Back to Journals » Clinical Pharmacology: Advances and Applications » Volume 8
A prospective, observational study comparing the PK/PD relationships of generic Meropenem (Mercide®) to the innovator brand in critically ill patients
Authors Mer M, Snyman JR, van Rensburg CEJ, van Tonder JJ, Laurens I
Received 18 February 2016
Accepted for publication 8 September 2016
Published 17 November 2016 Volume 2016:8 Pages 191—198
DOI https://doi.org/10.2147/CPAA.S106676
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Arthur E. Frankel
Mervyn Mer,1 Jacques Rene Snyman,2 Constance Elizabeth Jansen van Rensburg,2 Jacob John van Tonder,3 Ilze Laurens2
1Department of Medicine, Divisions of Critical Care and Pulmonology, University of the Witwatersrand, Johannesburg, South Africa; 2Office of the Dean, Faculty of Health Sciences, University of Pretoria, Pretoria, South Africa; 3Scientific Affairs Department, Triclinium Clinical Development (Pty) Ltd, Centurion, South Africa
Introduction: Clinicians’ skepticism, fueled by evidence of inferiority of some multisource generic antimicrobial products, results in the underutilization of more cost-effective generics, especially in critically ill patients. The aim of this observational study was to demonstrate equivalence between the generic or comparator brand of meropenem (Mercide®) and the leading innovator brand (Meronem®) by means of an ex vivo technique whereby antimicrobial activity is used to estimate plasma concentration of the active moiety.
Methods: Patients from different high care and intensive care units were recruited for observation when prescribed either of the meropenem brands under investigation. Blood samples were collected over 6 hours after a 30 minute infusion of the different brands. Meropenem concentration curves were established against United States Pharmacopeia standard meropenem (Sigma-Aldrich) by using standard laboratory techniques for culture of Klebsiella pneumoniae. Patients’ plasma samples were tested ex vivo, using a disc diffusion assay, to confirm antimicrobial activity and estimate plasma concentrations of the two brands.
Results: Both brands of meropenem demonstrated similar curves in donor plasma when concentrations in vials were confirmed. Patient-specific serum concentrations were determined from zones of inhibition against a standard laboratory Klebsiella strain ex vivo, confirming at least similar in vivo concentrations as the concentration curves (90% confidence interval) overlapped; however, the upper limit of the area under the curve for the ratio comparator/innovator exceeded the 1.25-point estimate, i.e., 4% higher for comparator meropenem.
Conclusion: This observational, in-practice study demonstrates similar ex vivo activity and in vivo plasma concentration time curves for the products under observation. Assay sensitivity is also confirmed. Current registration status of generic small molecules is in place. The products are therefore clinically interchangeable based on registration status as well as bioassay results, demonstrating sufficient overlap for clinical comfort. The slightly higher observed comparator meropenem concentration (4%) is still clinically acceptable due to the large therapeutic index and should ally fears of inferiority.
Keywords: bioequivalence, antimicrobial, multisource products, Meropenem, pharmacokinetics, pharmacodynamics
Introduction
Recent publications demonstrating differences in the quality of different generic products have made clinicians wary of using non-innovator brands (“generics”) when treating critically ill patients. Although bioequivalence testing and in vitro quality confirmation can go a long way in convincing the skeptics, the clinical outcome remains elusive in any argument.1 The skepticism remains, irrespective of the small molecule generic status with regulatory authorities.
