Back to Journals » Psychology Research and Behavior Management » Volume 8
A selective review of glutamate pharmacological therapy in obsessive–compulsive and related disorders
Authors Grados M, Atkins E, Kovacikova GI, McVicar E
Received 8 September 2014
Accepted for publication 10 December 2014
Published 28 April 2015 Volume 2015:8 Pages 115—131
DOI https://doi.org/10.2147/PRBM.S58601
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Dr Igor Elman
Marco A Grados,1 Elizabeth B Atkins,2 Gabriela I Kovacikova,3 Erin McVicar4
1Division of Child and Adolescent Psychiatry, Johns Hopkins University School of Medicine, Baltimore, MD, 2Department of Psychology, Smith College, Northampton, MA, 3Department of Psychology, Wellesley College, Wellesley, MA, 4Susquehanna University, Selinsgrove, PA, USA
Abstract: Glutamate, an excitatory central nervous system neurotransmitter, is emerging as a potential alternative pharmacological treatment when compared to gamma-aminobutyric acid (GABA)-, dopamine-, and serotonin-modulating treatments for neuropsychiatric conditions. The pathophysiology, animal models, and clinical trials of glutamate modulation are explored in disorders with underlying inhibitory deficits (cognitive, motor, behavioral) including obsessive–compulsive disorder, attention deficit hyperactivity disorder, Tourette syndrome, trichotillomania, excoriation disorder, and nail biting. Obsessive–compulsive disorder, attention deficit hyperactivity disorder, and grooming disorders (trichotillomania and excoriation disorder) have emerging positive data, although only scarce controlled trials are available. The evidence is less supportive for the use of glutamate modulators in Tourette syndrome. Glutamate-modulating agents show promise in the treatment of disorders of inhibition.
Keywords: glutamate, obsessive–compulsive disorder, attention deficit hyperactivity disorder, trichotillomania, excoriation disorder, modulation
Introduction
The use of glutamate-modulating drugs for neuropsychiatric conditions is emerging as an alternative to three decades of use of dopamine-, serotonin-, and gamma-aminobutyric acid (GABA)-modulating drugs in neuropsychiatry.1–4 While such use is still nascent in clinical settings, there is promise of significant impact on improved outcomes for patients. Research in genetic epidemiology and neurophysiology is pinpointing glutamate, the essential excitatory central nervous system (CNS) neurotransmitter, as a direct target for pharmacologic manipulation. In this selective review, we present current drug trial data in disorders that exhibit features of a lack of inhibitory control, including cognitive (obsessive–compulsive disorder [OCD]), motor (Gilles de la Tourette syndrome [TS]), behavioral (attention deficit hyperactivity disorder [ADHD]), and grooming (trichotillomania [TTM], excoriation disorder [ExD]) domains.
Obsessive–compulsive disorder
OCD is characterized by the recurrence of unwelcome and intrusive ideations, urges, or images (obsessions), as well as repetitive, rigid behaviors (compulsions).5 Obsessions can include fears related to contamination, religious, and sexual themes. Compulsions include behaviors such as washing, checking, counting, ordering, and hoarding. Obsessions and compulsions exist in symptom groupings or dimensions, with the four most commonly reported OCD symptom dimensions being: 1) contamination/washing; 2) aggressive/sexual/religious obsessions; 3) counting/repeating/checking compulsions; and 4) hoarding tendencies.6 The prevalence rate of OCD in adults in the United States is 2.3%,7 while it is lower in pediatric populations. In one study of 10,438 children in the United Kingdom, the prevalence rate of OCD was 0.25%.8 Standard pharmacologic treatment consists of serotonin reuptake inhibitors (SRIs), with moderate efficacy in children. A primary treatment approach in children with OCD is cognitive behavioral therapy (CBT) with exposure to OCD-related fears (exposure–response prevention [ERP]). The exposure paradigm is thought to be dependent upon glutamate-facilitated extinction learning.9 With medication and CBT/ERP, up to 54% of children with OCD experience clinical remission, but only 39% experience it with CBT alone and 21% with medication alone.10
A developmentally-mediated brain dysplasia in cortico–subcortico–cortical networks has been postulated to underlie pediatric OCD.11 Given that glutamate signaling is critical in early brain development through the facilitation of neuronal proliferation, migration, and differentiation, the formulation of the glutamate hypothesis of OCD relies on a dysregulation in developmental glutamate signaling homeostasis.12 A rich expression of glutamate and GABA neurotransmitter activity in the fetal and postnatal brain supports early neuronal growth and maturation. Metabotropic glutamate receptor (mGluR)1, for example, has high levels of expression in the fetal brain, especially in the striatum, with subsequent progressive decreases, while mGluR4 is expressed mostly in the adult brain.13 In a synchronized manner, both ionic and mGluRs are expressed in specific brain regions at specific times during brain development.14 The excitatory effect of glutamate neuronal networks makes it important for effective efferent activity, but it also creates the potential for excitotoxicity.15 In line with the glutamate hypothesis, functional imaging studies of OCD have shown an overactive cortico–striato–thalamo–cortical (CSTC) loop, with glutamate playing a central excitatory role (cortex, thalamus), while GABA and other inhibitory neurotransmitter systems are operant in the striatum and other subcortical regions.16 Overactivity of the right caudate in OCD and the limbic CSTC17,18 is postulated to be a parallel dysfunction in glutamate homeostatic systems, such as the glutamate transporter family. Among members of this family of transporters, SLC1A1 in chromosome region 9p24, has emerged as an intriguing positional candidate gene for OCD. Multiple studies support the association of OCD with SLC1A1 variants,19–21 including specific possible functional mutations,22 but a more recent meta-analytic study has lent less support to its impact.23 In the postsynaptic membrane, receptor conformation variants are plausibly linked to OCD. The NMDAR2B component of the N-methyl-D-aspartate (NMDA) receptor is codified by GRIN2B in chromosome region 12p12. GRIN2B variants have been linked to OCD24,25 and following the glutamate pathway’s downstream connections, variants in BDNF have also been linked to OCD. These BDNF variants include SNP rs2883187,26 SNP rs1519480,27 and the Met allele of Val66Met.28 The evidence is thus mounting for a direct role of glutamate in the pathophysiology of OCD (Table 1); however, it is too early to determine whether glutamate dysregulation is a sign of intracortical excitability due to aberrant CSTC aberrant inputs,29 another unknown glutamatergic abnormality, or a reactive effect of the disorder itself on brain function.30
 | Table 1 Glutamate-modulating pharmacotherapy of obsessive–compulsive disorder |
Animals have been heuristically useful in understanding the pathophysiologic mechanisms in OCD that may lead to drug discovery. The inhibition of marble-burying behavior has been used as a traditional model to test for anxiolytic drugs and OCD-related hoarding behaviors. In a test specific to anxiolytic effects, the mGluR5 (group 1) mediates marble-burying behavior in mice, and the mGluR5 antagonist, MPEP, is able to inhibit these behaviors.31 Similarly, the marble-burying behaviors are also mediated by group 2 mGluRs (mGluR2 and mGlur3).32 Additional support for glutamate-related animal models in OCD is provided by Sapap3 mutant mice, a knockout (KO) mouse that alters a synaptic density-associated protein. Sapap3 mutant mice demonstrate an excess of grooming behaviors, which is decreased significantly with the use of the selective SRI (SSRI) fluoxetine.33 Additionally, in these KO mice, the surface expression and activity of mGluR5s are increased, suggesting that glutamate-driven overactive mGluR5 transmission unfavorably alters synaptic plasticity. In effect, a positive allosteric modulator of mGluR5 reproduces this effect in wild-type mice.34 Interestingly, mGluR5 receptors have functional connections to BDNF, DLGAP1, and GRIN2B, as shown by a bioinformatics approach (http://www.string-db.org). In the rat attenuation model of OCD, decreasing rewards to lever pressing is used to identify those animals who “perseverate” in pressing behavior despite lower rewards. The excessive response can be abolished with fluoxetine and other serotonin-enhancing agents, but not by haloperidol or benzodiazepines.35 In a follow-up paradigm, the NMDA agonist D-cycloserine (DCS), but not an NMDA antagonist, helped attenuate the perseverative response, supporting a role for NMDA-facilitated learning of the extinction process.36
Clinical trials of glutamate-modulating drugs in OCD
Amantadine
Amantadine (1-adamantanamine hydrochloride) is an antiviral drug that was formerly, but no longer, used for influenza prophylaxis.37 Its current uses are to improve alertness and arousal in post-traumatic brain injury in children,38 improve executive dysfunction in patients with Alzheimer’s dementia,39 and treat the early stages of Parkinson’s.40 The mechanism of action is thought to consist of releasing dopamine from the presynapse, in addition to possible negative modulation of the NMDA receptor.41 While it can be neuroprotective due to its glutamate antagonism properties, amantadine has also resulted in acute neuropsychiatric side effects such as hallucinations and confusion.42 A case study reported that a treatment-refractory patient with OCD responded to amantadine (200 mg/day) added to clomipramine (225 mg/day).43 In an open-label study of amantadine in eight patients with OCD who had failed one SSRI trial,44 Yale–Brown Obsessive Compulsive Scale (Y-BOCS) scores improved for compulsions (15.3±3.2 versus 10.6±4.7; P<0.02; degrees of freedom [df] =7; t=2.36) and obsessions (12.7±3.3 versus 8.1±5; P<0.05; df=7; t=2.36).
