Back to Journals » International Journal of Nanomedicine » Volume 12
A simple reduction-sensitive micelles co-delivery of paclitaxel and dasatinib to overcome tumor multidrug resistance
Authors Li J, Xu R, Lu X, He J, Jin S
Received 3 August 2017
Accepted for publication 14 September 2017
Published 1 November 2017 Volume 2017:12 Pages 8043—8056
DOI https://doi.org/10.2147/IJN.S148273
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Prof. Dr. Dongwoo Khang
Jun Li,1,* Ruitong Xu,2,* Xiao Lu,3 Jing He,1 Shidai Jin1
1Department of Medical Oncology, 2Department of General Practice, The First Affiliated Hospital with Nanjing Medical University, Nanjing, 3Department of Medical Oncology, Changshu No 1 People’s Hospital, Changshu, People’s Republic of China
*These authors contributed equally to this work
Abstract: Multidrug resistance (MDR) is one of the major obstacles in successful chemotherapy. The combination of chemotherapy drugs and multidrug-resistant reversing agents for treating MDR tumor is a good strategy to overcome MDR. In this work, we prepared the simple redox-responsive micelles based on mPEG-SS-C18 as a co-delivery system to load the paclitaxel (PTX) and dasatinib (DAS) for treatment of MCF-7/ADR cells. The co-loaded micelles had a good dispersity and a spherical shape with a uniform size distribution, and they could quickly disassemble and rapidly release drugs under the reduction environment. Compared with MCF-7 cells, the DAS and PTX co-loaded redox-sensitive micelle (SS-PDNPs) showed stronger cytotoxicity and a more improving intracellular drug concentration than other drug formulations in MCF-7/ADR cells. In summary, the results suggested that the simple co-delivery micelles of PTX and DAS possessed significant potential to overcome drug resistance in cancer therapy.
Keywords: redox responsive, overcoming multidrug resistant, co-delivery, paclitaxel, dasatinib
Introduction
Currently, cancer is a leading cause of death worldwide.1 Hence, there is an urgent need to develop effective approaches for cancer therapy.2 Among these therapies, conventional chemotherapy is indispensable against cancer. However, the therapeutic efficacy of chemotherapy is usually limited by multidrug resistance (MDR), especially in advanced stages of cancer.3,4 Although the mechanism of MDR is complex, the overexpression of P-glycoprotein (P-gp) encoded by the MDR1 gene in cancer cells has been regarded as the major mechanism of MDR, which is able to pump various chemotherapeutics, such as doxorubicin (DOX) and paclitaxel (PTX), out of the cells, leading to low intracellular concentration of drug to kill the cells.5–8
PTX, a natural extractive compound from the Pacific yew tree (Taxus brevifolia), is one of the most effective chemotherapeutic agents, which has a great potency to treat a broad spectrum of solid tumors, such as breast cancer, ovarian cancer, lung cancer, and so on.9–12 However, the use of PTX alone in clinical trials always leads to MDR.13 Moreover, PTX resistance occurred in >70% of patients at the time of initial diagnosis and almost in all patients upon relapse.14,15 Therefore, researchers have tried to find agents against the MDR of cancer cells, such as verapamil, cyclosporine A, tamoxifen, quinidine, and so on.13,16 Dasatinib (DAS) is a tyrosine kinase inhibitor, which targets various kinases such as Src, BCR-Abl, FAK, c-Kit, and so on.17–19 This advantage of action by DAS suggests its potential application in combination chemotherapy with other anticancer drugs. For example, Xiao et al20 and Teoh et al21 both demonstrated that DAS had synergistic activity with PTX and carboplatin in ovarian cancer cells. This combination therapy had been used in clinical trials.22 Moreover, Chen et al23 have found that DAS could reverse the MDR of breast cancer MCF-7 cells to DOX by downregulating the expression of P-gp via inhibiting the activation of ERK signaling pathway. Hence, DAS has a profound potential role in reversing MDR.23 Otherwise, the extremely low solubility of PTX and DAS and different pharmacokinetic properties of the combined drugs will decrease the therapeutic efficacy. Besides, direct use of these drugs via systemic administration might result in severe systemic toxicities and poor biodistribution.24,25 Therefore, it is strongly recommended to develop safe and efficient delivery strategies that not only can co-deliver the two drugs to tumor tissues but also can increase the therapeutic efficacy and minimize the systemic toxicities.24,26,27 For example, Sun et al developed a simple PEGylated peptidic nanocarrier for effective co-delivery of DOX and DAS for combination chemotherapy.24 The results showed that this co-delivery system could increase the intracellular concentration of DOX and achieve a very high rate of tumor growth inhibition in vivo.
In the past few decades, stimuli-responsive polymeric micelles have attracted more attention as delivery vehicles for tumor drug. These nanocarriers can release drugs in response to internal or external stimuli, such as changes in pH value, enzyme level, light, temperature, and redox potential.28–32 Among these stimuli-responsive polymeric nanocarriers, reduction-sensitive polymeric micelles containing disulfide linkages have been extensively studied for intracellular anti-cancer drug delivery due to the existence of a high difference in the redox potential between the reducing intracellular fluids and the mildly oxidizing extracellular milieu.33 It has been reported that the intracellular concentrations (2–10 mM) of glutathione (GSH) are 100–1,000-fold higher than those in the extracellular compartment.34 In addition, the concentration of GSH in many tumors has been reported to be fourfold higher than that of normal tissues.34,35 Therefore, disulfide linkages can be stable in blood circulation and reversibly cleavable in response to reductive conditions. Moreover, the reduction-sensitive polymeric micelles containing disulfide linkages are usually designed to burst release the encapsulated agents in response to GSH. For example, Cui et al33 reported a reduction-sensitive micelle based on PEG-S-S-C16, which could efficiently internalize into HeLa cells and release DOX into the cytoplasm.
In this work, we designed and synthesized a simple reduction-sensitive polymeric micelle based on PEG-SS-C18 for co-delivery of PTX and DAS (SS-PDNPs) to overcome MDR. In SS-PDNPs, the disulfide bonds present in the main chain of the amphiphilic polymer could be immediately cleaved in response to the intracellular redox environment, leading to quick disassembly of the micelles and rapid drug release (Scheme 1). The release behavior of the co-delivery system was investigated. The in vitro experiments, including drug release, cellular internalization and drug accumulation in MDR cancer cells, and cell cytotoxicity induced by SS-PDNPs, were systematically investigated against the MDR cancer cells. The SS-PDNPs co-delivery system may serve as a potential candidate for reversing MDR treatment.