In general, the “generic” product is available at a lower acquisition cost than the innovator product due to the exclusion of some input costs associated with research and patent development.2 With the reduced cost, a phenomenal number of generic products are currently available on the market.2 This has led to many publications doubting the safety, efficacy, and quality of the different generic products, resulting in clinicians being wary of using “generic” brands when treating critically ill patients.3–6
One of the biggest concerns referred to in publications regarding the use of an intravenously administered “generic” product is that the “generic” product is not required to demonstrate therapeutic equivalence.1,4,7,8 According to the World Health Organization’s report on multisource (“generic”) products, the “generic” product needs to conform to the same standards as those required by the innovator product.7 Furthermore, the manufacturer of the “generic” product must provide “reasonable assurance” that the “generic” product is therapeutically equivalent to the innovator product. To demonstrate therapeutic equivalence in vivo, a clinical therapeutic equivalence study is needed that requires a large number of patients. It is, however, not economically viable or ethically justifiable to expose patients to unnecessary clinical trials, when regulatory bodies have already accepted similarity.7 Therefore, pharmacokinetic bioassays that demonstrate bioequivalence and pharmaceutical equivalence are preferred.7,9,10
An alternative to a typical clinical equivalence study is an ex vivo study where the treated patients’ plasma containing the active moiety (pharmacokinetic component) is used to establish a time-based ex vivo activity of the same against a known laboratory strain (pharmacodynamic component), comparing different brands. A study using the plasma levels and comparing this to in vitro antimicrobial activity was done to compare two meropenem brands in healthy volunteers.11 The issue for most clinicians, however, remains, “will this hold true in critically ill patients?”.10
In this current, in-practice study of meropenem, an ex vivo study was used for exploring the active pharmaceutical ingredient in serial dilutions of plasma after it has been injected intravenously into critically ill patients who required high or intensive care for their respective clinical condition. Compared to an in vitro study, drug concentrations determined at different time points, during the ex vivo study, directly relate to what can be expected in vivo.11 Therefore, performing ex vivo studies on “generic” products comparing them to the innovator product will further build confidence in the safety and efficacy of non-innovator brands. This process allows comparisons using similar lab strain organisms with time-matched plasma samples translating efficacy (zone of inhibition) to concentration of active moiety in the plasma, i.e., allowing for the disease entity and the patient-specific organ functions to have had an influence on the drugs kinetics as well as dynamics by taking into account possible degradation of active moiety, as was seen with vancomycin and possibly also with piperacillin/tazobactam combinations.5
The aim of the present study was therefore to explore the ex vivo dynamics of intravenously administered meropenem (i.e., generic or comparator Mercide® from Ranbaxy [Pty] Ltd and Meronem® the leading innovator brand available from Astra Zeneca, South Africa) in patients in high care or ICUs in an observational cohort study, i.e., data was collected from patients who were receiving the different meropenem brands due to their clinical need in different hospitals to be observed, i.e., timed blood samples were collected for analyses.
Materials and methods
In vitro validation of method
In order to establish the ex vivo assay sensitivity, concentrations and efficacy of the generic and innovator brands of meropenem products in vials had to be validated against pure chemical standards in vitro. A two-point agar diffusion test method was developed to determine the activity of innovator and comparator meropenem against the United States Pharmacopeia (USP) reference standard meropenem (Sigma-Aldrich). Two concentrations of the meropenem standard in a ratio of 4:1 were used, referred to as “standard high” and “standard low”. The antibiotics’ concentrations were selected on the basis that both should fall on the linear part of a standard curve graph when concentration is plotted against zone diameter., The exact levels also depended on the sensitivity of the test organism used for the specific test, i.e., Klebsiella pneumoniae (tested against 5 and 1.25 μg/mL) and Bacillus subtilis (1.25 and 0.31 μg/mL; Table 1). The two meropenem products were diluted to the same antibiotic concentrations referred to as “standard high or low”. All dilutions were tested on the same agar diffusion plate (manufactured as explained below under “Microbiological testing”) with a random Latin square design for 36 wells, resulting in 9 replicates for each (Table 2 and Figure 1). Growth medium agar, agar thickness, and test organism load condition stayed the same for USP standard and test products. Each brand name product was tested on its own individual agar diffusion plate.
