People’s Republic of China; 3Department of Ultrasound, Institute of Ultrasound Imaging, The Second Affiliated Hospital of Chongqing Medical University, Chongqing, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Jun Zhou, Tel +86 23 6288 7295, Email [email protected]
Background and Objective: Thrombosis is a common disease that poses a great threat to life and health. Most thrombolytic effects of traditional treatments or nanomedicine are not efficient or safe enough. Therefore, we designed a nanoparticle (NP) with a combination of a phase transition material and thrombolytic drugs for efficient and safe thrombolysis.
Methods: A thrombus fibrin-targeted and phase transition NP was designed and contained perfluorohexane (PFH) and the thrombolytic drug rtPA core, with CREKA polypeptides attached to the shell of the PLGA NPs. Characterization of the phase transition and ultrasound imaging of the NPs was carried out under low-intensity focused ultrasound (LIFU). LIFU-responsive drug release in vitro was also explored. Under the synergistic effect of PFH and rtPA, the efficient thrombolysis ability of the NPs was studied in vitro and in vivo. In vivo monitoring of thrombosis and biosafety were also verified.
Results: The PPrC NPs had good ultrasound imaging ability under LIFU irradiation and were related to the phase transition characteristics of the NPs. CREKA polypeptides can effectively increase the aggregation of the NPs on thrombi. Under static and dynamic conditions in vitro, the “liquid to gas” transformation effect of PFH can perform the destruction function of the excavator at the thrombus site and promote the specific release of rtPA, and the subsequent rtPA drug thrombolysis can further fully dissolve the thrombus. In vivo experiments showed that the NPs can monitor the formation of thrombi and have good thrombolytic effects, with significantly reduced bleeding side effects. The biochemical indexes of the rats were within normal limits after treatment.
Conclusion: PPrC NPs loaded with PFH and rtPA combining a mechanical way of blasting with thrombolytic drugs may be a promising new and reliable approach for thrombus monitoring and treatment.
Keywords: low-intensity focused ultrasound, thrombolysis, PFH, rtPA, phase transition
Introduction
Thrombosis is the pathological basis of three major cardiovascular diseases, namely, myocardial infarction, cerebral apoplexy and venous thromboembolism,1 which are serious threats to human health.2,3 At present, there are two main methods of clinical treatment for thrombosis: drug thrombolysis and interventional surgery. The most commonly used thrombolytic drug is recombinant tissue plasminogen activator (rtPA). However, rtPA has a low bioavailability and poor targeting ability and easily causes side effects such as bleeding. Interventional surgery is an invasive treatment method with high technical requirements, complex surgical conditions, and high price, making it difficult to use in large populations.4 Therefore, there is an urgent need to find a safer and more effective thrombolytic therapy.
Due to the disadvantages of traditional thrombolysis, researchers have turned their focus to nanomedicine. By targeting nanodrugs into thrombi, drugs can be accurately released in a controllable time and space to treat thrombi and reduce complications. It has been reported that under the action of an external magnetic field, magnetic NPs can be passively delivered to the thrombus site, and then the drug is released accurately.5–8 However, passively targeting thrombi is inconvenient, and the desired clinical outcome is difficult to achieve. Huang et al reported a liposome platform loaded with cRGD polypeptide, and this platform utilized the advantage of cRGD to actively target activated platelets; as a result, this platform achieved good thrombolytic effects in vitro.9 To improve the biocompatibility of NPs, Chen et al modified the surface of NPs with a cell membrane, which prolonged their blood circulation time and effectively enhanced their thrombi-targeting ability.10 In short, various approaches have been attempted to increase the enrichment of thrombolytic mediators at the thrombus site, reduce the dose of thrombolytic drugs and destroy the thrombus structure to the greatest extent.
In addition to improving the thrombolytic efficiency of drugs by enhancing the targeting ability of nanomedicine, researchers have also developed various new thrombolysis methods. Among them, the use of photothermal, magnetothermal and ultrasound-assisted thrombolysis has been widely studied. Walker et al developed gold NPs that produce free radicals under photothermal excitation, which causes fibrin to collapse and the thrombus structure to loosen.11 The laser can trigger gold nanorods to generate heat at the thrombus, which can quickly and accurately treat thrombi without drugs.12 However, inorganic gold NPs are difficult to degrade in vivo, and their safety needs to be improved.1 Ximendes et al prepared magnetic NPs with real-time thermal feedback that could be quickly and accurately located in tissue.13 Even though this kind of magnetic hyperthermia seems promising, the magnetocalorimeter cannot be focused, which makes it easy to burn the surrounding healthy tissues, and is deeply affected by the intensity and frequency of the magnetic field.14 As a routine examination method in clinical applications, ultrasound has the advantages of safety, noninvasiveness and simple operation. Wang et al used ultrasound to remotely activate drug release, deepen the penetration of drugs in the thrombus and accelerate the dissolution of thrombi.5 To further enhance the penetration of the thrombolytic medium, Cao et al integrated ultrasound-driven micromotors to realize ultrasound-driven thrombus penetration and ultrasound dynamic treatment without using any thrombolytic agent and observed good biosafety.15 Our team recently developed a new drug-free thrombolysis method based on the liquid-to-gas phase transition of perfluorohexane (PFH) and achieved good results through blasting mechanical thrombolysis under ultrasound irradiation.16,17 Other scholars also confirmed the feasibility of phase transition thrombolysis in a model of coronary microthrombosis.18 However, the harm of thrombus debris after mechanical thrombolysis needs to be considered. rtPA was noted to activate the transformation of plasminogen to plasmin. We assume that by loading rtPA into phase transition NPs, rtPA can better penetrate into interior areas and act on exposed fibrin based on the loosening of the fibrin structure caused by the phase transition. Based on the effectiveness of physical thrombolysis, rtPA completely eliminates the risk of fragmentation after thrombolysis and performs thrombolysis safely in vivo.
Herein, a synergistic and efficient thrombolytic nanoplatform combining mechanical blasting with thrombolytic drugs was proposed for the first time in this study (Figure 1). In this platform, PFH, as a phase transition material, changes from liquid to gas after being irradiated by a low-intensity focused ultrasound (LIFU). The droplets of PFH become bubbles, and as the bubbles oscillate and collapse, various physical and chemical effects are produced, resulting in the disintegration of blood clots.19,20 On the other hand, in the process of NPs phase transition, the thrombolytic drug rtPA is released, which causes plasminogen activation to plasmin and further promotes the thrombolytic process. Furthermore, the phase transition of NPs act as the destruction function of the excavator, resulting in a looser thrombus structure, which is more conducive to the full penetration of rtPA. Under the synergistic effect of the two methods, the thrombus disintegrates rapidly. At the same time, CREKA, a polypeptide that can target fibrin, is reliably bound to the nanocarriers. Once thrombosis occurs, the NPs can accurately target the fibrin in the thrombi. This new method combining mechanical and drug thrombolysis provides a promising strategy for the diagnosis and treatment of thrombosis and may become a safe clinical transformation method.
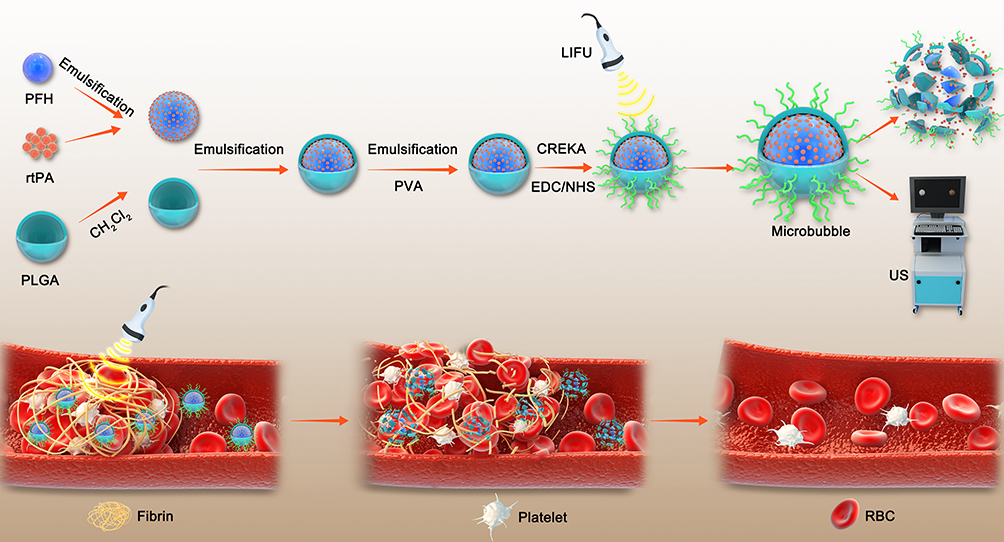 |
Figure 1 Schematic diagram of PPrC NPs synthesis and thrombus treatment with LIFU. |
Materials and Methods
Materials
PLGA-COOH (50:50, MV:12000Da) was purchased from Jinan Daigang Bioengineering Co., Ltd. (Shandong, China). PFH was purchased from J&K Scientific Ltd. (Beijing, China). rtPA was purchased from Boehringer Ingelheim Pharma GmbH & Co. KG (Germany). CREKA and FITC-CREKA were purchased from China Peptides Co., Ltd. (Jiangsu, China). DiI was purchased from Beyotime Biotechnology (Jiangsu, China). FITC was purchased from BioFroxx (Germany). DiR was purchased from MedChemExpress (America). 2-(N-Morpholino) ethanesulfonic acid (MES), N-hydroxysuccinimide (NHS), and 1-ethyl-3-(3-(dimethylamino) propyl) carbodiimide hydrochloride (EDC) were purchased from Sigma–Aldrich (St. Louis, MO, USA). All other chemicals used were at least analytical grade and were used directly without further treatment.