D-cycloserine
DCS is an analog of D-alanine and an NMDA receptor partial agonist at the glycine-binding site. It has been well established that DCS has the ability to contribute to fear extinction learning.45 DCS facilitates conditioned fear extinction in rats when injected in the amygdala or systemically, suggesting that treatments that activate NMDA receptors can promote the learning of extinction learning.46 The sum of preclinical studies thus supports the blocking of extinction learning by NMDA antagonists47 and its facilitation by agonists such as DCS.48 In the treatment of OCD, DCS has been combined with ERP as a treatment option for nonresponders to traditional ERP. DCS administered 2 hours prior to ERP can produce an accelerated decrease in obsession-related fear ratings in comparison to the placebo across four ERP therapy sessions.49 This acceleration of response was also seen in another study of 16 adults with OCD, nine of whom received DCS + ERP, while seven received placebo. Midtreatment, the active group had significantly lower Y-BOCS scores (t=2.87; df=21; P=0.009) with a large effect size (Cohen’s d =1.17). At 1-month follow-up, differences were no longer apparent.50 In children with OCD, a negative study suggested that the differences in methodology may affect results.51 A meta-analysis of 13 studies, only one using pediatric OCD subjects, showed a small-to-moderate effect size for the use of DCS augmentation for exposure therapy (Cohen’s d =−0.34).52
Glycine
Glycine is biosynthesized from serine in a reversible folate-dependent reaction to conform a nonessential amino acid (NH2CH2COOH), which can act as a neurotransmitter.53 In the nervous system, glycine is inhibitory in the spinal cord, playing a role in inhibitory postsynaptic potentials, but excitatory in the brain and cerebellum, where it is a glutamate coagonist at the NMDA receptors.54 Originally studied as a neurotransmitter for pain signaling in the spinal cord, it has gained importance in CNS disorders due to its NMDA coagonist properties and its possible role in determining the number and positioning of cortical interneurons. For example, activation of the NMDAR D-serine/glycine site or inhibition of glycine transport has been pharmacologically induced to benefit animal models and clinical trials in schizophrenia.55 In a placebo-controlled double-blind study of OCD,56 24 subjects were assigned glycine 60 g/day or placebo. Glycine noncompliance, partly due a lack of palatability of the compound, led to ten dropouts. Follow-ups at 4-weeks, 8-weeks, and 12-weeks showed a marginally greater decrease of Y-BOCS OCD severity (P=0.053) in the glycine group (6.04 mean decrease; number [n]=5) compared to the placebo group (1.0 mean decrease; n=9). There were two out of five responders in the glycine group and zero out of nine responders in the placebo group (P=0.11).56
Ketamine
Ketamine (2-[2-chlorophenyl]-2-[methylamino] cyclohexanone) is a noncompetitive, nonselective, high-affinity NMDA receptor antagonist with psychotomimetic properties. Related to the more potent phencyclidine, its profile additionally includes a short half-life and weak binding to the mu opiate receptor, monoamine transporter sites, and acetylcholinesterase receptors.57 Based on preclinical research and the emerging glutamate hypothesis of mental illness, ketamine was used at 0.5 mg/kg to treat refractory depression with positive results.58 More recent overviews of this use of ketamine overall confirm its utility, but unanswered questions still remain about the precise mechanism of action and long-term benefits.59
The treatment of OCD with ketamine is in the initial stages of validation. An isolated case report used two intravenous (IV) ketamine infusions (0.5 mg/kg) 1 week apart on a 24-year-old female with OCD. No symptom reduction was observed after placebo; on ketamine infusion, almost complete reduction of obsessions was observed up to 7 days postinjection.60 In an open-label trial of IV ketamine 0.5 mg/kg in ten patients with refractory OCD, the initial symptom reduction of OCD symptoms was observed, along with decreases in the Hamilton Depression Rating Scale-17, a clinician-administered Dissociative States Scale, and the Clinical Global Impression (CGI) scale. Though the decrease in symptoms was maintained at 50% in four out of seven depressed patients, none of the OCD patients (n=10) sustained significant improvements.61 In a recent randomized, double-blind, placebo crossover study, adults with OCD (n=15) received two IV ketamine infusion treatments. The comparison groups were saline (n=7) and low-dose ketamine (0.5 mg/kg) (n=8). Those in the full-dose experimental group reported substantial improvements in OCD symptom severity, with 50% meeting the criteria for treatment response (≥35% Y-BOCS reduction) using the OCD visual analog scale and the Y-BOCS. The residual effects of ketamine lasted longer than the expected 1-week postinfusion.62
Lamotrigine
Lamotrigine (6-[2,3-dichlorophenyl]-1,2,4-triazine-3,5-diamine) is an anticonvulsant, antiepileptic drug with voltage-gated Na-channel blocking properties.63 In addition, lamotrigine also facilitates GABA release and reduces the presynaptic release of glutamate.64 Its anticonvulsant and mood-stabilizing properties make it a treatment of choice for the management of seizures65 and manic–depressive illness, in particular through putative neuroprotective effects.66 It is especially effective in the prevention of the depressive component of bipolar disorder,67 bipolar relapse,68 and unipolar depression resistant to treatment.69 One double-blind trial has also shown preliminary efficacy in the treatment of the re-experiencing and numbing symptoms of post-traumatic stress disorder.70
The glutamate-modulating properties of lamotrigine, based on the property of inhibiting Na+ channel-opening glutamate release,71 suggests that it may have some utility in the treatment of OCD. In a single case study of a 59-year-old woman with treatment-resistant OCD,72 the combination of a stable dose of clomipramine (225 mg/day) and lamotrigine (up to 150 mg/day) yielded a significant reduction in OCD Y-BOCS severity scores after 10 weeks. A subsequent 16-week, double-blind, placebo-controlled study of lamotrigine in 33 SSRI-resistant OCD patients showed that lamotrigine, added to standard SSRI treatment, substantially improved obsession and compulsion severity (P<0.0001).73
Memantine
Memantine (3,5-dimethyladamantan-1-amine) is a noncompetitive NMDA receptor antagonist with neuroprotective properties in cortical neuron cultures74 and in humans.75 These properties propelled its use in Parkinson’s disease,76 Huntington’s disease,77 and Alzheimer’s disease.78 The same glutamate-modulating property has motivated the assessment of memantine in OCD.
A reported case study of a 15-year-old female with severe OCD used memantine 5 mg twice daily (bid) in addition to preexisting citalopram. Obsessions and rituals were soon controlled for the first time in 9 months.79 An additional case study yielded similar results in a 29-year-old male, wherein 3 weeks of 15 mg/day of memantine produced over a 40% reduction in the Y-BOCS severity score, although another patient did not benefit.80 Next, an open-label trial enrolled 15 SRI-resistant OCD patients for a 12-week augmentation using memantine 10 mg bid.81 Data from 14 of the 15 subjects yielded significantly lower Y-BOCS scores compared to baseline (P<0.05) and 42.9% of the participants were classified as responders (≥25% reduction in Y-BOCS scores and a CGI rating of “much” or “very much” improved).81 Another open-label trial82 in ten OCD and seven generalized anxiety disorder subjects also used memantine 10 mg bid as a standalone or add-on psychopharmacological treatment. Patients with OCD experienced a 41% reduction of Y-BOCS severity scores compared to baseline (P<0.001), with three of ten subjects classified as responders.82 Finally, an 8-week randomized, double-blind, placebo-controlled study of memantine 10 mg bid was conducted on 42 patients with moderate-to-severe OCD.83 Of 38 completers, memantine was titrated to 10 mg bid. By 8 weeks, 100% of those on memantine and 32% of those on placebo achieved partial or complete response (P<0.001).83
N-acetylcysteine
N-acetylcysteine (NAC), a derivative of cysteine, has been used as a mucolytic in bronchitis84,85 and as an anti-inflammatory antioxidant.86 Administration of NAC within 16 hours postingestion is a primary treatment of acute acetaminophen toxicity;87 it can also be useful in the management of toxicity when hepatic failure has been established.88,89 Based on its ability to replenish the endogenous active antioxidant glutathione and direct scavenging of reactive oxygen species, NAC has been targeted to decrease doxorubicin cardiotoxicity in rat animal models90 and the oxidative damage in acute respiratory distress syndrome.91 Its thiol properties have supported its use in the protection of chemotherapy-induced cytotoxicity, if administered shortly after cisplatin,92 and its microcirculatory effects and inhibition of neutrophil aggregation in ischemia–reperfusion cardiac injury for coronary artery bypass graft surgery.93 Finally, even dietary NAC can replenish the antioxidant glycine pool, with a resultant increase in cysteine, glutamine, and oxidized glutathione.94
In a single study that evaluated the total oxidant and antioxidant status in serum, there was increased oxidant status and lower antioxidant status, with a resulting high oxidative stress index in children with OCD.95 Another study in adults with OCD measured erythrocyte malondialdehyde, a product of lipid peroxidation, and found that OCD was associated with higher levels of malondialdehyde, suggesting that a higher oxidation level was present. In this study, the antioxidant vitamin E, but not vitamin C, was also significantly lower in patients with OCD.96 Clinically, only a single case study of a 58-year-old woman with SRI-refractory OCD, in whom 3 g/day of NAC augmented fluvoxamine, reported a decrease of her Y-BOCS score from 32 (severe OCD) to 9 (no OCD) over 12 weeks.97
Riluzole
Riluzole (2-amino-6-trifluoromethoxy benzothiazole) is a negative regulator of CNS glutamate activity.98 Animal models suggest that riluzole is not specific to glutamate – it also decreases the release of acetylcholine and dopamine-trough mechanisms independent of NMDA or AMPA receptors.99 Other cell experiments support that in cortical neurons, riluzole inhibits voltage-dependent Na+ channels, resulting in nonglutamate-mediated anticonvulsant properties.100 The neuroprotective properties of riluzole made it an attractive option for the treatment of amyotrophic lateral sclerosis (ALS), a neurodegenerative disease of cortical, brainstem, and spinal cord motor neurons.101 Based on successful double-blind trials, in 1996, riluzole was approved for the treatment of ALS.102 Clinical experience with riluzole in ALS suggests that it prolongs survival,103 along with an adequate long-term use safety profile.104
Riluzole has been used in open and randomized clinical trials for the management of OCD symptoms. An early open-label study on 13 treatment-resistant patients with OCD ages 18–65 years used riluzole 50 mg bid to augment therapy. Of the 13 patients, seven out of 13 (54%) reported a >35% reduction in the Y-BOCS severity score. Of these, five out of 13 (38%) were treatment responders. Depression and anxiety measures also improved, and no concerning side effects were reported.105 Another open-label study of six children with OCD demonstrated that four out of six (67%) had a mean 40% decrease in OCD symptom severity.106 However, a recent double-blind randomized trial on 60 children with OCD, using riluzole 100 mg/day for augmentation, did not demonstrate an advantage over placebo.107
Topiramate
Topiramate (C12H21NO8S) is an anticonvulsant agent with linear kinetics and low binding to plasma proteins.108 Its glutamate-modulating properties are mediated through AMPA receptor antagonism, in addition to the blockage of voltage-dependent Na+ channel and the potentiation of GABA-A neurotransmission.109 It also inhibits AMPA and kainate receptor facilitation of Ca2+ influx, thus providing a potential buffer to glutamatergic excitotoxicity.110 Topiramate’s anorectic properties have motivated its use for weight control in combination with phentermine,111 and its pain control properties support topiramate’s use in the prophylaxis of chronic migraine.112
A case report of a 45-year-old paroxetine-resistant patient with OCD highlighted the use of topiramate 150 mg/day.113 After 9 weeks, the Y-BOCS OCD severity score decreased significantly.113 A more extended review of 16 consecutive adult patients with OCD, who had only partial response to SRI with or without neuroleptic augmentation, were reviewed after topiramate was added. Upon topiramate addition at 250 mg/day over an average of 9 weeks, there was a positive response in eleven out of 16 (69%) patients.114 A 12-week, double-blind, placebo-controlled trial of topiramate was conducted in 36 patients with OCD.115 Eighteen patients received a 12-week SSRI trial in addition to topiramate (mean dose: 180 mg); the other 18 patients received placebo in addition to the SSRI. Only the compulsions subscale improved (P=0.01), not the obsessions or total Y-BOCS scales. However, many patients had discontinued treatment due to side effects in the active arm (5/18; 28%).115 One case study of OCD emerging after topiramate treatment remains as an isolated report.116
Pregabalin
Pregabalin, (S)-(+)-3-aminomethyl-5-methylhexanoic, was designed as a blood–brain barrier-permeable GABA analog related to gabapentin. Originally designed as an anticonvulsant, it has been found to be effective in the treatment of hyperalgesia and pathologic anxiety.117 In hippocampal neurons, pregabalin decreases exocytosis, which opens neurotransmitter vesicles from presynaptic sites, a process that appears to depend on NMDA activation.118 Based on its anxiolytic properties, pregabalin was used in an 8-week open-label trial in ten OCD SRI–neuroleptic-resistant patients.119 Adjunctive pregabalin was administered at 225–675 mg/day, resulting in a significant reduction of OCD Y-BOCS severity scores from 27.1 (standard deviation [SD] =6.5) to 12.3 (SD =7.1). Overall, a 35% symptom improvement was seen in eight of the patients’ Y-BOCS scores.119
Tourette syndrome
TS is a childhood onset neurobehavioral disorder characterized by >12 months of multiple motor tics and at least one vocal tic during the course of the disorder. Tics are stereotyped, suppressible, and suggestible repetitive movements of single muscle groups (simple tics) or multiple muscle groups (complex tics).120 Involuntary and detrimental to daily functioning, tics are commonly treated with behavioral therapy with an emphasis on tic suppression121 to improve psychiatric and psychosocial functioning in children122 and adults.123 Clinically, TS is often comorbid with ADHD,124 OCD,125 as well as anxiety and mood disorders. Comorbidities significantly impact the quality of life of TS patients.126
Pathophysiologically, TS has been associated with the CSTC with a key focus on synaptic neurotransmission abnormalities.127,128 One of the main proposed mechanisms in TS is the increased transmission of dopamine,129 possibly due to abnormally high dopamine receptor prefrontal density.130 An alternative view is illustrated by the putative involvement of glutamate in neurotransmission in CSTC circuits,131 extensive interaction of dopamine and glutamate systems (Table 2),132 and postmortem brain studies.133
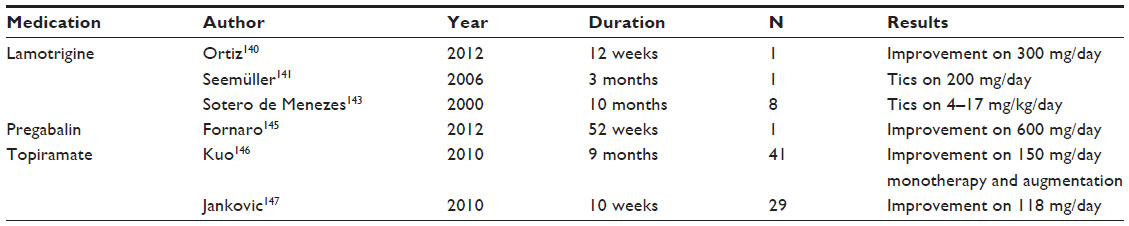 | Table 2 Glutamate-modulating treatments for Tourette syndrome |
Animal models in TS have been fraught with the innate complexity of modeling specific motor movements (tics) in nonhuman species. A working animal model will depend on the physiological assumptions about tics; for example, deficient gating has been proposed as a mechanism for propensity to tic expression based on premonitory urges associated with tics. The prepulse inhibition (PPI) of the acoustic startle reflex-deficit animal model is used to demonstrate deficient gating, and although not specific to TS, it can be used to explore pharmacologic options for tics.134 An alternate animal model relies on D1 receptor variants that drive glutamatergic neurocircuitry excitability; a mouse model displays compulsive-like repetitive behaviors.135,136 Interestingly, in these animal models, PPI is disrupted by the chemically-induced antagonism of D1 receptors in the medial prefrontal cortex (dopamine mechanism) and NMDA receptors in the ventral hippocampus (glutamate mechanism).137 These models suggest complex interactions in the cortical–subcortical neurocircuitry that involve dopamine and glutamate influences, along with a decrease in PPI.