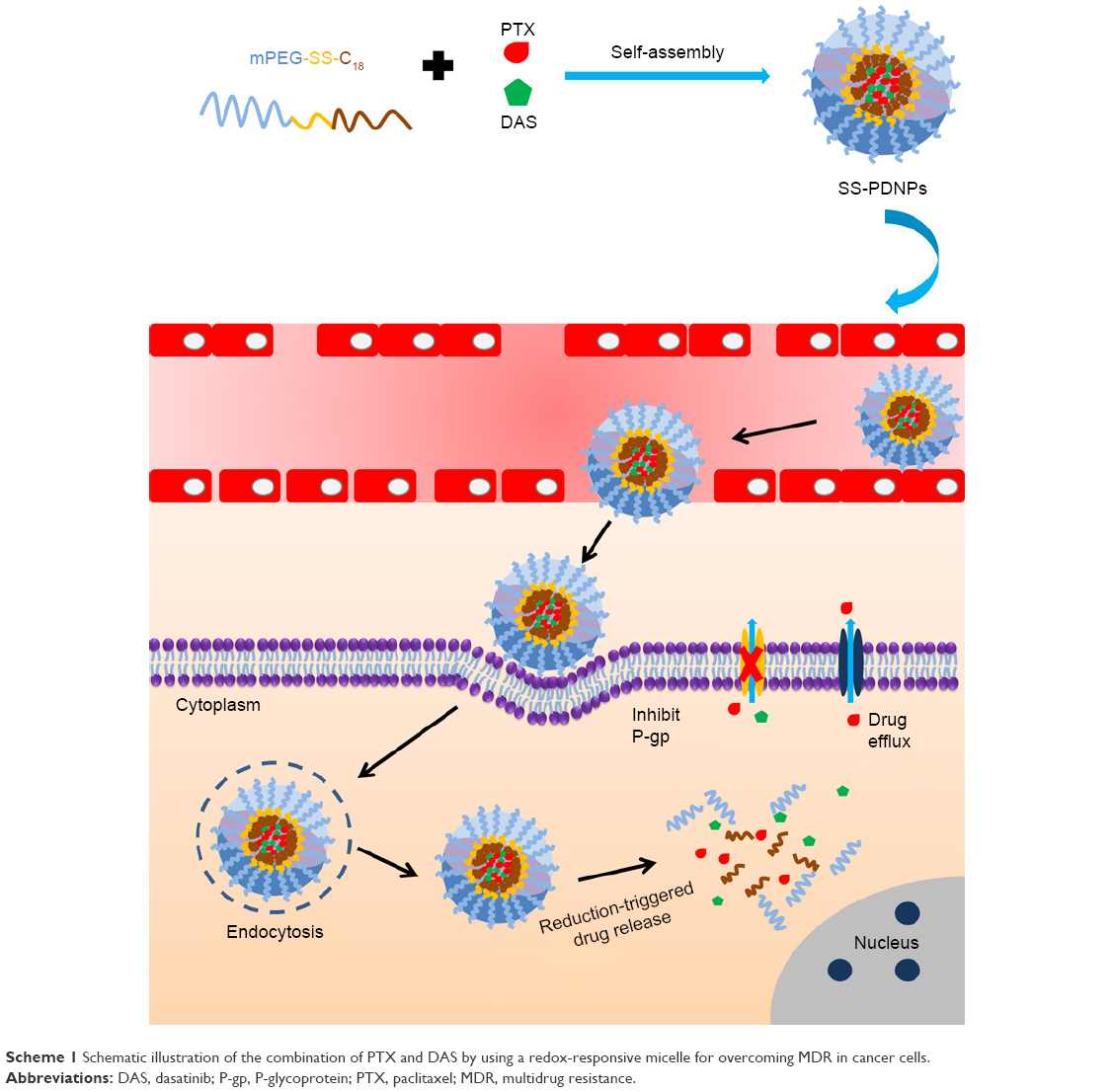 | Scheme 1 Schematic illustration of the combination of PTX and DAS by using a redox-responsive micelle for overcoming MDR in cancer cells. |
Experimental procedures
Materials
Monomethoxy-poly(ethylene glycol) (mPEG; Mn =2,000 Da) was purchased from Laysan Bio Co., Arab, AL, USA and dried in toluene by azeotropic distillation before use. DAS was purchased from Dalian Meilun Biotech Co., Ltd. (Dalian, People’s Republic of China). 3,3′-Dithiodipropionic acid (DTPA), octanedioic acid (OTDA), octadecylamine (C18-NH2), and GSH were purchased from Sigma-Aldrich Co. (St Louis, MO, USA) and used as received. PTX was purchased from Zelang Medical Technology Co., Ltd. (Nanjing, People’s Republic of China). MTT, Hoechst 33342, and BCA protein assay kit were all purchased from Beyotime (Shanghai, People’s Republic of China). RPMI 1640 medium, fetal bovine serum (FBS), and penicillin–streptomycin solution were purchased from Thermo Fisher Scientific (Waltham, MA, USA). All the other chemical reagents were of analytical grade and used as received.
Synthesis of mPEG-SS-C18 and mPEG-CC-C18
mPEG-SS-C18 was synthesized by two steps with a typical amidation reaction. First, mPEG was reacted with DTPA to obtain mPEG-SS-COOH. In a simple procedure, DTPA (15.75 mg, 0.075 mM), EDC·HCl (19.2 mg, 0.1 mM), and NHS (11.5 mg, 0.1 mM) were dissolved in 10 mL of dry N,N-dimethylformamide (DMF) under an argon atmosphere, and the reaction mixture was maintained under stirring for 4 h at room temperature. Subsequently, mPEG-NH2 (100 mg, 0.05 mM) and triethylamine (TEA) (42.8 mg, 0.4 mM) were dissolved in 20 mL of dry DMF and then added into the abovementioned solution. The coupling reaction was maintained at room temperature for 48 h. The excess solvent was removed by distillation under reduced pressure and then dialyzed (molecular weight cutoff [MWCO] 1 kDa) against distilled water, and the product mPEG-SS-COOH was obtained by lyophilization.
Then, mPEG-SS-COOH was reacted with C18-NH2 to obtain PEG-SS-C18. Typically, mPEG-SS-COOH (100 mg, 0.05 mM), 1-(3-Dimethylaminopropyl)-3-ethylcarbodiimide hydrochloride (EDC) (19.2 mg, 0.1 mM), and N-hydroxysuccinimide (NHS) (11.2 mg, 0.1 mM) were dissolved in 10 mL of dry dichloromethane under an argon atmosphere, and the reaction mixture was maintained under stirring for 4 h at room temperature. Subsequently, C18-NH2 (20 mg, 0.075 mM) and TEA (42.8 mg, 0.4 mM) were dissolved in 5 mL of dry dichloromethane and then added into the abovementioned solution. The coupling reaction was maintained at room temperature for 48 h. The excess solvent was removed by distillation under reduced pressure, the product was dissolved in DMSO and then dialyzed (MWCO 1 kDa) against DMSO and distilled water, and the product mPEG-SS-C18 was obtained by lyophilization.
As a control, mPEG-CC-C18 was also synthesized by a similar method, expect that DTPA was replaced by OTDA.
Preparation of blank and drug-loaded micelles
The blank redox-sensitive micelles (named SS-NPs) and redox-nonsensitive micelles (named CC-NPs), PTX-loaded redox-sensitive micelles (named SS-PNP), PTX and DAS co-loaded redox-sensitive micelles (named SS-PDNP) and redox-non-sensitive micelles (named CC-PDNP) were prepared by a dialysis method. In brief, 10 mg of PTX, 6 mg of DAS, and mPEG-SS-C18 (45 mg) were dissolved in 10 mL of DMSO, and the mixture was stirred at room temperature for 4 h. After that, the mixture solution was placed into a dialysis bag (MWCO 1 kDa) and dialyzed against distilled water for 48 h. The solution in the dialysis bag was filtered through a 0.45 μm filter and then lyophilized to obtain SS-PDNPs. The other micelles were prepared by the similar method except that no DAS or both drugs.
The concentrations of PTX and DAS were determined by the high performance liquid chromatography (HPLC) method. The drug loading content (DLC) and drug encapsulation efficiency (DEE) were calculated according to the following formulae:
|
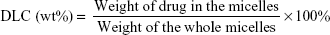
|

Characterization
1H NMR spectra were examined on a Varian-400 FT-NMR spectrometer at 500 MHz by using D2O or DMSO-d6 as the solvent depending on polymer solubility. The size distribution and zeta potential of all micelles were investigated by dynamic light scattering (DLS) (Zs90; Malvern Instruments, Malvern, UK). The morphology was observed by transmission electronic microscopy (TEM) (Hitachi Ltd., Tokyo, Japan).
Critical micelle concentration (CMC) of SS-NP and CC-NP micelles
The CMC values of SS-NP and CC-NP micelles were evaluated using Nile red as the fluorescence probe.24 The polymer solution with a concentration ranging from 1×10−5 to 5×10−1 mg/mL was added into the Nile red solution, and the final concentration of Nile red was fixed to be 6×10−7 M. The fluorescence spectra of the polymer solutions were measured by a fluorescence spectrometer (F-7000; Hitachi). The emission spectra were recorded from 570 to 720 nm. Subsequently, the CMC value of dual micelles was determined as the cross-point when extrapolating the intensity ratio at the polymer concentration regions.
Disassembly of blank micelles triggered by GSH
The size changes of redox-sensitive SS-NP micelles in response to various concentrations of GSH in PBS (10 mM, pH 7.4) were monitored by DLS measurement at different time intervals. Typically, 2 mL of SS-NP micellar solution in PBS containing GSH (0, 10 μM, and 10 mM) was placed in a shaking bed at 37°C at a rotation speed of 120 rpm. At different time intervals, the size of SS-NPs was detected by DLS. As a control, CC-NP micelles with and without 10 mM GSH were also measured, respectively.
In vitro reduction-triggered drug release
The release behaviors of PTX and DAS from SS-PDNP micelles at various concentrations of GSH were measured using a dialysis method. Briefly, 4 mL of SS-PDNPs containing 1 mg PTX and 0.5 mg DAS was transferred to a clamped dialysis bag (MWCO 3.5 kDa) and then immersed in 20 mL of PBS (0.1 mM, pH 7.4) containing 1% (w/v) Tween 80 or PBS with various concentrations of GSH (0, 10 μM, and 10 mM) in a shaking bed at 37°C at 120 rpm. At predetermined time intervals, 0.5 mL of external buffer was withdrawn and replaced with the same volume of fresh medium. From comparison, the release profile of PTX and DAS from CC-PDNPs with and without 10 mM GSH was studied as control. PTX and DAS released from drug-loaded micelles were measured using the HPLC method.
Cell culture
Human breast cancer cells MCF-7 and MCF-7/ADR were purchased from Institute of Biochemistry and Cell Biology, Shanghai Institutes for Biological Sciences, Chinese Academy of Sciences (Shanghai, People’s Republic of China). These cells were cultured in RPMI 1640 medium with 10% FBS, 100 U/mL penicillin G sodium, and 100 μg/mL streptomycin sulfate at 37°C and 5% CO2 in a humidified incubator. The drug resistance of MCF-7/ADR cells was maintained by addition of DOX (2 μg/mL) in the medium.