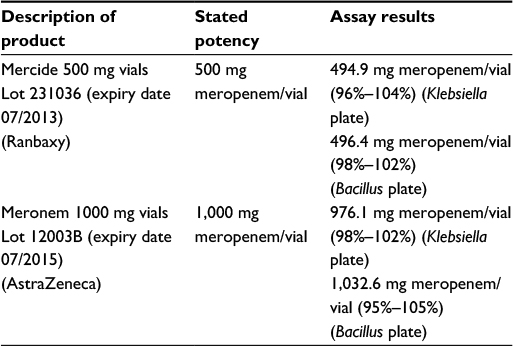 | Table 1 Comparator meropenem (Mercide®) and innovator meropenem (Meronem®) concentrations in vials |
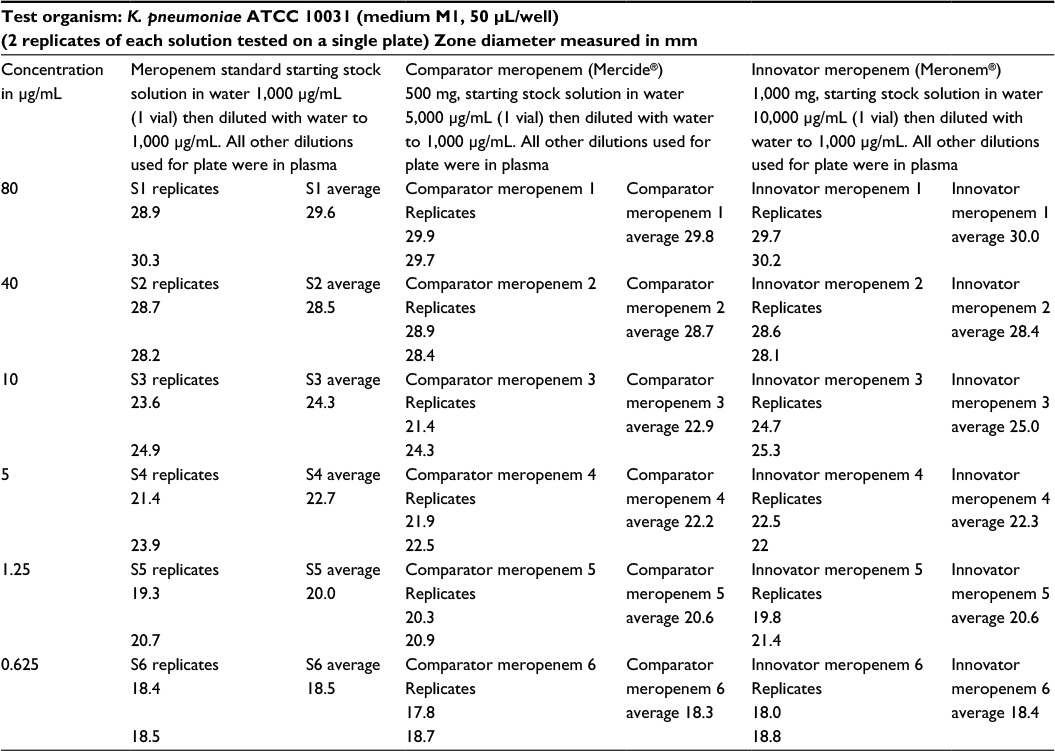 | Table 2 Test results of products and USP standard in human donor plasma against Klebsiella pneumoniae Notes: Meropenem – inhibition zone diameter readings. Parallelism observed between reference standard, meropenem, comparator meropenem 500 mg/vial and innovator meropenem 1000 mg/vial, all diluted in plasma over a range of 0.625–80 µg/mL Abbreviation: USP, United States Pharmacopeia; S, sample. |
Microbiological testing
Ex vivo assay: Nutrient agar/M1 slopes were inoculated with K. pneumoniae (ATCC 10031) and incubated at 30°C overnight. Growth was washed off with sterile saline and the suspension vortex and diluted in saline to a predetermined opacity to contain ~107/mL of organisms. This suspension was used to inoculate 160 mL antibiotic medium 1 (Bio-Lab Chemicals; seed agar), which was poured into a 356 mm by 356 mm sterile Nunc™ (Thermo Scientific) Bioassay dish with a 25 mm rim12 and placed on a level slab to solidify. Forty-nine wells, with a diameter of ~5 mm were made with a punch with an approximate diameter of 6.5 mm and the agar plug removed.
