Back to Journals » Pharmacogenomics and Personalized Medicine » Volume 16
A Retrospective Analysis of Clinically Focused Exome Sequencing Results of 372 Infants with Suspected Monogenic Disorders in China
Authors Jia A, Lei Y, Liu DP, Pan L, Guan HZ, Yang B
Received 5 September 2022
Accepted for publication 12 January 2023
Published 2 February 2023 Volume 2023:16 Pages 81—97
DOI https://doi.org/10.2147/PGPM.S387767
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Martin H Bluth
An Jia,1,* Yi Lei,2,* Dan-Ping Liu,2 Lu Pan,2 Hui-Zhen Guan,2 Bicheng Yang1,2
1Medical School, Huanghe Science and Technology College, Zhengzhou, People’s Republic of China; 2Jiangxi Key Laboratory of Birth Defect Prevention and Control, Jiangxi Maternal and Child Health Hospital, Nanchang, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Bicheng Yang, Jiangxi Provincial Key Laboratory of Birth Defect for Prevention and Control, Jiangxi Provincial Maternal and Child Health Hospital, Nanchang, 330006, People’s Republic of China, Tel +86 15350402147, Email [email protected]
Objective: The context was designed to optimize the diagnostic utility of clinically focused exome sequencing (CFES) and shorten the diagnostic odyssey among pediatric patients suspected of monogenic disorders (MDs).
Methods: Here, we retrospectively analyzed the clinical notes of 372 patients from different areas in the Jiangxi province that were referred for a diagnostic CFES and analysis from June 2018 to March 2022 with symptoms suggestive of MDs. In our study, preliminary tests using the proband-only clinical exome sequencing as a cost-effective first-tier diagnostic test for pediatric patients with unidentified MDs, supplemented by family segregation studies for targeted variants when indicated.
Results: Probands with confirmed diagnostic (CD) or likely diagnostic (LD) genetic influences accounted for 12% of all cases, whereas those with an uncertain diagnosis accounted for 48%. We also found that systemic primary carnitine deficiency (CDSP) (SLC22A5 gene) and phenylketonuria (PAH gene) were relatively more prevalent, and these patients with CDSP had the most frequent c.1400C > G variant (p.S467C) and c.51C > G variant (p. F17L) in this study. In addition, statistical analysis revealed that the estimates of diagnostic yields varied across certain phenotypic features of patients, and patients with specific phenotypic traits tended to benefit more from CFES.
Conclusion: The CFES may be a first-line genetic test for diagnosing young children with suspected genetic conditions, as it validates the identification of molecular genetics alterations and facilitates comprehensive medical management. Moreover, we found that infants exhibiting metabolism/homeostasis abnormalities, craniofacial /otolaryngology/ ophthalmologic abnormalities, and/or the integument were significantly more likely to receive a genetic diagnosis via CFES than infants without such features. However, due to the current study’s low diagnostic yield and inherent limitations, high-quality clinical studies with larger sample sizes are still needed to provide more likely results and confirm our findings.
Keywords: clinical exome sequencing, monogenic diseases, diagnostic rate, phenotypic features, clinical management
Introduction
Over the last few decades, the ever-increasing levels of rare diseases (RDs) worldwide reached about 7000, based on the National Institutes of Health.1 Nearly 4500 of these have been identified with apparent causative genes.2 Yet, genetic predisposition accounts for more than 80% of RDs,3 and a quarter of pediatric patients were hospitalized for this situation,1 which has even become a significant cause of infant mortality in neonatal intensive care units.4 NGS (next-generation sequencing) has been potentially valuable for infants with critical illnesses suspected of monogenic disorders (MDs). The early clinical diagnosis of MDs would allow for better disease management and point-of-care for patients to positively impact overall child prognosis and survival. Research showed that NGS could allow covering multiple types of variation, including small insertions and deletions of single nucleotide variants (SNVs), copy number variants (CNVs), deletions (INDELs), significant structural variants (SVs), an increasingly comprehensive utilization in first-line clinical diagnosis detection for inpatient infants.5–7 It permits the evaluation of genetic pathogenicity in non-phenotype-driven patterns, which is especially crucial for those patients diagnosed until later in childhood or even adulthood. Compared to traditional testing methods, such as tandem mass spectrometry (MS/MS) and biochemical tests, genetic screening offers significant advantages, including screening for diseases without reliable biochemical markers and higher throughput, potentially providing more rapid and accurate diagnoses.8 The diagnostic yield, however, might be affected by numerous elements such as patient inclusion criteria, analytical method (only probands/parent-offspring trios), morbidity age, family history, and disease type.9 Rapid and cost-effective testing in neonatal and early childhood were essential factors for the diagnostics utility of NGS for the single-gene disorder. Moreover, the findings contributed to diagnosis, disease management, and genetic counseling.
Clinically focused exome sequencing (CFES) should concentrate on testing diseases early in infancy and early intervention in cases to improve immediate or prolonged clinical outcomes. Combined with years of practice experience by trained geneticists and clinicians, in this study, our team was committed to investigating the clinical utility of CFES within young infants with suspected MDs. In addition, only genetic variants associated with newborn and young children of congenital disease onset were sequenced and analyzed by CFES. Accordingly, the selection for including genes in this assay was based on the following criteria: sufficient evidence for gene-disease correlation and high penetrance, early age of onset of symptoms, and the importance of early intervention. Contemporary stepwise approaches can be time-consuming, costly, and often insensitive, involving chromosomal microarray analysis, targeted sequencing, and Sanger sequencing.10 Although exome sequencing (ES)/genome sequencing (GS) appears to have high diagnostic value in individuals with suspected genetic disorders, given the fact that these genetic methods are not always available and are expensive.4,11–19 This suggests that we consider cost-benefit in addition to clinical benefit. In our preliminary research, CFES enables the detection of many genes at one time, creating higher clinical value at a relatively lower testing cost. There is growing evidence of its value in clinical diagnosis and discovering new genetically pathogenic genes. Therefore, the objective was to optimize the diagnosis workflow, reduce clinical workload and improve the diagnostic yield of NGS while minimizing the cost of sequencing and the time to a definitive diagnosis.
Materials and Methods
Patients’ Recruitment and Clinical Information Collection
The research involved 372 consecutive probands (from 368 unrelated families, including four sets of twins) who were referred by physicians from various districts in Jiangxi Province. Patients who were aged between 1 day and 270 days were enrolled from the pediatric clinic or inpatient in Jiangxi Maternal and Child Health Hospital. All patients were referred for diagnostic NGS from June 2018 to March 2022 with suspicion of monogenic disorders. Study participation was required with permission of law and patients/at least one legal guardian explaining the interests and risks of clinical NGS testing. The Ethical Commission approved this study of the faculty of Jiangxi Maternal and Child Health Hospital.
Identified medical information was assembled by a trained and senior pediatrician through a customized online platform for transmission to the sequencing laboratory and researchers. All patients’ medical information was checked jointly by senior geneticists and experienced medical experts. We classified patients’ phenotypes in the light of the Human Phenotype Ontology (HPO) terms20 based on previously collected clinical information.
Inclusion and Exclusion Criteria
To be enrolled in the research, a patient had to be an inpatient of Neonatal Intensive Care Unit/ Pediatric Intensive Care Unit (NICU/PICU) (327 patients) and no- NICU/PICU (45 patients) or clinic from Jiangxi Maternal and Child Health Hospital (Table 1). (I) If patients met the following criteria, patients were incorporated into the current cohort. The paternal and maternal sides were biologically associated with the probands and were from Jiangxi Province. (II) Eligible patients had one or more of the following clinical conditions: 1. The proband’s parents have a family history of the single-gene disorder or a consanguineous marriage; 2. Respiratory system anomalies including severe dyspnoea, respiratory distress, respiratory failure, pneumothorax, pulmonary hypoplasia, congenital central hypoventilation syndrome; 3. Metabolism/homeostasis anomalies, including patients suspected of an inborn error of metabolism by tandem mass spectrometry analyses, neonatal hyperbilirubinemia, acidosis, hyperammonaemia, and thyroid dysfunction, abnormal glucose metabolism, severe internal electrolyte disturbance; 4. Musculoskeletal system abnormality including osteogenesis imperfecta, limbs or spine malformation, dystonia abnormalities, increased muscle enzymes;5. Nervous system anomalies, including congenital hydrocephalus, cerebral dysplasia, Lateral ventricular dilatation, encephalopathy, seizure/epilepsy, intracranial hemorrhage, convulsions, mental retardation, and central nervous system infection; 6. Growth and development abnormalities, including speech and motor development delay; 7. Digestive system abnormalities, including digestive malformations, abdominal clefts, feeding difficulties, hepatosplenomegaly, cholestasis, recurrent diarrhea/vomiting/gastrointestinal bleeding/abdominal distention, congenital intestinal atresia, and abdominal hernia; 8. Hematologic abnormalities, including coagulation disorders, anemia/erythrocytosis, thrombocytopenia, disseminated intravascular coagulation, phagocytic syndrome), congenital neutropenia, severe anemia; 9. Blood and blood-forming tissue abnormalities, including coagulation abnormalities, anemia, polycythemia, thrombocytopenia, Congenital neutropenia, and disseminated intravascular coagulation;10. Immune system abnormalities, including immunodeficiency and sepsis;11. Genitourinary system abnormalities, including external genital deformities, urinary stones, renal dysplasia, ureter abnormality, recurrent urinary tract infections, hypospadias, cryptorchidism, or renal insufficiency; 12. Cardiovascular stem abnormalities, including congenital heart disease, arrhythmias, and hereditary cardiomyopathy;13. Anomalies of the integument and hair include albinism, cyanotic episodes, severe skin lesions, eczema, and abnormal hair color;14. Craniofacial abnormalities include abnormal pupil color, visual/auditory abnormalities, craniofacial/ear/nose deformities, cleft palate, cataracts, optic nerve atrophy, and nystagmus. 15. Endocrine system including precocious puberty, dwarfism, congenital adrenocortical hyperplasia, diabetes mellitus, and abnormal thyroid function. (III) Stillborn infants who died soon after parturition were also recruited for inclusion.