In vitro cytotoxicity assay
Optimization of the drug combination
The cytotoxicities of free drug combinations were studied by using the MTT assay. For this purpose, MCF-7/ADR cells were seeded in 96-well plates at a density of 1×104 cells/well and incubated overnight at 37°C. After that, the cells were treated with free PTX+DAS (mass ratio of PTX:DAS =10:1, 5:1, 2:1, 1:1, 1:2, and 1:5, respectively) for 48 h, and the final concentration of drug was uniform at 5 μg/mL. The cell viability was determined via MTT assay as described subsequently.
MTT assay
The different micelles against MCF-7 and MCF-7/ADR cells were evaluated in vitro by the MTT assay. Briefly, MCF-7 and MCF-7/ADR cells were seeded in 96-well plates at a density of 1×104 cells/well and incubated overnight to allow cell attachment. Then, the cells were treated with PTX, DAS, PTX+DAS, SS-PNPs, DAS+SS-PNPs, SS-PDNPs, and CC-PDNPs at various drug concentrations (mass ratio of PTX to DAS was fixed at 2:1) for 48 h. After that, the cell viability was measured via MTT assay as reported earlier,36 and the untreated cells were included as control. The cytocompatibility of the blank micelles was also evaluated by MTT assay, and the concentration of SS-NPs and CC-NPs ranged from 6.3 to 800 μg/mL.
Cellular uptake and subcellular localization
Rh B was used as a probe, and the cellular uptake was studied by fluorescence microscopy. Typically, MCF-7/ADR cells and MCF-7 cells were seeded in six-well plates at the density of 1×105 cells/well and incubated for 24 h to allow cell attachment. Then, the cells were treated with free Rh B+PTX, free Rh B+DAS, Rh B-loaded SS-PNPs, DAS+Rh B-loaded SS-PNPs, and Rh B-loaded SS-PDNPs (10 μg Rh B/mL, 2 μg PTX/mL, and 1 μg DAS/mL) in FBS-free culture medium for 1 and 3 h, respectively. After that, the cells were treated with Hoechst 33342 for 10 min, followed by washing three times with ice-cold PBS and fixed with 4% paraformaldehyde for 15 min. Then, the cells were observed under an inverted fluorescence microscope system (Nikon Eclipe Ti; Nikon Corporation, Tokyo, Japan).
Laser scanning confocal microscope (LSCM) was used to study the subcellular localization of SS-NPs. Briefly, MCF-7/ADR cells were seeded in six-well plates (involved a slides) at the density of 1×105 cells/well and incubated for 24 h to allow cell attachment. Then, the cells were treated with coumarin-6-labeled SS-NP micelles in FBS-free culture medium for 1 and 3 h, respectively. After that, the cells were stained with LysoTracker Red for 30 min and Hoechst 33342 for 10 min, followed by washing three times with ice-cold PBS and fixed with 4% paraformaldehyde for 15 min. Then, the cells were mounted on glass slides and visualized using a confocal microscope (LSCM, Fluoview 1000; Olympus Corporation, Tokyo, Japan).
Quantitative analysis of PTX intracellular accumulation
To investigate the PTX accumulation in cells, MCF-7 and MCF-7/ADR cells were seeded in 24-well plates at the density of 5×104 cells/well and incubated overnight to allow cell attachment. Then, the cells were treated with PTX, DAS, PTX+DAS, DAS+SS-PNPs, SS-PDNPs, and CC-PDNPs (PTX, 2 μg/mL and DAS, 1 μg/mL) for 1, 2, and 4 h at 37°C. After incubation, the cells were washed three times with cold PBS, and then 100 μL of cell lysis buffer (1% of Triton X-100) was added and incubated for 30 min. Subsequently, cell supernatants were collected by centrifuging at 10,000 rpm for 10 min, and the concentration of PTX was measured by HPLC. The BCA protein assay kit (Beyotime) was used for quantifying the protein amount, and the determined PTX amount was normalized for protein concentrations of cell lysates.
Inhibition of P-gp efflux test
To assay the function of P-gp efflux pump in drug-resistant cells, we employed Rhodamine 123 (Rh 123) as the model agent for evaluating P-gp transport activity as described previously.37,38 Briefly, MCF-7 and MCF-7/ADR cells were seeded into 24-well plates at the density of 5×104 cells/well and incubated overnight to allow cell attachment. Then, the cells were treated with Rh 123 (1 μg/mL) for 4 h. Then, the medium was replaced with PTX, PTX+DAS, SS-PNPs, DAS+SS-PNPs, SS-PDNPs, and CC-PDNPs (PTX, 2 μg/mL and DAS, 1 μg/mL) for 1 h. Eventually, the Rh 123 accumulation in cells was determined by flow cytometry. A total of 10,000 cells were recorded for each sample, and all experiments were repeated three times.
Apoptosis analysis
Qualitative apoptosis analysis by nucleus morphology observation
The MCF-7/ADR cell nucleus morphology after different treatments was investigated by Hoechst 33342 staining. In brief, MCF-7/ADR cells were seeded into 24-well plates and incubated for 24 h. After that, the cells were exposed to a medium containing Taxol, SS-PNPs, SS-PDNPs, and CC-PDNPs (5 μg/mL PTX and 2.5 μg/mL DAS) for 12 h, while the control cells were treated with drug-free medium. Then, the cells were washed with PBS for three times, fixed with 4% paraformaldehyde solution, stained with Hoechst 33342, and observed by fluorescence microscope.
Quantitative apoptosis analysis by annexin V-FITC/PI double staining
Quantitative apoptosis assay was evaluated by FCM using V-FITC/PI (Beyotime) staining by the manufacturer’s instructions. Briefly, MCF-7/ADR cells were incubated in six-well plates for 12 h and exposed to a medium containing Taxol, SS-PNPs, SS-PDNPs, and CC-PDNPs (5 μg/mL PTX and 2.5 μg/mL DAS) for 12 h. The cells incubated with drug-free medium were used as a control. At the end of the incubation time, the cells were trypsinized, collected, and suspended in binding buffer. Then, the cells were stained by a mixture of annexin V-FITC and PI for 30 min in the dark and analyzed by flow cytometry.
Statistical analysis
The results were given as mean ± SD. One-way single factorial analysis of variance (ANOVA) was performed to determine statistical significance of the data. In all statistical analyses, p<0.05 was considered to be statistically significant, and p<0.01 was considered to be highly statistically significant.
Results and discussion
Synthesis and characterization of PEG-SS-C18
mPEG-SS-C18 and mPEG-CC-C18 were obtained through two-step coupling reaction illustrated in Scheme S1.
First, mPEG-NH2 was reacted with DTPA to obtain mPEG-SS-COOH, and the product was confirmed by 1H NMR spectra. As shown in Figure 1A, the mPEG-SS-COOH was successfully synthesized, and the signals at δ 2.99 ppm contributed to the methylene peaks of DTPA (−CH2−CH2−SS−CH2−CH2−) and those at δ 3.78 ppm contributed to the methylene peaks of mPEG. Furthermore, the number of disulfides was calculated by comparing the integrals of signals at δ 2.99 and 3.45 ppm demonstrating an equivalent coupling of mPEG with DTPA.
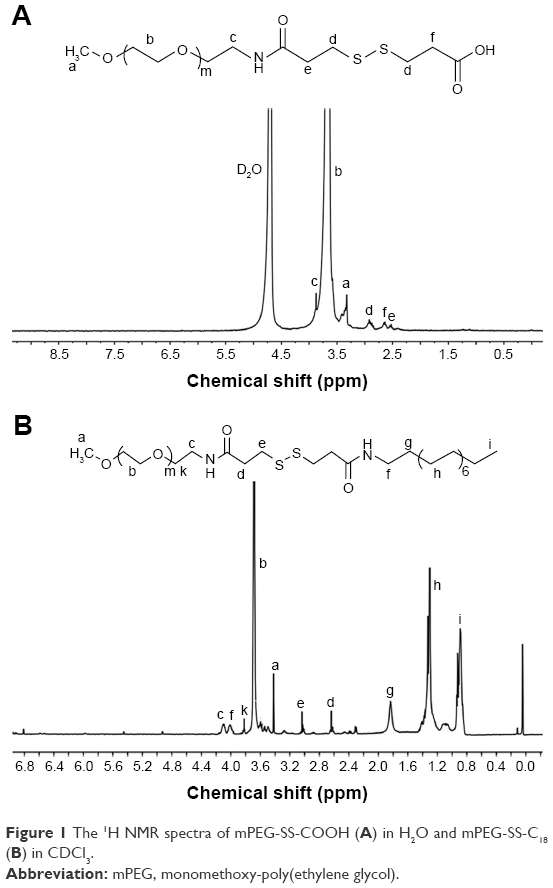 | Figure 1 The 1H NMR spectra of mPEG-SS-COOH (A) in H2O and mPEG-SS-C18 (B) in CDCl3. |
Then, mPEG-SS-C18 was obtained through the amidation reaction at room temperature. The product was confirmed by 1H NMR. As shown in Figure 1B, all the peaks in the spectra were well assigned. The signals at δ 3.78 and 3.45 ppm were contributed to mPEG and those at δ 0.88, 1.27, and 1.65 ppm were contributed to C18. Comparison of the integrals of signals at δ 3.78 and 1.27 ppm showed an equivalent coupling of mPEG-SS-COOH with C18.
