Back to Journals » Clinical Ophthalmology » Volume 13
A randomized controlled trial of povidone-iodine/dexamethasone ophthalmic suspension for acute viral conjunctivitis
Authors Pepose JS ![]() , Narvekar A, Liu W, Haque R
, Narvekar A, Liu W, Haque R ![]()
Received 31 October 2018
Accepted for publication 31 January 2019
Published 21 March 2019 Volume 2019:13 Pages 535—544
DOI https://doi.org/10.2147/OPTH.S191275
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Scott Fraser
Jay S Pepose,1,2 Abhijit Narvekar,3 Wenlei Liu,4 Reza Haque3
1Pepose Vision Institute, Chesterfield, MO, USA; 2Department of Ophthalmology and Visual Sciences, Washington University School of Medicine, St Louis, MO, USA; 3Ophthalmics, Shire, A Takeda Company, Lexington, MA, USA; 4Biostatistics and Programming, Shire, A Takeda Company, Lexington, MA, USA
Purpose: To evaluate the clinical safety and efficacy of povidone-iodine (PVP-I) 0.6%/dexamethasone (DEX) 0.1% ophthalmic suspension vs vehicle in patients with clinically suspected acute viral conjunctivitis.
Patients and methods: This was a randomized, double-masked, parallel-group, vehicle-controlled study. Adults with a clinical diagnosis of suspected acute viral conjunctivitis were randomized 1:1 to PVP-I/DEX ophthalmic suspension or vehicle bilaterally four times daily for 5 days (Days 1–5). Evaluation was on Days 1, 3 (+1-day window), and 6 (+1). Patients with signs of acute viral conjunctivitis at the Day 6 visit received open-label PVP-I/DEX for five additional days and were evaluated on Day 11–14. The primary efficacy endpoint was clinical resolution of acute viral conjunctivitis in the study eye at the Day 6 visit.
Results: Overall, 132 patients were randomized and received treatment (PVP-I/DEX, n=66; vehicle, n=66); 38 patients continued into the open-label portion of the study. Not enough patients with confirmed adenoviral conjunctivitis (n=32/132) were enrolled to assess the primary endpoint, although there were some efficacy trends in the PVP-I/DEX group for global clinical score (sum of watery conjunctival discharge and bulbar conjunctival redness). There were no serious treatment-emergent adverse events (TEAEs) and no patients discontinued due to a TEAE. In the masked phase, 56.1% of patients receiving PVP-I/DEX experienced at least one TEAE vs 43.9% in the vehicle group; 78.9% of patients in the open-label phase experienced at least one TEAE. Most TEAEs were mild in severity.
Conclusion: PVP-I/DEX ophthalmic suspension administered for ≤14 days had a favorable safety profile and was generally well tolerated.
Keywords: adenoviral conjunctivitis, dexamethasone, povidone-iodine, randomized controlled trial
Introduction
Infectious conjunctivitis is a common eye condition mainly caused by viruses and bacteria.1 Adenovirus is a frequent cause of infectious conjunctivitis, with reported rates of adenoviral conjunctivitis varying widely from 40% to 75% of all cases of infectious conjunctivitis.2–4 Adenoviral conjunctivitis is a public health concern due to its highly contagious nature.5 It is also associated with significant patient discomfort, lost productivity, and in rare cases can lead to complications such as subepithelial corneal infiltrates and permanent compromise of vision.6,7
Currently, there are no approved treatments for adenoviral conjunctivitis, with therapeutic options being limited to supportive therapies and palliative measures.7 A novel ophthalmic suspension of povidone-iodine (PVP-I) 0.6% and dexamethasone (DEX) 0.1% is currently under clinical investigation. PVP-I is an antiseptic with bactericidal, virucidal, and fungicidal properties.8 DEX is a corticosteroid routinely used as a topical ophthalmic suspension for the treatment of ocular inflammation.9 Both components are approved for use in other indications and have been shown to be safe and efficacious for use on the ocular surface in humans.10–13
Different ophthalmic formulations of PVP-I in combination with DEX have been investigated for the treatment of conjunctivitis in early-stage studies. In an in vivo study conducted in rabbits, PVP-I 0.4%/DEX 0.1% significantly improved clinical scores and viral titers vs control treatments.14 In a randomized controlled trial of 122 patients with presumed viral conjunctivitis, treatment with ophthalmic PVP-I 0.4%/DEX 0.1% four times daily (QID) significantly reduced the duration of conjunctivitis vs patients treated with artificial tears.15 Similar results were obtained in a randomized controlled trial of 74 patients with adenoviral keratoconjunctivitis confirmed with polymerase chain reaction (PCR). In that study, significantly faster improvement of clinical signs and adenoviral eradication (in 5–7 days) vs the control groups were observed in patients treated with PVP-I 1.0%/DEX 0.1%.16 In a recent Phase II randomized controlled study of 176 patients with acute adenoviral conjunctivitis, ophthalmic administration of PVP-I 0.6%/DEX 0.1% for 5 days demonstrated statistical superiority to vehicle for clinical resolution, adenoviral eradication, global clinical score, and expanded clinical cure.17
This report presents the results of a Phase II randomized controlled study conducted in Brazil to evaluate the clinical safety and efficacy of PVP-I 0.6%/DEX 0.1% ophthalmic suspension compared with vehicle in the treatment of patients with clinical suspicion of acute viral conjunctivitis.