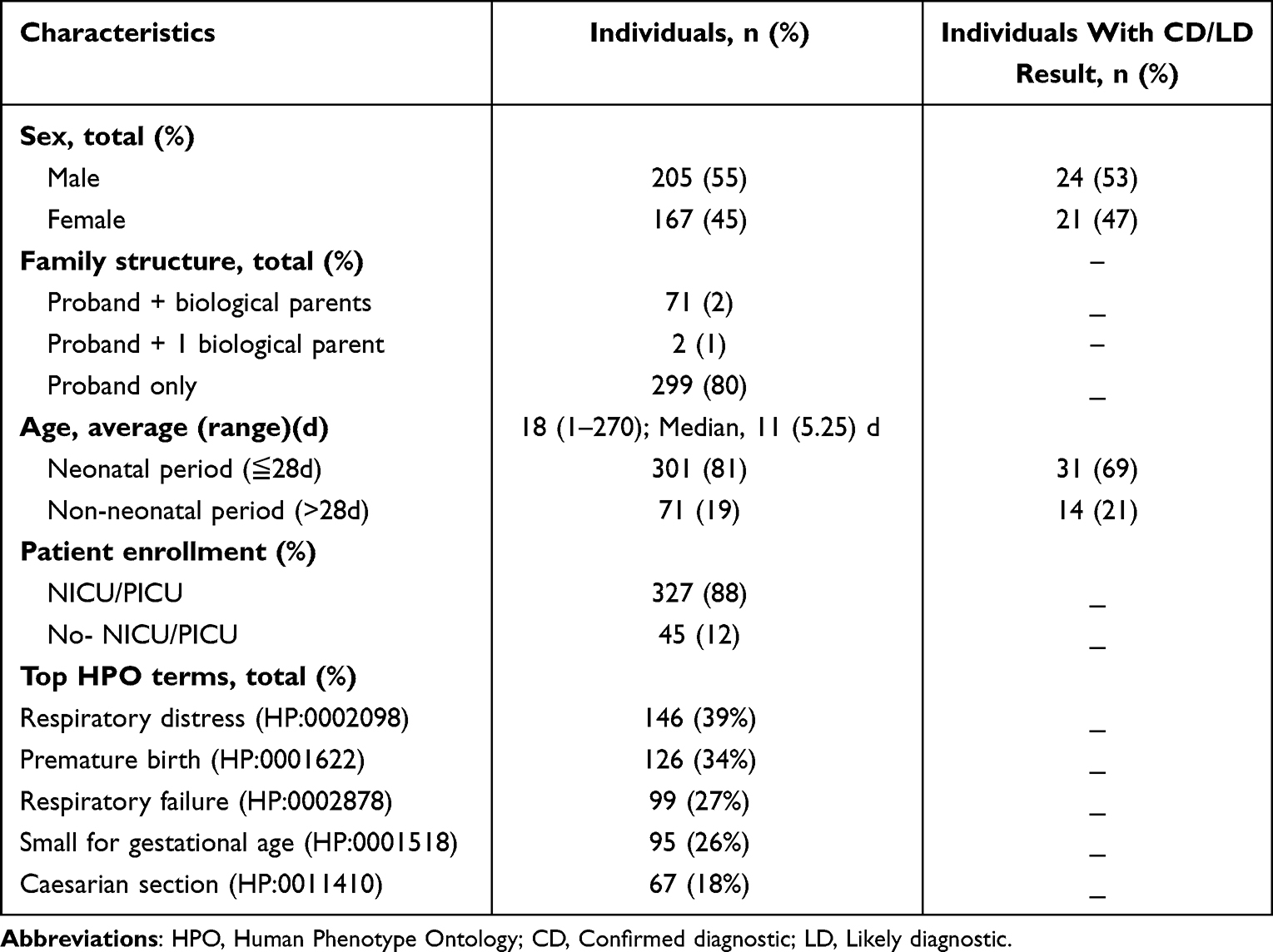 |
Table 1 The Main Demographics of Patients Included in This Study |
Patients with a definitive diagnosis of genetic disorders by clinical signs or methods other than clinically targeted genetic testing were excluded from this research. For example, the number of chromosome abnormalities such as Edwards syndrome and trisomy 21/13 syndrome, considering somatic gene variants in conjunction with the medical history, and congenital malformations caused by external environmental factors, such as toxins, drugs, rays, viruses, or parasites. This research also excluded that the pathogenic genes of the target disease were not covered by clinical focused exome sequencing.
2.3 Clinical Focused Exome Sequencing, Bioinformatics Analysis, and Variations Classification
DNA samples of probands and their parents were extracted from 2 mL EDTA peripheral blood. Sequencing libraries were constructed from genomic DNA using the lab’s custom library preparation protocol. Besides, Agilent Clear Seq Inherited Disease kits, comprising 2742 known pathogenic genes (Santa Clara, California, USA), were used for enrichment in accordance with the instructions of the manufacturer.
These resulting library products were performed paired-terminal sequencing on the Illumina HiSeqX device. The sequence data were analyzed by BWA software21 and read alignment of the human reference genome (UCSCHg19). Multi-step bioinformatics procedures were conducted using custom-designed sequence processing tools from raw data (FASTQ sequence files) to VCF files. In addition, the sequencing depth of each base and variation prediction were obtained from all genome sequencing data. The mean depth of coverage of the target region for exome sequencing was ≥ 90X, with 98% of the target capture regions reaching a depth of at least 20X.
The functional annotation of variations was performed using the Ensembl Variant Effect Predictor (VEP)22 and ANNOVAR algorithm.23 The emphasis of data analyses was the list of case-associated candidate genes, as denoted by HPO,20 Online Mendelian Inheritance in Man (OMIM) algorithm,24 and/or complemented by selected known genetic defects from previous studies. Large-scale population sequencing databases excluded variants with high frequency in normal populations, including the 1000 Genomes Sequencing Project and/or the Genome Aggregation Database (gnomAD) and/or the Exome Aggregation Consortium (ExAC). The subsequent filtering for the known pathogenic variants utilized three major databases containing known or suspected pathogenic variants, including Clin Var, OMIM, and HGMD (Human Gene Mutation Database). Meanwhile, various tools were used to predict, such as the function of missense variants and the annotation of non-coding regulatory sequences. Furthermore, variant sequences were named by the Human Genome Variation Society (HGVS). The clinical exome sequencing (CES) workflow of cases presented is shown in Figure 1.
