Back to Journals » Biologics: Targets and Therapy » Volume 11
A practical guide about biosimilar data for health care providers treating inflammatory diseases
Authors Markenson J, Alvarez DF, Jacobs I, Kirchhoff C
Received 14 September 2016
Accepted for publication 14 December 2016
Published 24 February 2017 Volume 2017:11 Pages 13—21
DOI https://doi.org/10.2147/BTT.S122335
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Doris Benbrook
Joseph Markenson,1,2 Daniel F Alvarez,3 Ira Jacobs,4 Carol Kirchhoff5
1Department of Rheumatology, 2Department of Medicine, Hospital for Special Surgery, New York, NY, 3Pfizer Inc, Collegeville, PA, 4Pfizer Inc, New York, NY, 5Pfizer Inc, Chesterfield, MO, USA
Abstract: To make informed decisions about the safety, efficacy, and clinical utility of a biosimilar, health care providers should understand the types and be able to analyze data generated from a biosimilar development program. This article reviews the biosimilar guidelines, the biosimilar development process to provide education and context about biosimilarity, and uses examples from infliximab biosimilars to review the terminology and potential types of analyses that may be used to compare potential biosimilars to the originator biologic. A biosimilar is a biologic product that is highly similar to an approved (originator) biologic, notwithstanding minor differences in clinically inactive components, and with no clinically meaningful differences in terms of the safety, purity, and potency of the product. Due to their complex nature and production in living systems, it is not possible to exactly duplicate the approved originator biologic. To ensure biosimilars provide consistent, safe, and effective treatment comparable to the originator biologic, extensive analyses of the potential biosimilar are conducted, including side-by-side analytical, nonclinical, and clinical comparisons. A key goal is to determine whether there are sufficient relevant similarities in chemical composition, biologic activity, and pharmacokinetic aspects between the potential biosimilar and the originator. Regulatory approvals and marketing authorizations for biosimilars are made on a case-by-case and agency-by-agency basis after evaluating the totality of the evidence generated from the entire development program. Understanding how regulatory agencies review data for approval can help health care providers make appropriate decisions when biosimilars become available for use in the treatment of inflammatory diseases, and therefore they should review the literature to gain further information about specific biosimilars.
Keywords: biosimilar, data analysis, inflammatory disease, rheumatoid arthritis
Introduction
Biologic drugs such as abatacept, adalimumab, anakinra, certolizumab pegol, etanercept, golimumab, infliximab, rituximab, secukinumab, and tocilizumab have made significant impact in the treatment of rheumatoid arthritis and other inflammatory diseases.1 These therapies specifically target key modulators of the inflammatory response, such as proinflammatory cytokines, lymphocytes (eg, T- or B-cells), and cell surface receptors.2 Although biologic therapies have demonstrated clinical efficacy, availability and patient access to these therapies may be limited, which can impact patient outcomes.3 Legislation and guidelines from regulatory agencies that allow development and approval of biosimilars may help increase patient access and improve disease outcomes.
Recently, the first biosimilars for inflammatory diseases have been approved by the European Medicines Agency (EMA), the US Food and Drug Administration (FDA), and regulatory agencies from other countries (Table 1).4–9 As other patents and data protection periods expire, additional biosimilars of biologic drugs currently used in inflammatory diseases are expected.
 | Table 1 Recently approved biosimilars for inflammatory diseases Note: aRemsima is also marketed as Inflectra (Hospira, Maidenhead, UK) in the EU. Abbreviation: EU, European Union. |
To make well-informed decisions about appropriate clinical use of a biosimilar, it is important for health care providers to understand and be able to analyze data generated from a biosimilar development program objectively. To help in this process, we use the biosimilar guidelines and examples from infliximab biosimilars to review the terminology and potential types of analyses that may be used to compare potential biosimilars to the originator biologic to demonstrate similarity.