Synthesis of the NPs
PLGA-PFH-rtPA (termed PPr NPs) was synthesized by a three-step emulsification.16 Initially, 0.1 mg rtPA was dissolved in 100 μL injection water. Then, a mixed solution of 200 μL of PFH and 100 μL of rtPA as the internal aqueous phase was emulsified in an ice bath with an ultrasonic vibrometer at a power of 52 W for 2.5 min. The above solution was poured into 2 mL of CH2Cl2, which contained 50 mg of PLGA, and was then emulsified (52 W, 2.5 min) in an ice bath to produce a white emulsion. After adding 6 mL of 4% PVA solution as the external aqueous phase, the third emulsification (52 W, 2.5 min) was continued. Finally, 10 mL of 2% isopropanol solution was added and magnetically stirred in a fume hood for 2 h to solidify the surface of the NPs and volatilize the organic solvent. The PPr NPs were centrifuged at 10000 rpm for 6 min; the precipitate was washed 3 times to collect the PPr NPs for further use.
PLGA-PFH-rtPA-CREKA (termed PPrC NPs) were synthesized through the carbodiimide method. First, the PPr NPs were resuspended with an appropriate amount of 0.1 M MES buffer (pH = 5.2). EDC and NHS were added at a molar ratio of 2:1. After being placed in an ice bath on a shaking table for 3 h, the samples were centrifuged and washed to remove the unreacted EDC and NHS. After resuspension in 0.1 M MES buffer (pH = 8.0), a small amount of CREKA polypeptide was added and incubated in an ice bath on a shaking table overnight. Finally, the PPrC NPs were centrifuged, the precipitates were washed 3 times to remove unreacted polypeptides, and the collected PPrC NPs were stored at 4℃ for further use. PPC NPs and PrC NPs were all prepared at the same time, but 100 μL or 200 μL double distilled water was used instead of rtPA or PFH. The preparation method for the DiI and DiR-labeled NPs was the same as described above except that a small amount of DiI and DiR, respectively, were added to the CH2Cl2 solution.
Characteristics of the NPs
The particle size, polymer dispersity index (PDI) and zeta potential of the PPrC, PPC, PrC and PPr NPs were measured by dynamic light scattering (DLS, Brookhaven Instruments ZEN3600, USA). The morphology and structure of the PPrC NPs were observed using transmission electron microscopy (TEM, Hitachi S-3400N, Hitachi, Ltd, Tokyo, Japan). To study the stability of the PPrC NPs, the particle size of the PPrC NPs in phosphate-buffered saline (PBS) was examined at different durations (0, 3, 6 and 9 days). In addition, the gross appearance of the NPs suspension was recorded. The amide bond formed by dehydration condensation of the amino group of CREKA polypeptide and the carboxyl group of PLGA was detected by Fourier transform infrared spectrometry (FTIR, Nicolet iS50, Thermo Scientific Co. Ltd, MA, USA). To further verify the carrier rate of the CREKA polypeptide on the PLGA shell, we labeled the PLGA shell with DiI and CREKA with FITC, and the carrier rate was detected by flow cytometry (FCM, FACS Vantage SE, Becton Dickinson, San Jose, CA, USA). The encapsulation efficiency of rtPA in the PPrC NPs was detected using a TPA ELISA kit. To determine whether the activity of rtPA was affected after the preparation of the NPs, the chromogenic substrate S-2288 was used to study this property. Briefly, rtPA or PPrC NPs solution with the same rtPA content was incubated with S-2288 solution. Finally, the reaction was terminated with acetic acid, and the absorbance was detected at 405 nm wavelength.
Dual Modal Imaging in vitro
Ultrasound Imaging and LIFU-Induced Phase Transition Performance of the NPs in vitro
A Vevo LAZR PA imaging system (VEVO 2100, FUJIFILM Visual Sonics, Inc., USA) was used to study the ultrasound imaging performance of the PPrC and PrC NPs. PPrC and PrC NPs (2.5 mg/mL) were added to the gel model, which was prepared in advance and irradiated for 1 h with LIFU (1 W/cm2), and B-mode and contrast-enhanced ultrasound (CEUS) images were collected at different time points (0, 10, 20, 30, 45 and 60 min). To more intuitively detect the process of NPs phase transitions, an optical microscope (Leica DMi1, Leica Co. Ltd., Heidelberg, Germany) was used to observe the morphological changes in the NPs during LIFU irradiation.
Fluorescence Imaging in vitro
DiR-labeled PPrC NPs were used to verify the fluorescence imaging ability of the NPs in vitro. The NPs were diluted to different concentrations (0.196, 0.391, 0.781, 1.563, 3.125, 6.25, 12.5 mg/mL), and their fluorescence imaging performance was measured by a Xenogen IVIS Spectrum imaging system (IVIS Lumina III; PerkinElmer, Waltham, MA, USA).
LIFU-Responsive Drug Release in vitro
To study the rtPA release ability in vitro, fluorescently labeled rtPA was prepared according to a previously reported method,9 and its release was detected based on the change in fluorescence intensity. FITC-rtPA was prepared as follows: FITC powder was dissolved in dimethyl sulfoxide at a concentration of 1 mg/mL, and 0.1 mg rtPA was dissolved in 100 μL injection water. Then, 100 μL of FITC solution was added to 100 μL of rtPA solution in PBS solution (pH = 8.0) and shaken in an ice bath overnight. Next, the liquid was transferred to a dialysis bag (MWCO = 3500 Da) and underwent dialysis for 8 h in PBS buffer (pH = 7.4); the dialysate was changed every 4 h. The final FITC-rtPA solution was used as soon as possible. The method for preparing the FITC-rtPA-containing NPs was the same as above. PPrC, PPr and PrC NPs containing FITC rtPA were termed F-PPrC, F-PPr and F-PrC NPs, respectively.
Four groups of NPs (namely, F-PPrC+LIFU, F-PPr+LIFU, F-PrC+LIFU and F-PPrC without LIFU) were used to verify the rtPA drug release properties of the NPs under LIFU irradiation. F-PPrC, F-PPr and F-PrC NPs were resuspended in 2 mL of PBS buffer and then irradiated for 2 h by LIFU with an acoustic power density of 1 W/cm2. Another group of F-PPrC NPs was not irradiated with LIFU. Then, the samples were centrifuged (10,000 rpm, 6 min) at predetermined time points (0, 10, 20, 30, 45, 60, 90, and 120 min); the supernatant was collected, the samples were supplemented with the same amount of PBS buffer, and finally, the fluorescence intensity of the supernatant was read on a microplate reader (Bio-Tek Instrument, Inc., USA). At the same time, the temperature value was recorded using a thermal imager.
Assessment of the Targeting Ability of the NPs in vitro
Sprague–Dawley (SD) rats (250~300 g) were obtained from the animal center of Chongqing Medical University. All animal procedures were performed in conformity with the guidelines of the National Institutes of Health Guide for the Care and Use of Laboratory Animals (NIH publication No. 85–23, revised 1996) and were approved by the Animal Ethics Committee of Chongqing Medical University. After the SD rats were anesthetized with 1% pentobarbital sodium through the abdominal cavity, blood was collected from the hearts of the rats and placed in a coagulation booster tube at room temperature for 6 h. Then, the blood was rinsed 3 times with PBS, cut into small round shapes and placed into 12-well plates. Two milliliters of DiR-labeled PPrC and PPr NPs at a concentration of 2.5 mg/mL was added to the plate as targeted and nontargeted groups, followed by incubation for 60 min. In addition, excess CREKA polypeptide was incubated with blood clots in the targeted group for 60 min in advance as a blocking group. The blood clots were removed at different time points (15, 30, 45 and 60 min) and cleaned with PBS. Fluorescence images were immediately collected by a Xenogen IVIS Spectrum imaging system, and their fluorescence intensity was analyzed.
Assessment of the Thrombolysis Efficacy of the NPs in vitro
Assessment of the Thrombolysis Efficacy of the NPs Under Static Conditions
The methods of preparing the blood clots were the same as those above. PPrC, PPC, PrC, and PPr NPs at a concentration of 2.5 mg/mL and saline were added to a 24-well plate (n = 3). During the incubation process, the dissolution of blood clots was photographed at predetermined time points (0, 15, 30, 45, and 60 min). All NP groups were irradiated with LIFU for 60 min (1 W/cm2), the weight changes before and after blood clot treatment were recorded, and the thrombolytic rate was calculated. The calculation formula is as follows: (weight before thrombolysis - weight after thrombolysis)/weight before thrombolysis × 100%. In addition, after 60 min of treatment, 200 μL of supernatant of blood clot lysis was collected, and the absorbance of the supernatant was measured at 540 nm using a microplate reader to detect the content of hemoglobin.
Assessment of the Thrombolysis Efficacy of the NPs Under Dynamic Conditions
The BE-FLOW microfluidic chip (width, 1.5 mm; height, 0.375 mm; and length, 43 mm; Beijing E-science CO., LTD, China) was covered with type I collagen fibers overnight, and the nonadherent proteins were washed off with PBS before perfusion.
The citrated blood was mixed with thrombin and calcium chloride, and the blood was perfused into the chip at a shear rate of 200 s−1. After the formation of a stable nonocclusive thrombus, the channel was gently rinsed with saline. Then, PPrC, PPC, PrC, and PPr NPs at a concentration of 2.5 mg/mL and saline were perfused into the channel at the same shear rate. All NP groups were irradiated with LIFU (1 W/cm2) for 1 h. Thrombolysis was observed through optical microscopy at predetermined time points (0, 15, 30, 45 and 60 min). The thrombus area was analyzed by ImageJ software. The thrombolytic rate was determined by the ratio of the area of thrombus reduction to the area of initial thrombus.