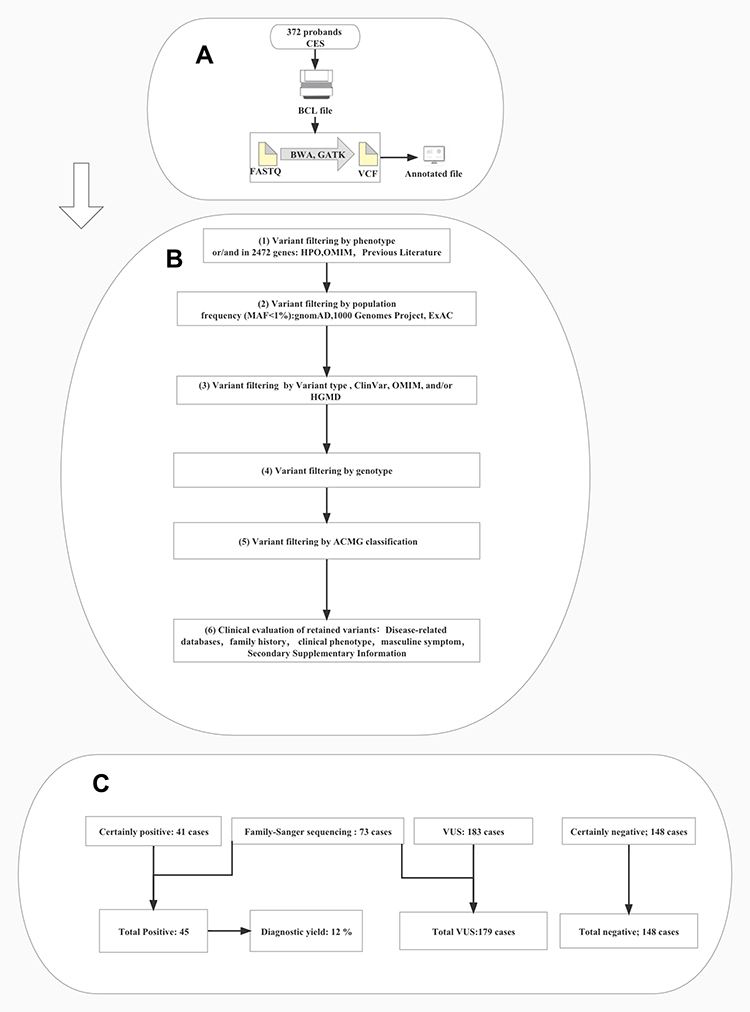 |
Figure 1 Clinical exome sequencing (CES) workflow of cases presented in this study.(A) Exome sequencing and bioinformatic analysis of 372 cases. (B) Using phenotype-driven strategy to filter and interpret variations: (1) variant filtering by phenotype and/or in silico gene list. (2) variant filtering by population frequency (minor allele frequency, MAF ≤1%. (3) variant filtering by variant type and/or ClinVar pathogenicity and/or OMIM and/or HGMD and/or internal pathogenic prediction tools. (4) variant filtering by genotype. (5) variant filtering by American College of Medical Genetics and Genomics (ACMG) classification. (6) clinical/phenotypic re-evaluation of retained variants. (C) Number of cases with identified causative variants [certainly, positive and variants in need for family segregation and targeted Sanger sequencing confirmation], no causative variant (certainly negative), and unsolved cases (further investigation for VUS was not applicable). Abbreviation: VUS, Variant of Uncertain Significance. |
In the absence of additional evidence such as genealogical analysis and genetic model, such variants in these congenital disabilities were potential to be preliminarily defined as benign, likely benign, pathogenic, likely pathogenic, variants of unknown significance (VUS) variants according to the American College of Medical Genetics and Genomics (ACMG) guidelines.25 Finally, the surviving inherited variants were reassessed and selected by multidisciplinary teamwork to ascertain their relevance to phenotype according to the above interpretation of bioinformatics data and available medical information. SNVs, INDELs, and CNVs findings were confirmed by Sanger sequencing/ multiplex ligation-dependent probe amplification (MLPA) in all CD/LD individuals. When available, parental samples were segregated for analysis to ascertain the mode of inheritance in the families. Additionally, the sequencing reports were redefined as three case-related classifications in the background of clinical phenotype compatibility, employing positive, uncertain, and negative in three clinical terminologies. Detection of pathogenic (PAT) and likely pathogenic (LP) variations entirely or partially explaining the clinical phenotypes were considered positive results which likely to assist clinicians in making the right and reasonable clinical decision; All detected variant was regarded as a priori VUS to reanalyze, which requires clinicians to make a comprehensive judgment combined with clinical information. VUS was a potential category until adequate evidence was available to classify it as benign or pathogenic reasonably. However, a previous study has shown that the majorities of variants classified as VUS in Clin Var were likely to be re-classify as harmless.26 In contrast, no currently known definite abnormalities within sequencing were classified as unfavorable. Clinical data and genotypes were re-checked by clinicians and geneticists in sequencing reports who jointly determine the molecular genetic diagnosis of patients. The phenotypes of patients were also organized and classified according to HPO terms.
Statistical Analysis
Correlativity test of diagnostic yield to differences between 14 HPO subcategories plus three common clinical elements, premature birth, cesarean section, and small for gestational age, were conducted using Fisher’s Exact test (two-tailed). P-values less than 5% were deemed statistical significance differences.
Results
The Demographics of Included Patients
This study involved 372 probands who included 205 (55%) males and 167 (45%) females (Table 1), and the gender structure difference has no statistical significance.
All patients were referred for a diagnostic NGS from June 2018 to March 2022, an almost 4-years period with suspicion of inherited metabolic disorders.
All parents of probands are native of Jiangxi Province and were no known consanguineous families.
Multiple systemic abnormalities may be involved in one patient, of which the four most affected systems were the respiratory system 51% (n=188/372), metabolic system 50% (n=187/372), neuromusculoskeletal /limb system, 41% (n=154/372), digestive system 24% (n=88/372). Available medical information was classified as 14 standard subcategories of HPO terms with the proposal from trained geneticists and experienced clinicians. In all probands, at least 179 of 278 cased-related HPO terms20 were observed in more than one baby (20 times per category). Among all probands, the four most frequent terms observed included 39% (n=146/372) had respiratory distress (HP: 0002098), 34% (n=126/372) with premature birth (HP: 0001622), 27% (n=99/372) had a respiratory failure (HP: 0002878), 26% (n=95/372) are small for gestational age (HP: 0001518), and 18% (n=67/372) via caesarian section (HP: 0011410) (Table 1).
All probands were CFES instead of ES or GS by design. 2 cases with biological father or mother were available for sequencing, 19% (n=71/372) with both biological father and mother (comprising 45 confirmed patients) were available for sequencing, 80% (n=299/ 372) with no parental sample was available for sequencing (Table 1).
The Utility for Diagnosis in Clinical Focused Exome Sequencing
Out of the 372 enrolled infants, 60% (n=224/372) probands detected variants of CD/LD or unknown clinical significance, whereas, for 40% (n=148/372), no finding of variation could interpret the case-related phenotype, at least according to the available research (sorted as report negative). Forty-one had definite or likely diagnostic pathogenic outcomes achieved by the current strategy (classified as report positive). Disease-targeted NGS findings were consistent with verifying Sanger sequencing/MLPA outcomes. Another 49% (n=183/372) cases received results of undetermined clinical significance. They were required to unite additional evidence, such as genealogical analysis and genetic model, to evaluate variation further, whose biological parental DNA samples were sequenced to assess and standardize the evaluation of potentially case-related variations when available. For 4 cases with VUS, the parental sanger sequencing findings were confirmed and supported the mode of inheritance (family segregation). Thus, the 4 cases with VUS had further to be pathogenic variants. In summary, 45 probands had a definite or likely diagnosis with a total diagnosis yield of 12% (n=45/372) and 48% (n=179/372) specified as VUS report. Among females, the diagnostic result was 21/167 (12.6%), which was a slightly higher yield than the males of 24/205 (11.7%) (Table 1). The mean TAT (turnaround time) from clinical samples to sequencing reports generation was 32 days, not including Sanger sequencing.
Genetic Variants Analysis Findings in Cohorts
A total of 368 clinically phenotypically relevant variant loci were detected in all patients. Remarkably, about 24.2% (n=89/368) of the identified variants were not previously described in any public database. Since this goes beyond the diagnostic setting, we do not include variants identified in genes not yet confirmed to be associated with human diseases. A total of findings involved 212 different genes, 13 only with PAT/LP results, 191 only with an undetermined effect, and eight with both above two developments, including G6PD, SLC22A5, PAH, PTS, ACADS, KCNQ2, GLDC, KMT2D. Of all detected genes, 174 were observed in only one patient (found in one infant), and 38 recurring genes were observed in at least one individual. We identified 56 pathogenic (PAT) or likely pathogenic (LP) variants accounting for 212 unique variants across 45 of the 372 cases.
The case-related diagnoses covered 21 unique genetic defects; 17 were identified in only one infant, with PAT/LP in 4 re-emerging genes, each detected in at least two infants. The four genes were SLC22A5 (11 patients), PAH (11 patients), PTS (4 patients), and G6PD (2 patients). (Table 2) The identified infants included 44 individuals diagnosed with SNV and/or Indel variations and one infant with an interpretive copy-number variant. One infant (NO.16) had both pathogenic SNV/Indel and CNV (Table 3). Beyond that, five infants harbored PAP/LP and VUS variants in each of at least two various genes (were found in one infant) (Table 3): (1) case NO.11: PTS, PUF60, and LDLR; (2) case NO.16: SLC25A13, IDS, and LMBRD1; (3) case NO.17: SLC22A5 and GJB1; (4) case NO.18: SLC22A5 and SEMA6B; (5) case NO.45: SLC22A5 and KMT2D. Combined with one positive CNV finding, IVS4ins6Kb variants (SLC25A13 genes) involving base large fragment insertion of the whole SLC25A13 gene. Significant SNV and/or Indel detected by NGS involved 14 different chromosomes across 45 positive cases (Table 3).
