Back to Journals » International Medical Case Reports Journal » Volume 13
A Rare Mutation in the MARVELD2 Gene Can Cause Nonsyndromic Hearing Loss
Authors Sadeghi Z ![]() , Chavoshi Tarzjani SP, Miri Moosavi RS, Saber S, Ebrahimi A
, Chavoshi Tarzjani SP, Miri Moosavi RS, Saber S, Ebrahimi A
Received 18 April 2020
Accepted for publication 10 July 2020
Published 27 July 2020 Volume 2020:13 Pages 291—296
DOI https://doi.org/10.2147/IMCRJ.S257654
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Ronald Prineas
Zahra Sadeghi,1,2 Seyedeh Parisa Chavoshi Tarzjani,1 Reyhaneh Sadat Miri Moosavi,2 Siamak Saber,2 Ahmad Ebrahimi2
1Department of Genetics, Tehran-North Branch, Islamic Azad University, Tehran, Iran; 2Jordan Medical and Genetic Laboratory, Tehran, Iran
Correspondence: Ahmad Ebrahimi
Jordan Medical and Genetic Laboratory, Tehran, Iran
Tel +98-912-4581801
Email [email protected]
Abstract: The MARVELD2 gene which is located on the 5q13.2 may cause nonsyndromic hearing loss (NSHL) with autosomal recessive inherited pattern. So far c.1331+1G>A (IVS4+1G>A); NM_001038603.3, variant in deafness, has only reported previously in one Pakistani family in 2008 and it is reported for the first time in Iran and second time in the world. The case is a 21-year-old Iranian woman who has NSHL referred for genetic consultation, and her parents had a consanguineous marriage. To study the responsible genes for the mentioned disorder, whole exome sequencing (WES) was performed for the case. The result of WES analysis revealed a transition at the splice donor variant site of the MARVELD2 gene. The NGS result was confirmed by Sanger sequencing.
Keywords: MARVELD2 gene, nonsyndromic hearing loss, whole exome sequencing
Introduction
Nonsyndromic hearing loss (NSHL) is one of the most common congenital impairments and a genetically extremely heterogeneous disease.1,2 More than 70% of hereditary deafness is nonsyndromic.3 Recent studies have shown that the frequency of hearing loss in Iran was estimated to be two to three times higher compared with other populations of the world.4 As it is considered a potential problem in developing countries, early diagnosis might be highly recommended.5 Consanguineous marriage is prevalent in Iran, and it has been shown that 61.54% of parents with deaf children experienced consanguineous marriage.6 The prevalence of bilateral sensory deafness was 62.90% in seven cities in Iran. Consanguineous marriages are associated with a higher incidence of sensorineural deafness.7,8 Genetic factors play a key role in nonsyndromic hearing impairment (NSHI) and more than 140 genes have been identified to be engaged in deafness.9 It has been clearly shown that different loci might be responsible for deafness. Genetic heterogeneity of deafness might be transferred either autosomal recessive (AR) loci or by engagement of several loci which results in hearing loss.10,11
At least 140 genes9 and 150 loci have been discovered to play key roles in deafness; 58 loci for autosomal dominant (AD) genes, 87 loci for autosomal recessive genes, and six loci for X-linked genes.12 Mutations in the MARVELD2 gene which may lead to NSHL, autosomal recessive inheritance, is at the DFNB49 locus.1,13 MARVELD2 encodes tricellulin and is located on chromosome 5q13.2.1 Tricellulin is a tricellulin tight-junction (tTJ) protein in the epithelial cells. The inner ear epithelial cells has several tight junction proteins.5,14 Due to the presence of many ethnic groups and the high frequency of consanguineous marriages, the Iranian population is a valuable genetic resource to study NSHL with autosomal recessive inheritance.5
In the present study, we reported a case of nonsyndromic hearing loss with a previously reported rare mutation in the MARVELD2 gene which was evaluated by Next Generation Sequencing (NGS).
Materials and Methods
Case Presentation
A 21-year-old Iranian woman with deafness was referred to the Jordan Medical and Genetics lab for premarital genetic counseling with an unrelated affected man (Figure 1). Her parents had a consanguineous marriage with no deafness, but her brother was affected with NSHL.
 |
Figure 1 Pedigree of two families with nonsyndromic hearing loss (autosomal recessive pattern). Filled symbols represent deafness individuals. Clear symbols represent no-deafness individuals. The arrow shows Proband. Dialognal line indicates deceased mal and deseased female. |
A second family is a 23-year-old man with deafness, whose parents did not have any sensorial problems, but his step-brother also suffered from deafness. All audio-metric parameters confirmed NSHL in all mentioned cases.