Six concentrations of a meropenem USP reference standard of known potency were prepared using pooled serum taken from healthy donors who were not on any antibiotic treatment. All serum samples were thawed just before use. A volume of 100 µL of the various meropenem dilutions as well as the patients’ serum samples were added to the wells at a random distribution format suggested by Bennett et al.12 Four replicates of each dilution as well as the patient samples were tested. The plates were incubated at 37°C for 24 h. The plates were then inverted and the inhibition zones measured with calipers.
Laboratory assessment of ex vivo activity was determined by investigators blinded to the collected sample’s brand type. The laboratory; Ampath (Pty) Ltd. was process and quality accredited (SANAS).
Patients observed
This was an open label observational study where samples were collected from preidentified patients according to their treatment regimen, including meropenem but no other antimicrobial drug with the same spectrum. Patients had to be hemodynamically stable for the duration of sample collection, with no overt kidney or liver failure. A washout period equivalent to 7 times the half-life of prior antibiotics was allowed when patients received other antimicrobials before commencement of sample collection. Patients were sourced from different clinical sites in South Africa where both brands of the medicine being studied were used. Since blinded allocation and collection was not possible, each site, however, had specific numbers allocated in a block design (4 per block) to ensure equal numbers of patients per product were observed per site, i.e., if the numbers for one product was reached only then were those on the other product recruited into the study for observation.
Patients in high care or ICUs of hospitals in South Africa, who were prescribed either of the specific brand names, i.e., Meronem or Mercide, were recruited into the study. Only patients >18 years of age who provided written informed consent to data use were included. This study was approved by the Independent Review Board for Clinical Trials (South African Medical Association Research Ethics Committee) and was conducted according to Good Clinical Practice guidelines and the World Medical Association Declaration of Helsinki’s Ethical Principles of Medical Research Involving Human Subjects. The researcher had no influence over product choice as this was an observation study.
Six blood samples were collected from each patient in 5 mL ethylenediaminetetraacetic acid vacutainer tubes at times 0 (before drug administration), and again at 30 (calculated from start of iv bolus), 60, 120, 240, and 360 minutes. Serial dilutions were done in vitro to test the “serum” activity against a standard laboratory pathogen. The innovator brand was compared to the generic brand with respect to growth inhibition (inhibition zone) at the various time points. This is indicative of clinical efficacy in vivo and also reinforces the pharmacokinetics of each product without the need for plasma level determinations by typical chemical analyses, e.g., high-performance liquid chromatography. This is based on the assumption that the serial dilutions at the different time points should result in the same inhibitory patterns if the same concentrations are available in in vivo bioassays, i.e., zone of inhibition of standard laboratory strain of K. pneumoniae. The plasma concentrations in patients were calculated based on the inhibition zones ex vivo.11,12 This is, however, a standard technique to demonstrate quality and outcome when testing the contents of vials.
Sample collection
Clotted blood samples were centrifuged and serum collected and divided into two 5 mL marked tubes and frozen at −70°C until tested.
Statistical analyses
The primary outcome was equivalence between the comparator and generic brand products. The pharmacokinetic (PK) parameters of area under the concentration-time curve to last time point t (AUC0–t) and area under the concentration time curve to infinity (AUC0–inf) for the two treatment groups were estimated by non-compartmental analysis.13 Bootstrapped estimates were used (1,000 replicates). Equivalence of AUC was assessed by estimating confidence intervals (CIs) for the ratio of the two AUC0–t estimates. The observed peak plasma concentration (Cmax) was used. Since Cmax was not estimated, equivalence of Cmax was determined using the two one-sided test approach on the log-transformed observations, with a region of similarity of ±0.223 (0.8–1.25 on linear scale) and α =0.05. Pharmacodynamic (PD) data (zone of inhibition) was modeled using a random intercept mixed effects model, with innovator meropenem as reference. Equivalence of ex vivo efficacy, i.e., inhibition diameter, was evaluated using a standard CI approach, using bootstrapped CI estimates of the treatment effect (1,000 replicates).14 Statistical analyses were performed using Microsoft R Open 3.2.4. (default CRAN mirror snapshot taken on 2016-04-01).15
Results
In vitro assay validation
The biological activity of both test products was verified against international reference standards (zones of inhibition against Gram-positive and Gram-negative organisms) and good comparisons within prespecified range, i.e., limits of error being 95%–105% of the standards response were obtained (Figure 1 and Table 2). Both the test products showed parallelism compared to the USP meropenem standard over the complete range of dilutions in the test method to be used for ex vivo testing of the same products.