Definition of a biosimilar
As defined by the FDA, a biosimilar is a biologic product that is “highly similar to a reference product (a licensed biologic product [hereafter also referred to as an originator or originator biologic]) notwithstanding minor differences in clinically inactive components.”10 In addition, there must be demonstration that there are “no clinically meaningful differences between the [biosimilar] biological product and the [originator] reference product in terms of the safety, purity, and potency of the product.”10 In contrast to a generic copy of a small-molecule (chemical) drug, which can be fully defined structurally and reproduced with an identical chemical structure, a biosimilar cannot be identical to the originator biologic. This is because biologic drugs are made in living cells, and the proprietary manufacturing processes are specialized and never fully disclosed by the manufacturer of the originator.11 Therefore, the biosimilar developer will use its own manufacturing processes that may involve different biologic systems.10 These different processes and systems may result in minor differences between the products.10 A key goal is to determine whether there are sufficient relevant similarities in chemical composition, biologic activity, and pharmacokinetic aspects between the potential biosimilar and the originator.11 Differences between the biosimilar and the originator biologic must be understood and justified as to their relevance to safety and efficacy of the product.10
The goal of the regulatory approval pathway for biosimilars is to develop a molecule with similar physicochemical properties, efficacy, and safety as the originator biologic. To ensure that biosimilars provide a safe and effective treatment that is similar to the originator, extensive quality control measures are in place, including a direct, comprehensive, comparative analysis of the potential biosimilar to the originator. There are some biologic products developed with the intention of being similar to an originator product that do not undergo a comprehensive side-by-side analytical, nonclinical, or clinical comparison established by the EMA, the FDA, and the World Health Organization (WHO). These products are considered “intended copies” or “noncomparable biologics” rather than biosimilars.12 For those products that will undergo comprehensive comparison, the demonstration of biosimilarity is conducted via a stepwise approach integrating multiple measures to characterize these complex products.11
Overview of regulatory approval of a biosimilar
As described, due to the complex nature of biologics, biosimilars are not considered generic equivalents of the originator biologic.10,13,14 Therefore, regulatory approval for a potential biosimilar requires more data than for approval of a small-molecule generic.10,14,15 Regulatory agencies worldwide, such as the EMA and the FDA, as well as the WHO, have developed guidelines for the approval of biosimilar products (Table 2).10,14,15 Most countries have adopted the principles from one or more of these regulatory guidelines (outside the USA, the majority follow the EMA or WHO guidelines), but some countries are not as strict and allow licensing of products without strict evaluation of efficacy and safety.16
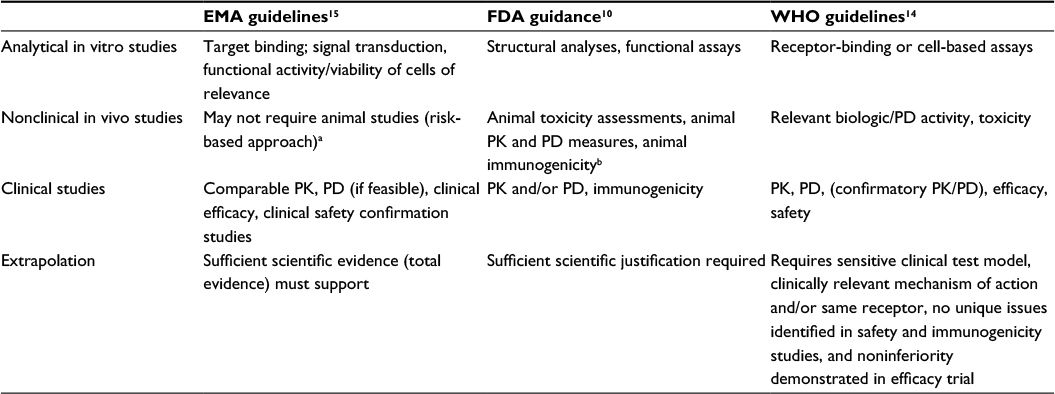 | Table 2 Overview of regulatory requirements for biosimilars Notes: Adapted from Socinski MA, Curigliano G, Jacobs I, Gumbiner B, MacDonald J, Thomas D. Clinical considerations for the development of biosimilars in oncology. MAbs. 2015;7(2):286–293.37 aIf in vitro comparability is satisfactory and does not identify any factors that would block direct entry to humans. bDiscussions between the FDA and the developer may potentially indicate that animal studies are not necessary. Abbreviations: EMA, European Medicines Agency; FDA, US Food and Drug Administration; PD, pharmacodynamics; PK, pharmacokinetics; WHO, World Health Organization. |