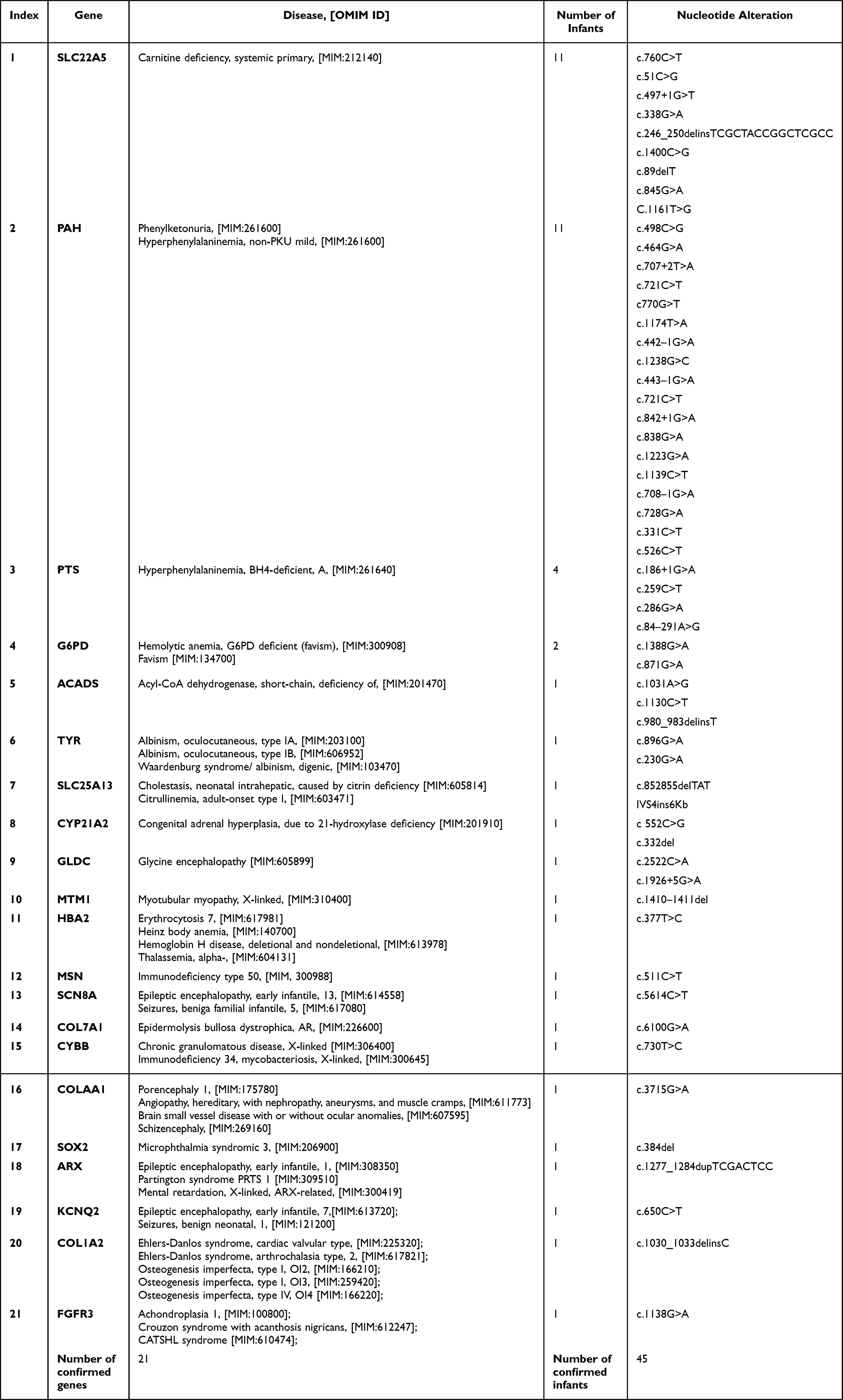 |
Table 2 The Spectrum of Diseases Associated with Genetic Defects Found in Infants with Definitive Molecular Diagnoses |
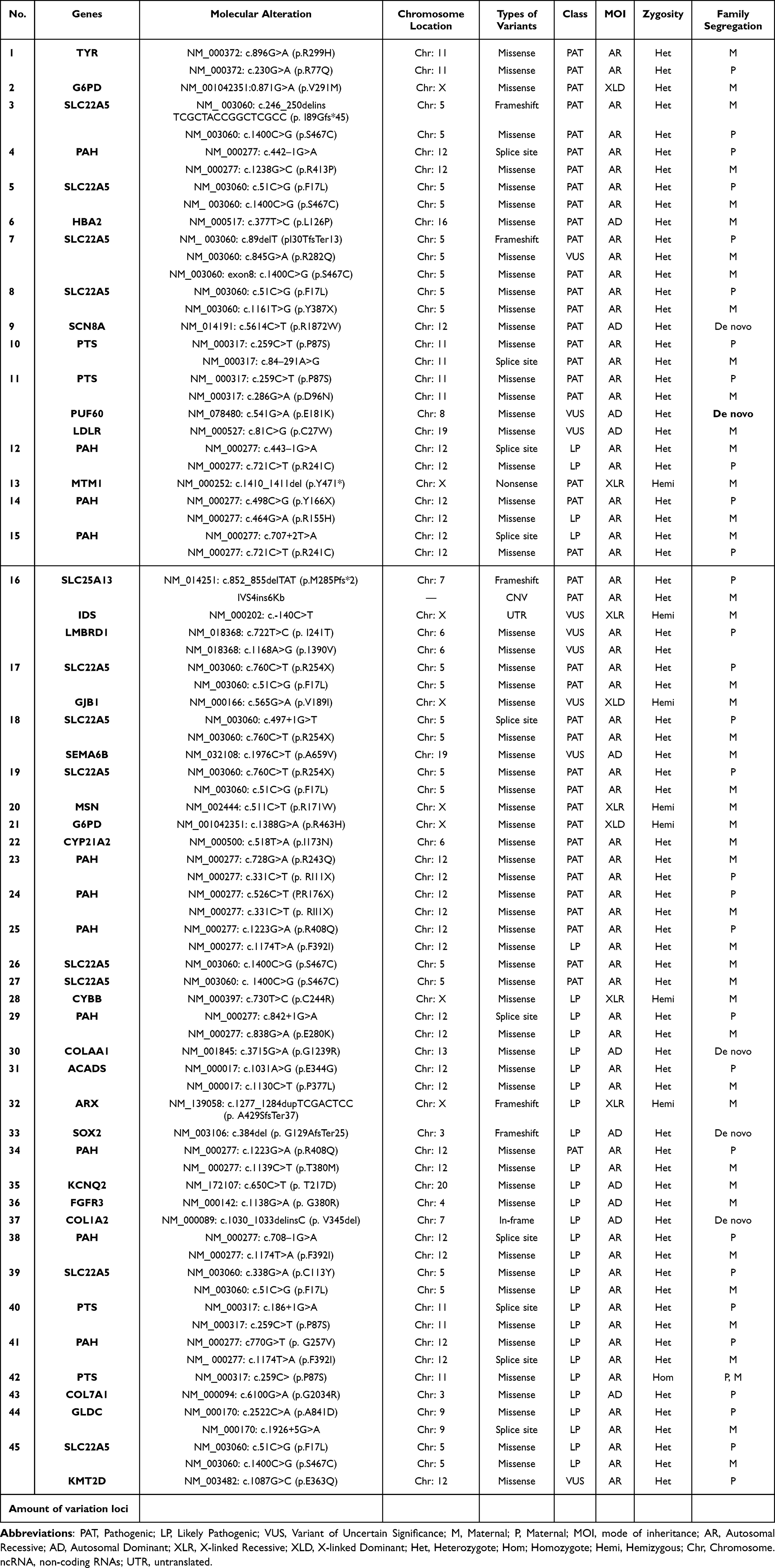 |
Table 3 Variant and Genetic Spectrum of Positive Patients in Our Cohort |
Parental samples were available for testing from 45 families with positive results. In 6% of definitive and likely diagnosed probands, the PAT/LP variant was identified as de novo within an autosomal dominant disorder gene, while 7% of standard and potentially diagnosed participants with definitive and likely diagnosed outcomes inherited from biparental in an autosomal dominant disorder gene. The majority were heterogeneous in 77% of inherited homozygous or compounded heterozygous variants within an autosomal recessive disorder gene (Figure 2). Furthermore, in X-linked recessive findings, 10% of definitive and likely diagnosed infants were inherited from maternal (Figure 2). Patients who obtained the definitive molecule harbored a total of 81 variant loci and represent a variety of molecular consequences [77% missense, 1% nonsense, 6% frameshifts, 12% disrupt splicing (at or near a canonical splice site), 1% in-frame deletions, 1% CNV, 1% UTR (untranslated)] (Figure 3). Most PAT/LP variants were non-protein truncated, containing missense variants or in-frame deletions.