Clinical trials of glutamate-modulating drugs in TS
Ketamine
Ketamine, a high-affinity NMDA receptor antagonist, has been found to increase PPI levels, making it a plausible drug to ameliorate tics. No direct trials of ketamine in TS are available; therefore, data on the effect of ketamine on PPI are reviewed. To measure the cognitive and PPI effects of ketamine, 20 ketamine-naïve subjects with negligible baseline startle responses were given either a placebo or a dose of ketamine of 0.23 mg/kg over 5 hours, followed by ketamine 0.5 mg/kg over 1 hour. A significant increase in PPI levels was detected in the active drug group.138 In another study with 16 healthy patients, researchers found that subanesthetic doses of ketamine significantly increased PPI.139 In both trials, experienced cognitive effects resembled those seen in schizophrenia, but PPI levels were increased by ketamine rather than decreased, as seen in schizophrenic patients, suggesting utility in disorders with abnormal levels of PPI.
Lamotrigine
The efficacy of lamotrigine in TS has been variable. A single case study reported decreased TS symptoms in a 26-year-old woman with a clinical diagnosis of bipolar II disorder, TS, migraine, and complex partial seizures.140 An initial Yale Global Tic Severity Scale (YGTSS) score of 39/50 was decreased over 6 months after a 12-week titration of lamotrigine to 300 mg/day.140 Another case report documented increased tics after lamotrigine 200 mg/day for mood stabilization in a 55-year-old female with bipolar disorder.141 Tics included shrugging her right shoulder, wagging her hips, pawing her feet on the ground, and vocal tics. After tapering lamotrigine dosage, her tic symptoms faded and disappeared in 2 weeks’ time.141 TS symptom emergence, which improved on lamotrigine withdrawal, has been reported in five children using lamotrigine for the control of seizure disorders.142,143
Pregabalin
Motor tics that accompany TS are self-injurious ~4%–43% of the time.144 In one case study, a 54-year-old man with TS with self-injury presented with fibromyalgia, comorbid OCD, and self-injurious behaviors (tongue self-mutilation).145 The patient was resistant to SSRIs, clomipramine, and haloperidol. Pregabalin monotherapy 600 mg/day was administered given the history of fibromyalgia and failure of other approaches; it was successful in improving self-injurious behaviors over 52 weeks of use.145
Topiramate
A retrospective chart review146 of 41 patients with TS who were administered topiramate (mean dose: 150 mg/day) for an average of 9 months, suggested that this drug could be effective for tic management. In this case series, topiramate was used as the primary monotherapy in 21 of 41 patients, while concomitant medications included neuroleptics (11/41), clonidine or guanfacine (6/41), tetrabenazine (3/41), and botox (3/41). Over 75% of subjects had “moderate to marked improvement” of tics. The main adverse effects were those common to topiramate: cognitive–language problems (25%) and aggression/mood swings (10%).146 A single 10-week, randomized, double-blind, placebo-controlled, parallel group study compared topiramate versus placebo in 29 patients with TS.147 The main outcome, “total tic score”, significantly improved in the active drug group (14.3 [SD =10.5]) compared to placebo (5.0 [SD =9.9]) (P<0.05). YGTSS motor, vocal, and total tic scores were significantly improved in the active drug group compared to placebo. No difference in adverse events was noted between the two groups.147 To date, topiramate constitutes a second-line agent in the management of clinically impairing tics.
Attention-deficit hyperactivity disorder
ADHD is a neurobehavioral disorder characterized by clinically impairing inattention and/or hyperactivity–impulsivity, which arise during early development. ADHD is categorized into three subtypes: 1) predominantly inattentive; 2) predominantly hyperactive–impulsive; and 3) combined.5 However, the full characterization of the syndrome should also emphasize related core deficits: an inability to inhibit the prepotent response;148,149 working memory developmental lags;150 and executive dysfunction.151 In a 2011 US survey, up to 11% of children and adolescents 4–17 years of age were affected, and less than two-thirds of diagnosed children were taking medication.152
Anatomical abnormalities in ADHD include reductions in total cerebral, prefrontal cortex, basal ganglia, dorsal anterior cingulate cortex, corpus callosum, and cerebellar volumes.153–155 Functional magnetic resonance imaging (fMRI),156,157 magnetic resonance spectroscopy (MRS),158 and resting-state fMRI also detect abnormalities in ADHD when compared to normotypicals.159 Specific to glutamate, a proton MRS ([1H]MRS) study of 40 adults with ADHD (24 medication-naïve), found that glutamate + glutamine (Glx) was lower in the basal ganglia and dorsolateral prefrontal cortex when compared to a control region (medial parietal cortex). Medication status did not alter the results, and the level of symptoms significantly correlated with Glx levels in the basal ganglia.160 Adults with autism spectrum disorder have also displayed decreased Glx concentrations in the basal ganglia compared to a parietal cortex control region.161 Other target regions in adults with ADHD, the right anterior cingulate cortex (decreased Glx)162 and the left cerebellar hemisphere (increased Glx),163 also have alterations in Glx (glutamate/glutamine) resonance. An additional study in children with ADHD found that particularly Glx – and not N-acetylaspartate, creatine/phosphocreatine (Cr), or choline – was increased in the right prefrontal cortex and left striatum.164 An increased Glx ratio was seen in the right prefrontal cingulate in another study of patients with ADHD aged 8–54 years compared to controls.165 The alterations in the Glx balance in different brain regions in ADHD are still of unknown valence. Although ADHD is known to be highly influenced by dopamine-linked neurocircuitry,159 extensive interactions between the glutamate and dopamine neural systems support a role for glutamate in ADHD pathophysiology (Table 3).148,149 For example, in the prefrontal cortex, D1 and D2 receptors modulate glutamate transmission at the NMDA and AMPA receptor sites. In turn, the downregulation of glutamate transmission for NMDA receptors is affected by GABAergic interneurons via D1 receptors.166 Following the demonstration of increased Glx content in ADHD,167 the effect of medication on brain regions has been examined. Methylphenidate and atomoxetine effect a 56% reduction in Glx in the left striatum in parallel to decreasing ADHD symptoms.168 In an analogous subsequent study in 13 treatment-naïve children with ADHD compared to controls, both Glx and Cr were increased pretreatment in the left striatum.169 After methylphenidate treatment, symptoms were improved and only striatal creatine was found to be reduced.169 A more recent MRS study in adolescents with ADHD found increased ratios of Glutamine/Inositol, Glutamate/Inositol, and Glx/Ino in the anterior cingulate – a brain region targeted for its role in attention in relation to error detection170 – in subjects with ADHD compared to controls.171 However, these differences were not statistically significant. Additionally, no significant differences were seen in these metabolite ratios pre- and post-treatment with a methylphenidate long-acting preparation.171
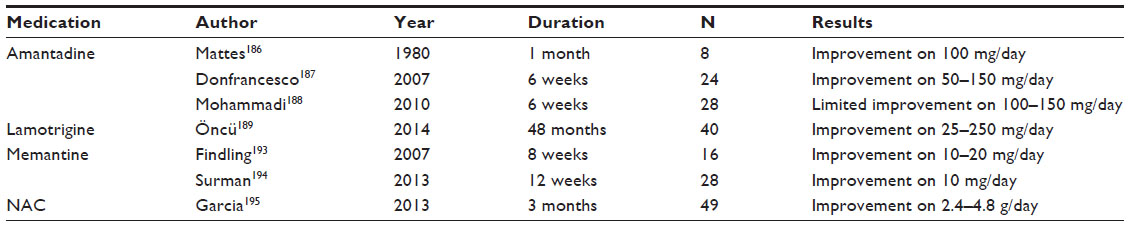 | Table 3 Glutamate-modulating pharmacotherapy for attention deficit hyperactivity disorder |
Animal models have been instrumental in understanding the biological underpinnings of ADHD, as well as the interaction of the dopamine and glutamate systems influencing attentional and hyperactivity behavioral domains. An early model located the dopamine receptor (DR)D1 as a potential target for genetic manipulation. The DRD1 KO mouse exhibits excessive motor activity, as well as reduced striatum dynorphin.172 Later experiments reaffirm that DRD1 plays a role in attentional performance with D1 agonists improving, and D1 antagonists worsening, attentional performance in mouse models. D2 antagonists have no effect on attentional performance in these models.173 A direct measure of the glutamate-stimulated release of dopamine in key ADHD-related brain regions used the spontaneously hypertensive rat model of ADHD (SHR). Compared to control rats, SHR showed increased glutamate-stimulated dopamine release in the substantia nigra, implicating altered glutamate regulation of dopamine neurons in this ADHD rodent model.174 A recent extension of these findings confirmed increased evoked glutamate release in the cingulate and infralimbic cortices, as well as in the striatum, using the SHR model of ADHD.175
Genetic lines of research are also pointing to the influence of glutamate in ADHD. A recent genome-wide study implicates glutamatergic synaptic adhesion molecules and mGluRs as etiologic in ADHD.176 Another genome-wide study using copy-number variants identified copy-number variants spanning GRM genes.177 DRD4 variants have also been associated with ADHD risk.159 It had been shown earlier that DRD4-deficient mice display cortical hyperexcitability, suggesting that DRD4 stimulation negatively regulates glutamate activity, thereby curbing neurotoxicity.178 In addition, manipulation of DRD4 activity in pyramidal cortical neurons resulted in reversible decreases of NMDA receptor-mediated currents, in association with inhibition of protein kinase A (PKA), activation of protein phosphatase 1, and inhibition of calmodulin-dependent protein kinase II (CAMKII).179 Conversely, in DRD4-deficient mice, there was increased expression of DRD1 and NMDA receptors in the nucleus accumbens and hippocampal CA1 tissue, suggesting a functional relationship between DRD4, DRD1, and NMDA receptors.180 These relationships carry over into the striatum, following more recent experiments with DRD4-deficient mice, which show that resting extracellular levels of glutamate are increased in the striatum, and glutamate clearance kinetics are significantly decreased in the dorsal striatum.181 PKA, an intracellular target of DRD1 stimulation, is also implicated in attentional performance. Inhibition of PKA both reduces attentional performance and increases locomotor activity. In turn, PKA activates cyclic adenosine monophosphate (cAMP) responsive element binding protein (CREB), a transcription factor that binds to DNA “responsive elements” intracellularly, increasing the transcription of c-Fos, BDNF, somatostatin, VGF, enkephalin, and corticotrophin releasing hormone, among others.182 Inhibition of PKA reduces the accuracy of a serial reaction time task in rodents, a test that parallels the continuous performance test in humans. The same effect is observed with the disruption of CREB.165 The interplay of glutamate signaling and CREB influences attentional and locomotor systems in animal models. Among these models, glutamate homeostasis in the ventromedial prefrontal cortex (vmPFC) is an important framework for exploring the symptoms of ADHD. Infusion of rat vmPFC with the NMDA antagonist 3-(R)-2-carboxypiperazin-4-propyl-1-phosphonic acid ([R]-CPP) decreases attentional performance, while increasing impulsivity and perseverative responses. These changes are accompanied by increased excitotoxic glutamate efflux in the vmPFC and changes in CREB expression in the vmPFC (increased) and striatum (decreased). Counteraction of the glutamate imbalance produced by (R)-CPP is achieved with the mGluR2–3 agonist agent, LY379268, which restores attentional performance and impulsivity, but not perseverative responses. This model suggests that the NMDA and mGluR2–3 pathways may underlie the core components of ADHD.183 Another rodent model of ADHD is the Naples high-excitability rat, which is bred to display behavioral arousal to novelty.184 Forebrain levels of glutamate, as well as d- and l-aspartate, are elevated in Naples high-excitability rats, suggesting that these levels may be causing neurotoxicity in the frontostriatal pathways.185
Clinical trials of glutamate-modulating drugs in ADHD
Amantadine
Recent trials of amantadine in pediatric populations have included its use in autism and ADHD.41 In an early open-label trial of amantadine, eight children with ADHD 10–13 years of age were taken off stimulant medication and provided with amantadine 100 mg bid for 1 month. While two children had similar responses as they had to methylphenidate, most only had an intermediate response compared to methylphenidate.186 A 6-week, open-label trial187 using amantadine 50–150 mg on 24 stimulant-naïve children with ADHD aged 5–13 years showed superiority over placebo. Only one child could not complete the trial due to headaches, and another had a transient decrease in appetite. Up to 60% of children had a >25% response in parent ADHD ratings, and 50% by teacher ratings.187 A head-to-head 6-week comparison trial188 of amantadine (100–150 mg/day) versus methylphenidate (20–30 mg/day) among 28 boys and 12 girls with ADHD aged 6–14 years showed no difference between the two drugs. Parent ratings showed 55% were responders on methylphenidate and 50% on amantadine; the teacher ratings were 35% responders for methylphenidate and 30% for amantadine.188 From these trials, amantadine has a modest effect in treating ADHD. Given its primary glutamate-modulating action, amantadine may be useful in treating symptoms of ADHD in specific subgroups of affected children.