As a control, the redox non-sensitive mPEG-CC-C18 polymer was also prepared (1H NMR spectra shown in Figure S1).
Characterization of the SS-PDNP micelles
The blank micelles were prepared by a dialysis method, and the micelles’ main properties are summarized in Table 1. The CMC values of SS-NPs and CC-NPs were surveyed by using Nile red as a fluorescence probe. As shown in Table 1, SS-NP micelles showed decreased CMC value (29.5 μg/mL) compared to CC-NP micelles, as reported previously.33 The redox sensitivity of SS-NP micelles was confirmed by measuring the size changes in response to GSH with CC-NPs as a control. As shown in Figure 2E and F, the CC-NP micelles were stable in the presence of 10 mM GSH for 3 h, while the size of SS-NP micelles changed from 130 to 1,209 nm after being incubated with 10 mM GSH for 3 h, indicating quick disassembly of SS-NP micelles. Furthermore, the stability of SS-NP micelles under 10 μM GSH (mimics the extracellular redox environment) was also evaluated. There is no obvious size change after being exposed to 10 μM GSH for 3 h, indicating that SS-NP micelles could be stable in the presence of mildly redox environment in the blood circulation.
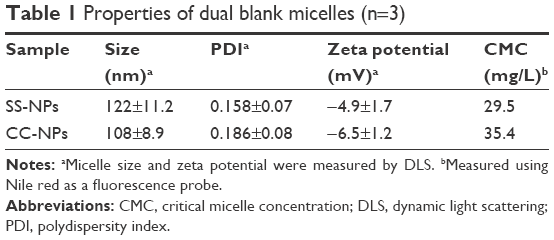 | Table 1 Properties of dual blank micelles (n=3) |
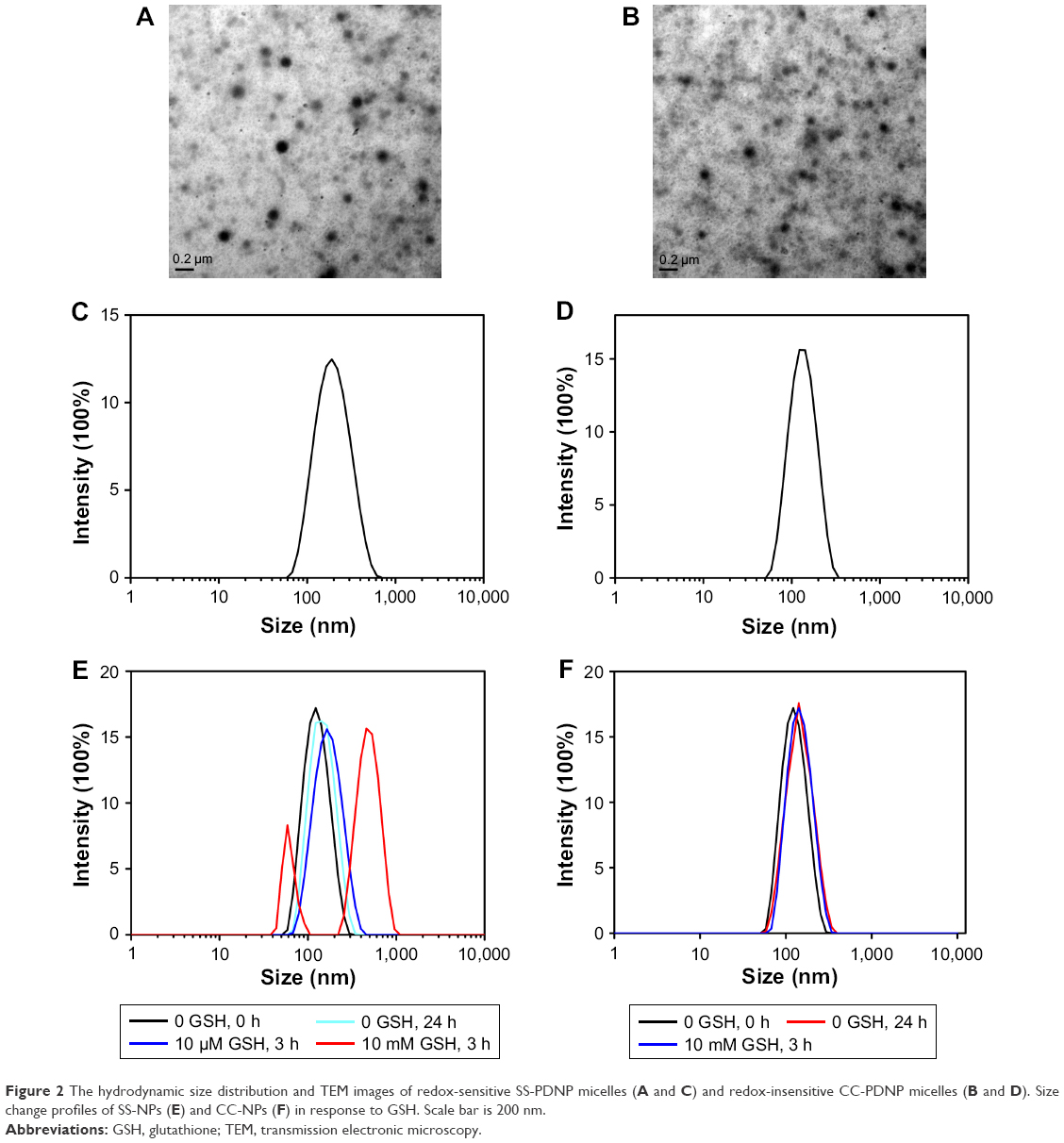 | Figure 2 The hydrodynamic size distribution and TEM images of redox-sensitive SS-PDNP micelles (A and C) and redox-insensitive CC-PDNP micelles (B and D). Size change profiles of SS-NPs (E) and CC-NPs (F) in response to GSH. Scale bar is 200 nm. |
To measure the optimal combination ratio of PTX and DAS, the in vitro toxicity of MCF-7/ADR cells treated with free PTX, free DAS, and free drug combination at various mass ratios was evaluated by MTT assay. Maximum cytotoxicity profile of MCF-7/ADR cells was achieved at a PTX/DAS mass ratio of 2:1 (Figure S1). Maybe in this mass ratio, the DAS could effectively inhibit the function of P-gp. Therefore, this mass ratio was selected as the optimal ratio for the following studies.
The particle size distribution and TEM image of dual drug-loaded redox-sensitive SS-PDNP micelles and redox-insensitive CC-PDNP micelles are displayed in Figure 2A–D, and their main properties are summarized in Table 2. The results showed that dual micelles had a good dispersity and a spherical shape with a uniform size distribution. The average size of redox-sensitive SS-NP micelles was 158±15 nm, which favored passive tumor targeting by enhanced permeability and retention effect.34
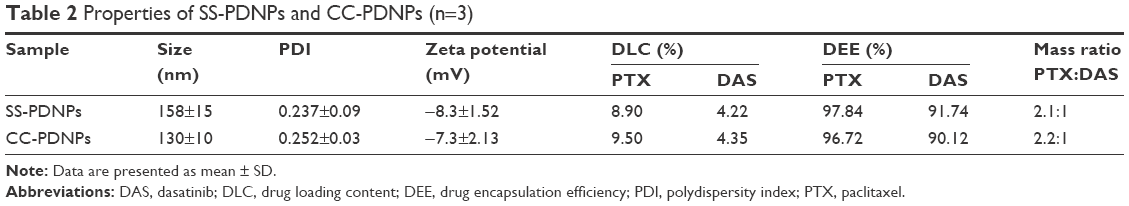 | Table 2 Properties of SS-PDNPs and CC-PDNPs (n=3) |
In vitro redox-responsive drug release
The aim of this work was to design and develop redox-responsive drug delivery vehicle, which could burst release of two drugs under GSH richer environment. To verify this potency, the in vitro release profiles of each drug were evaluated in PBS solution with various GSH concentrations. As shown in Figure 3B, little DAS release was observed from CC-PDNP micelles within 24 h in both the absence and presence of 10 mM GSH. Similarly, little DAS was released from SS-PDNPs incubated without GSH or with 10 μM GSH, further indicating the excellent stability of SS-PDNPs in intracellular environment, which was consistent with the result shown in Figure 2C. However, ~81.2% of DAS was released from SS-PDNPs within 24 h in the presence of 10 mM GSH, indicating that the SS-PDNP micelles could quickly disassemble in GSH richer environment.