Materials and methods
Study design
This was a randomized, double-masked, parallel-group, vehicle-controlled study that was planned across two medical centers, although all patients were enrolled at a single site in Brazil. Patients were randomized 1:1 to receive either PVP-I 0.6%/DEX 0.1% ophthalmic suspension or vehicle, instilled as a single drop in both eyes QID for 5 days. The study consisted of four visits over 11–14 days. Visit 1 occurred on Day 1, Visit 2 on Day 3+1-day window, Visit 3 on Day 6+1-day window, and Visit 4 on Days 11–14 (ie, visit 3+5–7 days; Figure 1). Visit 4 was only required for patients with signs of acute viral conjunctivitis in the study eye at Visit 3, with those patients receiving open-label PVP-I 0.6%/DEX 0.1% for an additional 5 days (Figure 1). All study treatments were supplied as sterile preserved suspensions or solutions and were colored, labeled and packaged identically. Noncompliance was recorded as a protocol deviation if >20% of doses during a given dosing period were missed.
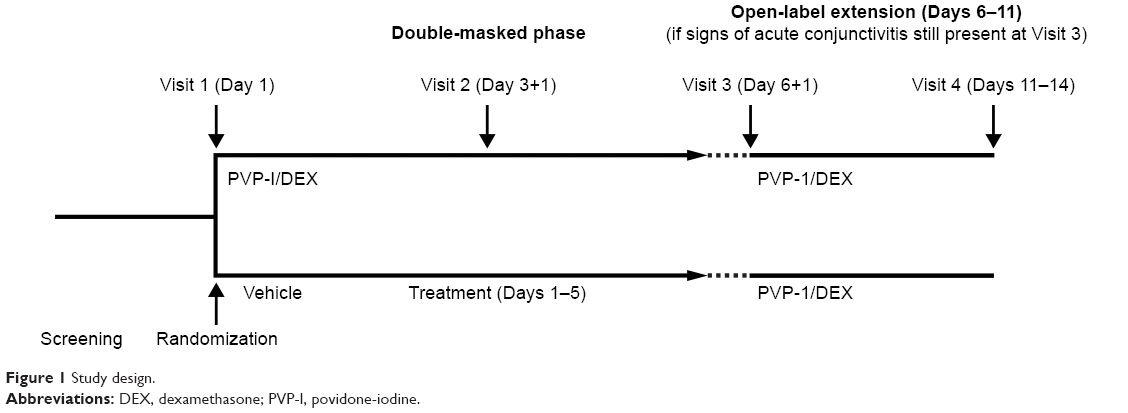 | Figure 1 Study design. |
Patients
Eligible patients were aged ≥18 years and had a best spectacle-corrected visual acuity (BSCVA) of 0.60 logarithm of the minimum angle of resolution (logMAR) or better in each eye, reported signs of viral conjunctivitis for ≤5 days before the study in at least one eye, clinical diagnosis of suspected acute adenoviral conjunctivitis in at least one eye, and the presence of both watery conjunctival discharge and bulbar conjunctival redness score of ≥1 in the same eye (0–3 scale; 0= absent/normal, 1= mild, 2= moderate, and 3= severe).
Exclusion criteria included the following: pregnancy or nursing a child; known sensitivity to any components of the investigational treatments; clinical signs, presence, or a history of herpes simplex keratitis; presence of ocular inflammation (eg, uveitis or iritis) or an ocular infection other than acute viral conjunctivitis; steroid responders with increased intraocular pressure or those with a history of glaucoma or elevated intraocular pressure >21 mmHg. In addition, patients with a history of recurrent corneal erosion syndrome or with active ulcerative keratitis, clinically significant optic nerve defects visible upon non-dilated fundus examination at baseline, uncontrolled systemic disease, autoimmune disease, or debilitating disease were excluded. The use of investigational devices, contact lenses, and the following treatments were not allowed in the study: corticosteroids (not including stable use of inhaled or nasal corticosteroids and topical dermal steroids, except around the eyes), topical ocular or systemic antivirals, or any other topical ophthalmic solutions including tear substitutes and diagnostics.
Efficacy assessments
The primary efficacy endpoint was clinical resolution of acute viral conjunctivitis in the study eye at the Day 6 visit. Clinical resolution was defined as the absence (score =0) of both bulbar conjunctival redness and watery conjunctival discharge. Additional efficacy measurements included individual measurement of watery conjunctival discharge and bulbar conjunctival redness in the study eye, expanded clinical cure (a score of 0 or 1 for both bulbar conjunctival redness and watery conjunctival discharge, with at least one sign having a score of 0), and global clinical score (sum of bulbar conjunctival redness and watery conjunctival discharge scores; total score 0–6). Crossover infection to a fellow eye was also recorded, based on the presence (scores >0) of both watery conjunctival discharge and bulbar conjunctival redness (only patients with one eye that did not show both signs of viral conjunctivitis at baseline were included).
All patients had a Rapid Pathogen Screening (RPS) Adeno-Detector Plus™ test (Rapid Pathogen Screening Inc., Sarasota, FL, USA) performed at the Day 1 visit on both eyes to identify, in office, whether they were RPS positive or negative. RPS-positive testing was not an inclusion criterion for the study. Conjunctival swabs of both eyes were taken at each visit using a flocked swab kit (Copan Diagnostics, Murrieta, CA, USA) and frozen at −70°C until analysis. Adenovirus testing with cell culture immunofluorescence assay (CC-IFA) and quantitative PCR (≥100 copies/mL was positive) was conducted on samples from eyes that were RPS positive at Visit 1.