We confirm that all participants gave informed consent with the genetic examination as they were referred to the genetic lab for genetic consultation. Also we confirm that all participants provided written informed consent to have their case details, and any accompanying images, published. Ethical approval was not required.
Investigations
A peripheral blood sample was taken from this case. Extracted DNA was sent to one of the companies which are service providers for next generation sequencing and have extended this portfolio by using various leading NGS sequencing technologies. The quality and quantity of each sample was defined by various and suitable methods such as agarose gel analysis, Qubit® Fluorometer, NanoDrop, and Agilent 2100 Bioanalyzer. For this patient, whole exome sequencing (WES), to access the majority of susceptible variants genes, was ordered and performed with an Illumina HiSeq 2500 machine (with SureSelect version6 kit) for ultra-deep sequencing with huge data output. After receiving row data as a fastq, FASTQC software was used for evaluation quality control analysis of read data by the company. The bioinformatics pipeline included Mapping of reads to human genome reference sequence using BWA-MEM, before accurate and annotated insertions-deletions (InDels) and single nucleotide polymorphisms (SNP) detection reports in Variant Calling Format (VCF) format. So SNP and InDel detection, based on the mapping results (VCF format) and annotation of all variants, was compared to known SNP lists (e.g. dbSNP, Clinvar), Using an online genome browser for interactive visualization of read mapping and variants, target coverage statistics were prepared for our Laboratory to analyze. After the data was received and analyzed, Sanger sequencing was used due to segregation in her family for evaluating founded variants. Peripheral blood samples were taken from related individuals and segregation analysis with Sanger sequencing was performed to confirm variants for all mentioned persons; II-5, III-2, III-5, III-6, III-8 (Figure 1).
Primer pairs were used to amplify the mentioned mutation (c.1331+1G>A) in the MARVELD2 gene. The sequence of designed primers was as follows: 5ʹ-CACCTGATCTTCTTCCTC-3ʹ as a forward primer and 5ʹ-GGGAAATCAGCTTATCTTAT-3ʹ as a reverse primer.
Results
We achieved annotated files from the European company (Testing and Laboratory Services) with one branch in India. The annotated file which had been sent was reported in two sheets. One of them was for SNPs and the other for InDels. According to the articles, two protocols were suggested to analyze in these cases. Data were sorted by a panel of deafness genes. The generated data was analyzed with the most well known pathogenic Nonsyndromic Hearing Loss (NSHL) genes; ABHD12, ACTG1, MSRB3, CCDC50, CEACAM16, CIB2, PCDH15, COL4A5, DFNA5, DFNB59, SLC17A8, EYA1, FGF3, FOXC1, TMPRSS3, GPSM2, GRXCR1, HGF, GJB2, KCNQ4, LOXHD1, MARVELD2, KCNE1, MYO1A, MYO6, OTOA, MYH14, RDX, SALL1, SERPINB6, PITX2, SMPX, TIMM8A, TMC1, SLC26A4, USH1G, WFS1, ADGRV1, TPRN, CDH23, ABHD5, CISD2, BSND, CRYM, CHD7, DIABLO, COCH, EYA4, DFNB31, FOXI1, DSPP, GRHL2, FGFR3, HSD17B4, GJB3, LHFPL5, HARS2, MASP1, KCNJ10, MYO3A, LRTOMT, OTOF, MYH9, RPS6KA3, MYO7A, ARSB, POU3F4, TECTA, SALL4, CLDN14, SLC26A5, USH2A, TJP2, DIAPH1, TRIOBP, KCNQ1, WHRN, GIPC3, BTD, MYO15A, ATP6V1B1, ILDR1, COL11A2, PRPS1, CLRN1, SIX1, ESRRB, SLC29A3, DNMT1, TMIE, GJB6, USH1C.
Some variants were filtered according to their functions (exonic/non-exonic). Exonic and splicing sites were selected and Synonymous were deleted. To have a wide evaluation and prevent data missing, we filtered frequencies of variants based on 1000 Genome <0.05 in spite of the fact that <0.01 should have been used. The data were analyzed and allele coverage, allele frequency, allele ratio, variant impact, and clinical significance were calculated. Data were checked by various databases including dbSNP, ClinVar, and also predictive databases such as SIFT, PolyPhen, MutationTaster, FATHMM, CADD, and Varsome scores. We should add that the predictive data defined was based on quality (etc. deleterious, non-deleterious, polymorphism, benign) and we could assess according to the quality.