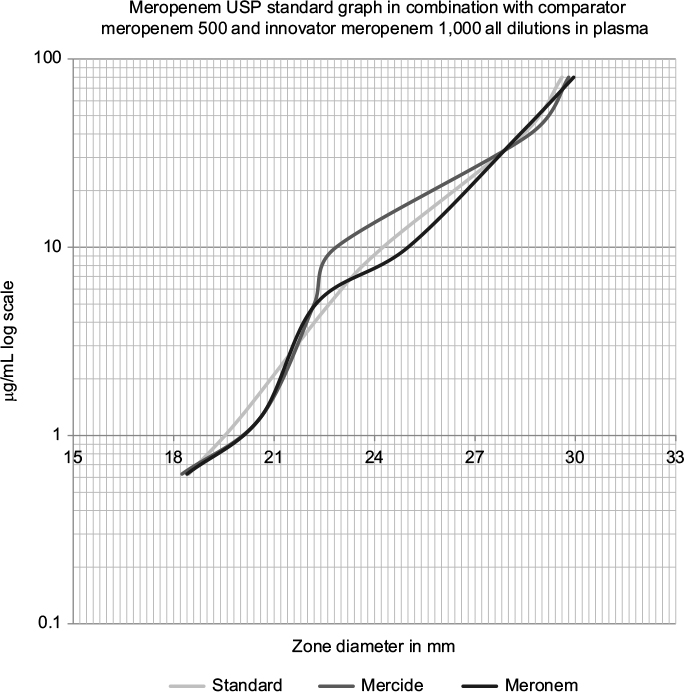 | Figure 1 Meropenem USP standard compared to comparator meropenem and innovator meropenem diluted in human donor plasma. Abbreviation: USP, United States Pharmacopeia. |
Ex vivo testing
Twenty-seven patients completed the study, of which data were complete for 24 patients for all time points (innovator meropenem n=13 and comparator meropenem n=14). The data of 3 patients were not utilized who had received an antimicrobial other than meropenem with a similar spectrum for K. pneumoniae just before sample collection or during sample collection, which then altered the zone of inhibition already at time zero, or demonstrated synergism at one of the other collection times, which would have confounded any scientific conclusions. One participant was excluded from PK and PD analysis with estimated plasma levels 2–3 times that of the remaining participants. This anomaly could not be explained other than possible faulty (duplicate) drug administration at the time of sample collection and hence the patient was excluded from analyses. PK and PD results are reported for the data from 20 participants (innovator meropenem n=9 and comparator meropenem n=11).
The estimated plasma concentrations from both products were comparable (Figure 2), with noticeable overlap of CIs at all the observation time points. The comparator meropenem treatment arm demonstrated 4% higher mean plasma AUC0–t compared with the innovator. AUC equivalence was assessed based on the 90% CI of the ratio of AUC0–t (comparator meropenem) / AUC0–t (innovator meropenem). By definition,16 bioequivalence could not be concluded for AUC0–t as the upper bound of the 90% CI exceeded 1.25, indicative of higher AUC0–t estimates for comparator meropenem, compared to innovator meropenem. The lower bound of the ratio of AUC0–t estimates was, however, well within the specified threshold of 0.8 (Table 3).