Lamotrigine
A retrospective case series of 40 adult patients aged 16–55 years with ADHD comorbid with bipolar II or recurrent depression was conducted to gauge the effect on ADHD symptoms.189 Most patients (38/40) were on methylphenidate as well. With lamotrigine doses ranging from 25–250 mg, CGI scores were improved over the 48-month follow-up span.189 Antiepileptics can worsen attentional performance, and surveys can been conducted to assess these detrimental effects. In one survey on newly diagnosed childhood absence epilepsy, valproate, but not lamotrigine or ethosuximide, worsened attentional performance.152 Overall, lamotrigine improved cognition and attention in 24 of 45 children receiving lamotrigine for seizure control in one study,190 and the improvement of ADHD symptoms with lamotrigine does not appear to be dependent on ameliorating seizure activity.158 Given its benign to favorable cognitive profile, clinicians have advocated using lamotrigine as the antiepilepitc of choice when seizures are comorbid with ADHD.191 Currently, only indirect evidence supports the use of lamotrigine for the management of ADHD symptoms in children.
Memantine
Memantine is related to amantadine, with both increasing striatal dopamine via NMDA antagonism.192 Memantine 20 mg/day was compared to placebo in 16 children ages 6–12 years to test for efficacy in ADHD. In this pilot study, memantine was found to be efficacious, with no significant adverse effects.193 A more recent open-label 12-week trial194 on 28 adults with ADHD or ADHD not otherwise specified used memantine 10 mg bid. Exclusion criteria decreased the subject pool to 28 participants. The Adult ADHD Investigator Symptom Report yielded a reduction in total symptoms (–17.5; P<0.001), inattentive symptoms (–10.6; P<0.001), and hyperactive symptoms (–6.9; P<0.01) at the 12-week assessment. A total of 44% of participants had CGI ratings of much or very much improved.194
N-acetylcysteine
A trial of NAC was conducted in 49 adults with systemic lupus erythematosis over 3 months.195 Using an ADHD rating scale, patients with systemic lupus erythematosis had significantly greater ADHD symptoms than controls. The ADHD symptoms significantly improved on NAC 2.4–4.8 g/day (P<0.001).195
Trichotillomania, excoriation disorder (OCD-related disorders)
TTM is an OCD-related disorder (OCD-RD) characterized by repetitive hair pulling resulting in noticeable hair loss. Individuals may develop rituals around the selection and removal of hair, and may engage in manipulations like rolling, examining, biting, or swallowing the hair after removal.164 Children with TTM engage in automatic hair pulling, while older children appear to have more focused pulling and are more aware of associated urges, with increased frequency of hair pulling.154
Dermatillomania or ExD is characterized by recurrent skin picking resulting in skin lesions. The age at onset can be in childhood and triggers include stress, boredom, or the feel or look of the skin.157 A striking observation is the high degree of comorbidity associated with skin-picking behaviors, including OCD, body dysmorphic disorder, substance use disorders, eating disorders, TTM, kleptomania, and compulsive buying.196 The prevalence of skin-picking disorder ranges from 1.4%–5.4% based on two recent community samples.197,198
TTM and ExD are classified by the Diagnostic and Statistical Manual of Mental Disorders, 5th edition, as OCD-RD. These body-focused repetitive behaviors may be preceded by a sense of increasing tension and followed by a sense of gratification, pleasure, or relief. A continuum of conscious awareness may be associated with TTM and ExD, and the behaviors may be preceded by stress, anxiety, or boredom.151
The pathophysiology of OCD-RD has been investigated; however, much work needs to be done to further clarify their relationship to OCD. There are indications of increased familiality in TTM199 and ExD,200 suggesting a genetic diathesis. A lack of motor inhibitory control, such as in the Go–NoGo task in TTM201 and in the stop–signal task in ExD,160 point to an underlying neural inhibitory network deficit. Positron emission tomography studies show an increase in mean global metabolism in TTM compared to controls, as well as normalization of increased orbitofrontal and anterior cingulate regions with clomipramine treatment.202 More recently, and paralleling neuropsychological inhibitory deficit findings, a white matter tract study of subjects with TTM found reduced fractional anisotropy (white matter disorganization) in the anterior cingulate, presupplementary motor area, and temporal cortices,161 as well as increased mean diffusivity in the fronto–striato–thalamic white matter tracts.203 In ExD, analogous white matter disorganization is also found in areas associated with inhibitory control, such as the tracts emerging from the inferior right prefrontal regions.204 These pathophysiologic changes suggest a neurocircuitry imbalance in inhibitory neural control systems (Table 4).
 | Table 4 Glutamate-modulating pharmacotherapy in grooming disorders and OCD-related disorders |
Animal models of OCD-RD have sought to simulate repetitive behaviors that include animal self-grooming motor sequences. Hox genes are developmental homeobox genes that direct somatic body structure,153 one of which is hoxb8, a transcription factor directing anterior limb development.205 The hoxb8 mutant mouse exhibits an abnormally high levels of grooming behaviors, causing skin lesions and hair removal.206 Another notable example of a mouse model of grooming behaviors is the SAP90/PSD95-associated protein 3 (SAPAP3) KO mouse, which exhibits heightened anxiety-like behaviors and excessive self-grooming behaviors leading to facial hair loss and skin lesions.33 SAPAPs are SAP90/PSD95-associated proteins that act as synaptic linker proteins between glutamate receptor-binding proteins and the cytoskeleton, while SAPAP3 is particularly highly expressed in glutamatergic synapses of the striatum.207 In humans, SAPAP3 is expressed in dendrites, and SAPAP3 genetic variants have been associated with TTM and ExD.208,209
Clinical trials of glutamate-modulating drugs in TTM and ExD
N-acetylycysteine
A 12-week, double-blind, placebo-controlled trial of NAC 1.2–2.4 g/day in 50 adults with TTM showed a greater reduction in hair-pulling behaviors in the active drug arm.163 Up to 56% of patients taking NAC were much or very much improved, while only 16% of subjects receiving placebo had the same benefit. NAC produced no adverse events in the treated group.163 Following the randomized trial, additional case reports of NAC in TTM have been reported. A case report of two females aged 23 years and 19 years showed benefit of NAC 1,200 mg/day, with complete hair regrowth at 3 months and 2 months, respectively, after long courses of illness.156 Another case report of a 58-year-old female with TTM used NAC 1,200 mg/day, with a gradual improvement of hair loss observed over a 10-week period.150 In contrast to positive trials in adults, a double-blind placebo randomized trial of NAC in TTM, which included 39 children ages 8–17 years, showed no difference between NAC (25% improved) when compared to placebo (21% improved).155
The use of NAC in ExD is highlighted by a case report of five patients with TTM or ExD who were given NAC for symptom management. One of the reported patients had ExD, which responded to NAC 1,800 mg/day after partial response to 600 mg/day, and then 1,200 mg/day.210 A more recent report highlighted the use of NAC in 35 patients with Prader–Willi syndrome (PWS) and skin-picking behaviors. Patients diagnosed with PWS were aged 5–39 years (23 females/12 males) and treatment consisted of NAC at 450–1,200 mg/day for 12 weeks.175 At treatment conclusion, all patients had reduced (n=10) or absent (n=25) skin-picking behaviors.175
Lamotrigine
An open-label trial of lamotrigine 25–300 mg/day for the management of skin-picking behaviors showed that 67% of subjects were much or very much improved, prompting a randomized controlled trial.211 A double-blind, placebo-controlled, randomized trial of lamotrigine for ExD showed a nonsignificant benefit of active drug (44% responders) compared to placebo (31% responders). Only those patients with difficulties in a test of shifting (cognitive flexibility) benefited maximally from the use of lamotrigine.162
Topiramate
An open-label, 16-week trial of topiramate 50–250 mg/day in 14 adults with TTM resulted in nine adults completing the study.212 Trial completers had a significant reduction of hair-pulling behaviors, with 6/9 classified as responders. Five patients dropped out due to adverse effects.212 In a model of ExD using patients with PWS, topiramate 50–150 mg/day was administered as an add-on for the management of self-injury. All three patients experienced a significant resolution of ExD within 8 weeks of initiating the topiramate trial.213 Another case report214 noted that two adolescents, aged 15 years and 16 years, with autism spectrum disorder and significant ExD symptoms benefited from topiramate addition to their psychotropic regimen in doses up to 200 mg/day for 12 weeks.