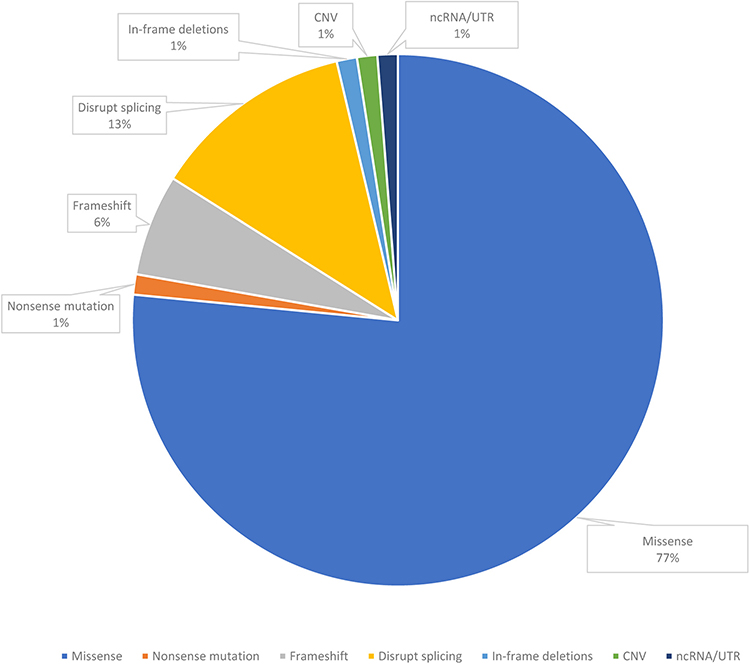 |
Figure 2 The molecular consequences of the patient with definite or likely diagnosis patients in gene defect. Abbreviations: CNV, copy number variants; UTR, untranslated. |
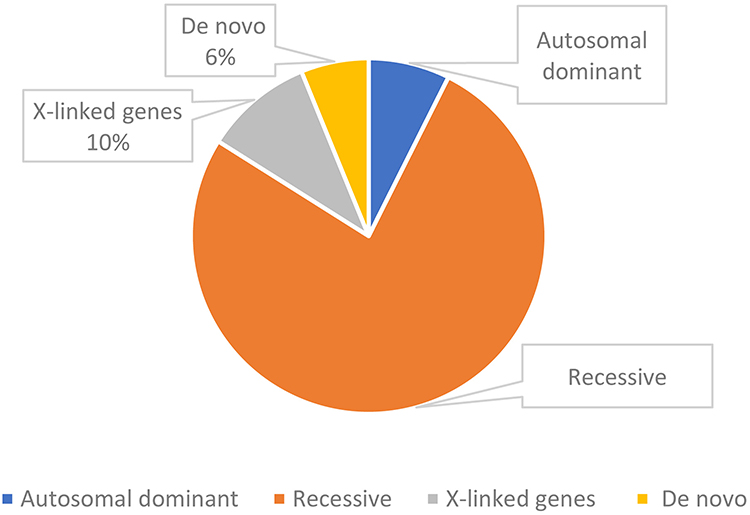 |
Figure 3 Zygosity of the identified variants of patients. |
Variations and Diseases Spectrum of MDs in Resolved Cases
Genetic sequencing was performed for 372 infants with suspected Mendelian disease, of which positive findings were obtained in 45 individuals. Among the findings of genetic testing of 372 infants with unknown genetic etiology, a total of 21 MDs within confirmed patients were detected, the top 4 of which with a relatively high proportion and accounted for 69% (n=28/45) (Table 2) including systemic primary carnitine deficiency (SLC22A5 deficiency; OMIM #212140) with 11 cases, phenylketonuria (PAH deficiency; OMIM #261600) with 11 cases, hyperphenylalaninemia (PTS deficiency; OMIM # 261640) with 4 cases, glucose-6-phosphate dehydrogenase deficiency/favism (G6PD deficiency; OMIM # 300908 /134700) with 2 cases (Table 2).
Infants with the suspected Mendelian disease of study-wide totals contain a significantly higher ratio in the neonatal period (81%, n=301) than in the non-neonatal period (19%, n=71). Of the newborns (age≦28d), 256 (85%) were from intensive care units. Similarly, for individuals with CD/LD results, a higher percentage was found in the neonatal period, 69% (n=31/45), whereas only 31% (n=14/45) during the non-neonatal period (Table 1). Among 56 deleterious variants recorded for the 45 positive patients, five novels one was detected, p.R1872W (SCN8A gene), p.E181K (PUF60 gene), p.G1239R (COLAAl gene), p.G129AfsTer25 (SOX2 gene), and p.V345del (COL1A2 gene). The following variants occurred in at least 2 positive patients (Table 2): c.760C>T (p.R254X), c.51C>G (p.F17L), c.1400C>G (p.S467C), c.1400C>G (p.S467C), c.1174T>A (p.F392I), c.1223G>A (p.R408Q), c.331C>T (p.R111X), of which the first three variants were harbored by CDSP (systemic primary carnitine deficiency, OMIM # 212140) patients and the latter one was carried by PKU (phenylketonuria, OMIM # 261600). Besides, the c.1400C > G (p. S467C) and c.51C > G (p. F17L) variants were the most frequent in these CDSP patients.
Correlation Analyses of Phenotypic Instruction and Diagnosis Yield
Available phenotypic information was classified as 14 standard subcategories of HPO terms with the proposal from trained geneticists and experienced clinicians. Correlation analyses of phenotypic instruction and diagnosis yielded that diagnostic outcome were strongly associated with multiple categories. Abnormality in metabolism/homeostasis, craniofacial/otolaryngology/ ophthalmologic, and the integument were positive predictive factors for producing diagnoses, whereas statistically significant negative indicators for abnormality in prenatal development or birth, respiratory system, neuromusculoskeletal/ limb, growth, digestive system, plus three clinical factors. Meanwhile, by clinical exome sequencing, neuromusculoskeletal/ limb abnormalities tend to acquire uncertain findings (VUS, variations of uncertain clinical significance). Moreover, three prevalent clinical features (premature birth, caesarian section, and small for gestational age) within the hospital setting were also linked to a substantially lower chance of molecular diagnoses (p<0.001, respectively) (Table 4).
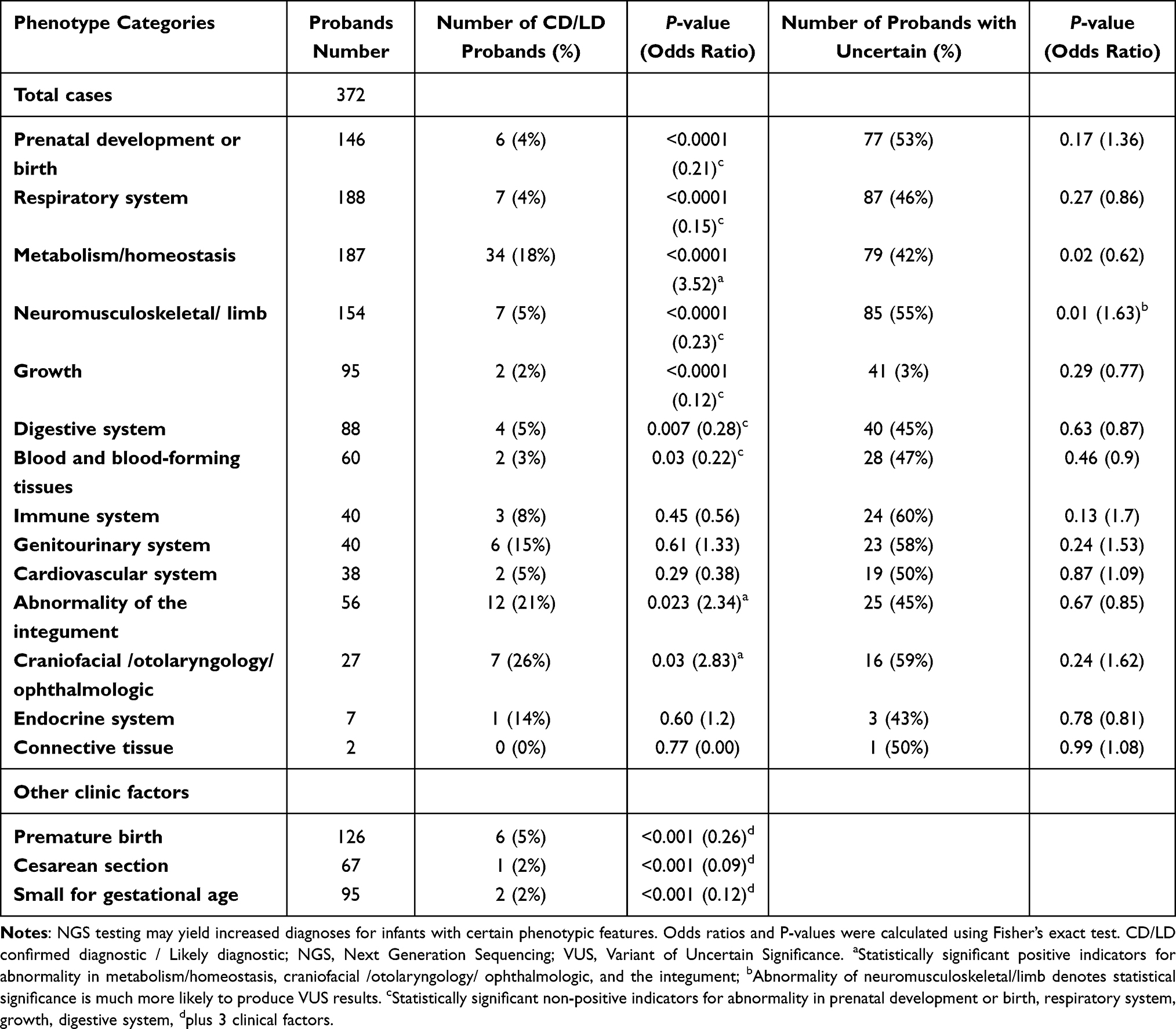 |
Table 4 Statistical Description of Clinical Information of Patients Who Underwent High-Throughput Sequencing |
Discussion
Genetic diseases resulting from alterations in gene function are usually associated with severe clinical phenotypes. A proper etiologic diagnosis is the foundation of precision medicine, and understanding the genetic elements of pre-documented individuals can help identify and improve potential treatment options. Thus, conducting immediate and accurate molecular diagnoses paramount for more effective management of these patients.9,27 However, traditional molecular genetic diagnostics based on single-gene sequencing frequently result in a negative or uncertain outcome.28–30 The potential merits of standardized and automated experimental processes in contrast to traditional sequencing include shortening the diagnostic turnaround to reduce the capital cost of sequencing significantly, accessibility to early and rational clinical treatment decisions, and the potential for providing patients with accurate treatment protocols.31 This study reported the clinical diagnostic utility of target sequencing for 372 consecutive cases with suspected Mendelian disorders, which was 12.0%. This was similar to previous positive results reported in China using the identical sequencing approach (approximately 10%).32,33 Nevertheless, it was a lower rate than that of previously reported exome/genome sequencing (ES/GS) participants for causative variants in rare disorders (25–50%).4,11–19 This discrepancy was probably attributed to variations in sample size, inclusive criteria, analytical manner, characteristics of the studied diseases, and the range of genes covered between our cohort and others. Pre-designed NGS panel only analyzed significant genes or gene regions correlated with disease or phenotype from literature and expert guidance. In contrast, the ES/GS covered fully known exome/genome genes containing newly identified sites. In other words, even if the platform had negative examination results, genetic disorders could not be excluded. Most previously reported study cohorts were rigorously screened for tangible signs of genetic diseases based on trained experts’ judgment smaller proportion of which met inclusion criteria than our study. Thus, the NGS panel’s positive detection levels correlated with the genetic etiologies’ prediction before testing. The cohort with more lenient inclusion criteria was linked to lower detection levels.34–36 Although these teams yield high diagnosis rates, it was unrealistic to expect such a process to be completed in the busy clinical setting. In comparison, we have more lenient inclusion criteria and produce low diagnosis rates for this retrospective analysis. Generally, the genome library of the NGS panel was relatively invariant for a period after design which was not overwriting the new genes discovered. New genes are continuously being uncovered, and researchers should regularly update the library’s data and elicit further genetic information, which is significant for assessing the sensitivity and accuracy of exome gene detection.