Evaluation of the Targeting Ability and Thrombolytic Effect in vivo
Jugular vein thrombosis was induced according to a previous protocol.16,21 Briefly, after the rats were anesthetized with 1% pentobarbital sodium through the abdominal cavity, the jugular vein was separated with a scalpel and tweezers. Then, the blood vessels were wrapped with filter paper that was dipped in 10% ferric chloride for 2 min. After thrombosis, the filter paper was removed, and the surgical site was washed repeatedly with normal saline.
Targeted Fluorescence Imaging in vivo
Considering the good fluorescence imaging ability of the NPs in vitro, the thrombus model rats were injected with DiR-labeled PPrC or PPr NPs (2.5 mg/mL, 1 mL) via the tail vein to observe their targeting ability. The images were acquired at predetermined points in time (0, 30, 60, 90, 120 min). After fluorescence imaging in vivo, the rats were sacrificed, and the injured jugular vessels were collected to acquire fluorescence images ex vivo (n = 3).
Ultrasound Imaging and Assessment of Thrombolysis in vivo
To evaluate the ultrasound imaging ability of the NPs in vivo, PPrC or PrC NPs (2.5 mg/mL, 1 mL) were injected into the thrombus model rats. Ninety minutes after injection, the thrombus model rats were vertically irradiated with LIFU with a power of 1 W/cm2 for 30 min, and ultrasound images were collected after LIFU irradiation using a PA imaging system. At the same time, the echo intensity was analyzed by DFY software (Chongqing Medical University, China).
To evaluate the effect of targeted thrombolysis in vivo, the rats were divided into 5 groups (n = 3): PPrC, PPC, PrC, PPr NPs (2.5 mg/mL, 1 mL) and saline. These NPs or saline was injected into the tail vein of rats. Ninety minutes after the injection, all NP groups were irradiated with LIFU at a power of 1 W/cm2 for 30 min. After being sacrificed, the jugular vessels of the rats were removed, and histological analysis was performed. The area of the thrombus was measured, and the thrombolytic efficiency was determined based on the area ratio of the remaining thrombus to the total blood vessels.
Biodistribution and Biosafety Evaluation in vivo
The anesthetized SD rats were injected with PPrC NPs (5 mg/mL, 1 mL) or saline through their tail veins. After 3 h of circulation, whole blood and plasma were collected for routine blood tests, liver and kidney function tests and coagulation tests. At 24 h after the injection, the main organs of the rats, including the heart, liver, spleen, lung and kidney, were collected for histological analysis.
A tail bleeding assay was used to further verify the biosafety of the NPs. This assay was carried out according to a previously reported method.22 The SD rats were anesthetized with 1% sodium pentobarbital through the abdominal cavity. Subsequently, PPrC, PPC, PrC, PPr NPs (2.5 mg/mL), free rtPA (0.9 mg/kg, 1 mg/mL) and saline were administered. Two hours after injection, 1 cm was cut from the distal tail, and the injured tail was placed into saline at 37℃. The hemostasis time was recorded as the time when the bleeding had stopped for at least 1 min.
To evaluate the biological distribution of the NPs in vivo, SD rats were injected with 1 mL of DiR-labeled PPrC NPs (2.5 mg/mL) via the tail vein (n = 3). The rats were sacrificed at predetermined time points (6, 12, 24, and 48 h). The main organs (heart, liver, spleen, lung and kidney) were collected, and then ex vivo fluorescence imaging was performed immediately.
Statistical Analysis
Statistical analyses were performed with GraphPad Prism 8 software. All data are expressed as the mean ± standard deviation (SD). The significance of the differences was evaluated with the Student’s t-test and one-way ANOVA. Differences were considered significant at *p < 0.05, **p < 0.01, ***p < 0.001 and ****p < 0.0001.
Results and Discussion
Characterization of the NPs
PLGA NPs were synthesized by a three-step emulsification process. The morphology, particle size, PDI, zeta potential and coupling of CREKA peptides on the NPs were characterized. As shown in Figure 2A, TEM revealed that the PPrC NPs were uniform and spherical and did not exhibit aggregation. The particle sizes of the PPrC, PPC, PrC and PPr NPs, measured by DLS, were 209.09 ± 3.01 nm, 187.28 ± 3.75 nm, 178.56 ± 1.25 nm and 193.70 ± 3.18 nm, respectively, and the PDI of the PPrC NPs was 0.066 ± 0.051 (Figure 2B), which confirmed that the NPs were relatively uniform. The size of the NPs is an important factor that affects their metabolism. Generally, NPs with particle sizes that are too small are not conducive to drug loading and are easily excreted by the kidney,23 while NPs with particle sizes that are too large have a risk of vascular embolism. The particle size of the prepared PPrC NPs was approximately 209 nm. It has been confirmed that NPs of this size can be loaded with sufficient PFH and rtPA, and that the particle size is uniform and the dispersion is good, which guarantees biosafety for in vivo applications. The zeta potential of the PPr NPs was −11.38 ± 3.55 mV, as CREKA itself is positively charged. After coupling with the CREKA polypeptide, the negative charge of the NPs slightly decreased and the zeta potential of the PPrC NPs changed to −6.37 ± 0.18 mV (Figure 2C). FTIR spectra showed that the PPrC NPs exhibited characteristic absorption peaks near 3352 and 1650 cm−1 compared with those of the PPr NPs, and these peaks were caused by stretching vibrations of amide bonds (Figure 2D). The result confirmed that the CREKA peptides were successfully coupled. In addition, FCM revealed that the carrier rate of the CREKA polypeptide was 83.45% (Figure 2E). In the 9-day stability experiment, it was found that the particle size of the PPrC NPs did not change much, which demonstrated that the NPs exhibited good stability (Figure 2F). The rtPA encapsulation efficiency, detected using a TPA ELISA kit and calculated from the standard curve (Figure 2G), was (66.14±1.76) %. The retention activity of the prepared PPrC NPs was (78 ± 0.06) % of that of the free rtPA.
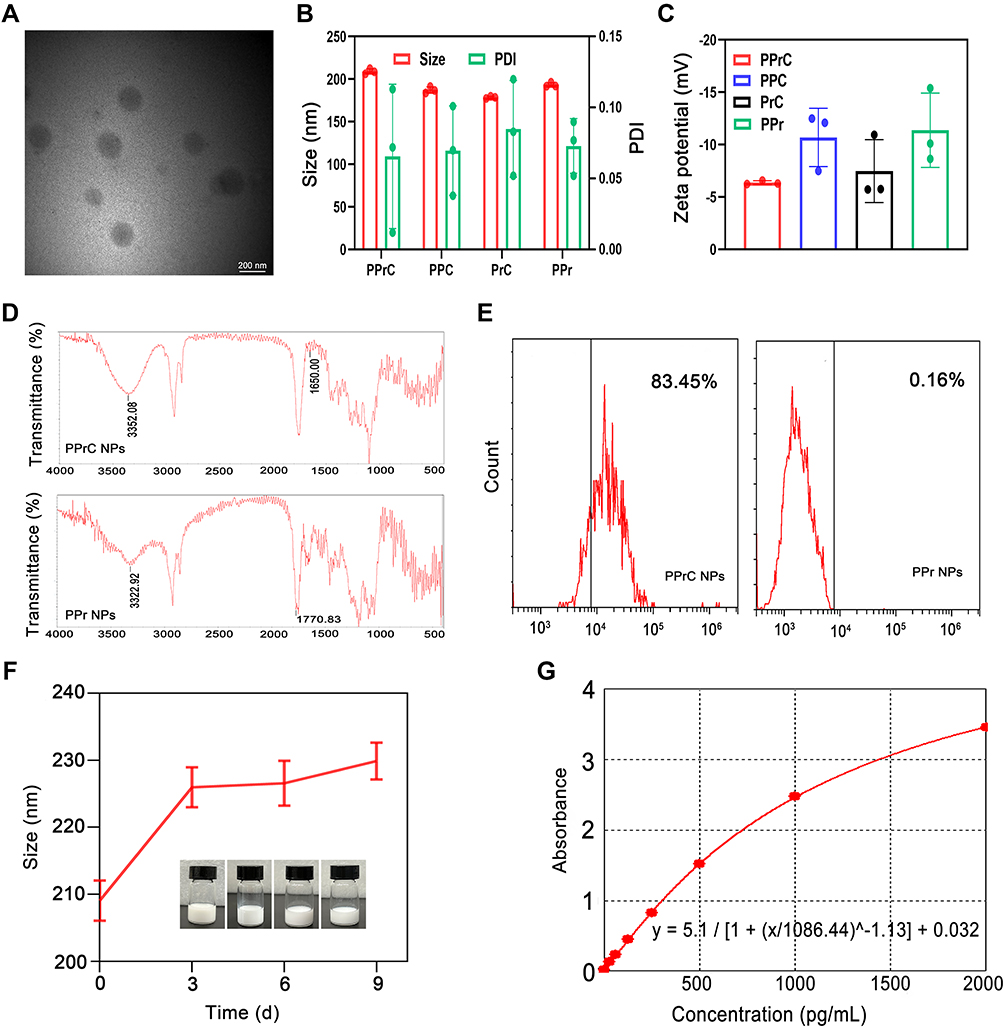 |
Figure 2 Characterization of the NPs. (A) TEM image of the PPrC NPs. (B) Average particle size and PDI of the PPrC, PPC, PrC, and PPr NPs. (C) Zeta potential of the PPrC, PPC, PrC, and PPr NPs. (D) FTIR of the PPrC NPs and PPr NPs. (E) FCM results of the PPrC NPs and PPr NPs. (F) Stability of the PPrC NPs over 9 days. (G) Standard curve for a TPA ELISA kit. |
Dual Modal Imaging in vitro
As reported in some studies, NPs that contain PFH can enhance ultrasound signals after LIFU irradiation.24,25 In our study, it was obvious that the NPs in B-mode and CEUS had poor echo signals before LIFU irradiation. After LIFU irradiation, the echo signal increased with time in B-mode and CEUS until 30 min and then decreased (Figure 3A). In contrast, the signal of the PFH-unloaded NPs did not change significantly. The corresponding echo signal intensity analysis of B-mode and CEUS is shown in Figure 3B and C. The echo signal intensity of the group loaded with PFH was the highest in B-mode and CEUS at 30 min, increasing to 130.67 and 59.67, respectively. In addition, from the optical microscope image, it can be seen that the particle size of the NPs continuously increased from nano to micron in the first 30 min, and the maximum particle size was tens of microns (Figure 3D), which was consistent with the ultrasound imaging results. It was further demonstrated that the increase in the ultrasound signal was due to an increase in the microbubble size caused by the phase transition of PPrC NPs. Additionally, we speculate that the reason for the weakening of the echo signal after 30 min is the rupture of microbubbles after irradiation for a period of time. In addition, in the in vitro fluorescence imaging results for the DiR-labeled PPrC NPs, the average fluorescence intensity increased with increasing NPs concentration (Figure 3E), which indicates that DiR dye is successfully labeled on the NPs and exhibits good performance in fluorescence imaging.