The EMA, FDA, and WHO guidelines have laid out generally similar requirements for approval of a biosimilar, including demonstration of biosimilarity, along with the use of a stepwise approach to develop the evidence and the provision that the recommendation for approval is based on the “totality of the evidence” for biosimilarity.10,14,15 The stepwise approach enables the biosimilar developer and the regulatory agencies to determine the extent of residual uncertainty of biosimilarity at each step of development and to identify additional relevant studies and analyses that may be needed to resolve this uncertainty.17 Totality of the evidence means that the application for regulatory approval for a potential biosimilar includes a comprehensive data package from all stages of development (ie, analytical and functional comparability, animal studies, and clinical data). For biosimilars, the primary focus of the comparability exercise is on analytical similarity and not on clinical efficacy and safety studies.10,14,15 This exercise is analogous to comparisons performed after manufacturing changes of the originator.18 However, the biosimilarity exercise is more extensive and addresses some fundamental differences between the proposed biosimilar and the originator, including use of a different cell line and manufacturing processes versus the originator; thus, more data are required to ensure that the biosimilar has similar clinical efficacy and safety.18 Although regulatory agencies acknowledge that some studies could be performed in parallel, and this would be permissible, in most cases, the recommendation is to follow the stepwise approach to allow the developer to incorporate feedback provided after agency review of the data and information collected at specific milestones and thus facilitate regulatory approval.10,15
To demonstrate biosimilarity, developers usually conduct several types of studies to compare safety, purity, and potency. The types of studies conducted may vary, and no firm set of requirements has been established, although the regulatory guidelines identify broad categories of types of data that may support the demonstration of biosimilarity. These categories include extensive analytical characterization and comparison demonstrating that the biological product is highly similar to the originator biologic; nonclinical animal studies, including assessment of toxicity; and a clinical study (or studies) to demonstrate similar pharmacokinetics (PK), safety, efficacy, and immunogenicity (Table 3).
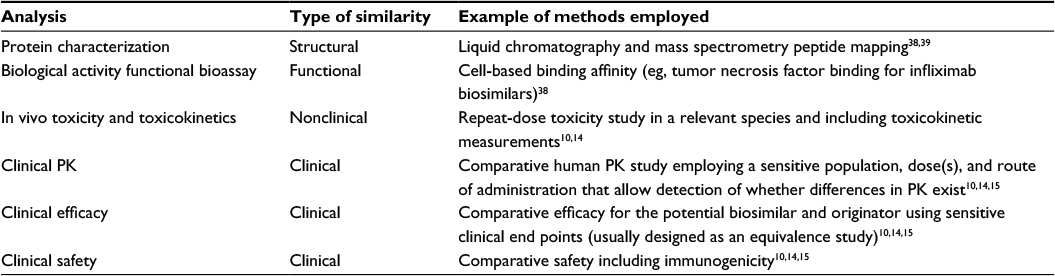 | Table 3 Examples of the types of analyses that may be used to demonstrate similarity of a potential biosimilar to the originator biologic Abbreviation: PK, pharmacokinetics. |
It is a common misconception that there are specific types of analyses or specific assays required for all biosimilar (or even for originator biologic) development programs, although developers may use similar methods for evaluating potential biosimilars. Instead, the choice of appropriate analyses will depend on the characteristics of the originator, and decisions are made on a case-by-case and agency-by-agency basis. In addition, it is likely that regulatory processes, types of analyses, and the amount of data required in approval applications will evolve as more biosimilars are evaluated.