Safety assessments
Adverse events (AEs; reported, elicited, and observed), slit-lamp biomicroscopy, and BSCVA were documented at all study visits. The definition of AEs included any pre-existing medical condition that worsened after administration of the study drug, for example, the significant worsening of viral conjunctivitis. Treatment-emergent AEs (TEAEs) were defined as events that occurred or worsened after the first dose. BSCVA was assessed using an Early Treatment Diabetic Retinopathy Study chart. Urine pregnancy test and nondilated fundus exam were performed at all visits except the Day 3 visit.
Statistical analyses
The primary population for the efficacy analysis was based on the modified intention-to-treat (mITT) population, consisting of patients in the intention-to-treat (ITT) population (randomized patients who received at least one dose of study medication) who had at least one visit after the Day 1 visit, and had a score ≥1 for watery conjunctival discharge and bulbar conjunctival redness scores in the same eye at the Day 1 visit. An additional analysis population was the viral-positive population, consisting of patients in the mITT population with a positive adenoviral test at the Day 1 visit in either eye by any method (RPS, CC-IFA, and/or quantitative PCR). The safety population consisted of randomized patients who received at least one dose of study medication.
A chi-square or Fisher’s exact test (for expected count <5) was used to compare treatment groups with respect to binary endpoints, and a 2-sample t-test was used to compare groups with respect to continuous endpoints. Testing was conducted at a two-sided 0.05 significance level.
Viral titer was assessed by quantitative PCR at each follow-up visit and summarized in the study eye by treatment group. The proportions of patients with a 3-point reduction from baseline in viral titer (log 10 transformed scale) in the two study arms were compared using a chi-squared test. If the viral test was negative (ie, <100 copies/mL), then the viral titer was considered to be 0. Due to the nature of the titer data, log base 10 transformations were performed on the raw data before any analyses were performed. To account for a value of 0 (ie, when the titer results are “negative”), 0.5 was added to all values before the log 10 transformations were taken.
The study eye was preliminarily designated by the investigator at the time of enrollment (baseline), based on signs alone. If both eyes had signs of viral conjunctivitis for ≤5 days before the baseline visit, the study eye was based on whichever eye had the greater cumulative score for watery conjunctival discharge and bulbar conjunctival redness at baseline. If both eyes had the same cumulative score, then the baseline-designated study eye was the right eye. For the analysis, in patients with only one eye with a positive adenoviral test by PCR and score ≥1 for watery conjunctival discharge and bulbar conjunctival redness at baseline, the infected eye was the study eye. If both eyes at baseline were positive for adenoviral testing and had sign scores of ≥1, the eye with the greater cumulative score for conjunctival discharge and redness at baseline was the study eye. If both eyes had the same cumulative score, then the study eye was the right eye. For patients with neither eye with a positive adenoviral test at baseline, the baseline-designated study eye was defined as the study eye.
Because this was a Phase II study, formal sample size calculations were not performed, but a sample size of 120 evaluable patients (60 per treatment arm) was deemed reasonable and appropriate for providing information for powering future studies.
Results
Study population
The study was conducted between May 2013 and March 2014. A total of 132 patients were randomized and 99 completed the study (Figure 2). The safety and ITT populations contained all 132 patients, while the mITT population contained 115 patients. A total of 38 patients (PVP-I/DEX, n=18; vehicle, n=20) continued into the open-label portion of the study.
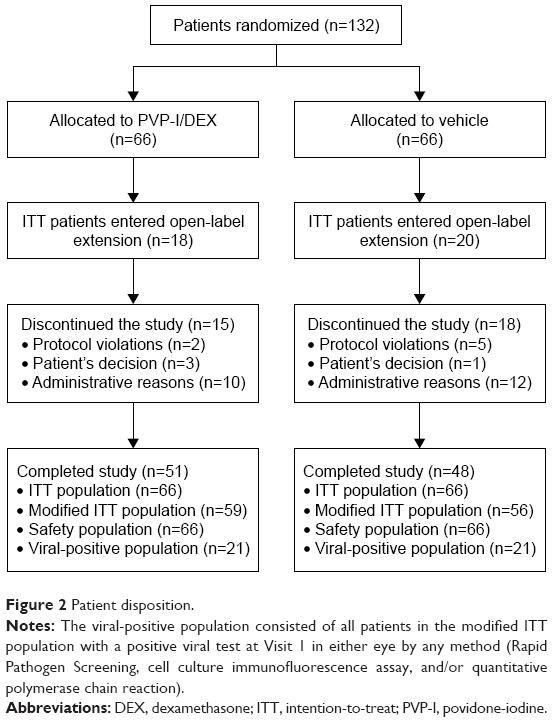 | Figure 2 Patient disposition. |
The mean (SD) age across all patients in the ITT population was 31.0 (9.87) years. Most patients were female (62.9%) and nearly all (97.0%) were of Hispanic or Latino ethnicity (Table 1). Four patients in the vehicle group and five in the PVP-I/DEX group were recorded as noncompliant with dosing. Altogether, 131 (99.2%) patients had a primary clinical diagnosis of viral conjunctivitis. With the exception of the primary diagnosis, the most common (>5%) occurrences in ocular medical history for all patients were eyelid edema (22.0%), optic nerve disorders (12.1%), keratitis (9.8%), corneal infiltrates (6.8%), and pinguecula (5.3%). The most common nonocular medical history included hypertension (5.3%), postmenopause (4.5%), female sterilization (3.0%), rhinitis (2.3%), gastritis (2.3%), and diabetes mellitus (2.3%).