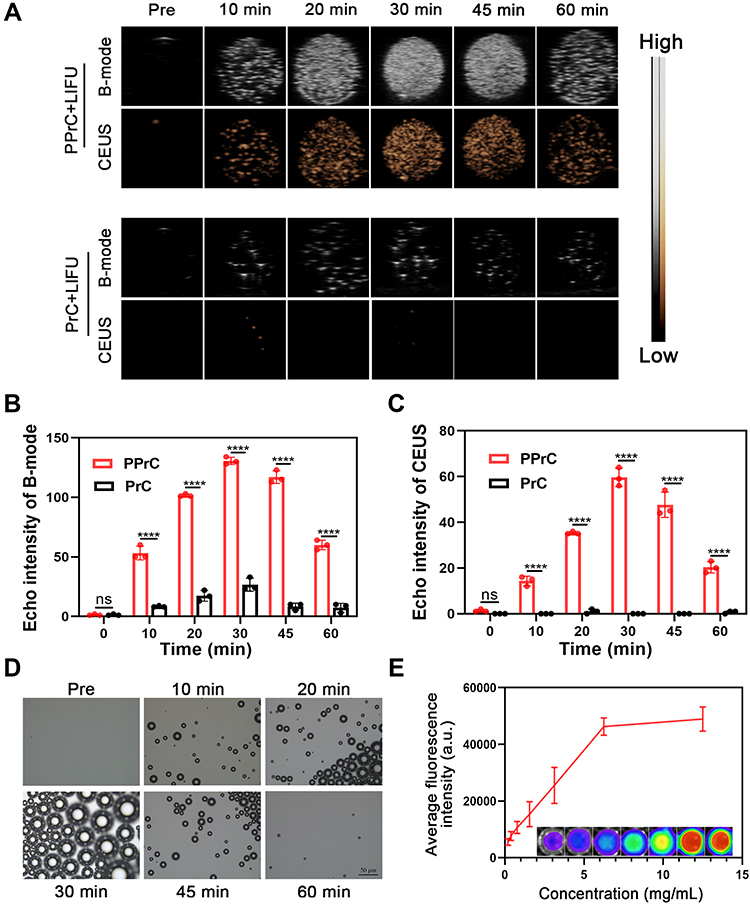 |
Figure 3 Dual modal imaging and LIFU-induced phase transition in vitro. (A) B-mode and CEUS images of PPrC and PrC NPs at different time points under LIFU irradiation. Quantitative analysis of the echo signal intensity in B-mode (B) and CEUS mode (C) (****p < 0.0001, ns = no significance). (D) Optical microscope images of PPrC NPs irradiated by LIFU at different time points. (E) Quantitative analysis of the fluorescence intensity of DiR-labeled PPrC NPs at different concentrations in vitro. |
LIFU-Responsive Drug Release in vitro
Drug release can be promoted by certain stimuli, such as ultrasounds, lasers and microenvironmental changes in vivo, to achieve site-specific release of targeted drugs.5,26–28 In this study, the phase transition material PFH is loaded in the PLGA shell. Under external LIFU irradiation, the droplets change from the nano scale to micron scale bubbles, and this is called acoustic droplet vaporization (ADV).19 This vaporization is an instantaneous, blasting effect of NPs and makes it possible for the rapid release of rtPA to occur. This precise and controllable drug release method can reduce the complications of bleeding caused by conventional drug thrombolysis. As shown in Figure 4A, rtPA in the F-PPrC+LIFU group was rapidly released in the first 60 min, especially in the first 30 min. It can be seen from the previous phase transition process that this rapid release may be due to the continuous expansion of the NPs in the first 30 min. Microbubbles contract and expand as they experience the compression and rarefaction cycles of passing acoustic waves. These volumetric oscillations can facilitate drug release, increased drug uptake and strong backscattered echoes that can be used for ultrasound imaging.29 The drug release rate in the 30–60 min period was slower than that in the first 30 min due to the blasting of some of the phase-transitioned NPs and a reduction of the number of NPs. After 60 min, the drug release entered the plateau phase; the reason may be that the vast majority of NPs undergo a phase transition, and the thrombolytic drug is almost completely released. The cumulative release rate of rtPA reached 77.75%. Interestingly, the release curve of the F-PPr+LIFU group, which was a nontargeted group, was similar to that of the F-PPrC+LIFU group at each time point, indicating that the coupling of CREKA polypeptides does not affect the release of rtPA. In contrast, the drug release of F-PPrC without LIFU irradiation was less than that with LIFU irradiation at each time point, and the release rate was slower. To further evaluate the effect of PFH on drug release, the F-PrC+LIFU group was set up for comparison. We found that the release of this group was slower than that of the F-PPrC+LIFU group. This indicates that in the NPs without PFH, there was no ADV effect to accelerate the release of rtPA, but the release rate of rtPA in the PrC+LIFU group was still higher than that in the PPrC without LIFU group, which may be because pure LIFU irradiation will slightly increase the temperature and promote drug release to a small extent. The above showed that the combination of LIFU and PFH could accelerate drug release through the expansion and blasting of NPs.
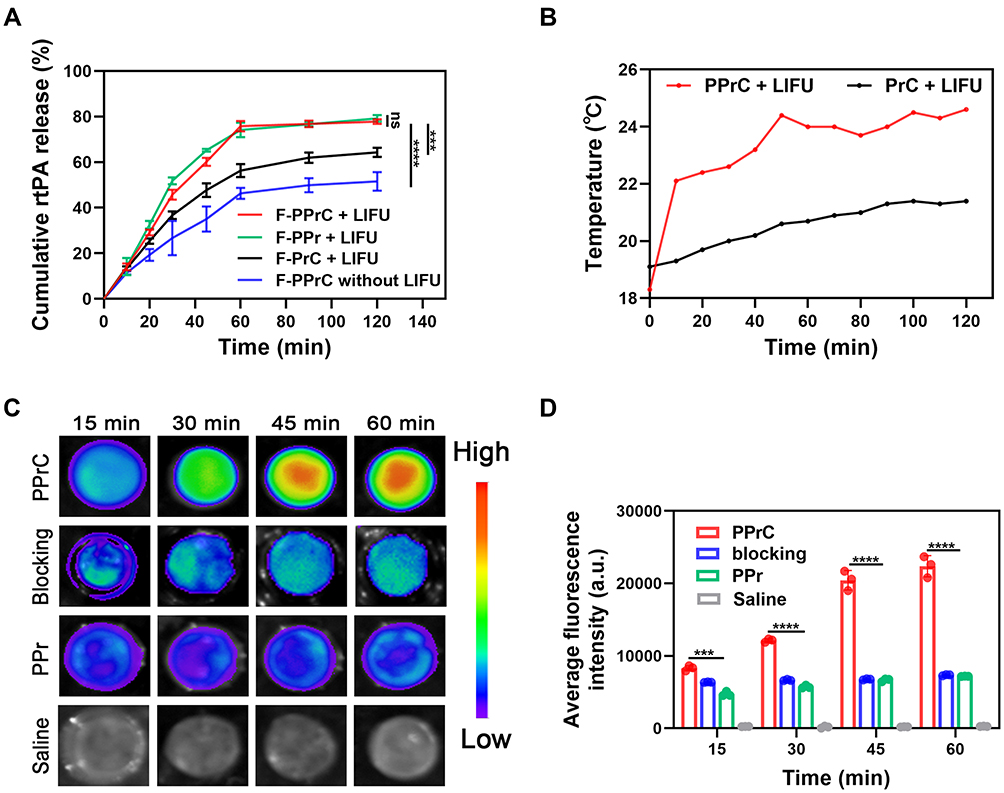 |
Figure 4 LIFU-responsive drug release and evaluation of targeting ability in vitro. (A) Cumulative release curves of the thrombolytic drug rtPA in different NPs at different time points (***p < 0.001, ****p < 0.0001, ns = no significance). (B) Time course of the temperature change. (C) Fluorescence images at different time points after coincubation with blood clots in the PPrC, blocking, PPr and saline groups. (D) Quantitative analysis of the fluorescence intensity of blood clots after different treatment methods (***p < 0.001, ****p < 0.0001). |
In addition, rtPA is essentially a serine protease,30 and the temperature is an important factor that affects its activity.31,32 Denaturation of protein molecules at a high temperature is a common feature leading to the loss of biological activity. Generally, proteins undergo thermal denaturation when the temperature exceeds 45℃,33 which is usually irreversible. In our experiment, the temperature of the PPrC NPs irradiated with LIFU was not higher than 24.6℃, and the temperature increase was 6.3℃, while the temperature increase of the PrC+LIFU group was only 2.3℃ (Figure 4B). These results showed that the temperature increase caused by LIFU did not affect protein denaturation, which is similar to previous research.16 Therefore, in this elevated temperature range, we believe that the increase in the local temperature does not cause the loss of rtPA enzyme activity and does not affect thrombolytic activity.