The results analysis revealed a very rare mutation (c.1331+1G>A; rs762352115) in a splice donor site in the MARVELD2 gene. According to the bioinformatics analysis and annotation with databases, this variant was predicted as pathogenic with related MAF=0.00002. This mutation is reported for the first time in one Iranian family (Table 1).
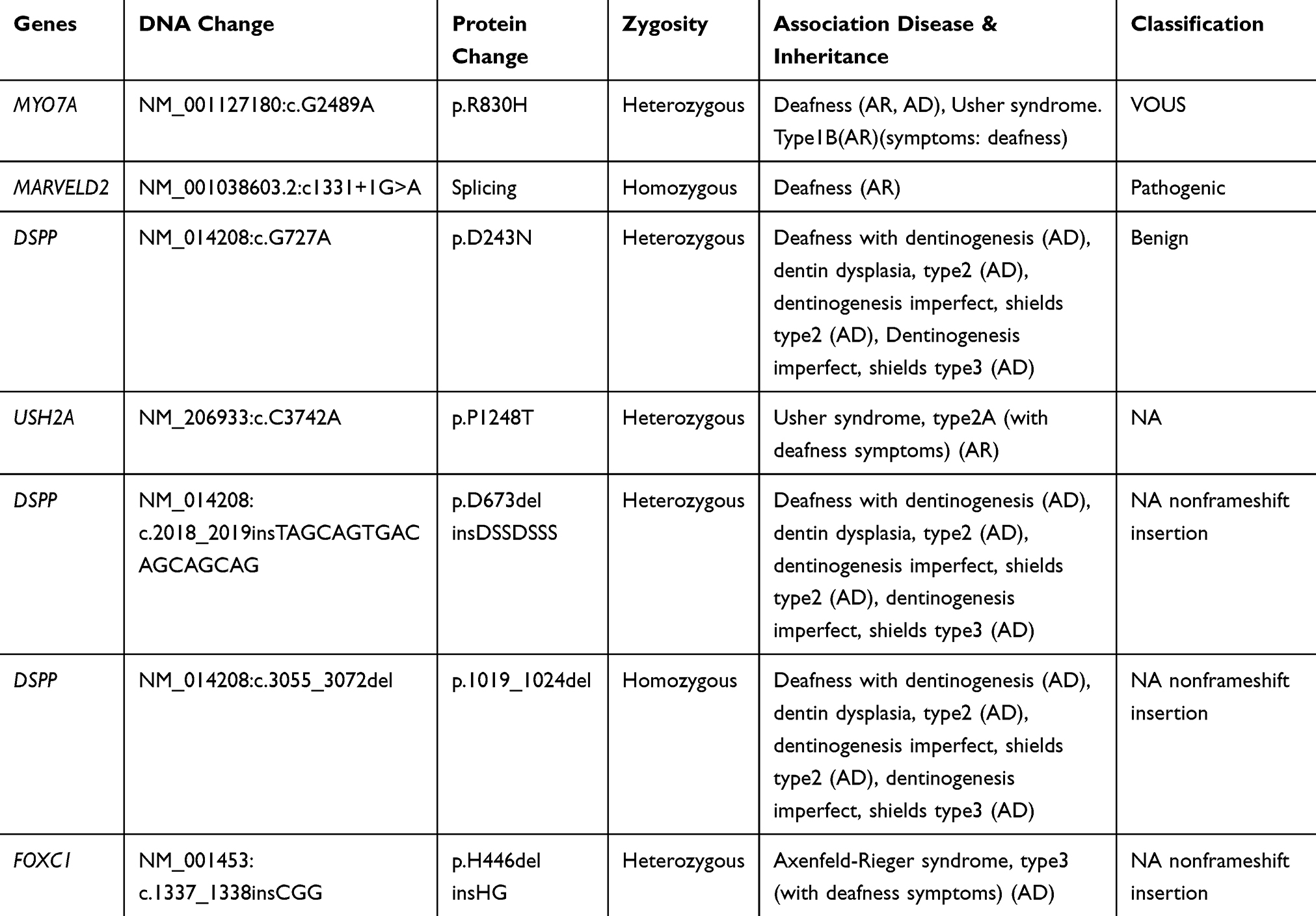 |
Table 1 All Changes in the Case Which Were Revealed by Next Generation Sequencing |
The proband; who is indicated in the pedigree (III-6), and her brother (III-8), were homozygote for mentioned mutation (c.1331+1G>A). Her mother, who has a consanguine marriage (II-5), was heterozygote for this variant. Other deaf patients from the second family (III-2 and III-5) were homo-normal about the mentioned mutation (Figures 1 and 2). The Sanger results are shown in Figure 2.