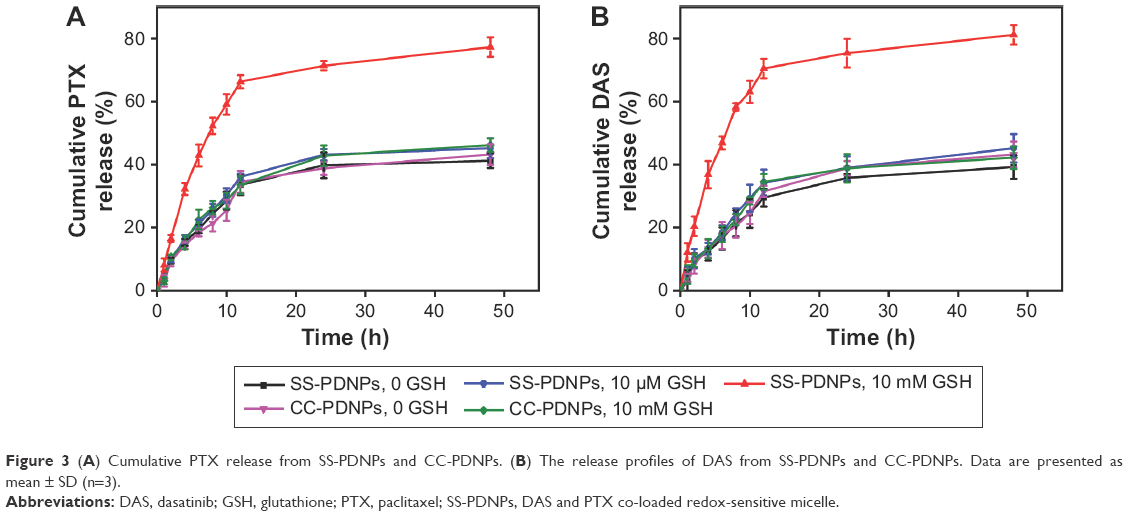 | Figure 3 (A) Cumulative PTX release from SS-PDNPs and CC-PDNPs. (B) The release profiles of DAS from SS-PDNPs and CC-PDNPs. Data are presented as mean ± SD (n=3). |
The release profiles of PTX from SS-PDNP and CC-PDNP micelles are shown in Figure 3A. In the absence of GSH, SS-PDNPs and CC-PDNPs showed a slow release of PTX and only 43.2% of the total PTX was released at 24 h. In comparison, no acceleration in PTX was observed for CC-PDNP micelles in the presence of GSH within 24 h, while PTX release from SS-PDNP micelles was significantly accelerated with 59.1% PTX being released at 10 h and 77.3% PTX being released after 24 h. This was attributed to the rapid disassembly of SS-PDNPs in response to the high concentration of GSH. Dramatically, the addition of 10 μM GSH did not obviously affect the release kinetics of SS-PDNPs. These results suggested that SS-PDNP micelles could be stable in the mildly redox environment in blood circulation, but could release PTX rapidly in response to GSH richer environment after being intracellular in cancer cells.
Cytotoxicity analysis
In vitro cytotoxicity was determined by MTT assay. As shown in Figure 4A and B, both SS-NP and CC-NP blank micelles did not show significant cytotoxic effect in drug-sensitive MCF-7 cells and drug-resistant MCF-7/ADR cells in the range of 6.3–800 μg/mL, which proved the non-cytotoxicity of the two micelles.
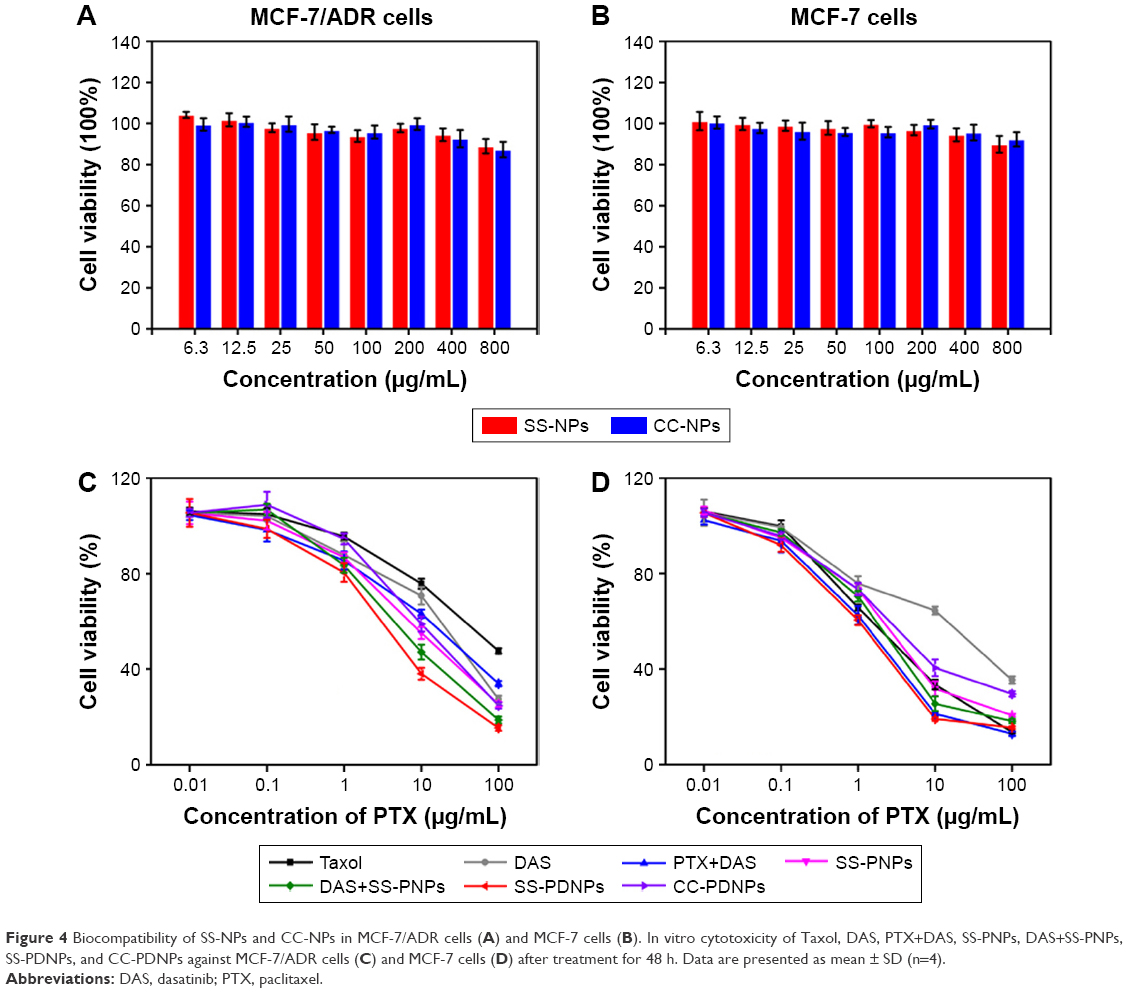 | Figure 4 Biocompatibility of SS-NPs and CC-NPs in MCF-7/ADR cells (A) and MCF-7 cells (B). In vitro cytotoxicity of Taxol, DAS, PTX+DAS, SS-PNPs, DAS+SS-PNPs, SS-PDNPs, and CC-PDNPs against MCF-7/ADR cells (C) and MCF-7 cells (D) after treatment for 48 h. Data are presented as mean ± SD (n=4). |
The in vitro cytotoxicity of PTX and DAS at different formulations was also measured by MTT, and the results are shown in Figure 4C and D, Table S1, and Figure S3. As known, MCF-7/ADR cell line was a typical resistant human breast cancer cell line of DOX with cross resistance to PTX.39 The IC50 value of Taxol (free PTX) was 53.25 and 2.28 μg/mL for MCF-7/ADR cells and MCF-7 cells, respectively, which verified the serious PTX resistance in MCF-7/ADR cell. After being encapsulated in SS-NPs, the cytotoxicity of PTX increased, maybe due to high cellular uptake. Fortunately, when PTX was combined with DAS, the cytotoxicity of PTX highly increased as a result of increased PTX intracellular accumulation by inhibiting the P-gp function of DAS. The IC50 values of PTX+DAS, SS-PNPs, SS-PDNPs, and CC-PDNPs in MCF-7/ADR were 6.5-, 5.6-, 12.5-, and 5.2-fold less than that treated with Taxol, respectively. Thereby, the PTX resistance in MCF-7/ADR cells could be greatly reduced by the co-delivery system. As a control, in MCF-7 cells, PTX could be internalized easily and accumulated in cells at high level due to low expression of P-gp (Figure 4D), hence, all forms of drugs could cause significant cytotoxicity. For CC-PDNPs and SS-PDNPs, the cell cytotoxicity was significantly different both in MCF-7 and MCF-7/ADR cells; this might be attributable to the fact that drug encapsulated in CC-PDNPs was difficult to be released.