Riluzole
A 52-year-old female with OCD, anorexia, and major depression, who also exhibited ExD, was treated with fluoxetine, to which riluzole 100 mg bid was added. Scores on OCD rating, eating disordered behaviors, and pathologic skin picking all improved on this regimen. It was noted that urges and compulsive behaviors had diminished notably with the addition of riluzole.215
Conclusion
CNS homeostasis of inhibition–disinhibition signaling ultimately depends on a well-regulated glutamate–GABA balance, in conjunction with other neurotransmitter systems that impact on this final effector pathway, which highly impacts neuronal health.216 Future drug design approaches will benefit from a better understanding of these pathways (which also impact on other biologic systems, including immune and developmental networks) in OCD and related disorders, tics and ADHD, in order to provide a paradigmatic framework to better understand the imbalance in inhibition–disinhibition from the molecular level (glutamate–GABA) to the macrobehavioral level (obsessions, compulsions, tics, hyperactivity, and grooming behaviors). The heuristic value of considering these cross-disorder clinical manifestations in toto in relation to glutamate awaits future drug discovery to address these disinhibitory phenomena. In summary, drugs that impact the glutamatergic balance in the CNS are emerging as a therapeutic alternative for neuropsychiatric disorders, which implicate abnormal inhibitory control in cognitive, motor, behavioral, and grooming domains. OCD has the most support at present for the use of glutamate modulators, with ADHD and grooming disorders also showing promise. TS appears to be less affected by glutamate modulators, but the research is quite sparse in this condition. Future research into the pathophysiology of glutamate actions, and resultant drug discovery, in the CNS should include its role in early development,217 neurogenesis,218 and trophic mechanisms.219
Disclosure
The authors report no conflicts of interest in this work.
References
Merritt K, McGuire P, Egerton A. Relationship between Glutamate Dysfunction and Symptoms and Cognitive Function in Psychosis. Front Psychiatry. 2013;4:151. | |
Hashimoto K, Malchow B, Falkai P, Schmitt A. Glutamate modulators as potential therapeutic drugs in schizophrenia and affective disorders. Eur Arch Psychiatry Clin Neurosci. 2013;263(5):367–377. | |
Musazzi L, Treccani G, Mallei A, Popoli M. The action of antidepressants on the glutamate system: regulation of glutamate release and glutamate receptors. Biol Psychiatry. 2013;73(12):1180–1188. | |
Coyle JT, Basu A, Benneyworth M, Balu D, Konopaske G. Glutamatergic synaptic dysregulation in schizophrenia: therapeutic implications. Handb Exp Pharmacol. 2012;(213):267–295. | |
American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders. 5th ed. Washington, DC: American Psychiatric Association; 2014. | |
Leckman JF, Grice DE, Boardman J, et al. Symptoms of obsessive-compulsive disorder. Am J Psychiatry. 1997;154(7):911–917. | |
Ruscio AM, Stein DJ, Chiu WT, Kessler RC. The epidemiology of obsessive-compulsive disorder in the National Comorbidity Survey Replication. Mol Psychiatry. 2010;15(1):53–63. | |
Heyman I, Fombonne E, Simmons H, Ford T, Meltzer H, Goodman R. Prevalence of obsessive-compulsive disorder in the British nationwide survey of child mental health. Br J Psychiatry. 2001;179:324–329. | |
Craske MG, Treanor M, Conway CC, Zbozinek T, Vervliet B. Maximizing exposure therapy: an inhibitory learning approach. Behav Res Ther. 2014;58:10–23. | |
Pediatric OCD Treatment Study (POTS) Team. Cognitive-behavior therapy, sertraline, and their combination for children and adolescents with obsessive-compulsive disorder: the Pediatric OCD Treatment Study (POTS) randomized controlled trial. JAMA. 2004;292(16):1969–1976. | |
Rosenberg DR, Keshavan MS. AE Bennett Research Award. Toward a neurodevelopmental model of obsessive – compulsive disorder. Biol Psychiatry. 1998;43(9):623–640. | |
Luján R, Shigemoto R, López-Bendito G. Glutamate and GABA receptor signalling in the developing brain. Neuroscience. 2005;130(3):567–580. | |
Martin LJ, Furuta A, Blackstone CD. AMPA receptor protein in developing rat brain: glutamate receptor-1 expression and localization change at regional, cellular, and subcellular levels with maturation. Neuroscience. 1998;83(3):917–928. | |
Furuta A, Martin LJ. Laminar segregation of the cortical plate during corticogenesis is accompanied by changes in glutamate receptor expression. J Neurobiol. 1999;39(1):67–80. | |
Sattler R, Tymianski M. Molecular mechanisms of glutamate receptor-mediated excitotoxic neuronal cell death. Mol Neurobiol. 2001;24(1–3):107–129. | |
Grados MA, Specht MW, Sung HM, Fortune D. Glutamate drugs and pharmacogenetics of OCD: a pathway-based exploratory approach. Expert Opin Drug Discov. 2013;8(12):1515–1527. | |
Rosenberg DR, MacMaster FP, Keshavan MS, Fitzgerald KD, Stewart CM, Moore GJ. Decrease in caudate glutamatergic concentrations in pediatric obsessive-compulsive disorder patients taking paroxetine. J Am Acad Child Adolesc Psychiatry. 2000;39(9):1096–1103. | |
MacMaster FP, O’Neill J, Rosenberg DR. Brain imaging in pediatric obsessive-compulsive disorder. J Am Acad Child Adolesc Psychiatry. 2008;47(11):1262–1272. | |
Arnold PD, Sicard T, Burroughs E, Richter MA, Kennedy JL. Glutamate transporter gene SLC1A1 associated with obsessive-compulsive disorder. Arch Gen Psychiatry. 2006;63(7):769–776. | |
Stewart SE, Fagerness JA, Platko J, et al. Association of the SLC1A1 glutamate transporter gene and obsessive-compulsive disorder. Am J Med Genet B Neuropsychiatr Genet. 2007;144B(8):1027–1033. | |
Dickel DE, Veenstra-VanderWeele J, Cox NJ, et al. Association testing of the positional and functional candidate gene SLC1A1/EAAC1 in early-onset obsessive-compulsive disorder. Arch Gen Psychiatry. 2006;63(7):778–785. | |
Shugart YY, Wang Y, Samuels JF, et al. A family-based association study of the glutamate transporter gene SLC1A1 in obsessive-compulsive disorder in 378 families. Am J Med Genet B Neuropsychiatr Genet. 2009;150B(6):886–892. | |
Stewart SE, Mayerfeld C, Arnold PD, et al. Meta-analysis of association between obsessive-compulsive disorder and the 3′ region of neuronal glutamate transporter gene SLC1A1. Am J Med Genet B Neuropsychiatr Genet. 2013;162B(4):367–379. | |
Arnold PD, Macmaster FP, Richter MA, et al. Glutamate receptor gene (GRIN2B) associated with reduced anterior cingulate glutamatergic concentration in pediatric obsessive-compulsive disorder. Psychiatry Res. 2009;172(2):136–139. | |
Alonso P, Gratacós M, Segalàs C, et al. Association between the NMDA glutamate receptor GRIN2B gene and obsessive-compulsive disorder. J Psychiatry Neurosci. 2012;37(4):273–281. | |
Tükel R, Ozata B, Oztürk N, Ertekin BA, Ertekin E, Direskeneli GS. The role of the brain-derived neurotrophic factor SNP rs2883187 in the phenotypic expression of obsessive-compulsive disorder. J Clin Neurosci. 2014;21(5):790–793. | |
Márquez L, Camarena B, Hernández S, Lóyzaga C, Vargas L, Nicolini H. Association study between BDNF gene variants and Mexican patients with obsessive-compulsive disorder. Eur Neuropsychopharmacol. 2013;23(11):1600–1605. | |
Tükel R, Gürvit H, Ozata B, et al. Brain-derived neurotrophic factor gene Val66Met polymorphism and cognitive function in obsessive-compulsive disorder. Am J Med Genet B Neuropsychiatr Genet. 2012; 159B(7):850–858. | |
Greenberg BD, Ziemann U, Corá-Locatelli G, et al. Altered cortical excitability in obsessive-compulsive disorder. Neurology. 2000;54(1):142–147. | |
Pittenger C, Krystal JH, Coric V. Glutamate-modulating drugs as novel pharmacotherapeutic agents in the treatment of obsessive-compulsive disorder. NeuroRx. 2006;3(1):69–81. | |
Nicolas LB, Kolb Y, Prinssen EP. A combined marble burying-locomotor activity test in mice: a practical screening test with sensitivity to different classes of anxiolytics and antidepressants. Eur J Pharmacol. 2006;547(1–3):106–115. | |
Shimazaki T, Iijima M, Chaki S. Anxiolytic-like activity of MGS0039, a potent group II metabotropic glutamate receptor antagonist, in a marble-burying behavior test. Eur J Pharmacol. 2004;501(1–3):121–125. | |
Welch JM, Lu J, Rodriguiz RM, et al. Cortico-striatal synaptic defects and OCD-like behaviours in Sapap3-mutant mice. Nature. 2007;448(7156):894–900. | |
Chen M, Wan Y, Ade K, Ting J, Feng G, Calakos N. Sapap3 deletion anomalously activates short-term endocannabinoid-mediated synaptic plasticity. J Neurosci. 2011;31(26):9563–9573. | |
Joel D, Avisar A. Excessive lever pressing following post-training signal attenuation in rats: a possible animal model of obsessive compulsive disorder? Behav Brain Res. 2001;123(1):77–87. | |
Albelda N, Bar-On N, Joel D. The role of NMDA receptors in the signal attenuation rat model of obsessive-compulsive disorder. Psychopharmacology (Berl). 2010;210(1):13–24. | |
Alves Galvão MG, Rocha Crispino Santos MA, Alves da Cunha AJ. Amantadine and rimantadine for influenza A in children and the elderly. Cochrane Database Syst Rev. 2008;(1):CD002745. | |
Williams SE. Amantadine treatment following traumatic brain injury in children. Brain Inj. 2007;21(9):885–889. | |
Drayton SJ, Davies K, Steinberg M, Leroi I, Rosenblatt A, Lyketsos CG. Amantadine for executive dysfunction syndrome in patients with dementia. Psychosomatics. 2004;45(3):205–209. | |
Hubsher G, Haider M, Okun MS. Amantadine: the journey from fighting flu to treating Parkinson disease. Neurology. 2012;78(14):1096–1099. | |
Hosenbocus S, Chahal R. Amantadine: a review of use in child and adolescent psychiatry. J Can Acad Child Adolesc Psychiatry. 2013;22(1):55–60. | |
Neagoe AD. Delirium with manic and psychotic features associated with amantadine. Gen Hosp Psychiatry. 2013;35(6):680. e7–e8. | |
Pasquini M, Berardelli I, Biondi M. Amantadine augmentation for refractory obsessive-compulsive disorder: a case report. J Clin Psychopharmacol. 2010;30(1):85–86. | |
Stryjer R, Budnik D, Ebert T, et al. Amantadine augmentation therapy for obsessive compulsive patients resistant to SSRIs-an open-label study. Clin Neuropharmacol. 2014;37(3):79–81. | |
Myskiw JC, Izquierdo I, Furini CR. Modulation of the extinction of fear learning. Brain Res Bull. 2014;105:61–69. | |