The Targeting Ability of the PPrC NPs to Thrombi in vitro
The effective accumulation of NPs in thrombi is the key step in thrombus treatment. To enhance the specific targeting of the NPs to thrombi, CREKA was covalently bound to the NP shell. As shown in Figure 4C, the fluorescence intensity of the nontargeted PPr group was slightly enhanced. In contrast, the PPrC group showed increasingly strong fluorescence at the thrombus over time due to the loading of the CREKA polypeptide on the surface of the NPs, which mediated the specific binding of the NPs to fibrin. The fluorescence intensity of the targeted group that was loaded with CREKA was approximately 3.1 times that of the nontargeted group at 60 min (Figure 4D). In the blocking group, the fluorescence intensity was lower than that of the targeted group at each time point, which may be due to the excessive polypeptide occupying the fibrin target on the blood clot in advance and forming a competitive inhibition of CREKA on the surface of the NPs. This demonstrates that these PPrC NPs exhibited a superior targeting ability and can become the basis for effective thrombolysis.
Assessment of Thrombolysis Efficacy in vitro
Assessment of the Thrombolysis Efficacy of the NPs Under Static Conditions
The in vitro effect of thrombolysis was evaluated by preparing blood clots from rat whole blood. Figure 5A shows representative images of different treated clot lysis samples that were collected at different time points. After 60 min, there was no significant change in the color of the supernatant in the saline group, which demonstrated the stability of the prepared blood clot. At each time point, the color of the blood clot supernatant treated with the PPrC NPs was darker than that in the other treatment groups (PPC, PrC, PPr and saline), and the difference became more obvious with the extension of time, suggesting the rupture of the fibrin network and the release of hemoglobin. In addition, by comparing the weights before and after the treatment, we found that the thrombolytic rate of the PPrC group was 51.57% and that of the PrC group was 25.32% (Figure 5B). To further compare the thrombolytic effect of each group, the absorbance of the supernatant at 540 nm was also detected. The absorbance of the blood clot supernatant that was treated with the PPrC NPs was higher than that of any other group (Figure 5C). This indicated that there was more hemoglobin released due to thrombolysis in the PPrC group, which is consistent with the representative imaging results. Therefore, the thrombolytic effect was more significant in the presence of PFH, and this may be due to the cavitation effect caused by phase transition, which loosens the thrombus structure through micro jets, shock waves and accelerated release of rtPA.34 Both PFH and rtPA have independent thrombolytic effects.9,17 After we performed the same treatment on different groups of NPs, it was found that the thrombolytic rate of the PPC group was 40.81% and that of the PrC group was 25.32%, indicating that PFH may play a more significant role in the thrombolysis of the NPs. We speculate that the result of this phenomenon may be that the PFH-containing NPs are thrombolytic through their expansion and rupture, which promotes deeper NPs penetration and improves the thrombolytic effect. At the same time, we found that the thrombolytic rate in the PPr group was 33.41%, which was because the PPr NPs could not target thrombi, and the aggregation of the NPs with thrombi was reduced, resulting in a much lower thrombolytic effect than that in the target group. The feasibility of phase transition thrombolysis has been preliminarily confirmed in previous research by our group.16 However, this mechanical thrombolysis method has difficulty achieving thrombolysis at the cellular and molecular levels and has the potential risk of inducing thrombus shedding. Considering the risk of re-embolization, the second-generation thrombolytic drug rtPA was added to the NPs in this study. As mentioned above, after irradiation with LIFU at a power of 1 W/cm2, the PFH underwent a process of swelling and gradual volume shrinkage within 60 min, and rtPA was also rapidly released within 60 min, so the in vitro thrombolysis experiment in this research was carried out for 60 min. rtPA can act on fibrin, activate plasminogen to plasmin and realize thrombolysis, but insufficient penetration of the drug into the thrombus is the key factor limiting the thrombolytic effect, while PPrC NPs can destroy the fibrin network structure after physical explosion so that the released rtPA can penetrate deeper into the thrombus. In vitro studies demonstrated that ultrasound waves can disrupt clots by inducing plasmin-mediated thrombolysis and that simultaneous use of ultrasound with thrombolytics is synergistic.35,36 These factors further improve the thrombolytic effect of the NPs.
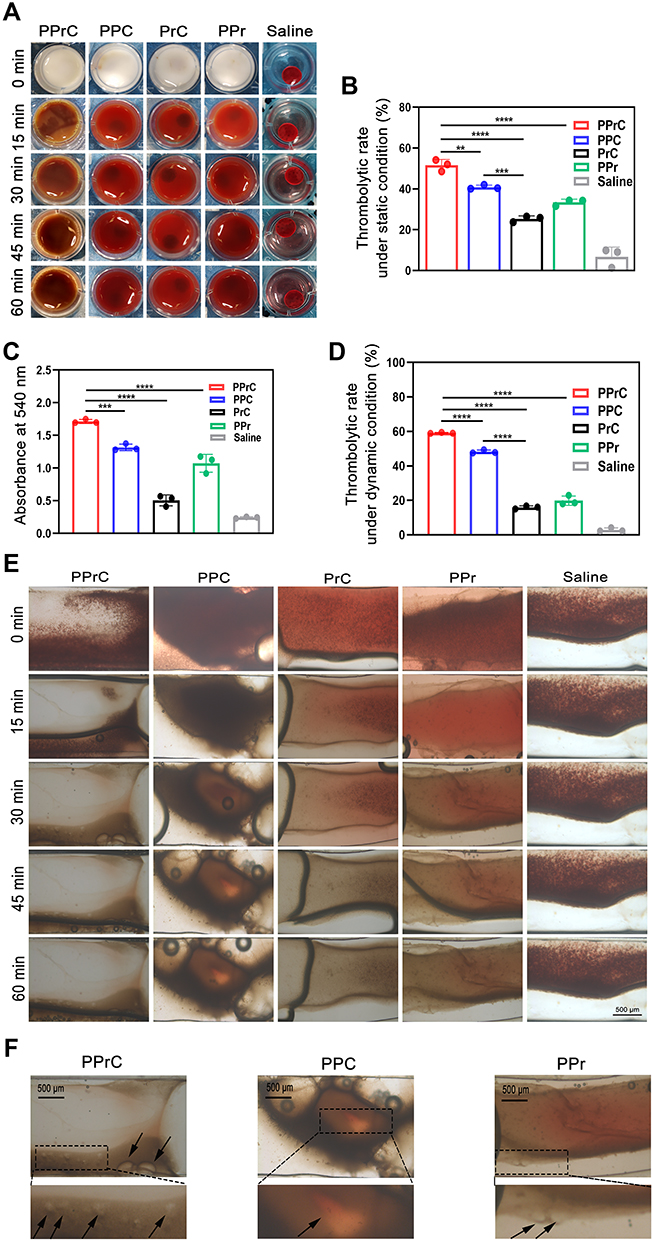 |
Figure 5 Thrombolytic effects of different treatment methods in vitro. (A) Representative images of blood clots at different time points of treatment under static conditions in the PPrC, PPC, PrC, PPr and saline groups. (B) Thrombolytic rate after different treatments under static conditions (**p < 0.01, ***p < 0.001, ****p < 0.0001). (C) Absorbance value at 540 nm of the supernatant after blood clot dissolution (***p < 0.001, ****p < 0.0001). (D) Thrombolytic rate after different treatments under dynamic conditions (****p < 0.0001). (E) Optical microscopic images of thrombolytic effects under dynamic conditions. (F) Local enlarged view of the thrombolytic effects of PPrC, PPC and PPr NPs under dynamic conditions (the black arrows represent the uneven pore structures). |
Assessment of the Thrombolysis Efficacy of the NPs Under Dynamic Conditions
Based on static thrombolysis, to further explore the thrombolytic efficiency of the PPrC NPs in a dynamic fluid environment, a microfluidic chip was used to simulate the thrombolytic ability under physiological conditions. The channel in the microfluidic chip was perfused with collagen in advance to promote the formation of thrombi and was then washed with normal saline. After the whole blood was perfused into the channel to form a thrombus, the thrombolysis process was observed under an optical microscope. As shown in Figure 5E, there were no significant changes in the thrombus area in the normal saline group, which demonstrated the stability of this thrombus. The thrombus in the PPrC group was significantly reduced, and the fastest reduction rate was observed in the first 30 min, and then the rate slowed. According to previous experimental results, this may be due to the phase transition, volume expansion and final disintegration and rupture of the PPrC NPs in the first 30 min. The mechanical impact produced by the PPrC NPs could destroy the thrombus structure. Based on the release curve, rtPA was rapidly released during this period and further acted on fibrin. These factors jointly affected the dissolution of the thrombus. In the area analyzed before and after thrombolysis, the thrombolytic rate in the PPrC group was significantly higher than that in the other groups (Figure 5D), similar to the thrombolytic rate under static conditions. In the PPr group, the thrombolytic effect under dynamic conditions was slightly worse than that under static conditions. The reason may be that the thrombus was soaked in the PPr NPs solution under static conditions, and the contact time between the NPs and blood clot was long. However, under dynamic fluid conditions, without the targeting assistance of CREKA, the contact time between the PPr NPs and the thrombus was very short, resulting in a lower thrombolysis efficiency, thus further illustrating the importance of targeting assistance under dynamic fluid conditions in vivo, especially for vascular lesions such as thrombi and plaques. At the edge of the thrombus, the groups containing PFH (PPrC, PPC and PPr groups) were rougher than the PrC group. In addition, the thrombus images of the PFH group (PPrC, PPC and PPr groups) were enlarged in Figure 5F, and we observed uneven pore structures on the thrombus, which may be caused by the mechanical destruction of the thrombus by the micro jet and shock wave that were produced by the blasting of the NPs. This effect could act as a nanoexcavator inside the thrombus and provide a channel for the penetration of subsequent thrombolytic drugs, solving the problem that traditional thrombolytic drugs cannot penetrate deep into the thrombus, which is consistent with a previous study.21
The Ability of PPrC NPs to Target Thrombi in vivo
In previous in vitro experiments, the ability of the NPs to be used in fluorescence imaging was verified. Therefore, DiR-labeled NPs were used for in vivo imaging to observe the targeting ability of the NPs in the thrombotic jugular vein of rats. Although the fluorescence signal in the PPr group increased slightly, it was not significant compared with that in the PPrC group (Figure 6A). By analyzing the fluorescence intensity of the jugular vein with the thrombus (Figure 6B), it was found that the fluorescence intensity of the PPrC group peaked at 90 min, and this peak was 14.92 times higher than that of the PPr group. This result indicates that the concentration of PPrC NPs in the jugular vein with the thrombus was the highest 90 min after injection, and this result was significant in that it provided a reference for subsequent experiments. After the rats were sacrificed, the thrombotic jugular vein was removed for fluorescence imaging (Figure 6A). As expected, the fluorescence intensity of the jugular vein in the PPrC group was significantly higher than that in the PPr group (Figure 6C).