Cell uptake of drug-loaded micelles
To explore the enhanced cytotoxicity of SS-PDNPs on MCF-7/ADR cells, the cellular uptake of various drug formulations was measured. Rh B, a substrate of P-gp,39,40 was used as a probe to test the ability of the cellular uptake of various drug formulations, and the cellular uptake was performed by fluorescence microscope in the MCF-7 cells (Figure S4) and MCF-7/ADR cells (Figure 5). As shown in Figure S2, all samples exhibited a significant red fluorescence throughout the cytoplasm after 3 h of exposure, which suggested that Rh B had been effectively taken up by MCF-7 cells. After DAS was added, the fluorescence did not obviously increase due to low expression of P-gp. However, it is hard for Rh B to accumulate in cytoplasm of drug-resistant MCF-7/ADR cells (Figure 5). As shown in Figure 5, after adding DAS, the red fluorescence highly increased by inhibiting the P-gp function. Since, the SS-PNPs and SS-PDNPs could be internalized into the cells through endocytic pathways like other nanoparticles and counteracted the P-gp effect of mediating drug efflux, leading to cellular fluorescence further increase.
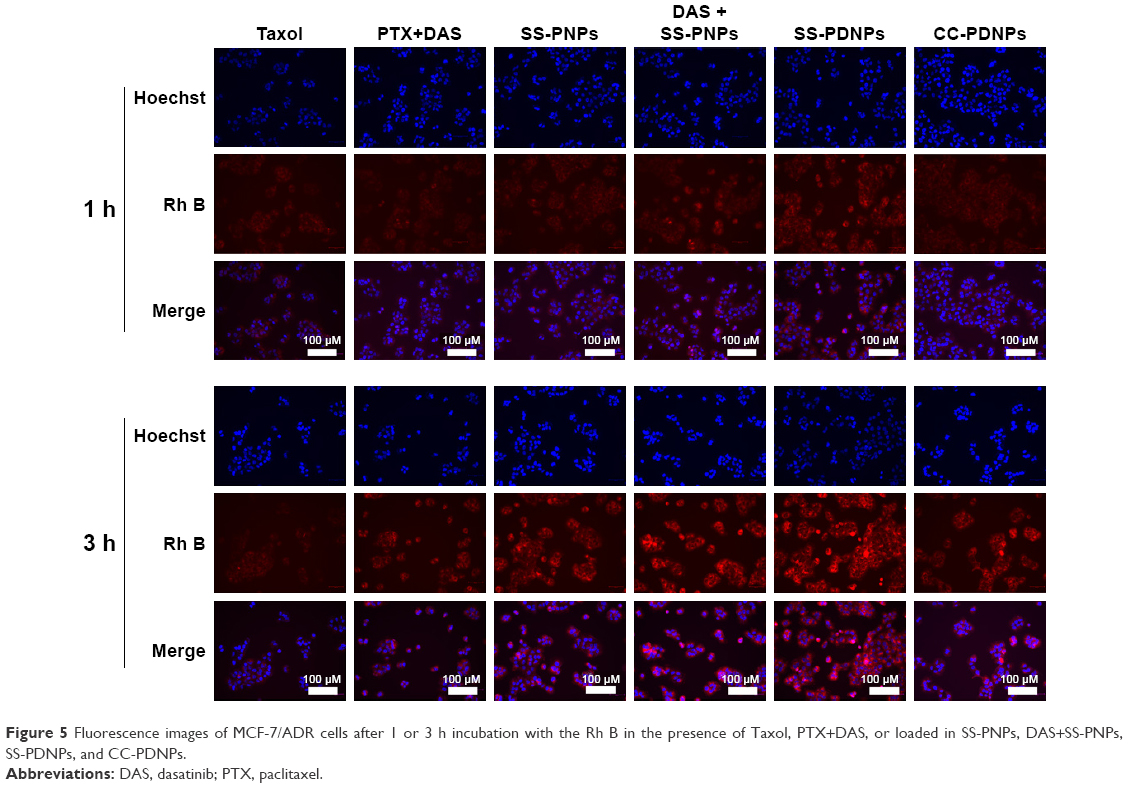 | Figure 5 Fluorescence images of MCF-7/ADR cells after 1 or 3 h incubation with the Rh B in the presence of Taxol, PTX+DAS, or loaded in SS-PNPs, DAS+SS-PNPs, SS-PDNPs, and CC-PDNPs. |
To further demonstrate the drug accumulation in MCF-7/ADR cells, the intracellular PTX concentration was quantitatively measured at different time intervals. As shown in Figure 6, the intracellular accumulation of all drug formulations was time-dependent. Compared with Taxol, much higher intracellular accumulation of free PTX can be detected in MCF-7/ADR cells when it is combined with DAS or loaded in SS-NPs (Figure 6A). As a comparison, the intracellular free PTX stayed at a low level due to the relative low hydrolysis rate of the ester bond in CC-PDNPs. These results further confirmed that the quick cleavage of “S-S” bonds led to the quick disassembly of polymer micelles and rapid release of drugs, eventually increased intracellular free PTX amount as expected. In MCF-7 cells, the intracellular PTX concentration of all drug formulations was obviously higher than that of MCF-7/ADR cells (Figure 6B). We found that the PTX concentration in MCF-7 cells treated with SS-PDNPs was significantly higher than that treated with CC-PDNPs, which may be caused by the rapid release of drug from SS-PDNPs in response to the reductive environment. This was consistent with the results (Figure 5).
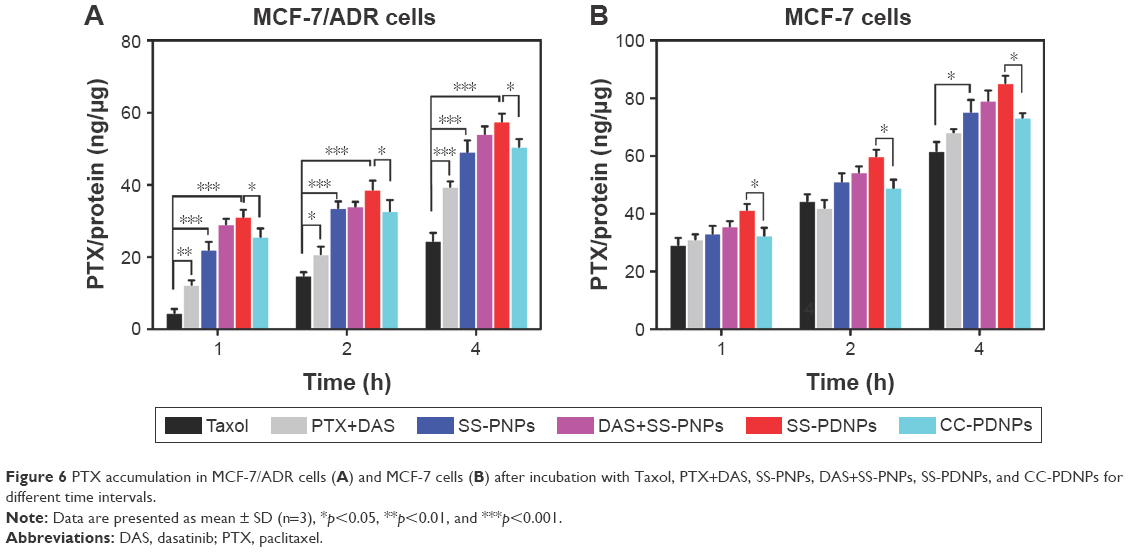 | Figure 6 PTX accumulation in MCF-7/ADR cells (A) and MCF-7 cells (B) after incubation with Taxol, PTX+DAS, SS-PNPs, DAS+SS-PNPs, SS-PDNPs, and CC-PDNPs for different time intervals. |
Moreover, the subcellular localization of coumarin-6-labeled SS-NPs in MCF-7/ADR was observed with LSCM. As shown in Figure 7, the endo/lysosomes were labeled with red fluorescent probe, and the yellow fluorescence which was the co-localization of green fluorescence (coumarin-6) and the red fluorescence (Lysosome red track) was observed after treating MCF-7/ADR cells with SS-NPs for 1 h, indicating that the endocytic pathway was involved. As the culture time increased to 3 h, more green fluorescence was observed in the cytoplasm and outside of the endo/lysosomes, which may be due to the escape of SS-NP micelles from the lysosomes. These phenomena suggested that the SS-NP micelles were internalized by endocytosis pathway as reported previously.33
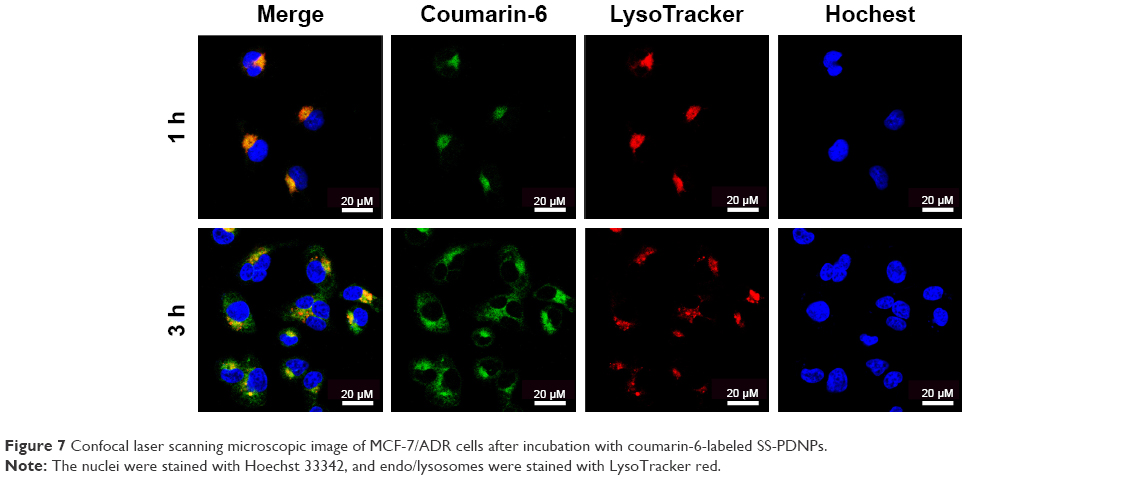 | Figure 7 Confocal laser scanning microscopic image of MCF-7/ADR cells after incubation with coumarin-6-labeled SS-PDNPs. |