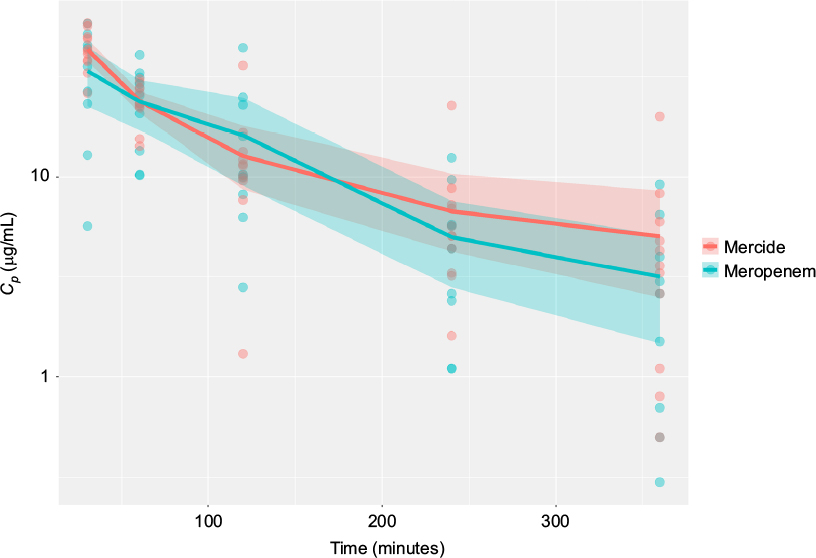 | Figure 2 Superimposed log-linear plots of plasma concentration time curves for meropenem present in patients’ serum over a 6-hour period. Notes: Cp = plasma concentration (log-scale y-axis). Lines and ribbons represent means and 95% confidence intervals, respectively, based on bootstrapped estimates (10,000 replicates). Points represent actual observations. |
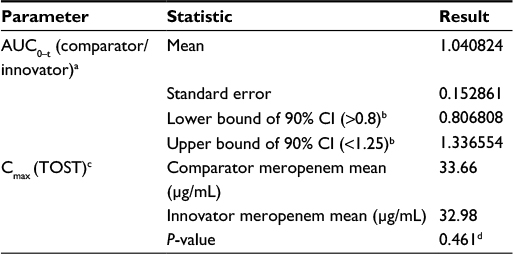 | Table 3 Results of statistical assessment of the equivalence of PK parameters Notes: aBootstrapped estimate, based on 1,000 replicates, bReference for bioequivalence16, cTOST approach performed on log-transformed data, dNull hypothesis not rejected, i.e., equivalence assumed. Abbreviations: AUC0–t, area under the concentration-time curve to last time point, t; CI, confidence interval; Cmax, observed peak plasma concentration; PK, pharmacokinetic; TOST, two one-sided test. |
Mean ± standard deviation estimates of Cmax for comparator and innovator meropenem were 33.66±15.31 µg/mL and 32.98±21.30 µg/mL, respectively. Equivalence was concluded with respect to Cmax, as the null hypothesis was not rejected for the two one-sided test hypothesis testing approach (P-value = 0.461). See Table 3.
Mean (95% CI) estimates of zone of inhibition for comparator meropenem and innovator meropenem were 18.2 (16.7; 19.6) mm and 18.5 (16.9; 20.1) mm, respectively. Results are graphically represented in Figure 3. Equivalence was concluded, based on the 90% CI of the coefficient of the effect of treatment on the zone of inhibition (−2.24; 1.39), which, statistically, did not differ from 0. Considering an intercept of 18.73, effects of −2.24 and 1.39 indicate a <±15% change in outcome (Table 4). This can easily be ascribed to variability inherent in experimentation in the biological sciences. For this reason, the effect of treatment can be considered clinically not relevant. Furthermore, statistically, the coefficient for treatment effect was not significantly different from 0 (P-value = 0.76).