 |
Figure 2 The results of Sanger sequencing of MARVELD2 gene mutation. Sequence analysis shows homo-mutant (A), hetero-mutant (B), and homo-normal (C) individuals of families. The splice-site mutation (c. 1331 + 1G > A) was found in the case and her family. |
The segregation screening revealed that the mentioned mutation (c.1331+1G>A) was seen only in the case (III-6) and two members of her family; (II-5) as a heterozygote and (III-8) who was affected as well.
Discussion
Nonsyndromic hearing loss is a common sensorineural disorder.15 More than 140 genes are known to cause hearing loss.9 NSHL is genetically heterogeneous, but in almost all cases has autosomal recessive inheritance.13 The MARVELD2 gene is an integral membrane protein, which contains seven exons and 558 amino acids.5,11 The human MARVELD2 gene at the DFNB49 locus, which has been shown to cause NSHL in different studies, is located on chromosome 5q13.2 (Chromosome 5: 68,710,939–68,740,157).5,16
Tight junctions (TJ) are composed of networks and many different proteins, and prevent leakage of transported solutes in epithelial cells.17 One of the integral membrane proteins; MARVELD2 or Tricellulin (TRIC), is concentrated in TJ strands of inner-ear epithelia.18
TRIC is necessary to form the epithelial barrier. Suppression of mouse TRIC showed disorganized tricellular contacts.17 Homozygous mutation in TRIC (MARVELD2; 610,572) causes deafness-49 with an autosomal recessive pattern (DFNB49).19
We evaluated a patient with NSHL by a panel of deafness with NGS and found a very rare mutation in the MARVELD2 gene (c.1331+1G>A). There was just one previous population in the world (Pakistani families) that had reported this mutation related to NSHL, and there were no other reports worldwide. Some other pathogenic variants of the MARVELD2 gene have been reported in different countries (Table 2).3,5,13,15,18,20,21,22
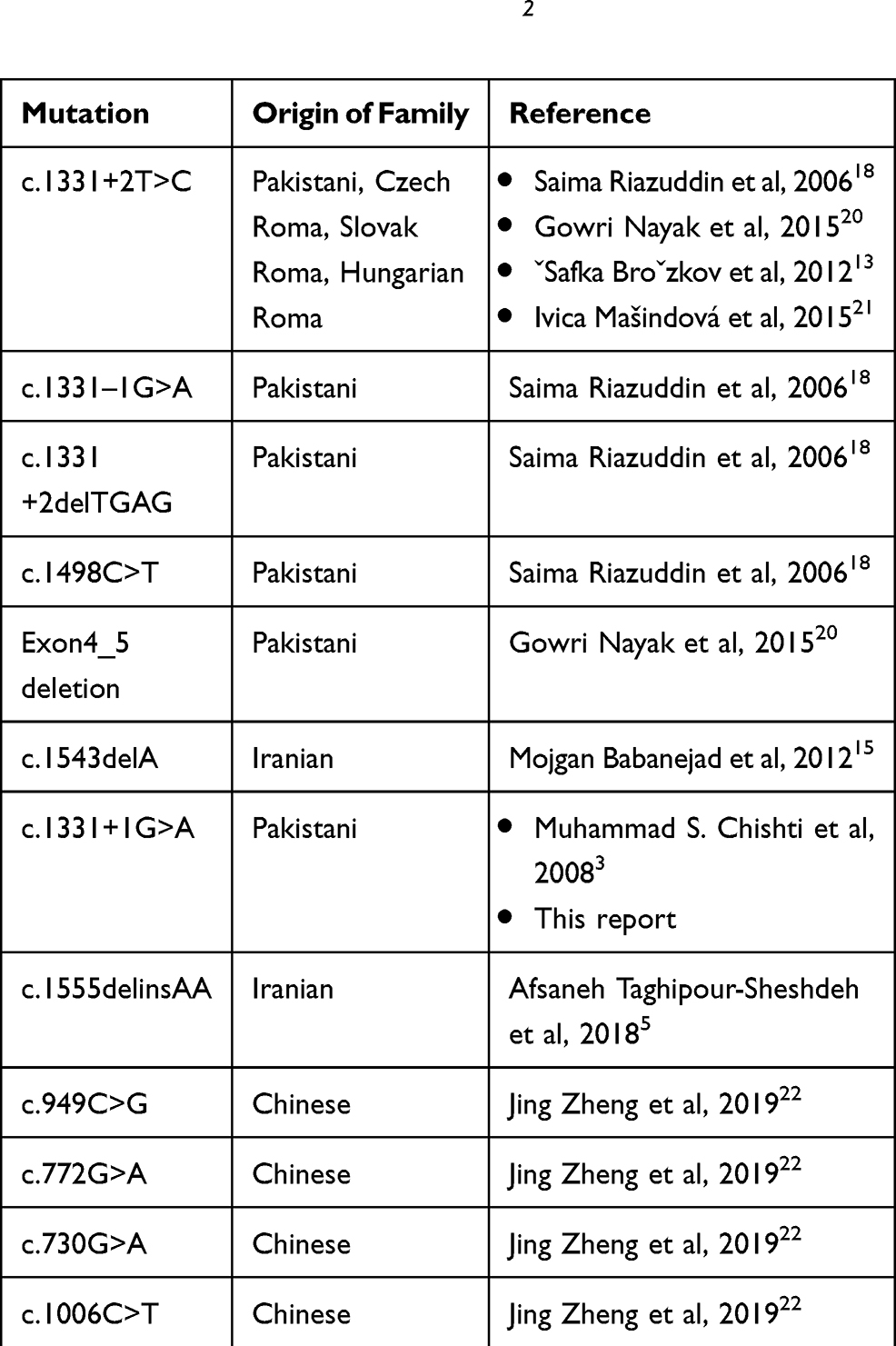 |
Table 2 All Variants of MARVELD2 Gene Which Previously Reported Were Caused of Hearing Loss |