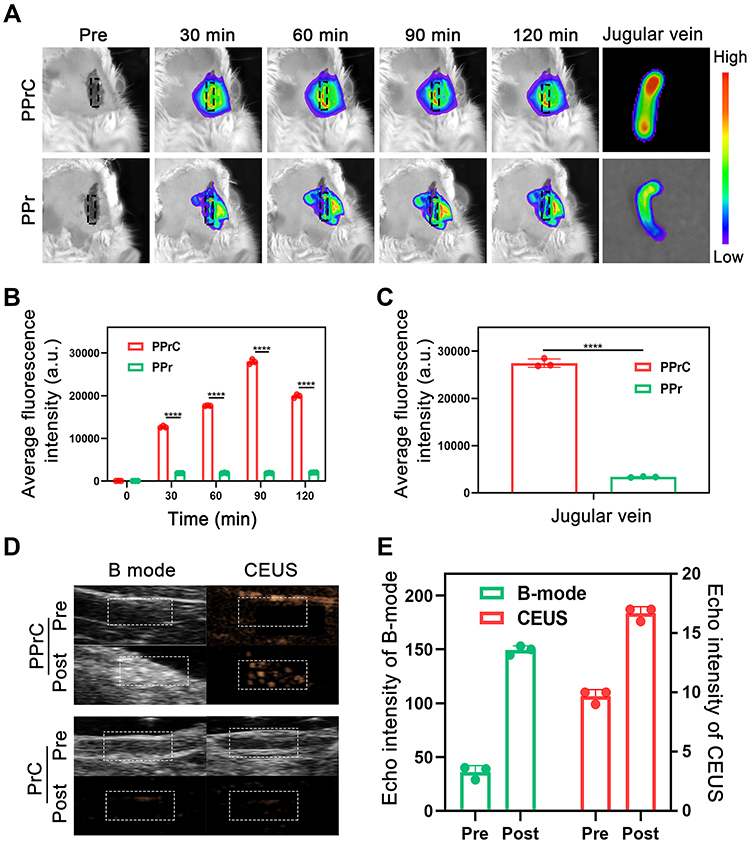 |
Figure 6 Fluorescence imaging and ultrasound imaging in vivo. (A) Jugular vein fluorescence images of the PPrC and PPr groups at different time points in vivo and jugular vein fluorescence images after resection. (B) Quantitative analysis of the fluorescence intensity of blood vessels at different time points (****p < 0.0001). (C) Quantitative analysis of the fluorescence intensity of the jugular vein after resection (****p < 0.0001). (D) Ultrasound imaging of the PPrC and PrC groups in vivo. (E) Quantitative analysis of the ultrasound echo intensity of the PPrC group in vivo. |
Ultrasound Imaging in vivo
Based on the good results for the ultrasound imaging in vitro, we also carried out this experiment in vivo. In the in vivo fluorescence imaging experiment, we inferred that the concentration of the PPrC NPs reached the highest level 90 min after injection. Therefore, in the ultrasound imaging experiment, when the NPs had circulated in vivo for 90 min, we began to perform LIFU irradiation. Before LIFU irradiation, the vascular structure was clearly visible in B-mode, but it was difficult to distinguish the thrombus from the surrounding tissue. However, the echo signal of the thrombus in the PPrC group was significantly enhanced after LIFU irradiation. For comparison, there was no significant change in the ultrasound signal in the PrC group (Figure 6D). In addition, the ultrasound signal intensity after irradiation was significantly higher than that before irradiation in B-mode and CEUS mode in the PPrC group (Figure 6E). This confirmed that the PPrC NPs successfully underwent liquid-to-gas phase transition in vivo, which has the potential to guide clinical diagnoses.
Assessment of Thrombolysis in vivo
Motivated by the excellent thrombolytic effect in vitro, we verified whether this new synergistic thrombolytic strategy is suitable for animals in vivo. The most commonly used model for inducing thrombosis in vivo was adopted, and this model was simple and rapid to establish. A stable noncompletely occlusive thrombus was observed in the saline group. Obviously, the percentage of the remaining thrombus area in the PPrC group (17.67%) was lower than that in the PPr group (46.27%) (Figure 7A and B), which may be because PPrC NPs can target fibrin components in thrombi, thereby increasing the effective accumulation of the NPs in the thrombus. Comparing the PPC group with the PrC group, the PPC group had a lower percentage of the thrombus area after LIFU irradiation than the PrC group, but both were still higher than that in the PPrC group. This finding indicates that under the experimental conditions, both PFH and rtPA have independent thrombolytic effects, but the thrombolytic effect of PFH is more significant. This is consistent with the results of in vitro studies. When the two functions are combined (in the PPrC group), the microbubbles formed after the phase transition of the NPs under ultrasound irradiation can be used as cavitation nuclei to reduce the cavitation threshold.37 The volume of the NPs first expanded and then collapsed and disintegrated under ultrasound irradiation. The resulting shock wave destroyed the thrombus structure, which also explains why the thrombus structure in the group containing PFH was looser than that of the group without PFH. After the thrombus structure was destroyed, more thrombus binding sites were exposed, which further promoted the targeted binding of the NPs. The release of rtPA was accelerated due to cavitation of the NPs and realized the benign cycle of PFH and rtPA synergistic therapy. Previous studies have shown that low-power ultrasound treatments do not produce acute side effects38 or damage vascular tissues.16 Similarly, in our experiment, the treated jugular vein wall remained intact and did not show signs of damage. In general, with a power of 1 W/cm2, LIFU is suitable for use in the biomedical field.