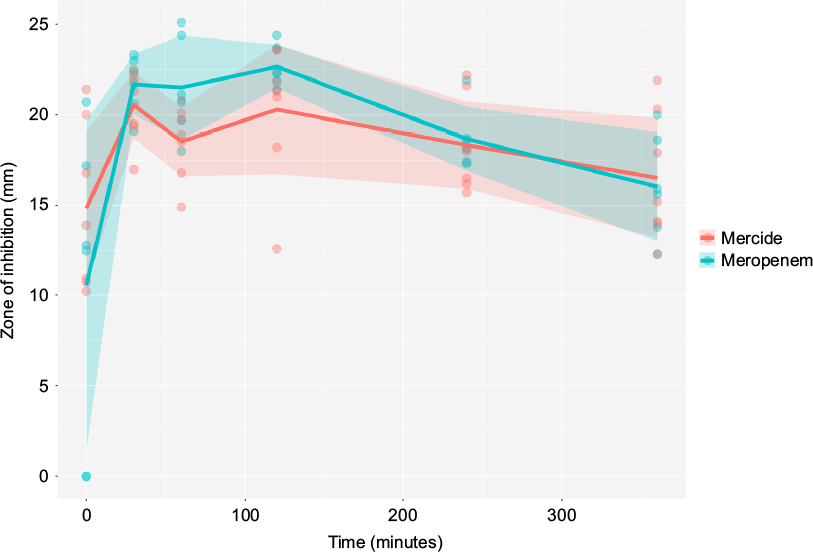 | Figure 3 Superimposed plots of zone of inhibition data collected over a 6-hour period. Lines and ribbons represent means and 95% confidence intervals, respectively. Points represent actual observations. |
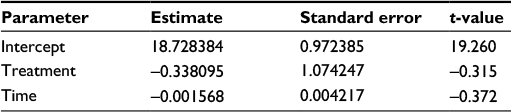 | Table 4 Parameter estimates of a random intercept mixed effects model of the equivalence of PD data (zone of inhibition data) Notes: No significant treatment × time effect was detected. Abbreviation: PD, pharmacodynamic. |
Discussion and conclusion
It is common to substitute innovator brand products with multisource generics in order to curtail cost. These latter products must, by international norms and regulations, be of the same quality and guarantee the same clinical outcomes.16–18 However, when using antimicrobials, especially in critically ill patients, the use of generic products is often met with resistance and sometimes with good reason when one considers the number of publications citing inferior test outcomes, i.e., quality, and in vitro and/or in vivo study results.5,6 Antimicrobials target microbes and therefore the effectiveness of products should be established in vivo to verify similarity when innovator brands are tested against generics.17 This antimicrobial efficacy should also be related to the products’ tested PKs.11
A recent study by Leelarasamee et al11 describes a similar type of ex vivo confirmation of antibacterial equivalence of two meropenem products at specific plasma concentrations. In the present study the biological test (zone of inhibition) was used to demonstrate clinical, i.e., time-point specific antibacterial effectiveness, after infusion of the test products in everyday clinical practice. The latter test outcome was then back extrapolated to plasma concentrations. This was done by constructing concentration curves based on known concentrations correlating with growth inhibition zones against two test organisms. As in the Leelarasamee et al11 study, the clinical effectiveness was assumed based on ex vivo antimicrobial activity. The latter served as a proxy for clinical outcomes, as doing a randomized controlled study with clinical endpoints would have been impractical. The aim of the present study was not to confirm bioequivalence of the test product but to demonstrate clinical similarity in everyday practice and therefore the study was not powered or designed as a bioequivalence study from the outset. The study achieved this object by demonstrating similar ex vivo inhibition zones at predetermined time points, which could be back extrapolated to concentrations in vivo.
In this study, it was clearly demonstrated that the two products under observation performed similarly in critically ill patients with respect to kinetic profile based on ex vivo inhibition of K. pneumoniae growth. Bioequivalence was not concluded in terms of AUC0–t, due to the AUC0–t estimates for the generic being too high, compared to the innovator, but the lower bound of the bioequivalence specifications was easily met. Equivalence was concluded for both Cmax and zone of inhibition data. This clinical activity suggests true similarity in vivo. Since the patients were so diverse in disease entity, i.e., severity and type, no meaning in such a small study could be assigned to true clinical outcome of each individual antimicrobial and brand. This was also not the aim of the study, but rather to demonstrate uniformity without exposing large number of vulnerable patients to products in a typical prospective double-blind treatment allocation study, as well as to assess real-life clinical practice using a non-invasive study design to demonstrate the same.
Acknowledgments
The authors hereby acknowledge the contributions of Clindev Pty Ltd as the CRO for the study, while also helping to conduct the study.
Funds for this study were provided by Ranbaxy (S.A) (Pty) Ltd; 3rd Floor Outspan House; 1006 Lenchen Avenue North; Centurion, Pretoria, Gauteng, South Africa.