Genetic sequencing was performed for 372 infants, of which positive findings were obtained in 45 individuals, and 11 cases of CDSP patients were confirmed by DNA analysis. The pathogenic variants c.1400C > G (p. S467C) and c.51C > G (p. F17L) located in gene SLC22A5 were the most frequent in these CDSP patients in our study. This finding was consistent with a previous report of a small cohort in southern China about a high-frequency variation of CDSP,37 which perhaps reflected its high-frequency variation more commonly observed in south China. Of note, there were 26 MDs, with the two most common, whether in the non-neonatal or neonatal periods, primary carnitine deficiency (SLCC22A5 genes) and phenylketonuria (PAH genes) in all MDs of 45 diagnosed cases accounting for 48% (n=22/45). In addition, the spectrum of disease in MDs has been inconsistent with previously published genetics reported from other hospitals in other regions of China, which counted Methylmalonic acidemia (MMA) as the most prevalent MDs.38 This discrepancy might be due to the following multi-dimensional causes: (I) It perhaps reflects that the higher frequency of primary carnitine deficiency and phenylketonuria exists in the population of the Jiangxi area. (II) Meanwhile, the onset age of MDs patients varies from birth to adulthood, a broad age span, mostly in infants, yet this study was limited to infants younger than 270 days. (III) Additionally, there seems to be primary carnitine deficiency, and phenylketonuria in certain areas consideration was not taken as an observed index.
Without prompt L-carnitine supplement and untreated, SLC22A5-related CDSP can suffer from acute metabolic dysfunction in the early stage, sudden skeletal and myocardial lesions, and even sudden cardiac death in the late.30,39,40 The early diagnosis and prompt treatment, hence for patients with CDSP patients to be conducted to improve survival and symptoms with a better long-term prognosis. For example, an ordinary full-term birth weight boy (patient NO.3) whose mother had a history of spontaneous abortion, 6-month-olds, with uneventful antenatal care to neonatal periods, was admitted to our institution because of hypoglycemia and dehydration. Abnormity of free carnitine levels was discovered in the routine inspection using clinical tandem mass spectrometry with a primary presumptive diagnosis of CDSP. By NGS panel and sanger validation, complex heterozygous deleterious variants, p. I89Gfs*45 from paternal and p.S467C from maternal, in the SLC22A5 gene, were detected. Immediately, he started medical treatment with L-carnitine after being diagnosed. After seven days of regular systemic therapy and after symptoms improved significantly, he was discharged. During followed up regular periods of 1 year, no weakness was observed, and he continued to be healthy during carnitine therapy.
In the study, 81% (n=301/372) of participants exhibited biochemical/clinical abnormalities in the neonatal period. Of them, 256 (85%) individuals were from intensive care units. These findings corroborated a prior study41 that the non-typical and unrecognized presentations of MDs were found in some young babies, challenging conventional first-line testing strategies in the NICU. The phenotype matching and pathogenicity interpretation in the genetic analysis were primarily performed manually by our multidisciplinary team, which was costly and time-consuming for the diagnostics of RDs. Nevertheless, the current clinical requirements vastly outstripped the number of trained geneticists in this domain, and the demand is expected to grow as sequencing technologies develop and costs diminish. New automated algorithms hold the potential to substantially streamline and expedite the phenotype matching and variant interpretation process by aligning forecast approaches with the increasing genetics expertise.42–44 Finally, real-time updating and expansion of the internal database were crucial for the correct diagnosis of genetic disorders. Compared to pediatric patients, the non-specific and unrecognized manifestation in the neonatal stage has challenged automatic interpretation based on phenotype.45 Future endeavors to maximize the intuitive understanding of test results and shorten diagnostic cycles could indirectly minimize the expense of sequencing. The charge for a case was roughly $380, including equipment and consumables in the lab ($160), bioinformatics analysis, and labor (approximately $220). In comparison, the mean fee of applying other single-gene diagnostic technologies for a patient was $480 [such as MLPA/Single-gene sequencing /Real-time Fluorescence Quantitative Polymerase Chain Reaction (qPCR), plus the labor costs]. Therefore, the cost of CFES for a case is lower than the average costs of other single-gene diagnostic technologies. Moreover, genetic tests are not covered by health insurance in China, meaning these costs are self-financed. In this context, this test is more affordable and reasonably cost-effective for a one-stop screening test to either pinpoint a specific disease or rule out most of the known genetic causes for conditions. Although more comprehensive, the application of ES or GS in routine testing will likely remain to be assessed and investigated, largely restricted principally by its prohibitive cost. Our team could afford this high-quality and economical detection by confining to a proband-only sequencing method and analyzing genes with undisputed clinical significance and correlation to newborns and young infants. In this case, the AI-assisted CFES should be considered a first-line diagnostic approach for young infants with suspected MDs to identify and exclude known genetic etiologies with good economic benefits.
Our team sorted probands into 14 upper phenotype categories based on 278 presupposed HPO terms, and the ratio of the finding of different categories were compared in propositus with and without specific phenotypic characteristics. This study showed that infants who exhibited abnormalities in metabolism/homeostasis, craniofacial/ otolaryngology/ ophthalmologic, and integument were approximately 3.5, 2.8, and 2.3 times, respectively, which are more prone to produce a positive outcome than infants who did not exhibit these phenotypic attributes. Meanwhile, it reflected that Mendelian etiology is often the foundation for such manifestation, and infants with those phenotypic features were more likely to be diagnosed. In comparison, infants with neuromusculoskeletal/ limb were 1.6 times more likely to produce an uncertain outcome, probably revealing the truth that some infantile neurologic signatures fail to be observed in the clinical setting.46 A relatively higher diagnostic yield of infants with combined metabolic/homeostatic abnormalities is revealed in this cohort. This might be since more referrals are from endocrinology and the smaller sample size of our study. Abnormalities in the metabolism/homeostasis accounted for a considerable portion of the pediatric ward burden. Some biomarkers of inborn errors of metabolism (IEMs) are non-specifically variable, challenging traditional diagnostic tools. The NGS panel covering the target genomic region has a more reliable etiological diagnosis.