In Pakistani families, the prevalence of NSHL, with autosomal recessive pattern, due to MARVELD2 mutations, is about 1.06%.19 This variant, the same as in a previously mentioned report, was considered to be pathogenic variant (MAF=0.00002) in this Iranian family, as had been reported previously in one Pakistani family in 2008.3 The prevalence of this variation is not clear in Iranian families due to no evaluated data in our population.
We imagine that it could be some connection between this Iranian family and the Pakistani region due to the same mutation appearing in these two populations, maybe there is a common old ancestor creating a possible origin of this mutation.23 So further large prospective studies are required to confirm, and we recommend more investigations in these populations. This is the first report of this mutation in the Iranian family with NSHL and the second report worldwide. These findings suggest that the MARVELD2 gene should be considered in Iranian patients affected with NSHL.
Conclusion
The segregation and data bases annotations revealed that the MARVELD2 gene c.1331+1G>A variant is probably a pathogenic mutation that can cause NSHL which shows autosomal recessive inheritance pattern.
Abbreviations
AD, Autosomal Dominant; AR, Autosomal Recessive; BWA, Burrows–Wheeler Aligner; CADD, Combined Annotation Dependent Depletion; ClinVar, Clinical Variant; dbSNP, single Nucleotide Polymorphism Database; DFNB, Deafness, Autosomal Recessive; FATHMM, Functional Analysis through Hidden Markov Models; InDel, Insertion-Deletion; MEM, Maximum Exact Matches; NGS, Next Generation Sequencing; NSHL, Nonsyndromic Hearing Loss; PolyPhen, Polymorphism Phenotyping; rs, refSNP; SIFT, Sorting Intolerant From Tolerant; SNP, Single Nucleotide Polymorphism; tTJ, tricellulin Tight-Junction; VCF, Variant Calling Format; WES, Whole Exome Sequencing; □, male; ○, female; ●, Affected female; ■, Affected male; □, Deceased male; ○, Deceased female; ═, Consanguineous Marriage; ──, Nonconsanguineous Marriage.
Acknowledgments
We appreciate the Genetic laboratory of Jordan, Tehran, Iran, who supported this research.