Data considerations for regulatory approval of biosimilars
It is important for health care providers to understand that the biosimilar approval pathway tends to place a greater emphasis on data from comparative analytical studies and less on clinical trials, which is in contrast to the approval pathway for a novel biologic. There are several reasons for this. First, biosimilars do not require proof of the mechanism of action because, by definition, this is the same as the originator biologic agent.15 Second, dose-finding studies are not required for a potential biosimilar since the optimal dose has been established for the originator biologic. In addition, the goal of confirmatory clinical trials is demonstration of similar efficacy and safety (including comparable potency, PK, pharmacodynamics [PD], and immunogenicity) between the biosimilar and the originator biologic.14,15 Finally, the goal of the clinical trial program is not to re-establish patient benefit, since this has already been demonstrated through the clinical studies conducted by the developer of the originator.
To adequately develop a product that is similar to an approved biologic therapy, developers of biosimilars must analyze the originator biologic extensively. Since manufacturing processes are proprietary, the developer of a potential biosimilar must employ a process of reverse engineering. This requires substantial knowledge and expertise regarding the development and manufacture of biologics to determine structural and functional properties of the originator and then mimic these properties as closely as possible in the potential biosimilar. Analyses used to fully characterize the originator can also be employed subsequently to demonstrate similarity of the biosimilar to the originator. Thus, it is likely that the number of analyses performed to demonstrate structural and functional biosimilarity may be greater than those included as part of the originator’s license application. This results in a significant amount of analytical similarity data for a potential biosimilar and the originator biologic for the regulatory approval process.
The similarity assessment conducted as part of the analytical evaluation includes an extensive structural and functional characterization and comparison between the proposed biosimilar and the originator using state-of-the-art analytical methods. For example, regulatory agencies expect detailed analysis of primary amino acid sequences for the potential biosimilar and the originator biologic.10,14 If minor differences in primary structure such as N- or C-terminal truncations are identified, the effects on safety and efficacy should be evaluated and explained by the sponsor to confirm that these differences do not change product performance.10,14 Additional data should also be provided from relevant characterization studies, including higher order structural analyses (assessed, eg, via circular dichroism among other techniques), posttranslational modifications (assessed, eg, as glycosylation analysis via mass spectrometry), biological activity (assessed by a number of bioassays, such as target-binding affinity or cell-based bioassays), and any other relevant characteristics.10,13–15,19–21
The demonstration of structural similarity should be completed before progression to the next step of development, but the exact nature of the analyses and data vary. There is no established set of required analyses, although most biosimilar developers will conduct several studies to confirm that the biosimilar has a similar quality target product profile as the originator. For example, if the data published for potential biosimilars to infliximab are compared, the types of analyses conducted vary (Table 4). It should be noted that the data published likely represent only a portion of the full analytical assessments conducted on a particular biologic and potential biosimilar; more data may be disclosed in future publications, documents released from regulatory agencies, or the product label.