Norberg MM, Krystal JH, Tolin DF. A meta-analysis of D-cycloserine and the facilitation of fear extinction and exposure therapy. Biol Psychiatry. 2008;63(12):1118–1126. | |
Lee H, Kim JJ. Amygdalar NMDA receptors are critical for new fear learning in previously fear-conditioned rats. J Neurosci. 1998;18(20):8444–8454. | |
Hofmann SG, Pollack MH, Otto MW. Augmentation treatment of psychotherapy for anxiety disorders with D-cycloserine. CNS Drug Rev. 2006;12(3–4):208–217. | |
Kushner MG, Kim SW, Donahue C, et al. D-cycloserine augmented exposure therapy for obsessive-compulsive disorder. Biol Psychiatry. 2007;62(8):835–838. | |
Wilhelm S, Buhlmann U, Tolin DF, et al. Augmentation of behavior therapy with D-cycloserine for obsessive-compulsive disorder. Am J Psychiatry. 2008;165(3):335–341; quiz 409. | |
Storch EA, Merlo LJ, Bengtson M, et al. D-cycloserine does not enhance exposure-response prevention therapy in obsessive-compulsive disorder. Int Clin Psychopharmacol. 2007;22(4):230–237. | |
Rodrigues H, Figueira I, Lopes A, et al. Does D-cycloserine enhance exposure therapy for anxiety disorders in humans? A meta-analysis. PLoS One. 2014;9(7):e93519. | |
Hernandes MS, Troncone LR. Glycine as a neurotransmitter in the forebrain: a short review. J Neural Transm. 2009;116(12):1551–1560. | |
Clements JD, Westbrook GL. Activation kinetics reveal the number of glutamate and glycine binding sites on the N-methyl-D-aspartate receptor. Neuron. 1991;7(4):605–613. | |
Labrie V, Roder JC. The involvement of the NMDA receptor D-serine/glycine site in the pathophysiology and treatment of schizophrenia. Neurosci Biobehav Rev. 2010;34(3):351–372. | |
Greenberg WM, Benedict MM, Doerfer J, et al. Adjunctive glycine in the treatment of obsessive-compulsive disorder in adults. J Psychiatr Res. 2009;43(6):664–670. | |
Krystal JH, Karper LP, Seibyl JP, et al. Subanesthetic effects of the noncompetitive NMDA antagonist, ketamine, in humans. Psychotomimetic, perceptual, cognitive, and neuroendocrine responses. Arch Gen Psychiatry. 1994;51(3):199–214. | |
Berman RM, Cappiello A, Anand A, et al. Antidepressant effects of ketamine in depressed patients. Biol Psychiatry. 2000;47(4):351–354. | |
Caddy C, Giaroli G, White TP, Shergill SS, Tracy DK. Ketamine as the prototype glutamatergic antidepressant: pharmacodynamic actions, and a systematic review and meta-analysis of efficacy. Ther Adv Psychopharmacol. 2014;4(2):75–99. | |
Rodriguez CI, Kegeles LS, Flood P, Simpson HB. Rapid resolution of obsessions after an infusion of intravenous ketamine in a patient with treatment-resistant obsessive-compulsive disorder. J Clin Psychiatry. 2011;72(4):567–569. | |
Bloch MH, Wasylink S, Landeros-Weisenberger A, et al. Effects of ketamine in treatment-refractory obsessive-compulsive disorder. Biol Psychiatry. 2012;72(11):964–970. | |
Rodriguez CI, Kegeles LS, Levinson A, et al. Randomized controlled crossover trial of ketamine in obsessive-compulsive disorder: proof-of-concept. Neuropsychopharmacology. 2013;38(12):2475–2483. | |
Xie X, Hagan RM. Cellular and molecular actions of lamotrigine: Possible mechanisms of efficacy in bipolar disorder. Neuropsychobiology. 1998;38(3):119–130. | |
Cunningham MO, Jones RS. The anticonvulsant, lamotrigine decreases spontaneous glutamate release but increases spontaneous GABA release in the rat entorhinal cortex in vitro. Neuropharmacology. 2000;39(11):2139–2146. | |
Burstein AH. Lamotrigine. Pharmacotherapy. 1995;15(2):129–143. | |
Ketter TA, Manji HK, Post RM. Potential mechanisms of action of lamotrigine in the treatment of bipolar disorders. J Clin Psychopharmacol. 2003;23(5):484–495. | |
Bowden CL, Calabrese JR, Sachs G, et al; Lamictal 606 Study Group. A placebo-controlled 18-month trial of lamotrigine and lithium maintenance treatment in recently manic or hypomanic patients with bipolar I disorder. Arch Gen Psychiatry. 2003;60(4):392–400. | |
Calabrese JR, Suppes T, Bowden CL, et al. A double-blind, placebo-controlled, prophylaxis study of lamotrigine in rapid-cycling bipolar disorder. Lamictal 614 Study Group. J Clin Psychiatry. 2000;61(11):841–850. | |
Thomas SP, Nandhra HS, Jayaraman A. Systematic review of lamotrigine augmentation of treatment resistant unipolar depression (TRD). J Ment Health. 2010;19(2):168–175. | |
Hertzberg MA, Butterfield MI, Feldman ME, et al. A preliminary study of lamotrigine for the treatment of posttraumatic stress disorder. Biol Psychiatry. 1999;45(9):1226–1229. | |
Waldmeier PC, Baumann PA, Wicki P, Feldtrauer JJ, Stierlin C, Schmutz M. Similar potency of carbamazepine, oxcarbazepine, and lamotrigine in inhibiting the release of glutamate and other neurotransmitters. Neurology. 1995;45(10):1907–1913. | |
Uzun O. Lamotrigine as an augmentation agent in treatment-resistant obsessive-compulsive disorder: a case report. J Psychopharmacol. 2010;24(3):425–427. | |
Bruno A, Micò U, Pandolfo G, et al. Lamotrigine augmentation of serotonin reuptake inhibitors in treatment-resistant obsessive-compulsive disorder: a double-blind, placebo-controlled study. J Psychopharmacol. 2012;26(11):1456–1462. | |
Erdö SL, Schäfer M. Memantine is highly potent in protecting cortical cultures against excitotoxic cell death evoked by glutamate and N-methyl-D-aspartate. Eur J Pharmacol. 1991;198(2–3):215–217. | |
Kornhuber J, Weller M, Schoppmeyer K, Riederer P. Amantadine and memantine are NMDA receptor antagonists with neuroprotective properties. J Neural Transm Suppl. 1994;43:91–104. | |
Emre M, Tsolaki M, Bonuccelli U, et al; 11018 Study Investigators. Memantine for patients with Parkinson’s disease dementia or dementia with Lewy bodies: a randomised, double-blind, placebo-controlled trial. Lancet Neurol. 2010;9(10):969–977. | |
Cankurtaran ES, Ozalp E, Soygur H, Cakir A. Clinical experience with risperidone and memantine in the treatment of Huntington’s disease. J Natl Med Assoc. 2006;98(8):1353–1355. | |
Tariot PN, Farlow MR, Grossberg GT, Graham SM, McDonald S, Gergel I; Memantine Study Group. Memantine treatment in patients with moderate to severe Alzheimer disease already receiving donepezil: a randomized controlled trial. JAMA. 2004;291(3):317–324. | |
Hezel DM, Beattie K, Stewart SE. Memantine as an augmenting agent for severe pediatric OCD. Am J Psychiatry. 2009;166(2):237. | |
Pasquini M, Biondi M. Memantine augmentation for refractory obsessive-compulsive disorder. Prog Neuropsychopharmacol Biol Psychiatry. 2006;30(6):1173–1175. | |
Aboujaoude E, Barry JJ, Gamel N. Memantine augmentation in treatment-resistant obsessive-compulsive disorder: an open-label trial. J Clin Psychopharmacol. 2009;29(1):51–55. | |
Feusner JD, Kerwin L, Saxena S, Bystritsky A. Differential efficacy of memantine for obsessive-compulsive disorder vs generalized anxiety disorder: an open-label trial. Psychopharmacol Bull. 2009;42(1):81–93. | |
Ghaleiha A, Entezari N, Modabbernia A, et al. Memantine add-on in moderate to severe obsessive-compulsive disorder: randomized double-blind placebo-controlled study. J Psychiatr Res. 2013;47(2):175–180. | |
Blumstein CG. Drug treatment in bronchial asthma. Semin Drug Treat. 1973;2(4):385–401. | |
Stey C, Steurer J, Bachmann S, Medici TC, Tramèr MR. The effect of oral N-acetylcysteine in chronic bronchitis: a quantitative systematic review. Eur Respir J. 2000;16(2):253–262. | |
Sadowska AM, Verbraecken J, Darquennes K, De Backer WA. Role of N-acetylcysteine in the management of COPD. Int J Chron Obstruct Pulmon Dis. 2006;1(4):425–434. | |
Rumack BH. Acetaminophen overdose in children and adolescents. Pediatr Clin North Am. 1986;33(3):691–701. | |
Kearns GL, Leeder JS, Wasserman GS. Acetaminophen intoxication during treatment: what you don’t know can hurt you. Clin Pediatr (Phila). 2000;39(3):133–144. | |
Heard K, Green J. Acetylcysteine therapy for acetaminophen poisoning. Curr Pharm Biotechnol. 2012;13(10):1917–1923. | |
Arica V, Demir IH, Tutanc M, et al. N-acetylcysteine prevents doxorubucine-induced cardiotoxicity in rats. Hum Exp Toxicol. 2013;32(6):655–661. | |
Soltan-Sharifi MS, Mojtahedzadeh M, Najafi A, et al. Improvement by N-acetylcysteine of acute respiratory distress syndrome through increasing intracellular glutathione, and extracellular thiol molecules and anti-oxidant power: evidence for underlying toxicological mechanisms. Hum Exp Toxicol. 2007;26(9):697–703. | |
Wu YJ, Muldoon LL, Neuwelt EA. The chemoprotective agent N-acetylcysteine blocks cisplatin-induced apoptosis through caspase signaling pathway. J Pharmacol Exp Ther. 2005;312(2):424–431. | |
Orhan G, Yapici N, Yuksel M, et al. Effects of N-acetylcysteine on myocardial ischemia-reperfusion injury in bypass surgery. Heart Vessels. 2006;21(1):42–47. | |
Borges-Santos MD, Moreto F, Pereira PC, Ming-Yu Y, Burini RC. Plasma glutathione of HIV+ patients responded positively and differently to dietary supplementation with cysteine or glutamine. Nutrition. 2012;28(7–8):753–756. | |
Kandemir H, Abuhandan M, Aksoy N, Savik E, Kaya C. Oxidative imbalance in child and adolescent patients with obsessive compulsive disorder. J Psychiatr Res. 2013;47(11):1831–1834. | |
Ersan S, Bakir S, Erdal Ersan E, Dogan O. Examination of free radical metabolism and antioxidant defence system elements in patients with obsessive-compulsive disorder. Prog Neuropsychopharmacol Biol Psychiatry. 2006;30(6):1039–1042. | |
Lafleur DL, Pittenger C, Kelmendi B, et al. N-acetylcysteine augmentation in serotonin reuptake inhibitor refractory obsessive-compulsive disorder. Psychopharmacology (Berl). 2006;184(2):254–256. | |
Wikke J. Riluzole. Lancet. 1996;348(9030):795–799. | |
Jehle T, Bauer J, Blauth E, et al. Effects of riluzole on electrically evoked neurotransmitter release. Br J Pharmacol. 2000;130(6):1227–1234. | |
Urbani A, Belluzzi O. Riluzole inhibits the persistent sodium current in mammalian CNS neurons. Eur J Neurosci. 2000;12(10):3567–3574. | |
Sreedharan J, Brown RH Jr. Amyotrophic lateral sclerosis: problems and prospects. Ann Neurol. 2013;74(3):309–316. | |
Clark W, Kendall MJ. Therapeutic advances: riluzole for the treatment of motor neurone disease. J Clin Pharm Ther. 1996;21(6):373–376. | |
Gordon PH. Amyotrophic lateral sclerosis: an update for 2013 clinical features, pathophysiology, management and therapeutic trials. Aging Dis. 2013;4(5):295–310. | |