Author Contributions
All authors made substantial contributions to conception and design, acquisition of data, or analysis and interpretation of data; took part in drafting the article or revising it critically for important intellectual content; and agree to be accountable for all aspects of the work.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Najmabadi H, Kahrizi K. Genetics of non-syndromic hearing loss in the Middle East. Int J Pediatr Otorhinolaryngol. 2014;78(12):2026–2036. doi:10.1016/j.ijporl.2014.08.036
2. Smith RJ, Bale JF
3. Chishti MS, Bhatti A, Tamim S, et al. Splice-site mutations in the TRIC gene underlie autosomal recessive nonsyndromic hearing impairment in Pakistani families. J Hum Genet. 2008;53(2):101–105. doi:10.1007/s10038-007-0209-3
4. Mahdieh N, Rabbani B, Wiley S, Akbari MT, Zeinali S. Genetic causes of nonsyndromic hearing loss in Iran in comparison with other populations. J Hum Genet. 2010;55(10):639–648. doi:10.1038/jhg.2010.96
5. Taghipour-Sheshdeh A, Nemati-Zargaran F, Zarepour N, et al. A novel pathogenic variant in the MARVELD2 gene causes autosomal recessive non-syndromic hearing loss in an Iranian family. Genomics. 2018. doi:10.1016/j.ygeno.2018.05.008
6. Nemati S, Afrooz GA, Asgari A, Bonab BG. Prevalence of consanguineous marriage among parents of deaf and normal children in Ardabil, North Western Iran. Audiology. 2012;21(2):66–70.
7. Lotfi Y, Mehrkian S. Hearing impairments in consanguineous marriage. Iran Rehabil J. 2004;2(1):9–14.
8. Jehangir W, Iqbal MJ, Khan AUH. Sensorineural hearing loss seen in siblings of consanguineous marriages at multan. Paki J Med Health Sci. 2012;6(3):577–579.
9. Li H, Chai R. Hearing Loss: Mechanisms, Prevention and Cure. Springer; 2019.
10. Reardon W. Genetic deafness. J Med Genet. 1992;29(8):521. doi:10.1136/jmg.29.8.521
11. Bayazit YA, Yılmaz M. An overview of hereditary hearing loss. ORL J. 2006;68(2):57–63. doi:10.1159/000091090
12. Beheshtian M, Babanejad M, Azaiez H, et al. Heterogeneity of hereditary hearing loss in Iran: a comprehensive review. Arch Iran Med. 2017;19(10):720.
13. Šafka Brožková D, Laštůvková J, Štěpánková H, et al. DFNB49 is an important cause of non-syndromic deafness in Czech Roma patients but not in the general Czech population. Clin Genet. 2012;82(6):579–582. doi:10.1111/j.1399-0004.2011.01817.x
14. Kitajiri S-I, Katsuno T. Tricellular tight junctions in the inner ear. Biomed Res Int. 2016;2016:1–5. doi:10.1155/2016/6137541
15. Babanejad M, Fattahi Z, Bazazzadegan N, et al. A comprehensive study to determine heterogeneity of autosomal recessive nonsyndromic hearing loss in Iran. Am J Med Genet A. 2012;158(10):2485–2492. doi:10.1002/ajmg.a.35572
16. Ramzan K, Shaikh RS, Ahmad J, et al. A new locus for nonsyndromic deafness DFNB49 maps to chromosome 5q12. 3-q14. 1. Hum Genet. 2005;116(1–2):17–22. doi:10.1007/s00439-004-1205-8
17. Ikenouchi J, Furuse M, Furuse K, Sasaki H, Tsukita S, Tsukita S. Tricellulin constitutes a novel barrier at tricellular contacts of epithelial cells. J Cell Biol. 2005;171(6):939–945. doi:10.1083/jcb.200510043
18. Riazuddin S, Ahmed ZM, Fanning AS, et al. Tricellulin is a tight-junction protein necessary for hearing. Am J Hum Genet. 2006;79(6):1040–1051. doi:10.1086/510022
19. OMIM. 2019. Available from: www.omim.org.
20. Nayak G, Varga L, Trincot C, et al. Molecular genetics of MARVELD2 and clinical phenotype in Pakistani and Slovak families segregating DFNB49 hearing loss. Hum Genet. 2015;134(4):423–437. doi:10.1007/s00439-015-1532-y
21. Mašindová I, Šoltýsová A, Varga L, et al. MARVELD2 (DFNB49) mutations in the Hearing Impaired Central European Roma Population-prevalence, clinical impact and the common origin. PLoS One. 2015;10(4):e0124232. doi:10.1371/journal.pone.0124232
22. Zheng J, Meng W-F, Zhang C-F, et al. New SNP variants of MARVELD2 (DFNB49) associated with non-syndromic hearing loss in Chinese population. J Zhejiang Univ Sci B. 2019:1–6. doi:10.1631/jzus.B1700185
23. Mehrjoo Z, Fattahi Z, Beheshtian M, et al. Distinct genetic variation and heterogeneity of the Iranian population. PLoS Genet. 2019;15(9):9. doi:10.1371/journal.pgen.1008385
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.