The molecular diagnostics made by the CFES immediately influenced clinical treatment and prognosis for resolved patients after the report outcome was available. Terms of this immediate impact can be classified into four main aspects: (I) Diet and lifestyle management. Firstly, a specially formulated diet was administered to 67% (n=30/45) of patients with G6PD, SLC25A13, ACADS, SLC22A5, PTS, and PAH gene defects. Here, we reported a 21-day-old asymptomatic female diagnosed with Short-chain acyl-CoA dehydrogenase deficiency (SCADD) by newborn screening. Tandem mass spectrometry screening for IEMs revealed an elevated butyrylcarnitine (C4) concentration of 1.427 µmol/L in the initial test 1.281µmol/L was Still elevated in the re-examination. Furtherly, molecular diagnostics with c.1031A>G (p.E344G) and c.1130C>T (p.P377L) on chromosome 12 were received by CFES. So, immediate initiation of treatment by limiting fat intake, avoiding hunger, and appropriate supplementation of carnitine or vitamin B2 as a prophylactic measure for this patient with a genetic defect in ACADS and monitoring this patient’s condition. During follow-up, she remains symptomless with metabolic disease. This child is growing up without health-related quality of life compromised by symptoms such as seizures and hypotonia. (II) Implementation of targeted medication. For these patients, the confirmatory molecular diagnosis made it possible to select a more targeted drug therapy in 20% (n=9/45) of patients in whom the detection made the diagnosis of PAT/LP variants in CYP21A2, GLDC, KCNQ2, ARX, SOX2, MTM1, MSN, CYBB, and SCN8A gene defects. (III) Application of medical techniques and clinical procedures, including organ or tissue transplantation, surgical removal or repair, and surgical correction of deformities. In our cohort, three patients with FGFR3, COL1A2, and COLAA1 gene variants, respectively, had a severe illness trajectory, underwent surgical procedures, and were admitted to the NICU; Hemopoietic stem cell transplantation (HSCT) was conducted in one patient with PAT/LP variants of HBA2 gene defects. (IV) Practical aspects and prospects of gene therapy in MDs. Recent advances in gene therapy were expected for a patient with RDEB (recessive dystrophic epidermolysis bullosa) in our study to correct COL7A1 gene defects.47 This can potentially help patients with RDEB to reduce genetic defects and gain an improved quality of life. However, there also is no effective treatment for certain diseases caused by congenital disabilities. Early identification of genetic defects and timely treatment by the above approaches also was a profound clinical meaning in preventing disease progression. Such as, a patient with P/LP variants of TYR gene defect was confirmed by genetic sequencing in our cohort who avoided intense sunlight radiation and wore sunscreens to protect against skin aging and lesions. Collectively, a precise molecular diagnosis of patients enables enhanced recognition in clinicians to furnish more comprehensive and practical medical management strategies for therapy and prognosis.
Our research also has some restrictions that should be regarded. First, the sample size of the current study is too small. Furthermore, the extent to which the findings can be generalized to a general population calls for future population-based research with larger samples. As a result, a subgroup analysis based on different characteristics of patients was not conducted. Second, this single-center study involved patients aged between 1 day and 270 days who were enrolled from pediatric inpatient in our region, and the risk for selection bias would appear owing to the demographics of our geographical site. Finally, concerning the retrospective study, some cases may develop other genetic diseases after NGS during our data collection period, which tends to skew the results.
Conclusion
Conclusively, CFES as a potentially effective diagnostic strategy for 1st-line testing of newborns or young children at high risk for MDs could enable a cost-effective molecular diagnostic that could benefit patients in clinically under-resourced settings, especially those with poor economic status. Moreover, with the development of artificial intelligence technology and medical advancement, the decreasing cost of exome sequencing will continue. It will gradually become a routine test for patients with suspected monogenic genetic diseases in clinical settings. However, realizing the advantages of CFES in the future would also require highly coordinated clinical and laboratory turnaround conditions, such as timely patient identification and sophisticated evaluation systems to prevent the selection of inappropriate genetic test ordering and “misinterpretation” of results.
Declaration
This study complies with the Declaration of Helsinki.
Data Sharing Statement
The original contributions presented in the study are included in the article; further inquiries can be directed to the corresponding author.
Funding
This study is supported by the Superior scientific and technological innovation team of Jiangxi Province (No. 20181BCB24014). Moreover, this study received funding from Zhengzhou Key Laboratory of Antitumor Traditional Chinese Medicine Research.
Disclosure
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Boycott KM, Vanstone MR, Bulman DE, MacKenzie AE. Rare-disease genetics in the era of next-generation sequencing: discovery to translation. Nat Rev Genet. 2013;14(10):681–691. PMID: 23999272. doi:10.1038/nrg3555
2. Amberger JS, Bocchini CA, Schiettecatte F, Scott AF, Hamosh A. OMIM.org: Online Mendelian Inheritance in Man (OMIM), an Online catalog of human genes and genetic disorders. Nucleic Acids Res. 2014;43:1. doi:10.1093/nar/gku1303
3. Haijes HA, Molema F, Langeveld M, et al. Retrospective evaluation of the Dutch pre-newborn screening cohort for propionic acidemia and isolated methylmalonic acidemia: what to AIM, expect, and evaluate from newborn screening? J Inherit Metab Dis. 2020;43:3. doi:10.1002/jimd.12193
4. Farwell KD, Shahmirzadi L, El-Khechen D, et al. Enhanced utility of family-centered diagnostic exome sequencing with inheritance model-based analysis: results from 500 unselected families with undiagnosed genetic conditions. Genet Med. 2015;17(7):578–586. PMID: 25356970. doi:10.1038/gim.2014.154
5. Krumm N, Sudmant PH, Ko A, et al. Copy number variation detection and genotyping from exome sequence data. Genome Res. 2012;22(8):1525–1532. PMID: 22585873; PMCID: PMCPMC3409265. doi:10.1101/gr.138115.112
6. Retterer K, Juusola J, Cho MT, et al. Clinical application of whole-exome sequencing across clinical indications. Genet Med. 2016;18(7):696–704. PMID: 26633542. doi:10.1038/gim.2015.148
7. Rossi M, El-Khechen D, Black MH, Farwell Hagman KD, Tang S, Powis Z. Outcomes of diagnostic exome sequencing in patients with diagnosed or suspected autism spectrum disorders. Pediatr Neurol. 2017;70:34–43.e2. PMID: 28330790. doi:10.1016/j.pediatrneurol.2017.01.033
8. Tong F, Wang J, Xiao R, et al. application of next generation sequencing in the screening of monogenic diseases in China, 2021: a consensus among Chinese newborn screening experts. World J Pediatr. 2022;18(4):235–242. PMID: 35292922. doi:10.1007/s12519-022-00522-8
9. Taylor JC, Martin HC, Lise S, et al. Factors influencing success of clinical genome sequencing across a broad spectrum of disorders. Nat Genet. 2015;47(7):717–726. PMID: 25985138; PMCID: PMCPMC4601524. doi:10.1038/ng.3304
10. Lionel AC, Costain G, Monfared N, et al. Improved diagnostic yield compared with targeted gene sequencing panels suggests a role for whole-genome sequencing as a first-tier genetic test. Genet Med. 2018;20(4):435–443. doi:10.1038/gim.2017.119
11. Makrythanasis P, Nelis M, Santoni FA, et al. Diagnostic exome sequencing to elucidate the genetic basis of likely recessive disorders in consanguineous families. Hum Mutat. 2014;35(10):1203–1210. PMID: 25044680. doi:10.1002/humu.22617.