Disclosure
The authors report no conflicts of interest in this work.
References
Vesga O, Agudelo M, Salazar BE, Rodriguez CA, Zuluaga AF. Generic Vancomycin products fail in vivo despite being pharmaceutical equivalents to the innovator. Antimicrob Agent Chemother. 2010;54(8):3271–3279. | ||
Kogut SJ, Spooner LM. Resistance to in-office dispensing of generic antibiotic samples. J Manag Care Pharm. 2009;15(1):62–65. | ||
Snyman JR. Bioequivalence, should we trust generic antibiotics? South Afr J Epidemiol Infect. 2005;20(3):78–79. | ||
Finch R. Generic antibiotics, antibiotic resistance and drug licensing. Lancet Infect Dis. 2010;10(11):754–754. | ||
Dunne SS, Dunn CP. What do people really think of generic medicines? A systematic review and critical appraisal of literature on stakeholder perceptions of generic drugs. BMC Medicine. 2015;13(1):1–27. | ||
Tattevina P, Crémieux AC, Rabaude C, Gauzitf R. Efficacy and quality of antibacterial generic products approved for human use: a systematic review. Clin Infect Dis. 2014;58(4):458–469. | ||
World Health Organization. Multisource (generic) pharmaceutical products: guidelines on registration requirements to establish interchangeability. WHO Tech Rep Ser. 2006;937:347–390. | ||
Zuluaga AF, Agudelo M, Rodriguez CA, Vesga O. Application of microbiological assay to determine pharmaceutical equivalence of generic intravenous antibiotics. BMC Clin Pharmacol. 2009;9(1):1–11. | ||
Liebowitz LD, Slabbert M. Comparative in vitro activity of generic injectable cephalosporins. South Afr J Epidemiol Infect. 2005;20(3):82–84. | ||
Richards GA, Elliott E, Shaddock EJ, et al. A comparison of the pharmacokinetics of aspen ceftriaxone and rocephin in community-acquired meningitis. S Afr Med J. 2013;103(12):906–909. | ||
Leelarasamee A, Sathirakul K, Trakulsomboon S, Pongpech P, Naenna P. Bioequivalence study with comparative antibacterial activity of a generic meropenem. J Infect Dis Antimicrob Agents. 2008;25:63–72. | ||
Bennett JV, Brodie JL, Benner EJ, Kirby WM. Simplified, accurate method for antibiotic assay of clinical specimens. Appl Microbiol. 1966;14(2):170–177. | ||
Jaki T, Wolfsegger MJ. Estimation of pharmacokinetic parameters with the R package PK. Pharm Stat. 2011;10(3):294–288. | ||
Bates D, Maechler M, Bolker B, Walker S. Fitting linear mixed-effects models using lme4. J Stat Softw. 2015;67(1):1–48. | ||
R-project.org [homepage on the internet]. Austria: The R Foundation for Statistical Computing;c2016. Available from: http://r-project.org/. Accessed April 01, 2016. | ||
U.S. Department of Health and Human Services, Food and Drug Administration, Center for Drug Evaluation and Research (CDER). Approved drug products with therapeutic equivalence evaluations [serial on the internet]; 2015 [31 December]: 1320 pp. Available from: http://www.fda.gov/downloads/Drugs/DevelopmentApprovalProcess/UCM071436.pdf. Accessed January 26, 2016. | ||
U.S. Department of Health and Human Services, Food and Drug Administration, Center for Drug Evaluation and Research (CDER). Guidance for industry: Microbiological data for systemic antibacterial drug products - Development, analysis, and presentation [serial on the internet]; 2009 Sep [36 p]. Available from: http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/ Guidances/UCM182288.pdf. Accessed May 10, 2015. | ||
U.S. Department of Health and Human Services, Food and Drug Administration, Center for Drug Evaluation and Research (CDER). Guidance for Industry: Submission of summary bioequivalence data for ANDAs [serial on the internet]; 2011 [May] 9 pp. Available from: http://www.fda.gov/downloads/Drugs/.../Guidances/UCM134846.pdf. Accessed May 10, 2015. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.