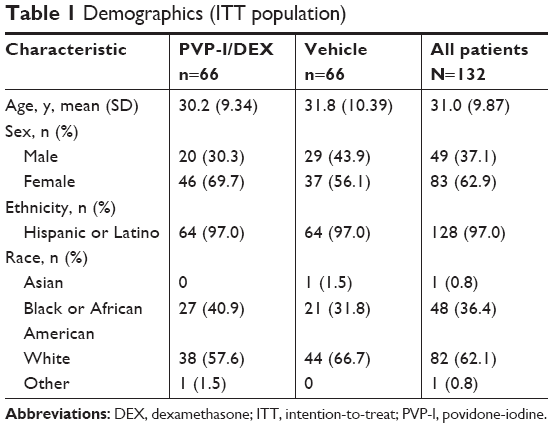 | Table 1 Demographics (ITT population) |
Viral status
Confirmatory adenovirus testing was planned to be carried out on baseline RPS-positive eyes at each visit using CC-IFA and quantitative PCR. At baseline, adenovirus testing was conducted on 27 and 23 mITT patients in the PVP-I/DEX and vehicle groups, respectively. Of these, adenovirus was detected by CC-IFA and/or quantitative PCR in either eye of 59.3% (16/27) and 69.6% (16/23) of patients in the PVP-I/DEX and vehicle groups, respectively. At the Day 3 visit, 73.7% (14/19) and 77.8% (14/18) of patients in those groups were adenovirus positive. At the Day 6 visit, 70.6% (12/17) and 77.8% (14/18) of patients were adenovirus positive. At the Day 11–14 visit, 50.0% (6/12) and 45.5% (5/11) of patients were adenovirus positive, respectively. The two treatment groups were not statistically different from each other in viral status at any visit.
The agreement between the adenovirus tests was as follows: 65.45% (36/55) of eyes tested for both RPS and CC-IFA at baseline had consistent results between the two tests. The agreement between the RPS and quantitative PCR tests was 73.85% [48/65] of eyes tested at baseline; and between the CC-IFA and quantitative PCR tests, the agreement was 72.68% [133/183] of eyes tested across all visits.
Efficacy
Clinical resolution
Analysis in the mITT population with last observation carried forward (LOCF) showed no statistical difference between the PVP-I/DEX and vehicle groups at the Day 6 visit for clinical resolution (66.1% [39/59] PVP-I/DEX vs 58.9% [33/56] vehicle; P=0.4268). The proportions of patients with resolution of individual conjunctival signs in the study eye at the Day 6 visit were numerically higher in the PVP-I/DEX group compared with the vehicle group, but the difference was not statistically significant (watery conjunctival discharge, 76.3% [45/59] PVP-I/DEX vs 62.5% [35/56] vehicle; P=0.1087; bulbar conjunctival redness, 67.8% [40/59] PVP-I/DEX vs 60.7% [34/56] vehicle; P=0.4280). Less than one-quarter (24.2%; 32/132) of patients had confirmed adenoviral conjunctivitis in either eye at baseline, but an attempt was made to analyze the data in these patients. At the Day 6 visit, in the viral-positive population with LOCF, there were no statistically significant differences between treatment groups in clinical resolution (33.3% [7/21] PVP-I/DEX vs 28.6% [6/21] vehicle; P=0.7385), or in resolution of watery conjunctival discharge (42.9% [9/21] PVP-I/DEX vs 33.3% [7/21] vehicle; P=0.5251), or resolution of bulbar conjunctival redness (33% [7/21] PVP-I/DEX vs 33.3% [7/21] vehicle; P=0.9999).
In the extension phase of the study (during which all patients received open-label PVP-I/DEX after the Day 6 visit), 60.0% (9/15) of patients in the ITT population who had received PVP-I/DEX before the Day 6 visit achieved clinical resolution compared with 44.4% (8/18) who were in the vehicle-treated group before the Day 6 visit; the between-group difference was not statistically significant (P=0.3733). As with the masked part of the study, there were few patients in the extension with confirmed adenoviral conjunctivitis, but an attempt was made to analyze these data. In this limited population, a positive adenoviral test did not appear to have an impact on the response rate (54.5% [6/11] PVP-I/DEX vs 50.0% [5/10] vehicle; P=0.8350).
Other efficacy endpoints
Viral titers were similar between the two treatment groups at all visits in the viral-positive population. In this population, at the Day 6 visit, 12.5% (2/16) of patients in the PVP-I/DEX group achieved a 3-point reduction (log 10 transformed scale) from baseline in viral titer in the study eye compared with 17.6% (3/17) of patients in the vehicle-treated group; the difference was not statistically significant (P=0.9999). In the mITT population, there were no reductions in viral conjunctivitis (study and fellow eyes) between the PVP-I/DEX and vehicle treatment groups at any visit as measured by adenovirus (detected by CC-IFA or quantitative PCR) (Figure 3).