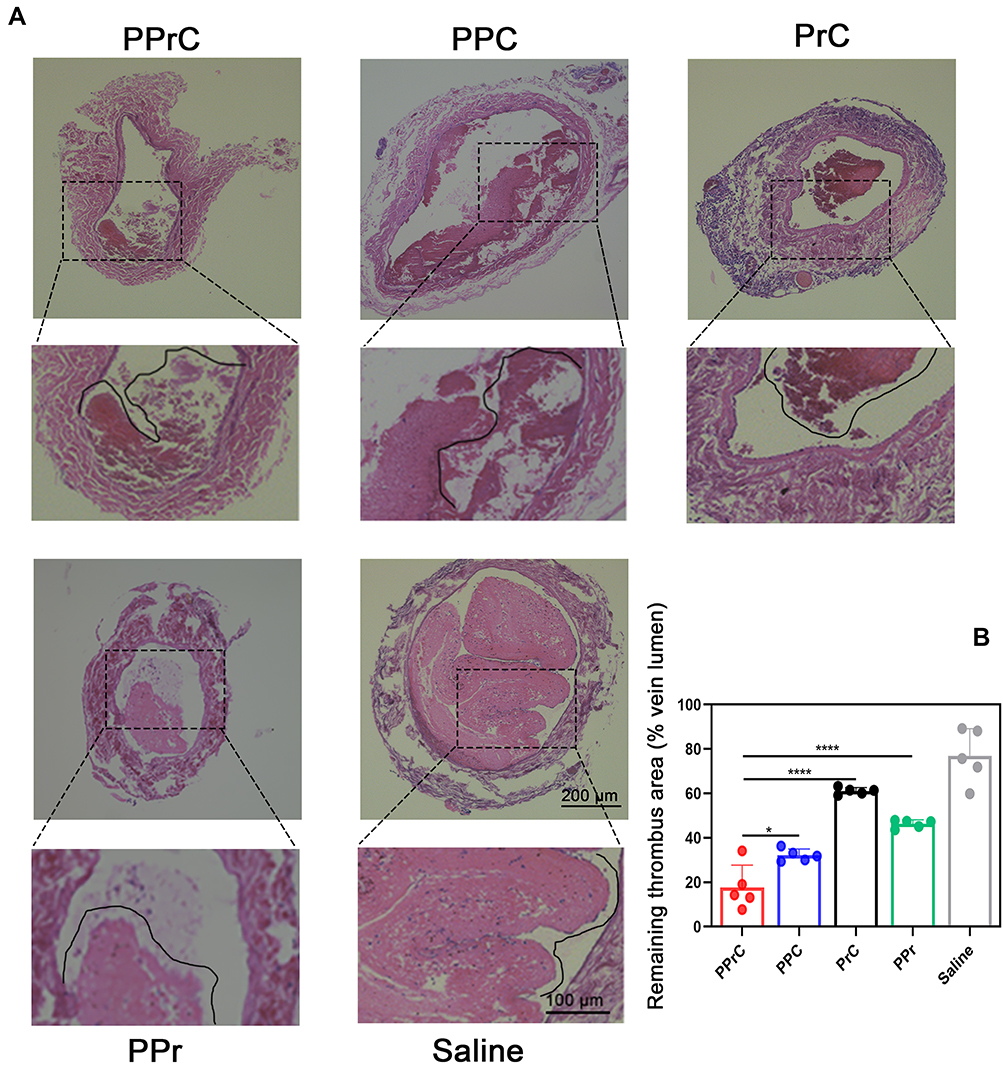 |
Figure 7 Histological analysis of jugular vein thrombosis in SD rats after the corresponding treatment. (A) H&E staining of the jugular veins treated by different methods. (B) Quantitative analysis of the remaining thrombus area (% vein lumen) in the jugular veins in the different treatment groups (*p < 0.05, ****p < 0.0001). |
Biodistribution and Biosafety Evaluation in vivo
Regarding the biodistribution of the NPs, as shown in Figure 8A and B, the PPrC NPs were mainly distributed in the liver and spleen. Drug carriers larger than 20 nm are mainly cleared from the circulation through the reticuloendothelial system (including the liver, spleen and lymph nodes).23 Furthermore, the average fluorescence intensity of each organ gradually decreased over time, which could help avoid potential long-term toxicity threats.
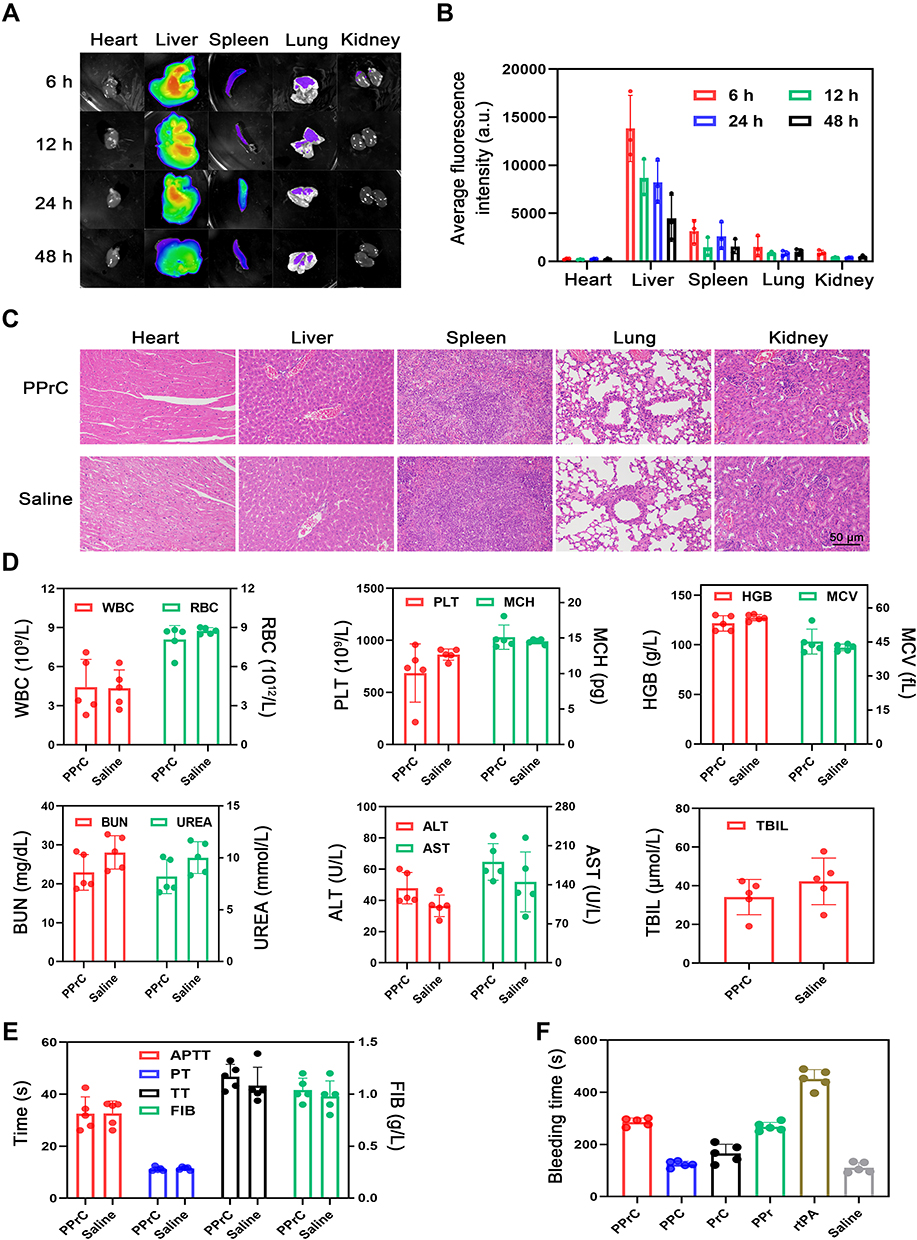 |
Figure 8 Biological metabolism and safety assessment of the NPs in vivo. (A) Fluorescence images of the isolated heart, liver, spleen, lung and kidney at different time points. (B) Quantitative analysis of the fluorescence intensity in Figure (A). (C) H&E staining of the main organs (heart, liver, spleen, lung and kidney) after treatment with the PPrC NPs or normal saline. (D) Routine blood examination and blood biochemical indexes after treatment with the PPrC NPs or normal saline. (E) Results of the four items of the coagulation tests. (F) Hemostatic time of severed tails in different treatment groups. |
The biosafety of the NPs was evaluated by histological examination and whole blood and plasma examinations. Figure 8C provides a histological overview of the main organs. It was obvious that there was no difference between the saline group and the PPrC group, and H&E staining revealed no histopathological abnormalities. In addition, the results of the routine blood examination and plasma biochemical examination showed no abnormalities (Figure 8D). The above data indicate that the PPrC NPs have good biocompatibility and safety. In addition, four items of coagulation (prothrombin time: PT; activated partial thromboplastin time: APTT; thrombin time: TT; and fibrinogen: FIB) were examined for thrombotic diseases, and the results showed that all indexes were normal (Figure 8E). In addition, considering that many thrombolytic methods involve the risk of bleeding complications, the bleeding time of the rat tail was recorded. The tail bleeding time of the rats treated with free rtPA was almost 4 times that of the saline-treated group, indicating that the coagulation system was abnormal (Figure 8F). In contrast, the bleeding time in the PPC, PPr, PrC and PPrC groups was significantly less than that in the rtPA group, indicating that the prepared NPs have a significant effect on reducing the risk of bleeding.
Conclusion
In summary, phase transition thrombolysis and drug thrombolysis were integrated into an NP to achieve synergistic and efficient thrombolysis. First, modification of CREKA polypeptides on the NP surface enables it to target the fibrin component of the thrombus and accumulate at the thrombus site. Then, under LIFU irradiation, the volume expansion, collapse and disintegration of the NPs played the function of the excavator inside the thrombus, destroyed the thrombus structure, and accelerated the release and deep penetration of the thrombolytic drug rtPA, thus further improving its thrombolytic effect; sequential drug thrombolysis overcomes the potential risks of phase transition thrombolysis in return. In conclusion, compared with conventional thrombolysis, the PPrC NPs can more effectively dissolve thrombi and reduce the side effects of drug thrombolysis and have good biocompatibility and biosafety. We believe that this will be a promising thrombolytic strategy. We will conduct real-time monitoring and evaluation of this combined thrombolysis method (mechanical thrombolysis and drug thrombolysis) in the future to further determine the safety and efficacy of in vivo application.
Acknowledgments
This work was financially supported by the National Natural Science Foundation of China (grant Nos. 81971608, 81901807, 81701650, 82102063), the Chongqing Natural Science Foundation (cstc2021jcyj-msxmX0210) and the Kuanren Talents Program of the Second Affiliated Hospital of Chongqing Medical University (2020–7, 2021–24). Liu Hu and Jie Xu are co-first authors for this study.
Disclosure
The authors declare no conflicts of interest in this work.