P-gp inhibition assay
Overexpressed P-gp on the membrane of MCF-7/ADR cells was one of the major causes of the MDR phenomenon.41 The expression level of P-gp of MCF-7 and MCF-7/ADR cells used in this work was first detected by Western blot shown in Figure S5. Compared with MCF-7 cells, MCF-7/ADR cells exhibited significant P-gp expression. Rh-123 retention fluorescence intensity was used to evaluate the inhibition effect of DAS on P-gp. As shown in Figure 8, the fluorescence intensity of free Rh-123 was quite low in MCF-7/ADR cells, because Rh-123 was a kind of P-gp substrate, which could easily be pumped out by MCF-7/ADR cells. After treating with verapamil and DAS, increased fluorescence intensity was observed, which suggested that the function of P-gp efflux pump has decreased. The intensity of SS-PDNPs was markedly higher than others. It is possible that DAS amount could be increased in cells after encapsulation in micelles and eventually the RH-123 retention increased in MCF-7/ADR cells. Therefore, it might be concluded that DAS could effectively inhibit the function of P-gp and avoid the “pumping-out” of the drug, resulting in an increase in chemosensitivity on MCF-7/ADR cells. Moreover, the DAS and PTX co-loaded redox-sensitive micelles could effectively overcome MDR in MCF-7/ADR cells.
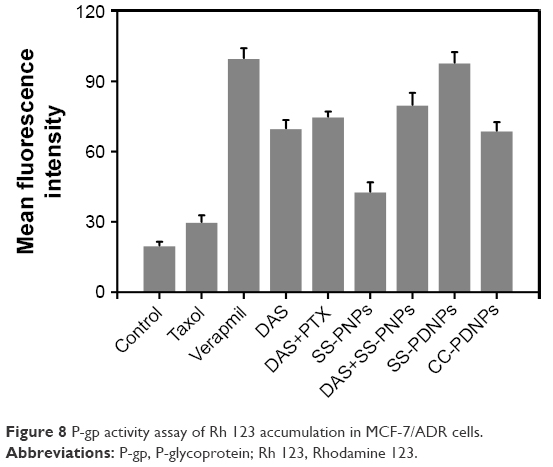 | Figure 8 P-gp activity assay of Rh 123 accumulation in MCF-7/ADR cells. |
Apoptosis induction of SS-PDNPs
The MDR cell apoptosis caused by the different formulations of PTX was first qualitatively analyzed by Hoechst staining and observed by fluorescence microscope. As shown in Figure 9A, the nuclei in the control cells were homogeneously stained blue and similar to the cytoplasm. In comparison to some typical apoptosis features, such as cell shrinkage, chromatin condensation, and fragmentation of the nucleus, the apoptotic body appeared in the cells treated with different PTX formulations. This was in harmony with the results of MTT analysis. To quantitatively verify the extent of apoptosis caused by SS-PDNPs and other forms, annexin V-FITC/PI double staining assay was performed and evaluated. As shown in Figures 9B and S6, the apoptosis percentage in MCF-7/ADR cells treated with SS-PDNPs was 26.2%, which significantly improved 5.6-fold than that of Taxol (4.6%), and was also higher than that of SS-PNPs and CC-PDNPs, respectively. All these results could demonstrate that the DAS has the potential for reversing the resistance of PTX.
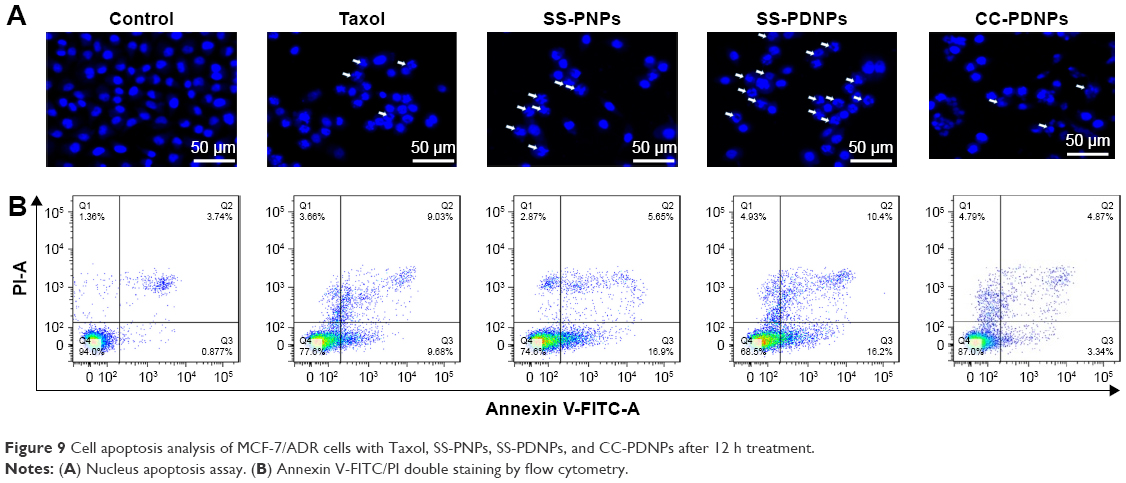 | Figure 9 Cell apoptosis analysis of MCF-7/ADR cells with Taxol, SS-PNPs, SS-PDNPs, and CC-PDNPs after 12 h treatment. |
Conclusion
A simple redox-sensitive polymer was successfully synthesized and applied in anticancer drug delivery. The broad-spectrum anticancer drug PTX and P-gp inhibitor DAS were co-encapsulated into these simple polymeric micelles to treat drug-resistant breast cancer. The SS-PDNPs co-delivery system showed rapid drug release in response to GSH and significantly increased cytotoxicity and intracellular concentration in drug-resistant MCF-7/ADR cells by inhibiting the P-gp function in cell membranes. Thereby, the co-delivery of chemotherapy drugs and P-gp inhibitor together by redox-responsive polymeric micelles may provide potential to treat MDR breast cancer in clinical trials.
Acknowledgment
This research was supported by the Oncology Translational Medicine Central of Jiangsu Province (BL20122008), the Priority Academic Program Development of Jiangsu Higher Education Institutions (JX10231801).
Disclosure
The authors report no conflicts of interest in this work.