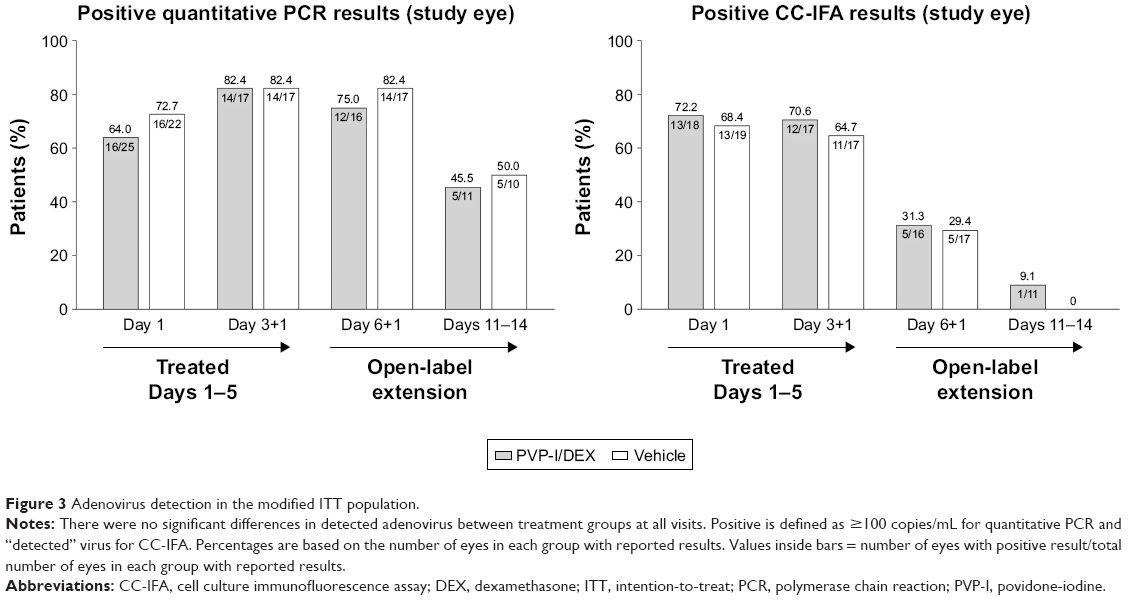 | Figure 3 Adenovirus detection in the modified ITT population. |
In the mITT population with LOCF, the percentage of patients who met the definition of expanded clinical cure in the study eye at the Day 6 visit was numerically higher in the PVP-I/DEX group (76.3%; 45/59) compared with the vehicle group (62.5%; 35/56); the difference was not statistically significant (P=0.1087).
The proportion of patients with crossover infection, defined as the presence (scores >0) of both watery conjunctival discharge and bulbar conjunctival redness, was lower in the PVP-I/DEX group compared with the vehicle group (Table 2).
 | Table 2 Crossover infection to the fellow eye (mITT population) |
In patients with a positive adenoviral test among the ITT population, at the Day 3 visit, a reduction from baseline in global clinical score (mean change, −1.5 vs 0.1; P=0.0078) or the proportion of patients with any improvement from baseline in global clinical score (75.0% [15/20] PVP-I/DEX vs 30.0% [6/20] vehicle; P=0.0044) were significantly greater with PVP-I/DEX compared with vehicle. Between-group differences for the proportions of patients with a 3-point reduction (25% [5/20] PVP-I/DEX vs 10% [2/20] vehicle) or ≥50% reduction (40% [8/20] PVP-I/DEX vs 15% [3/20] vehicle) from baseline in global clinical score were not statistically significant at this time point. At the Day 6 visit in this population, of the various measures of change from baseline in global clinical score shown in Table 3, only the proportion of patients in the PVP-I/DEX group with a reduction of ≥50% from baseline was significantly greater compared with the vehicle group (Table 3).
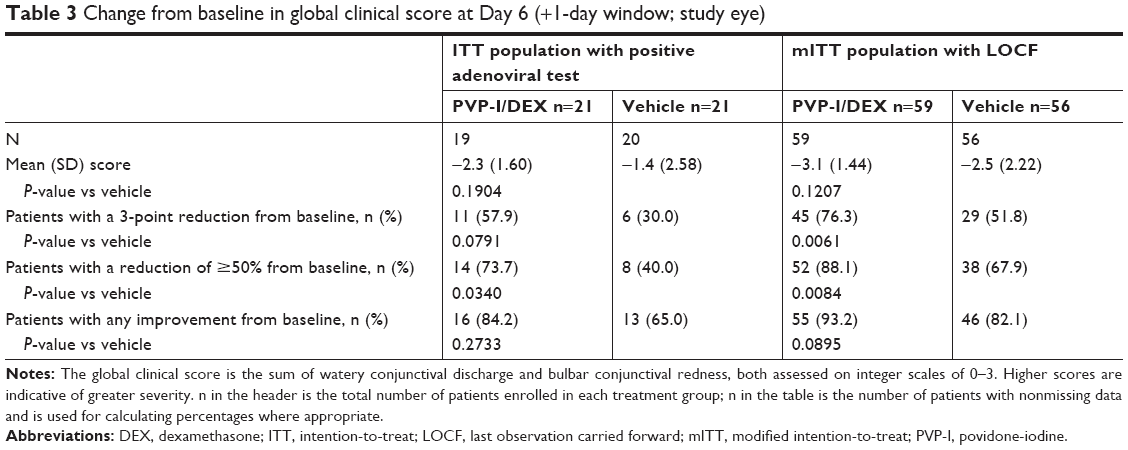 | Table 3 Change from baseline in global clinical score at Day 6 (+1-day window; study eye) |
In the mITT population with LOCF, at the Day 6 visit, a reduction from baseline in global clinical score or the proportion of patients with any improvement from baseline in global clinical score were numerically greater with PVP-I/DEX vs vehicle, although the differences were not significantly different (Table 3). Between-group differences for the proportion of patients with a 3-point or ≥50% reduction from baseline in global clinical score significantly favored PVP-I/DEX at this time point (Table 3).