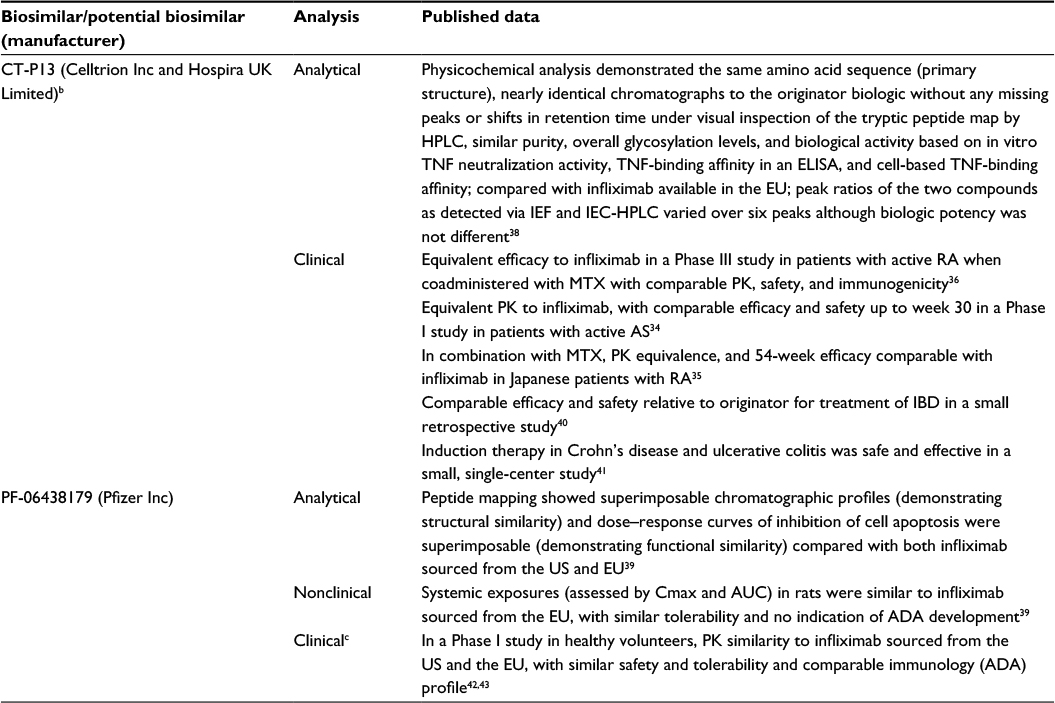 | Table 4 Samples of the types of data that may support demonstration of biosimilarity (using potential biosimilars approved or in development for infliximab as an example)a Notes: aPublications identified using the search term “biosimilar infliximab” on PubMed or Web of Science databases. All data for a given potential biosimilar may not have been published at the time PubMed and Web of Science databases were searched. bMarketed as Remsima (Celltrion Inc, Incheon, Republic of Korea) and Inflectra (Hospira UK Limited, Warwickshire, UK). cA comparative clinical study is ongoing at the time of this manuscript development. Abbreviations: ACR20, American College of Rheumatology 20% improvement criteria; ADA, antidrug antibody; AS, ankylosing spondylitis; AUC, area under the plasma concentration–time curve; Cmax, maximum plasma concentration; ELISA, enzyme-linked immunosorbent assay; EU, European Union; HPLC, high-performance liquid chromatograph; IBD, irritable bowel disease; IEC, ion exchange chromatography; IEF, isoelectric focusing; MTX, methotrexate; PK, pharmacokinetics; RA, rheumatoid arthritis; TNF, tumor necrosis factor. |
Nonclinical studies are conducted to demonstrate that the biosimilar acts on the same target or physiologic process and has similar toxicity as the originator (Table 4).10,13,14 Generally, nonclinical studies include animal studies to assess similarity of in vivo activity and toxicokinetics. However, animal studies need to be justified and, in some cases, may not be necessary if no issues that would require additional characterization before the potential biosimilar can be assessed in clinical studies (eg, factors that could impact PK, such as extensive glycosylation) are identified or if a relevant in vivo animal model is not available.10,13,14 When appropriate, these studies may parallel or replicate studies conducted for the originator biologic.