12. Bureau A, Parker MM, Ruczinski I, et al. Whole exome sequencing of distant relatives in multiplex families implicates rare variants in candidate genes for oral clefts. Genetics. 2014;197(3):1039–1044. PMID: 24793288; PMCID: PMCPMC4096358. doi:10.1534/genetics.114.165225
13. Fahiminiya S, Almuriekhi M, Nawaz Z, et al. Whole exome sequencing unravels disease-causing genes in consanguineous families in Qatar. Clin Genet. 2014;86(2):134–141. PMID: 24102521. doi:10.1111/cge.12280
14. Lee H, Deignan JL, Dorrani N, et al. Clinical exome sequencing for genetic identification of rare Mendelian disorders. JAMA. 2014;312(18):1880–1887. PMID: 25326637; PMCID: PMCPMC4278636. doi:10.1001/jama.2014.14604
15. Yang Y, Muzny DM, Xia F, et al. Molecular findings among patients referred for clinical whole-exome sequencing. JAMA. 2014;312(18):1870–1879. PMID: 25326635; PMCID: PMCPMC4326249. doi:10.1001/jama.2014.14601
16. Atwal PS, Brennan ML, Cox R, et al. Clinical whole-exome sequencing: are we there yet? Genet Med. 2014;16(9):717–719. PMID: 24525916. doi:10.1038/gim.2014.10
17. Yang Y, Muzny DM, Reid JG, et al. Clinical whole-exome sequencing for the diagnosis of Mendelian disorders. N Engl J Med. 2013;369(16):1502–1511. PMID: 24088041; PMCID: PMCPMC4211433. doi:10.1056/NEJMoa1306555
18. Alfares A, Alfadhel M, Wani T, et al. A multicenter clinical exome study in unselected cohorts from a consanguineous population of Saudi Arabia demonstrated a high diagnostic yield. Mol Genet Metab. 2017;121(2):91–95. doi:10.1016/j.ymgme.2017.04.002
19. Al-Dewik N, Mohd H, Al-Mureikhi M, et al. Clinical exome sequencing in 509 middle eastern families with suspected Mendelian diseases: the Qatari experience. Am J Med Genet A. 2019;179(6):927–935. PMID: 30919572; PMCID: PMCPMC6916397. doi:10.1002/ajmg.a.61126
20. Groza T, Köhler S, Doelken S, et al. Automatic concept recognition using the human phenotype ontology reference and test suite corpora. Database. 2015;2015:bav005–bav005. PMID: 25725061; PMCID: PMCPMC4343077. doi:10.1093/database/bav005
21. Li H, Handsaker B, Wysoker A, et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009;25(16):2078–2079. PMID: 19505943; PMCID: PMCPMC2723002. doi:10.1093/bioinformatics/btp352
22. Cunningham F, Pritchard B, Rios D, Chen Y, Flicek P, Cunningham F. Deriving the consequences of genomic variants with the Ensembl API and SNP effect predictor. Bioinformatics. 2010;26(16):2069–2070. doi:10.1093/bioinformatics/btq330
23. Wang K, Li M, Hakonarson H, Wang K, Li M, Hakonarson H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nuc Acids Res. 2010;38(16):e164. doi:10.1093/nar/gkq603
24. Mckusick VA. Mendelian inheritance in man and its online version, OMIM. Am J Hum Genet. 2007;80(4):588–604. doi:10.1086/514346
25. Richards S, Aziz N, Bale S, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17(5):405–424. PMID: 25741868; PMCID: PMCPMC4544753. doi:10.1038/gim.2015.30
26. Whiffin N, Minikel E, Walsh R, et al. Using high-resolution variant frequencies to empower clinical genome interpretation. Genet Med. 2017;19(10):1151–1158. PMID: 28518168; PMCID: PMCPMC5563454. doi:10.1038/gim.2017.26
27. Gyngell C, Newson AJ, Wilkinson D, Stark Z, Savulescu J. Rapid challenges: ethics and genomic neonatal intensive care. Pediatrics. 2019;143:S14–s21. PMID: 30600266; PMCID: PMCPMC6379057. doi:10.1542/peds.2018-1099D
28. Yoon PW. Contribution of birth defects and genetic diseases to pediatric hospitalizations. Arch Pediatr Adolesc Med. 1997;151(11):1096. doi:10.1001/archpedi.1997.02170480026004
29. McCandless SE, Brunger JW, Cassidy SB. The burden of genetic disease on inpatient care in a children’s hospital. Am J Hum Genet. 2004;2004:1.
30. Wang Y, Kelly MA, Cowan TM, Longo N. A missense mutation in the OCTN2 gene associated with residual carnitine transport activity. Hum Mutat. 2000;15(3):238–245. PMID: 10679939. doi:10.1002/(sici)1098-1004(200003)15:3<238::aid-humu4>3.0.Co;2-3
31. Hess JF, Kohl TA, Kotrová M, et al. Library preparation for next generation sequencing: a review of automation strategies. Biotechnol Adv. 2020;41:107537. PMID: 32199980. doi:10.1016/j.biotechadv.2020.107537
32. Yang L, Wei Z, Chen X, et al. Use of medical exome sequencing for identification of underlying genetic defects in NICU: experience in a cohort of 2303 neonates in China. Clin Genet. 2022;101(1):101–109. PMID: 34671977. doi:10.1111/cge.14075
33. Hu X, Li N, Xu Y, et al. Proband-only medical exome sequencing as a cost-effective first-tier genetic diagnostic test for patients without prior molecular tests and clinical diagnosis in a developing country: the China experience. Genet Med. 2018;20(9):1045–1053. PMID: 29095814. doi:10.1038/gim.2017.195
34. French CE, Delon I, Dolling H, et al. Whole genome sequencing reveals that genetic conditions are frequent in intensively ill children. Intensive Care Med. 2019;45(5):627–636. PMID: 30847515; PMCID: PMCPMC6483967. doi:10.1007/s00134-019-05552-x
35. Gubbels CS, VanNoy GE, Madden JA, et al. Prospective, phenotype-driven selection of critically ill neonates for rapid exome sequencing is associated with high diagnostic yield. Genet Med. 2020;22(4):736–744. PMID: 31780822; PMCID: PMCPMC7127968. doi:10.1038/s41436-019-0708-6
36. Kingsmore SF, Cakici JA, Clark MM, et al. A randomized, controlled trial of the analytic and diagnostic performance of singleton and trio, rapid genome and exome sequencing in ill infants. Am J Hum Genet. 2019;105(4):719–733. PMID: 31564432; PMCID: PMCPMC6817534. doi:10.1016/j.ajhg.2019.08.009
37. Yang X, Li Q, Wang F, et al. Newborn screening and genetic analysis identify six novel genetic variants for primary carnitine deficiency in Ningbo Area, China. Front Genet. 2021;12:686137. PMID: 34249102; PMCID: PMCPMC8264545. doi:10.3389/fgene.2021.686137
38. Hong S, Wang L, Zhao D, et al. Clinical utility in infants with suspected monogenic conditions through next-generation sequencing. Mol Genet Genomic Med. 2019;7(6):e684. PMID: 30968598; PMCID: PMCPMC6565546. doi:10.1002/mgg3.684
39. Rose EC, Di San Filippo CA, Ndukwe Erlingsson UC, Ardon O, Pasquali M, Longo N. Genotype-phenotype correlation in primary carnitine deficiency. Hum Mutat. 2012;33(1):118–123. doi:10.1002/humu.21607
40. Longo N, Frigeni M, Pasquali M. Carnitine transport and fatty acid oxidation. Biochim Biophys Acta. 2016;1863(10):2422–2435. PMID: 26828774; PMCID: PMCPMC4967041. doi:10.1016/j.bbamcr.2016.01.023
41. Meng L, Pammi M, Saronwala A, et al. Use of exome sequencing for infants in intensive care units: ascertainment of severe single-gene disorders and effect on medical management. JAMA Pediatr. 2017;171(12):e173438–e. doi:10.1001/jamapediatrics.2017.3438
42. Vega FMDL, Chowdhury S, Moore B, Frise E, Kingsmore SF. Artificial intelligence enables comprehensive genome interpretation and nomination of candidate diagnoses for rare genetic diseases. Genome Med. 2021;13:1. doi:10.1186/s13073-020-00808-4
43. Daniela H, Markus S, Ellen K, et al. MutationDistiller: user-driven identification of pathogenic DNA variants. Nuc Acids Res. 2019;2019(W1):W1.
44. Clark MM, Hildreth A, Batalov S, et al. Diagnosis of genetic diseases in seriously ill children by rapid whole-genome sequencing and automated phenotyping and interpretation. Sci Transl Med. 2019;11(489):eaat6177. doi:10.1126/scitranslmed.aat6177
45. Wise AL, Manolio TA, Mensah GA, et al. Genomic medicine for undiagnosed diseases. Lancet. 2019;394(10197):533–540. PMID: 31395441; PMCID: PMCPMC6709871. doi:10.1016/s0140-6736(19)31274-7
46. Ceyhan-Birsoy O, Murry JB, Machini K, et al. Interpretation of genomic sequencing results in healthy and ill newborns: results from the babyseq project. Am J Hum Genet. 2019;104(1):76–93. PMID: 30609409; PMCID: PMCPMC6323417. doi:10.1016/j.ajhg.2018.11.016
47. Osborn MJ, Newby GA, McElroy AN, et al. Base editor correction of COL7A1 in recessive dystrophic epidermolysis bullosa patient-derived fibroblasts and iPSCs. J Invest Dermatol. 2020;140(2):338–47.e5. PMID: 31437443; PMCID: PMCPMC6983342. doi:10.1016/j.jid.2019.07.701
 © 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.