Safety
There were no serious AEs or deaths during the course of this study and no patients discontinued as a result of an AE. In addition, no patterns of AEs that suggested systemic toxicities or localized complications were identified. In the masked phase of this study, of patients receiving PVP-I/DEX, 51.5% (34/66) reported at least one ocular TEAE compared with 39.4% (26/66) in the vehicle group; 78.9% (30/38) of patients in the open-label phase experienced at least one ocular TEAE. Considerably fewer patients reported at least one nonocular TEAE [masked phase, 4.5% (3/66) PVP-I/DEX vs 12.1% (8/66) vehicle; open-label phase, 10.5% (4/38)]. The most frequently reported ocular (>5% of patients) or nonocular (>1 patient) TEAEs in either treatment group are presented in Table 4. In the masked phase, all ocular and nonocular TEAEs in the PVP-I/DEX group were considered to be mild in severity. In the open-label extension, two ocular TEAEs (reduced visual acuity and scarring) reported by 2.6% (1/38) of patients receiving PVP-I/DEX were classified as moderate; all other TEAEs in the extension were considered to be mild (Table 4).
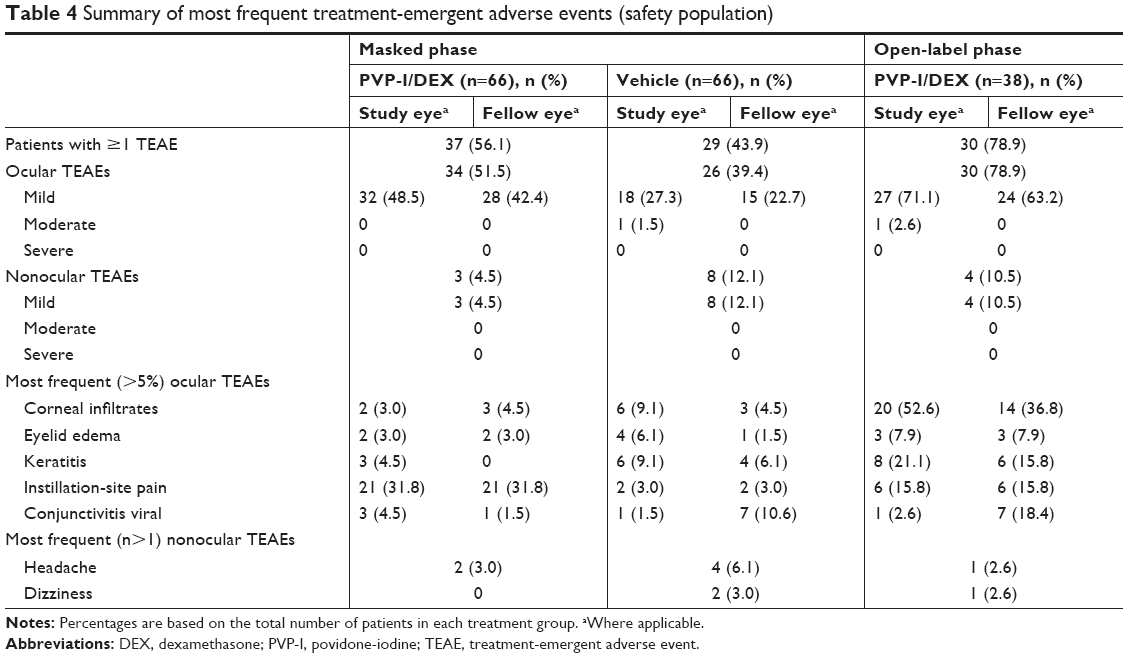 | Table 4 Summary of most frequent treatment-emergent adverse events (safety population) |
In the masked phase, 34.8% (23/66) of patients in the PVP-I/DEX group reported a total of 23 ocular TEAEs in the study eye that were suspected to be related to the study treatment. These were predominantly mild instillation-site pain (21/23 ocular TEAEs). The most frequently reported ocular TEAEs that were not suspected of being related to study treatment in the PVP-I/DEX group included corneal infiltrates (reported by 3.0% and 4.5% of patients in the study eye and fellow eye, respectively), ocular pruritus (reported by 3.0% of patients in both the study eye and fellow eye), and viral conjunctivitis (reported by 4.5% and 1.5% of patients in the study eye and fellow eye, respectively). No nonocular TEAEs were suspected to be related to treatment in the PVP-I/DEX treatment group during the masked phase of the study.
In the open-label phase, ocular TEAEs (mild corneal infiltrates, mild keratitis, and mild instillation-site pain) were reported in 78.9% (30/38) of patients. Four nonocular TEAEs were reported in 10.5% (4/38) of the open-label–treated patients. These were pyrexia, dizziness, headache, and dysmenorrhea. Only the dizziness (mild in severity) was suspected of being related to study treatment (Table 4).
In the masked and open-label phases of the study, the only nonocular TEAEs that were reported in more than one patient in any treatment group were headache and dizziness (Table 4).
Slit-lamp biomicroscopy examinations revealed several cases of shifts from normal at baseline to abnormal (clinically significant or nonclinically significant) at subsequent visits. These shifts were noted across treatment groups and visits predominantly for evaluations of the cornea. There were no clinically significant abnormal findings with fundoscopic examinations of the optic nerve after treatment with PVP-I/DEX or vehicle. Mean changes in BSCVA over the course of the study did not show any notable increases in score in any treatment group; however, there were some individuals who experienced a change in BSCVA of ≥0.22 logMAR (Day 3 visit, 0 PVP-I/DEX vs 7.4% [4/54] vehicle; Day 6 visit, 3.6% [2/55] PVP-I/DEX vs 3.7% [2/54] vehicle; Day 11–14 visit, 5.0% [1/20] PVP-I/DEX vs 10.0% [2/20] vehicle).