References
1. Zenych A, Fournier L, Chauvierre CJB. Nanomedicine progress in thrombolytic therapy. Biomaterials. 2020;258:120297. doi:10.1016/j.biomaterials.2020.120297
2. Li X, Guo T, Feng Q, et al. Progress of thrombus formation and research on the structure-activity relationship for antithrombotic drugs. Eur J Med Chem. 2022;228:114035. doi:10.1016/j.ejmech.2021.114035
3. Benjamin E, Blaha M, Chiuve S, et al. Heart disease and stroke statistics-2017 update: a report from the American Heart Association. Circulation. 2017;135(10):e146–e603. doi:10.1161/CIR.0000000000000485
4. Absar S, Gupta N, Nahar K, Ahsan FT. Engineering of plasminogen activators for targeting to thrombus and heightening thrombolytic efficacy. J Thromb Haemost. 2015;13(9):1545–1556. doi:10.1111/jth.13033
5. Wang S, Guo X, Xiu W, et al. Accelerating thrombolysis using a precision and clot-penetrating drug delivery strategy by nanoparticle-shelled microbubbles. Sci Adv. 2020;6(31):eaaz8204. doi:10.1126/sciadv.aaz8204
6. Liu Y, Chen D, Shang P. A review of magnet systems for targeted drug delivery. J Control Release. 2019;302:90–104. doi:10.1016/j.jconrel.2019.03.031
7. Liu C, Hsu H, Chen J, Wu T, Ma YN. Thrombolysis induced by intravenous administration of plasminogen activator in magnetoliposomes: dual targeting by magnetic and thermal manipulation. Nanomedicine. 2019;20:101992. doi:10.1016/j.nano.2019.03.014
8. Ma Y, Wu S, Wu T, Chang Y, Hua M, Chen JJB. Magnetically targeted thrombolysis with recombinant tissue plasminogen activator bound to polyacrylic acid-coated nanoparticles. Biomaterials. 2009;30(19):3343–3351. doi:10.1016/j.biomaterials.2009.02.034
9. Huang Y, Yu L, Ren J, et al. An activated-platelet-sensitive nanocarrier enables targeted delivery of tissue plasminogen activator for effective thrombolytic therapy. J Control Release. 2019;300:1–12. doi:10.1016/j.jconrel.2019.02.033
10. Chen K, Wang Y, Liang H, et al. Intrinsic biotaxi solution based on blood cell membrane cloaking enables fullerenol thrombolysis in vivo. ACS Appl Mater Interfaces. 2020;12(13):14958–14970. doi:10.1021/acsami.0c01768
11. Walker JM, Zaleski JM. Non-enzymatic remodeling of fibrin biopolymers via photothermally triggered radical-generating nanoparticles. Chem Mater. 2014;26(17):5120–5130. doi:10.1021/cm5024713
12. Dong L, Liu X, Wang T, et al. Localized light-au-hyperthermia treatment for precise, rapid, and drug-free blood clot lysis. ACS Appl Mater Interfaces. 2019;11(2):1951–1956. doi:10.1021/acsami.8b20616
13. Ximendes E, Marin R, Shen Y, et al. Infrared-emitting multimodal nanostructures for controlled in vivo magnetic hyperthermia. Adv Mater. 2021;33(30):e2100077. doi:10.1002/adma.202100077
14. Chang D, Lim M, Goos J, et al. Biologically targeted magnetic hyperthermia: potential and limitations. Front Pharmacol. 2018;9:831. doi:10.3389/fphar.2018.00831
15. Cao W, Liu Y, Ran P, et al. Ultrasound-propelled janus rod-shaped micromotors for site-specific sonodynamic thrombolysis. ACS Appl Mater Interfaces. 2021;13(49):58411–58421. doi:10.1021/acsami.1c19288
16. Zhong Y, Zhang Y, Xu J, et al. Low-Intensity focused ultrasound-responsive phase-transitional nanoparticles for thrombolysis without vascular damage: a synergistic nonpharmaceutical strategy. ACS Nano. 2019;13(3):3387–3403. doi:10.1021/acsnano.8b09277
17. Xu J, Zhou J, Zhong Y, et al. Phase transition nanoparticles as multimodality contrast agents for the detection of thrombi and for targeting thrombolysis: in vitro and in vivo experiments. ACS Appl Mater Interfaces. 2017;9(49):42525–42535. doi:10.1021/acsami.7b12689
18. Bai S, Liao J, Zhang B, et al. Multimodal and multifunctional nanoparticles with platelet targeting ability and phase transition efficiency for the molecular imaging and thrombolysis of coronary microthrombi. Biomater Sci. 2020;8(18):5047–5060. doi:10.1039/d0bm00818d
19. Aliabouzar M, Kumar K, Sarkar K. Effects of droplet size and perfluorocarbon boiling point on the frequency dependence of acoustic vaporization threshold. J Acoust Soc Am. 2019;145(2):1105. doi:10.1121/1.5091781
20. Xu T, Cui Z, Li D, et al. Cavitation characteristics of flowing low and high boiling-point perfluorocarbon phase-shift nanodroplets during focused ultrasound exposures. Ultrason Sonochem. 2020;65:105060. doi:10.1016/j.ultsonch.2020.105060
21. Yang A, Qiao B, Strohm E, et al. Thrombin-responsive engineered nanoexcavator with full-thickness infiltration capability for pharmaceutical-free deep venous thrombosis theranostics. Biomater Sci. 2020;8(16):4545–4558. doi:10.1039/d0bm00917b
22. Zhang F, Liu Y, Lei J, et al. Metal-organic-framework-derived carbon nanostructures for site-specific dual-modality photothermal/photodynamic thrombus therapy. Adv Sci. 2019;6(17):1901378. doi:10.1002/advs.201901378
23. Myerson J, Anselmo A, Liu Y, Mitragotri S, Eckmann D, Muzykantov VR. Non-affinity factors modulating vascular targeting of nano- and microcarriers. Adv Drug Deliv Rev. 2016;99:97–112. doi:10.1016/j.addr.2015.10.011
24. Tang H, Guo Y, Peng L, et al. In vivo targeted, responsive, and synergistic cancer nanotheranostics by magnetic resonance imaging-guided synergistic high-intensity focused ultrasound ablation and chemotherapy. ACS Appl Mater Interfaces. 2018;10(18):15428–15441. doi:10.1021/acsami.8b01967
25. Qin H, Teng R, Liu Y, Li J, Yu M. Drug release from gelsolin-targeted phase-transition nanoparticles triggered by low-intensity focused ultrasound. Int J Nanomedicine. 2022;17:61–71. doi:10.2147/IJN.S341421
26. Chang L, Chuang E, Cheng T, et al. Thrombus-specific theranostic nanocomposite for codelivery of thrombolytic drug, algae-derived anticoagulant and NIR fluorescent contrast agent. Acta Biomater. 2021;134:686–701. doi:10.1016/j.actbio.2021.07.072
27. Mei T, Kim A, Vong L, et al. Encapsulation of tissue plasminogen activator in pH-sensitive self-assembled antioxidant nanoparticles for ischemic stroke treatment - Synergistic effect of thrombolysis and antioxidant. Biomaterials. 2019;215:119209. doi:10.1016/j.biomaterials.2019.05.020
28. Mei T, Shashni B, Maeda H, Nagasaki YJB. Fibrinolytic tissue plasminogen activator installed redox-active nanoparticles (t-PA@iRNP) for cancer therapy. Biomaterials. 2020;259:120290. doi:10.1016/j.biomaterials.2020.120290
29. Sirsi S, Borden MA. State-of-The-art materials for ultrasound-triggered drug delivery. Adv Drug Deliv Rev. 2014;72:3–14. doi:10.1016/j.addr.2013.12.010
30. Amiri M, Jalali-Javaran M, Haddad R. In silico and in vivo analyses of the mutated human tissue plasminogen activator (mtPA) and the antithetical effects of P19 silencing suppressor on its expression in two Nicotiana species. Sci Rep. 2018;8(1):14079. doi:10.1038/s41598-018-32099-6
31. Vashishth A, Ram S, Beniwal VJB. Cereal phytases and their importance in improvement of micronutrients bioavailability. Biotech. 2017;7(1):42. doi:10.1007/s13205-017-0698-5
32. Devendran S. Microwave assisted enzymatic kinetic resolution of (±)-1-phenyl-2-propyn-1-ol in nonaqueous media. Biomed Res Int. 2014;2014:482678. doi:10.1155/2014/482678
33. Bischof J. Thermal stability of proteins. Ann N Y Acad Sci. 2005;1066:12–33. doi:10.1196/annals.1363.003
34. Hennerici M, Kern R, Szabo KN. Non-pharmacological strategies for the treatment of acute ischaemic stroke. Lancet Neurol. 2013;12(6):572–584. doi:10.1016/S1474-4422(13)70091-7
35. Tang J, Tang J, Zhu Q, et al. A minimally invasive strategy to evacuate hematoma by synergy of an improved ultrasonic horn with urokinase: an in vitro study. Med Phys. 2022;49(3):1333–1343. doi:10.1002/mp.15453
36. Furfaro D, Stephens R, Streiff M. Catheter-directed thrombolysis for intermediate-risk pulmonary embolism. Ann Am Thorac Soc. 2018;15(2):134–144. doi:10.1513/AnnalsATS.201706-467FR
37. Petit B, Bohren Y, Gaud E, et al. Sonothrombolysis: the contribution of stable and inertial cavitation to clot lysis. Ultrasound Med Biol. 2015;41(5):1402–1410. doi:10.1016/j.ultrasmedbio.2014.12.007
38. Lu S, Zhao P, Deng Y, Liu YJP. Mechanistic Insights and therapeutic delivery through micro/nanobubble-assisted ultrasound. Pharmaceutics. 2022;14(3). doi:10.3390/pharmaceutics14030480
 © 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.