References
Lee AL, Ng VW, Gao S, Hedrick JL, Yang YY. Injectable hydrogels from triblock copolymers of vitamin e-functionalized polycarbonate and poly (ethylene glycol) for subcutaneous delivery of antibodies for cancer therapy. Adv Funct Mater. 2014;24(11):1538–1550. | ||
Hamdy FC, Donovan JL, Lane JA, et al. 10-year outcomes after monitoring, surgery, or radiotherapy for localized prostate cancer. N Engl J Med. 2016;375(15):1415–1424. | ||
Guo X, Wei X, Jing Y, Zhou S. Size changeable nanocarriers with nuclear targeting for effectively overcoming multidrug resistance in cancer therapy. Adv Mater Deerfield. 2015;27(41):6450–6456. | ||
Yin M, Bao Y, Gao X, et al. Redox/pH dual-sensitive hybrid micelles for targeting delivery and overcoming multidrug resistance of cancer. J Mater Chem B. 2017;5(16):2964–2978. | ||
Ramos P, Bentires-Alj M. Mechanism-based cancer therapy: resistance to therapy, therapy for resistance. Oncogene. 2015;34(28):3617–3626. | ||
Gottesman MM, Fojo T, Bates SE. Multidrug resistance in cancer: role of ATP-dependent transporters. Nat Rev Cancer. 2002;2(1):48. | ||
Meng H, Mai WX, Zhang H, et al. Codelivery of an optimal drug/siRNA combination using mesoporous silica nanoparticles to overcome drug resistance in breast cancer in vitro and in vivo. ACS Nano. 2013;7(2):994–1005. | ||
Zhang R, Gao S, Wang Z, et al. Multifunctional molecular beacon micelles for intracellular mRNA imaging and synergistic therapy in multidrug-resistant cancer cells. Adv Funct Mater. 2017;27(31):1701027. | ||
Dong X, Mattingly CA, Tseng MT, et al. Doxorubicin and paclitaxel-loaded lipid-based nanoparticles overcome multidrug resistance by inhibiting P-glycoprotein and depleting ATP. Cancer Res. 2009;69(9):3918–3926. | ||
Chadderton A, Villeneuve DJ, Gluck S, et al. Role of specific apoptotic pathways in the restoration of paclitaxel-induced apoptosis by valspodar in doxorubicin-resistant MCF-7 breast cancer cells. Breast Cancer Res Treat. 2000;59(3):231–244. | ||
Bao Y, Guo Y, Zhuang X, et al. D-alpha-tocopherol polyethylene glycol succinate-based redox-sensitive paclitaxel prodrug for overcoming multidrug resistance in cancer cells. Mol Pharm. 2014;11(9):3196–3209. | ||
Megerdichian C, Olimpiadi Y, Hurvitz SA. nab-Paclitaxel in combination with biologically targeted agents for early and metastatic breast cancer. Cancer Treat Rev. 2014;40(5):614–625. | ||
Wang N, He T, Shen Y, et al. Paclitaxel and tacrolimus coencapsulated polymeric micelles that enhance the therapeutic effect of drug-resistant ovarian cancer. ACS Appl Mater Interfaces. 2016;8(7):4368–4377. | ||
Yusuf R, Duan Z, Lamendola D, Penson R, Seiden M. Paclitaxel resistance: molecular mechanisms and pharmacologic manipulation. Curr Cancer Drug Targets. 2003;3(1):1–19. | ||
Gong C, Xie Y, Wu Q, et al. Improving anti-tumor activity with polymeric micelles entrapping paclitaxel in pulmonary carcinoma. Nanoscale. 2012;4(19):6004–6017. | ||
Wu H, Hait WN, Yang J-M. Small interfering RNA-induced suppression of MDR1 (P-glycoprotein) restores sensitivity to multidrug-resistant cancer cells. Cancer Res. 2003;63(7):1515–1519. | ||
Lombardo LJ, Lee FY, Chen P, et al. Discovery of N-(2-chloro-6-methyl-phenyl)-2-(6-(4-(2-hydroxyethyl)-piperazin-1-yl)-2-methylpyrimidin-4-ylamino) thiazole-5-carboxamide (BMS-354825), a dual Src/Abl kinase inhibitor with potent antitumor activity in preclinical assays. J Med Chem. 2004;47(27):6658–6661. | ||
Araujo J, Logothetis C. Dasatinib: a potent SRC inhibitor in clinical development for the treatment of solid tumors. Cancer Treat Rev. 2010;36(6):492–500. | ||
Shor AC, Keschman EA, Lee FY, et al. Dasatinib inhibits migration and invasion in diverse human sarcoma cell lines and induces apoptosis in bone sarcoma cells dependent on SRC kinase for survival. Cancer Res. 2007;67(6):2800–2808. | ||
Xiao J, Xu M, Hou T, Huang Y, Yang C, Li J. Dasatinib enhances antitumor activity of paclitaxel in ovarian cancer through Src signaling. Mol Med Rep. 2015;12(3):3249–3256. | ||
Teoh D, Ayeni TA, Rubatt JM, et al. Dasatinib (BMS-35482) has synergistic activity with paclitaxel and carboplatin in ovarian cancer cells. Gynecol Oncol. 2011;121(1):187–192. | ||
Secord AA, Teoh DK, Barry WT, et al. A phase I trial of dasatinib, an SRC-family kinase inhibitor, in combination with paclitaxel and carboplatin in patients with advanced or recurrent ovarian cancer. Clin Cancer Res. 2012;18(19):5489–5498. | ||
Chen T, Wang C, Liu Q, et al. Dasatinib reverses the multidrug resistance of breast cancer MCF-7 cells to doxorubicin by downregulating P-gp expression via inhibiting the activation of ERK signaling pathway. Cancer Biol Ther. 2015;16(1):106–114. | ||
Sun J, Liu Y, Chen Y, et al. Doxorubicin delivered by a redox-responsive dasatinib-containing polymeric prodrug carrier for combination therapy. J Control Release. 2017;258:43–55. | ||
Poon C, He C, Liu D, Lu K, Lin W. Self-assembled nanoscale coordination polymers carrying oxaliplatin and gemcitabine for synergistic combination therapy of pancreatic cancer. J Control Release. 2015;201:90–99. | ||
Gupta B, Ramasamy T, Poudel BK, et al. Development of bioactive PEGylated nanostructured platforms for sequential delivery of doxorubicin and imatinib to overcome drug resistance in metastatic tumors. ACS Appl Mater Interfaces. 2017;9(11):9280–9290. | ||
Duan X, Xiao J, Yin Q, et al. Smart pH-sensitive and temporal-controlled polymeric micelles for effective combination therapy of doxorubicin and disulfiram. ACS Nano. 2013;7(7):5858–5869. | ||
Yan T, Li D, Li J, et al. Effective co-delivery of doxorubicin and curcumin using a glycyrrhetinic acid-modified chitosan-cystamine-poly(epsilon-caprolactone) copolymer micelle for combination cancer chemotherapy. Colloids Surf B Biointerfaces. 2016;145:526–538. | ||
Shen M, Xu YY, Sun Y, Han BS, Duan YR. Preparation of a thermosensitive gel composed of a mPEG-PLGA-PLL-cRGD nanodrug delivery system for pancreatic tumor therapy. ACS Appl Mater Interfaces. 2015;7(37):20530–20537. | ||
Yan Y, Ochs CJ, Such GK, Heath JK, Nice EC, Caruso F. Bypassing multidrug resistance in cancer cells with biodegradable polymer capsules. Adv Mater Deerfield. 2010;22(47):5398–5403. | ||
Fan J, He Q, Liu Y, et al. Light-responsive biodegradable nanomedicine overcomes multidrug resistance via NO-enhanced chemosensitization. ACS Appl Mater Interfaces. 2016;8(22):13804–13811. | ||
He YJ, Xing L, Cui PF, et al. Transferrin-inspired vehicles based on pH-responsive coordination bond to combat multidrug-resistant breast cancer. Biomaterials. 2017;113:266–278. | ||
Cui C, Xue YN, Wu M, et al. Cellular uptake, intracellular trafficking, and antitumor efficacy of doxorubicin-loaded reduction-sensitive micelles. Biomaterials. 2013;34(15):3858–3869. | ||
Zhu J, Niu Y, Li Y, et al. Stimuli-responsive delivery vehicles based on mesoporous silica nanoparticles: recent advances and challenges. J Mater Chem B. 2017;5(7):1339–1352. | ||
Deng B, Ma P, Xie Y. Reduction-sensitive polymeric nanocarriers in cancer therapy: a comprehensive review. Nanoscale. 2015;7(30):12773–12795. | ||
Li S-X, Liu L, Zhang L-J, et al. Synergetic enhancement of antitumor efficacy with charge-reversal and reduction-sensitive polymer micelles. Polym Chem. 2016;7(32):5113–5122. | ||
Bu H, He X, Zhang Z, Yin Q, Yu H, Li Y. A TPGS-incorporating nanoemulsion of paclitaxel circumvents drug resistance in breast cancer. Int J Pharm. 2014;471(1–2):206–213. | ||
Chen F, Wu J, Zheng C, et al. TPGS modified reduced bovine serum albumin nanoparticles as a lipophilic anticancer drug carrier for overcoming multidrug resistance. J Mater Chem B. 2016;4(22):3959–3968. | ||
Altenberg GA, Vanoye CG, Horton JK, Reuss L. Unidirectional fluxes of rhodamine 123 in multidrug-resistant cells: evidence against direct drug extrusion from the plasma membrane. Proc Natl Acad Sci U S A. 1994;91(11):4654–4657. | ||
Tai J, Cheung S, Wu M, Hasman D. Antiproliferation effect of Rosemary (Rosmarinus officinalis) on human ovarian cancer cells in vitro. Phytomedicine. 2012;19(5):436–443. | ||
Kathawala RJ, Gupta P, Ashby CR Jr, Chen ZS. The modulation of ABC transporter-mediated multidrug resistance in cancer: a review of the past decade. Drug Resist Updat. 2015;18:1–17. |
 © 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.