Discussion
The present study intended to evaluate the superiority of PVP-I/DEX ophthalmic suspension compared with vehicle for clinical resolution of acute viral conjunctivitis, but not enough patients with confirmed adenoviral conjunctivitis were enrolled to assess the primary efficacy endpoint for PVP-I/DEX. An attempt was made to analyze the data on patients with confirmed adenoviral conjunctivitis; however, there was very little remaining statistical power to assess efficacy. Overall, some trends toward efficacy were observed for PVP-I/DEX, and the drug combination had a favorable safety profile and was generally well tolerated in this study, with no serious ocular TEAEs reported and no patients withdrawn from the study due to an AE.
Less than one-quarter (n=32/132) of patients in this study had confirmed adenoviral conjunctivitis at baseline by CC-IFA or quantitative PCR. A positive RPS Adeno-Detector Plus test was not an inclusion criterion in this study, which could explain the low number of adenoviral-positive patients enrolled, because an accurate diagnosis of viral conjunctivitis based on clinical signs is known to be difficult.18 In contrast, in a Phase II efficacy and safety trial conducted in India that included only RPS-positive patients, sufficient numbers of patients with confirmed adenoviral conjunctivitis were enrolled (81.8% of randomized patients) and PVP-I/DEX demonstrated statistical superiority to vehicle for clinical resolution, adenoviral eradication, global clinical score, and expanded clinical cure.17 In the present study, although we were not able to assess the primary efficacy endpoint, some improvements in global clinical scores were observed following treatment with PVP-I/DEX in patients with a positive adenoviral test among the ITT population and in the mITT population with LOCF. PVP-I/DEX also appeared to have a benefit relative to crossover infection rates, with lower proportions of patients with crossover infection compared with vehicle treatment.
The observed safety profile of PVP-I/DEX in this study appeared to be consistent with the well-characterized safety profiles of PVP-I and DEX, and no patterns of AEs indicative of systemic toxicities or localized complications were identified. Overall, no serious AEs were reported and the majority of TEAEs were ocular in nature and mild in severity. Most of the AEs with a suspected relationship to treatment with PVP-I/DEX consisted of mild instillation-site pain. All nonocular TEAEs were mild and the only nonocular TEAEs that occurred in more than one patient in any treatment group were headache and dizziness. Importantly, no patients in this study discontinued due to AEs. The safety and tolerability results of the present study are consistent with results from the study conducted in India.17 In both studies, there were no increases in AEs that have been previously associated with the use of topical ocular corticosteroids, ie, increases in intraocular pressure, cataract development, or glaucoma.19–21
Infectious conjunctivitis is a clinically challenging condition due to the overlap of symptoms between bacterial and viral causes and because no treatment is approved for viral conjunctivitis. A drug that treats both adenoviral and bacterial conjunctivitis would mitigate the negative effects of misdiagnosis, which can occur in up to 50% of cases.22 Phase III clinical trials are ongoing in adults and children to evaluate the efficacy and safety of PVP-I 0.6%/DEX 0.1% in adenoviral conjunctivitis (NCT02998541 and NCT02998554).
Potential limitations of this study are that the study was conducted in a single country and that serotyping was not performed, which may limit the generalizability of the results. In addition, according to the instructions of the RPS Adeno-Detector Plus test used, the sensitivity of the test was 84%, so negative test samples that were not subsequently tested by quantitative PCR or CC-IFA could have been positive for adenovirus or other viruses.
Future studies evaluating the efficacy of PVP-I/DEX could be improved by modifying study eligibility criteria to increase the likelihood of enrolling subjects with confirmed adenoviral conjunctivitis. This may be achieved by the use of the AdenoPlus® test as a screening test and/or better clinical assessment criteria. A study with an adequate number of subjects with adenoviral conjunctivitis confirmed by culture would enable a robust assessment of efficacy.
Conclusion
The QID dosing of PVP-I 0.6%/DEX 0.1% ophthalmic suspension for ≤14 days was generally well tolerated, with no unexpected TEAEs, and a safety profile that was consistent with the known pharmacological profile of PVP-I and DEX. Overall, some trends toward efficacy were observed for PVP-I/DEX, but large Phase III trials enrolling a high proportion of patients with confirmed adenoviral conjunctivitis are needed to further evaluate the efficacy of this product for adenoviral conjunctivitis. Such studies are ongoing and should provide important information.
Ethics approval and informed consent
This trial was compliant with the principles of the Declaration of Helsinki and the International Conference on Harmonization guidelines for Good Clinical Practice; it was registered at ClinicalTrials.gov (identifier, NCT01461954). Written informed consent was obtained from each patient prior to any study-related procedures at Visit 1. The study protocol and its amendments, and the informed consent form, were reviewed and approved by Alpha Independent Review Board (San Clemente, CA, USA) and Comitê de Ética em Pesquisa da Universidade Federal de São Paulo (São Paulo, Brazil).
Data sharing statement
The datasets generated and/or analyzed during the current study are not publicly available due to the need to minimize risk to the privacy and confidentiality of research participants and ensure compliance with legal requirements for privacy and data protection but are available from the corresponding author on reasonable request.
Abbreviations
AE, adverse event; BSCVA, best spectacle-corrected visual acuity; CC-IFA, cell culture immunofluorescence assay; DEX, dexamethasone; ITT, intention-to-treat; LOCF, last observation carried forward; logMAR, logarithm of the minimum angle of resolution; mITT, modified intention-to-treat; PCR, polymerase chain reaction; PVP-I, povidone-iodine; QID, four times daily; RPS, Rapid Pathogen Screening; TEAE, treatment-emergent adverse event.
Acknowledgments
The authors thank Nasser Malik, PhD, of Excel Scientific Solutions, who provided medical writing assistance funded by Shire. This study was funded by Shire. The sponsor participated in the design of the study and in the collection, analysis and interpretation of the data.