Lacomblez L, Bensimon G, Leigh PN, et al; ALS Study Groups I and II. Long-term safety of riluzole in amyotrophic lateral sclerosis. Amyotroph Lateral Scler Other Motor Neuron Disord. 2002;3(1):23–29. | |
Coric V, Taskiran S, Pittenger C, et al. Riluzole augmentation in treatment-resistant obsessive-compulsive disorder: an open-label trial. Biol Psychiatry. 2005;58(5):424–428. | |
Grant P, Lougee L, Hirschtritt M, Swedo SE. An open-label trial of riluzole, a glutamate antagonist, in children with treatment-resistant obsessive-compulsive disorder. J Child Adolesc Psychopharmacol. 2007;17(6):761–767. | |
Grant PJ, Joseph LA, Farmer CA, et al. 12-week, placebo-controlled trial of add-on riluzole in the treatment of childhood-onset obsessive-compulsive disorder. Neuropsychopharmacology. 2014;39(6):1453–1459. | |
Garnett WR. Clinical pharmacology of topiramate: a review. Epilepsia. 2000;41 Suppl 1:S61–S65. | |
Bauer J, Schwalen S. [Topiramate (Topamax). Pharmacological characteristics and current use in epilepsy treatment]. Nervenarzt. 2000;71(6):495–501. German. | |
Poulsen CF, Simeone TA, Maar TE, Smith-Swintosky V, White HS, Schousboe A. Modulation by topiramate of AMPA and kainate mediated calcium influx in cultured cerebral cortical, hippocampal and cerebellar neurons. Neurochem Res. 2004;29(1):275–282. | |
Manning S, Pucci A, Finer N. Pharmacotherapy for obesity: novel agents and paradigms. Ther Adv Chronic Dis. 2014;5(3):135–148. | |
Linde M, Mulleners WM, Chronicle EP, McCrory DC. Topiramate for the prophylaxis of episodic migraine in adults. Cochrane Database Syst Rev. 2013;6:CD010610. | |
Hollander E, Dell’Osso B. Topiramate plus paroxetine in treatment-resistant obsessive-compulsive disorder. Int Clin Psychopharmacol. 2006;21(3):189–191. | |
Van Ameringen M, Mancini C, Patterson B, Bennett M. Topiramate augmentation in treatment-resistant obsessive-compulsive disorder: a retrospective, open-label case series. Depress Anxiety. 2006;23(1):1–5. | |
Berlin HA, Koran LM, Jenike MA, et al. Double-blind, placebo-controlled trial of topiramate augmentation in treatment-resistant obsessive-compulsive disorder. J Clin Psychiatry. 2011;72(5):716–721. | |
Ozkara C, Ozmen M, Erdogan A, Yalug I. Topiramate related obsessive-compulsive disorder. Eur Psychiatry. 2005;20(1):78–79. | |
Lauria-Horner BA, Pohl RB. Pregabalin: a new anxiolytic. Expert Opin Investig Drugs. 2003;12(4):663–672. | |
Micheva KD, Taylor CP, Smith SJ. Pregabalin reduces the release of synaptic vesicles from cultured hippocampal neurons. Mol Pharmacol. 2006;70(2):467–476. | |
Oulis P, Mourikis I, Konstantakopoulos G. Pregabalin augmentation in treatment-resistant obsessive-compulsive disorder. Int Clin Psychopharmacol. 2011;26(4):221–224. | |
Rampello L, Alvano A, Battaglia G, Bruno V, Raffaele R, Nicoletti F. Tic disorders: from pathophysiology to treatment. J Neurol. 2006;253(1):1–15. | |
Specht MW, Nicotra CM, Kelly LM, et al. A Comparison of urge intensity and the probability of tic completion during tic freely and tic suppression conditions. Behav Modif. 2014;38(2):297–318. | |
Woods DW, Piacentini JC, Scahill L, et al. Behavior therapy for tics in children: acute and long-term effects on psychiatric and psychosocial functioning. J Child Neurol. 2011;26(7):858–865. | |
Wilhelm S, Peterson AL, Piacentini J, et al. Randomized trial of behavior therapy for adults with Tourette syndrome. Arch Gen Psychiatry. 2012;69(8):795–803. | |
Rizzo R, Gulisano M. Clinical pharmacology of comorbid attention deficit hyperactivity disorder in Tourette syndrome. Int Rev Neurobiol. 2013;112:415–444. | |
Neri V, Cardona F. Clinical pharmacology of comorbid obsessive-compulsive disorder in Tourette syndrome. Int Rev Neurobiol. 2013;112:391–414. | |
Rizzo R, Gulisano M, Pellico A, Calì PV, Curatolo P. Tourette syndrome and comorbid conditions: a spectrum of different severities and complexities. J Child Neurol. 2014;29(10):1383–1389. | |
Berardelli A, Currà A, Fabbrini G, Gilio F, Manfredi M. Pathophysiology of tics and Tourette syndrome. J Neurol. 2003;250(7):781–787. | |
Singer HS, Minzer K. Neurobiology of Tourette’s syndrome: concepts of neuroanatomic localization and neurochemical abnormalities. Brain Dev. 2003;25 Suppl 1:S70–S84. | |
Nomura Y, Segawa M. Neurology of Tourette’s syndrome (TS) TS as a developmental dopamine disorder: a hypothesis. Brain Dev. 2003; 25 Suppl 1:S37–S42. | |
Yoon DY, Gause CD, Leckman JF, Singer HS. Frontal dopaminergic abnormality in Tourette syndrome: a postmortem analysis. J Neurol Sci. 2007;255(1–2):50–56. | |
Udvardi PT, Nespoli E, Rizzo F, Hengerer B, Ludolph AG. Nondopaminergic neurotransmission in the pathophysiology of Tourette syndrome. Int Rev Neurobiol. 2013;112:95–130. | |
Singer HS, Morris C, Grados M. Glutamatergic modulatory therapy for Tourette syndrome. Med Hypotheses. 2010;74(5):862–867. | |
Anderson GM, Pollak ES, Chatterjee D, Leckman JF, Riddle MA, Cohen DJ. Postmortem analysis of subcortical monoamines and amino acids in Tourette syndrome. Adv Neurol. 1992;58:123–133. | |
Swerdlow NR, Sutherland AN. Using animal models to develop therapeutics for Tourette Syndrome. Pharmacol Ther. 2005;108(3):281–293. | |
McGrath MJ, Campbell KM, Parks CR, Burton FH. Glutamatergic drugs exacerbate symptomatic behavior in a transgenic model of comorbid Tourette’s syndrome and obsessive-compulsive disorder. Brain Res. 2000;877(1):23–30. | |
Nordstrom EJ, Burton FH. A transgenic model of comorbid Tourette’s syndrome and obsessive-compulsive disorder circuitry. Mol Psychiatry. 2002;7(6):617–625, 524. | |
Shoemaker JM, Saint Marie RL, Bongiovanni MJ, Neary AC, Tochen LS, Swerdlow NR. Prefrontal D1 and ventral hippocampal N-methyl-D-aspartate regulation of startle gating in rats. Neuroscience. 2005;135(2):385–394. | |
Abel KM, Allin MP, Hemsley DR, Geyer MA. Low dose ketamine increases prepulse inhibition in healthy men. Neuropharmacology. 2003;44(6):729–737. | |
Duncan EJ, Madonick SH, Parwani A, et al. Clinical and sensorimotor gating effects of ketamine in normals. Neuropsychopharmacology. 2001;25(1):72–83. | |
Ortiz A, Alda M, O’Donovan C. Tourette syndrome with comorbid bipolar disorder and migraine: can lamotrigine monotherapy help? J Clin Psychopharmacol. 2012;32(1):140–142. | |
Seemüller F, Dehning S, Grunze H, Müller N. Tourette’s symptoms provoked by lamotrigine in a bipolar patient. Am J Psychiatry. 2006; 163(1):159. | |
Lombroso CT. Lamotrigine-induced tourettism. Neurology. 1999; 52(6):1191–1194. | |
Sotero de Menezes MA, Rho JM, Murphy P, Cheyette S. Lamotrigine-induced tic disorder: report of five pediatric cases. Epilepsia. 2000; 41(7):862–867. | |
Robertson MM, Eapen V, Cavanna AE. The international prevalence, epidemiology, and clinical phenomenology of Tourette syndrome: a cross-cultural perspective. J Psychosom Res. 2009;67(6):475–483. | |
Fornaro M, Maremmani AG, Colicchio MG, et al. A case of severe oral self-injurious Tourette’s syndrome alleviated by pregabalin. Gen Hosp Psychiatry. 2012;34(3):321. e1–e4. | |
Kuo SH, Jimenez-Shahed J. Topiramate in treatment of tourette syndrome. Clin Neuropharmacol. 2010;33(1):32–34. | |
Jankovic J, Jimenez-Shahed J, Brown LW. A randomised, double-blind, placebo-controlled study of topiramate in the treatment of Tourette syndrome. J Neurol Neurosurg Psychiatry. 2010;81(1):70–73. | |
Underhill SM, Wheeler DS, Li M, Watts SD, Ingram SL, Amara SG. Amphetamine modulates excitatory neurotransmission through endocytosis of the glutamate transporter EAAT3 in dopamine neurons. Neuron. 2014;83(2):404–416. | |
Rolland B, Jardri R, Amad A, Thomas P, Cottencin O, Bordet R. Pharmacology of hallucinations: several mechanisms for one single symptom? Biomed Res Int. 2014;2014:307106. | |
Taylor M, Bhagwandas K. N-acetylcysteine in trichotillomania: a panacea for compulsive skin disorders? Br J Dermatol. 2014;171(5):1253–1255. | |
Van Ameringen M, Patterson B, Simpson W. DSM-5 obsessive-compulsive and related disorders: clinical implications of new criteria. Depress Anxiety. 2014;31(6):487–493. | |
Masur D, Shinnar S, Cnaan A, et al; Childhood Absence Epilepsy Study Group. Pretreatment cognitive deficits and treatment effects on attention in childhood absence epilepsy. Neurology. 2013;81(18):1572–1580. | |
Mallo M, Alonso CR. The regulation of Hox gene expression during animal development. Development. 2013;140(19):3951–3963. | |
Panza KE, Pittenger C, Bloch MH. Age and gender correlates of pulling in pediatric trichotillomania. J Am Acad Child Adolesc Psychiatry. 2013;52(3):241–249. | |
Bloch MH, Panza KE, Grant JE, Pittenger C, Leckman JF. N-Acetylcysteine in the treatment of pediatric trichotillomania: a randomized, double-blind, placebo-controlled add-on trial. J Am Acad Child Adolesc Psychiatry. 2013;52(3):231–240. | |
Rodrigues-Barata AR, Tosti A, Rodríguez-Pichardo A, Camacho-Martínez F. N-acetylcysteine in the treatment of trichotillomania. Int J Trichology. 2012;4(3):176–178. | |
Grant JE, Odlaug BL, Chamberlain SR, Keuthen NJ, Lochner C, Stein DJ. Skin picking disorder. Am J Psychiatry. 2012;169(11):1143–1149. | |
Schneebaum-Sender N, Goldberg-Stern H, Fattal-Valevski A, Kramer U. Does a normalizing electroencephalogram in benign childhood epilepsy with centrotemporal spikes abort attention deficit hyperactivity disorder? Pediatr Neurol. 2012;47(4):279–283. | |
Wu J, Xiao H, Sun H, Zou L, Zhu LQ. Role of dopamine receptors in ADHD: a systematic meta-analysis. Mol Neurobiol. 2012;45(3):605–620. | |
Grant JE, Odlaug BL, Chamberlain SR. A cognitive comparison of pathological skin picking and trichotillomania. J Psychiatr Res. 2011;45(12):1634–1638. | |
Chamberlain SR, Hampshire A, Menzies LA, et al. Reduced brain white matter integrity in trichotillomania: a diffusion tensor imaging study. Arch Gen Psychiatry. 2010;67(9):965–971. | |
Grant JE, Odlaug BL, Chamberlain SR, Kim SW. A double-blind, placebo-controlled trial of lamotrigine for pathological skin picking: treatment efficacy and neurocognitive predictors of response. J Clin Psychopharmacol. 2010;30(4):396–403. | |
Grant JE, Odlaug BL, Kim SW. N-acetylcysteine, a glutamate modulator, in the treatment of trichotillomania: a double-blind, placebo-controlled study. Arch Gen Psychiatry. 2009;66(7):756–763. | |