Clinical data considerations for biosimilars
The clinical trial program for a potential biosimilar includes assessments of PK, clinical efficacy, clinical safety, and immunogenicity with the aim to show the similarity of the potential biosimilar to the originator (Table 4).10,13,14 As described, clinical benefit of a biosimilar is expected, by definition, to be the same as that of the originator, so clinical trials evaluating efficacy of the biosimilar versus a placebo or dose-finding studies are not required.13,14 Instead, clinical trials are specifically designed as a final comparative evaluation step(s) and to confirm whether the product can be considered a biosimilar, as well as to address remaining residual uncertainty. Regulatory agencies advise that, whenever possible, the design and conduct of clinical studies should be performed in a way to maximize the contribution to demonstrating biosimilarity (ie, sensitive to detect clinical meaningful differences should they exist).10
The first step of a tailored clinical trial program usually begins with a clinical PK comparison to the originator biologic; however, PK and PD may be evaluated in a combined study if appropriate PD markers exist.10,13,14 Generally, PK assessments are conducted in healthy volunteers to reduce variability unrelated to differences between products. This may not always be appropriate, however, if the originator biologic causes pharmacologic effects that would be unacceptable for healthy volunteers but appropriate in a patient population.13,14
After PK similarity is demonstrated, a clinical comparative study (or studies) is conducted in patients.10,13,14 The study is conducted using a sensitive population and method to identify any potential differences in efficacy, safety, or immunogenicity. Thus, it is possible that the population in clinical trials evaluating a potential biosimilar may not be the same as that employed in pivotal clinical trials for the originator.10,13,14
Clinical trials to compare clinical efficacy end points between the biosimilar and the originator should usually be conducted as equivalence studies.10,14,15 In contrast to the design employed to evaluate a novel therapeutic, which is commonly a superiority trial (its primary objective is to show the investigational product is superior to an active or placebo control), equivalence trials are designed to evaluate differences in response between two treatments to support the hypothesis that the biosimilar and originator have no clinically meaningful differences and that neither agent is superior or inferior to the other.14,22 Equivalence of two treatments is demonstrated when the entire confidence interval of the treatment difference lies within a prespecified range of clinically acceptable differences (ie, the lower and upper equivalence margins established prior to the experiment or study).23 Occasionally, noninferiority studies, which evaluate whether a biosimilar is not clinically inferior to the originator (thus, using only one margin), may be employed for assessment of biosimilars if the study population and end point(s) are appropriate and sufficient scientific justification is provided.14,22 For example, establishing that the originator biologic reaches target saturation at the clinical dose used in the clinical trial may indicate that a noninferiority study design is sufficient.10
End points selected for demonstrating comparable efficacy and safety must allow determination of whether any clinically meaningful differences exist between the potential biosimilar and the originator.10 Since some end points (such as PD measures) are more sensitive than clinical end points, these may be selected as the primary end points of a biosimilar clinical study to enable more precise comparisons to the originator.10 This means that the primary end points employed may not be the same as those used in the pivotal clinical trials for the originator, although the regulatory guidelines recommend inclusion of some common end points at least as secondary end points to enable comparisons across products.10,13 End points that could be used to evaluate efficacy in potential biosimilars with inflammatory disease indications include the American College of Rheumatology 20% response rate, the Disease Activity in 28 joints score, the Crohn’s Disease Activity Index, the Ankylosing Spondylitis Disease Activity Score, and the Psoriasis Area and Severity Index score.24