Author contributions
All authors contributed to data analysis, drafting and revising the article, gave final approval of the version to be published, and agree to be accountable for all aspects of the work.
Disclosure
Jay S Pepose: consultant/advisor for AcuFocus, Inc., Bausch & Lomb, BRIM Biotech, Johnson & Johnson, Noveome, Shire, Sun Pharma, and TearLab; and has equity interest in AcuFocus, Inc., Mimetogen, Ocunexis, Okogen, and Stuart Pharmaceuticals. Wenlei Liu is an employee of Shire, a Takeda company, and owns stock/stock options in Takeda. Abhijit Narvekar and Reza Haque were employees of Shire, a Takeda company, and owned stock/stock options in Shire at the time of this work.
References
Azari AA, Barney NP. Conjunctivitis: a systematic review of diagnosis and treatment. JAMA. 2013;310(16):1721–1729. | ||
Li J, Lu X, Jiang B, et al. Adenovirus-associated acute conjunctivitis in Beijing, China, 2011–2013. BMC Infect Dis. 2018;18(1):135. | ||
Sambursky RP, Fram N, Cohen EJ. The prevalence of adenoviral conjunctivitis at the Wills Eye Hospital emergency room. Optometry. 2007;78(5):236–239. | ||
Woodland RM, Darougar S, Thaker U, et al. Causes of conjunctivitis and keratoconjunctivitis in Karachi, Pakistan. Trans R Soc Trop Med Hyg. 1992;86(3):317–320. | ||
O’Brien TP, Jeng BH, McDonald M, Raizman MB. Acute conjunctivitis: truth and misconceptions. Curr Med Res Opin. 2009;25(8):1953–1961. | ||
Ford E, Nelson KE, Warren D. Epidemiology of epidemic keratoconjunctivitis. Epidemiol Rev. 1987;9:244–261. | ||
AAO Cornea/External Disease PPP Panel, Hoskins Center for Quality Eye Care. Conjunctivitis PPP; 2018. Available from: http://www.aao.org/preferred-practice-pattern/conjunctivitis-ppp-2018. Accessed February 26, 2019. | ||
Lachapelle JM, Castel O, Casado AF, et al. Antiseptics in the era of bacterial resistance: a focus on povidone iodine. Future Med. 2013;10(5):579–592. | ||
Saraiya NV, Goldstein DA. Dexamethasone for ocular inflammation. Expert Opin Pharmacother. 2011;12(7):1127–1131. | ||
Berkelman RL, Holland BW, Anderson RL. Increased bactericidal activity of dilute preparations of povidone-iodine solutions. J Clin Microbiol. 1982;15(4):635–639. | ||
Isenberg SJ, Apt L, Yoshimori R, Pham C, Lam NK. Efficacy of topical povidone-iodine during the first week after ophthalmic surgery. Am J Ophthalmol. 1997;124(1):31–35. | ||
Mohan N, Gupta V, Tandon R, Gupta SK, Vajpayee RB. Topical ciprofloxacin-dexamethasone combination therapy after cataract surgery: randomized controlled clinical trial. J Cataract Refract Surg. 2001;27(12):1975–1978. | ||
Speaker MG, Menikoff JA. Prophylaxis of endophthalmitis with topical povidone-iodine. Ophthalmology. 1991;98(12):1769–1775. | ||
Clement C, Capriotti JA, Kumar M, et al. Clinical and antiviral efficacy of an ophthalmic formulation of dexamethasone povidone-iodine in a rabbit model of adenoviral keratoconjunctivitis. Invest Ophthalmol Vis Sci. 2011;52(1):339–344. | ||
Pinto RDP, Lira RPC, Abe RY, et al. Dexamethasone/povidone eye drops versus artificial tears for treatment of presumed viral conjunctivitis: a randomized clinical trial. Curr Eye Res. 2015;40(9):870–877. | ||
Kovalyuk N, Kaiserman I, Mimouni M, et al. Treatment of adenoviral keratoconjunctivitis with a combination of povidone-iodine 1.0% and dexamethasone 0.1% drops: a clinical prospective controlled randomized study. Acta Ophthalmol. 2017;95(8):e686–e692. | ||
Pepose JS, Ahuja A, Liu W, Narvekar A, Haque R. Randomized, controlled, phase 2 trial of povidone-iodine/dexamethasone ophthalmic suspension for treatment of adenoviral conjunctivitis. Am J Ophthalmol. 2018;194:7–15. | ||
Rietveld RP, van Weert HCPM, ter Riet G, Bindels PJE. Diagnostic impact of signs and symptoms in acute infectious conjunctivitis: systematic literature search. BMJ. 2003;327(7418):789. | ||
Bartlett JD, Horwitz B, Laibovitz R, Howes JF. Intraocular pressure response to loteprednol etabonate in known steroid responders. J Ocul Pharmacol. 1993;9(2):157–165. | ||
Clark AF, Wilson K, de Kater AW, Allingham RR, McCartney MD. Dexamethasone-induced ocular hypertension in perfusion-cultured human eyes. Invest Ophthalmol Vis Sci. 1995;36(2):478–489. | ||
El-Harazi SM, Feldman RM. Control of intra-ocular inflammation associated with cataract surgery. Curr Opin Ophthalmol. 2001;12(1):4–8. | ||
Visscher KL, Hutnik CM, Thomas M. Evidence-based treatment of acute infective conjunctivitis: breaking the cycle of antibiotic prescribing. Can Fam Physician. 2009;55(11):1071–1075. |
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.