Chamberlain SR, Odlaug BL, Boulougouris V, Fineberg NA, Grant JE. Trichotillomania: neurobiology and treatment. Neurosci Biobehav Rev. 2009;33(6):831–842. | |
Paine TA, Neve RL, Carlezon WA Jr. Attention deficits and hyperactivity following inhibition of cAMP-dependent protein kinase within the medial prefrontal cortex of rats. Neuropsychopharmacology. 2009;34(9):2143–2155. | |
Tseng KY, O’Donnell P. Dopamine-glutamate interactions controlling prefrontal cortical pyramidal cell excitability involve multiple signaling mechanisms. J Neurosci. 2004;24(22):5131–5139. | |
MacMaster FP, Carrey N, Sparkes S, Kusumakar V. Proton spectroscopy in medication-free pediatric attention-deficit/hyperactivity disorder. Biol Psychiatry. 2003;53(2):184–187. | |
Carrey N, MacMaster FP, Fogel J, et al. Metabolite changes resulting from treatment in children with ADHD: a 1H-MRS study. Clin Neuropharmacol. 2002;26(4):218–221. | |
Carrey N, MacMaster FP, Sparkes SJ, Khan SC, Kusumakar V. Glutamatergic changes with treatment in attention deficit hyperactivity disorder: a preliminary case series. J Child Adolesc Psychopharmacol. 2002;12(4):331–336. | |
Bush G, Luu P, Posner MI. Cognitive and emotional influences in anterior cingulate cortex. Trends Cogn Sci. 2000;4(6):215–222. | |
Hammerness P, Biederman J, Petty C, Henin A, Moore CM. Brain biochemical effects of methylphenidate treatment using proton magnetic spectroscopy in youth with attention-deficit hyperactivity disorder: a controlled pilot study. CNS Neurosci Ther. 2012;18(1):34–40. | |
Xu M, Moratalla R, Gold LH, et al. Dopamine D1 receptor mutant mice are deficient in striatal expression of dynorphin and in dopamine-mediated behavioral responses. Cell. 1994;79(4):729–742. | |
Granon S, Passetti F, Thomas KL, Dalley JW, Everitt BJ, Robbins TW. Enhanced and impaired attentional performance after infusion of D1 dopaminergic receptor agents into rat prefrontal cortex. J Neurosci. 2000;20(3):1208–1215. | |
Warton FL, Howells FM, Russell VA. Increased glutamate-stimulated release of dopamine in substantia nigra of a rat model for attention-deficit/hyperactivity disorder – lack of effect of methylphenidate. Metab Brain Dis. 2009;24(4):599–613. | |
Miller JL, Angulo M. An open-label pilot study of N-acetylcysteine for skin-picking in Prader-Willi syndrome. Am J Med Genet A. 2014;164A(2):421–424. | |
Lesch KP, Merker S, Reif A, Novak M. Dances with black widow spiders: dysregulation of glutamate signalling enters centre stage in ADHD. Eur Neuropsychopharmacol. 2013;23(6):479–491. | |
Elia J, Glessner JT, Wang K, et al. Genome-wide copy number variation study associates metabotropic glutamate receptor gene networks with attention deficit hyperactivity disorder. Nat Genet. 2012;44(1):78–84. | |
Rubinstein M, Cepeda C, Hurst RS, et al. Dopamine D4 receptor-deficient mice display cortical hyperexcitability. J Neurosci. 2001;21(11):3756–3763. | |
Wang X, Zhong P, Gu Z, Yan Z. Regulation of NMDA receptors by dopamine D4 signaling in prefrontal cortex. J Neurosci. 2003;23(30):9852–9861. | |
Gan L, Falzone TL, Zhang K, Rubinstein M, Baldessarini RJ, Tarazi FI. Enhanced expression of dopamine D(1) and glutamate NMDA receptors in dopamine D(4) receptor knockout mice. J Mol Neurosci. 2004;22(3):167–178. | |
Thomas TC, Grandy DK, Gerhardt GA, Glaser PE. Decreased dopamine D4 receptor expression increases extracellular glutamate and alters its regulation in mouse striatum. Neuropsychopharmacology. 2009;34(2):436–445. | |
Shaywitz AJ, Greenberg ME. CREB: a stimulus-induced transcription factor activated by a diverse array of extracellular signals. Annu Rev Biochem. 1999;68:821–861. | |
Pozzi L, Baviera M, Sacchetti G, et al. Attention deficit induced by blockade of N-methyl D-aspartate receptors in the prefrontal cortex is associated with enhanced glutamate release and cAMP response element binding protein phosphorylation: role of metabotropic glutamate receptors 2/3. Neuroscience. 2011;176:336–348. | |
Viggiano D, Vallone D, Welzl H, Sadile AG. The Naples High- and Low-Excitability rats: selective breeding, behavioral profile, morphometry, and molecular biology of the mesocortical dopamine system. Behav Genet. 2002;32(5):315–333. | |
Ruocco LA, Gironi Carnevale UA, Sadile AG, et al. Elevated forebrain excitatory L-glutamate, L-aspartate and D-aspartate in the Naples high-excitability rats. Behav Brain Res. 2009;198(1):24–28. | |
Mattes J. A pilot trial of amantadine in hyperactive children. Psychopharmacol Bull. 1980;16(3):67–69. | |
Donfrancesco R, Calderoni D, Vitiello B. Open-label amantadine in children with attention-deficit/hyperactivity disorder. J Child Adolesc Psychopharmacol. 2007;17(5):657–664. | |
Mohammadi MR, Kazemi MR, Zia E, Rezazadeh SA, Tabrizi M, Akhondzadeh S. Amantadine versus methylphenidate in children and adolescents with attention deficit/hyperactivity disorder: a randomized, double-blind trial. Hum Psychopharmacol. 2010;25(7–8):560–565. | |
Öncü B, Er O, Çolak B, Nutt DJ. Lamotrigine for attention deficit-hyperactivity disorder comorbid with mood disorders: a case series. J Psychopharmacol. 2014;28(3):282–283. | |
Uvebrant P, Bauzienè R. Intractable epilepsy in children. The efficacy of lamotrigine treatment, including non-seizure-related benefits. Neuropediatrics. 1994;25(6):284–289. | |
Schubert R. Attention deficit disorder and epilepsy. Pediatr Neurol. 2005;32(1):1–10. | |
Peeters M, Maloteaux JM, Hermans E. Distinct effects of amantadine and memantine on dopaminergic transmission in the rat striatum. Neurosci Lett. 2003;343(3):205–209. | |
Findling RL, McNamara NK, Stansbrey RJ, et al. A pilot evaluation of the safety, tolerability, pharmacokinetics, and effectiveness of memantine in pediatric patients with attention-deficit/hyperactivity disorder combined type. J Child Adolesc Psychopharmacol. 2007;17(1):19–33. | |
Surman CB, Hammerness PG, Petty C, et al. A pilot open label prospective study of memantine monotherapy in adults with ADHD. World J Biol Psychiatry. 2013;14(4):291–298. | |
Garcia RJ, Francis L, Dawood M, Lai ZW, Faraone SV, Perl A. Attention deficit and hyperactivity disorder scores are elevated and respond to N-acetylcysteine treatment in patients with systemic lupus erythematosus. Arthritis Rheum. 2013;65(5):1313–1318. | |
Arnold LM, Auchenbach MB, McElroy SL. Psychogenic excoriation. Clinical features, proposed diagnostic criteria, epidemiology and approaches to treatment. CNS Drugs. 2001;15(5):351–359. | |
Keuthen NJ, Koran LM, Aboujaoude E, Large MD, Serpe RT. The prevalence of pathologic skin picking in US adults. Compr Psychiatry. 2010;51(2):183–186. | |
Hayes SL, Storch EA, Berlanga L. Skin picking behaviors: An examination of the prevalence and severity in a community sample. J Anxiety Disord. 2009;23(3):314–319. | |
Swedo SE, Rapoport JL. Annotation: trichotillomania. J Child Psychol Psychiatry. 1991;32(3):401–409. | |
Neziroglu F, Rabinowitz D, Breytman A, Jacofsky M. Skin picking phenomenology and severity comparison. Prim Care Companion J Clin Psychiatry. 2008;10(4):306–312. | |
Bohne A, Savage CR, Deckersbach T, Keuthen NJ, Wilhelm S. Motor inhibition in trichotillomania and obsessive-compulsive disorder. J Psychiatr Res. 2008;42(2):141–150. | |
Swedo SE, Rapoport JL, Leonard HL, Schapiro MB, Rapoport SI, Grady CL. Regional cerebral glucose metabolism of women with trichotillomania. Arch Gen Psychiatry. 1991;48(9):828–833. | |
Roos A, Fouche JP, Stein DJ, Lochner C. White matter integrity in hair-pulling disorder (trichotillomania). Psychiatry Res. 2013;211(3):246–250. | |
Grant JE, Odlaug BL, Hampshire A, Schreiber LR, Chamberlain SR. White matter abnormalities in skin picking disorder: a diffusion tensor imaging study. Neuropsychopharmacology. 2013;38(5):763–769. | |
Stratford TH, Kostakopoulou K, Maden M. Hoxb-8 has a role in establishing early anterior-posterior polarity in chick forelimb but not hindlimb. Development. 1997;124(21):4225–4234. | |
Greer JM, Capecchi MR. Hoxb8 is required for normal grooming behavior in mice. Neuron. 2002;33(1):23–34. | |
Welch JM, Wang D, Feng G. Differential mRNA expression and protein localization of the SAP90/PSD-95-associated proteins (SAPAPs) in the nervous system of the mouse. J Comp Neurol. 2004;472(1):24–39. | |
Bienvenu OJ, Wang Y, Shugart YY, et al. Sapap3 and pathological grooming in humans: Results from the OCD collaborative genetics study. Am J Med Genet B Neuropsychiatr Genet. 2009;150B(5):710–720. | |
Züchner S, Wendland JR, Ashley-Koch AE, et al. Multiple rare SAPAP3 missense variants in trichotillomania and OCD. Mol Psychiatry. 2009;14(1):6–9. | |
Odlaug BL, Grant JE. N-acetyl cysteine in the treatment of grooming disorders. J Clin Psychopharmacol. 2007;27(2):227–229. | |
Grant JE, Odlaug BL, Kim SW. Lamotrigine treatment of pathologic skin picking: an open-label study. J Clin Psychiatry. 2007;68(9):1384–1391. | |
Lochner C, Seedat S, Niehaus DJ, Stein DJ. Topiramate in the treatment of trichotillomania: an open-label pilot study. Int Clin Psychopharmacol. 2006;21(5):255–259. | |
Shapira NA, Lessig MC, Murphy TK, Driscoll DJ, Goodman WK. Topiramate attenuates self-injurious behaviour in Prader-Willi Syndrome. Int J Neuropsychopharmacol. 2002;5(2):141–145. | |
Jafferany M, Shireen F, Ibrahim A. An open-label trial of topiramate in the treatment of skin picking in pervasive developmental disorder not otherwise specified. Prim Care Companion J Clin Psychiatry. 2010;12(2). pii: PCC.09l00829. | |
Coric V, Kelmendi B, Pittenger C, Wasylink S, Bloch MH, Green J. Beneficial effects of the antiglutamatergic agent riluzole in a patient diagnosed with trichotillomania. J Clin Psychiatry. 2007;68(1):170–171. | |
Rothman SM, Mattson MP. Activity-dependent, stress-responsive BDNF signaling and the quest for optimal brain health and resilience throughout the lifespan. Neuroscience. 2013;239:228–240. | |
Tanaka K. [Brain development and glutamate]. Brain Nerve. 2013;65(10):1121–1132. Japanese. | |
Nakamichi N, Takarada T, Yoneda Y. Neurogenesis mediated by gamma-aminobutyric acid and glutamate signaling. J Pharmacol Sci. 2009;110(2):133–149. | |
Mattson MP. Glutamate and neurotrophic factors in neuronal plasticity and disease. Ann N Y Acad Sci. 2008;1144:97–112. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.