In addition to the potential for differences in the patient population and study end points, the entire clinical program of the originator should not need to be replicated for approval of the biosimilar because the guidelines indicate that extrapolating efficacy and safety data from clinical studies of the potential biosimilar to other indications may be possible.10,14,15 Extrapolation is the approval of a biosimilar for use in an indication held by the originator that is not directly studied in a comparative clinical trial with the biosimilar through the use of all data collected (the totality of the evidence from all comparability analyses, not just the clinical data).10,14,15 Extrapolation is core to the concept of biosimilarity and reduces or eliminates the need for duplicative clinical studies for the biosimilar in multiple indications.10,13,14 The decision to grant approval in other indications is based on the totality of the evidence via extrapolation and conducted on an agency-by-agency and case-by-case basis.10,13,14 Thus, various regulatory agencies may arrive at different decisions as to whether to allow extrapolation of the data for a given potential biosimilar. This was the case of the recent regulatory approvals for the biosimilar infliximab products Remsima and Inflectra that were granted approval for the full range of indications of the originator biologic by EMA, whereas Health Canada initially did not grant approval across all indications due to functional differences between products, which correlated with pertinent differences in the mechanism of action that did not support extrapolation to Crohn’s disease and ulcerative colitis.25–28 More recently, Health Canada approved Remsima and Inflectra for Crohn’s disease, fistulizing Crohn’s disease, and ulcerative colitis.29 The additional indications were approved based on similarity between the biosimilar and originator in product quality, mechanism of action, disease pathophysiology, safety profile, dosage regimen, and on clinical experience with the originator.29 In addition, data from prospective studies in IBD are becoming available.30–32
At the time of regulatory approval of any drug, the data from clinical studies are usually too limited to identify all potential safety issues, particularly rare but potentially serious adverse events.10,14,15 Thus, as typically occurs with a novel biologic, ongoing postmarketing safety monitoring for an approved biosimilar will likely be needed to evaluate long-term safety and, thus, plans are included in applications for regulatory approval.10,14,15 These plans should be based on any known safety signals associated with the originator, known risks associated with the drug class, and any novel safety concerns arising during evaluation of the biosimilar, but typically do not require a specific number of subjects.10,14,15 In addition, although immunogenicity is evaluated as part of the clinical safety studies of the potential biosimilar, ongoing postapproval follow-up is expected to specifically monitor long-term immunogenicity.10,14,15
Some physician groups have expressed concerns about switching between the originator and biosimilars, particularly if this is conducted via automatic substitution at the pharmacy.33 However, several studies conducted have shown that patients who switched to an infliximab biosimilar had similar efficacy and tolerability to those who continued on the originator in patients with RA, AS, and IBD.30,34–36 This is an area that will continue to be examined closely as more data, and additional biosimilars become available.
Summary and conclusion
As more biosimilars are approved, the availability of additional treatment options may change the treatment of inflammatory and rheumatic diseases through increased access to biologics. The regulatory approval process for biosimilars relies on a considerable amount of data from all stages of development, particularly physicochemical and functional comparisons. Regulatory decisions are made on a case-by-case and agency-by-agency basis, and therefore not all biosimilars for a given originator may receive the same approvals worldwide. In addition, the amount of data available for health care providers to review to make prescribing decisions may differ and may not cover the full breadth of data supporting demonstration of biosimilarity. Health care providers should expect to see different types of (and likely less) clinical data than what they usually see for originator products. Understanding how regulatory agencies make decisions for approval and what data are available for review can help health care providers have increased confidence in biosimilars. In addition, because product labeling and prescribing information policies are not consistent across regulatory agencies globally and may provide varying amounts of data in the product label, which demonstrates similarity of the biosimilar to the originator, health care providers should review the literature to gain further information about specific biosimilars.
Acknowledgments
Medical writing support was provided by Christina McManus, PhD, of Engage Scientific Solutions and funded by Pfizer Inc.
Disclosure
Joseph Markenson has received personal fees for participation in speakers’ bureaus and advisory boards for Amgen, BMS, Jansen, Abbvie, Celgen, Pfizer, and IROKO and for participation in advisory boards for Lilly. Daniel Alvarez, Carol Kirchhoff, and Ira Jacobs are employees of Pfizer and hold stock or stock options. The authors report no other conflicts of interest in this work.
References
Scheinberg MA, Azevedo VF. Biosimilars in rheumatology: perspective and concerns. Rheumatology (Oxford). 2014;53(3):389–390. | ||
Quan LD, Thiele GM, Tian J, Wang D. The development of novel therapies for rheumatoid arthritis. Expert Opin Ther Pat. 2008;18(7):723–738. | ||
Desai RJ, Rao JK, Hansen RA, Fang G, Maciejewski ML, Farley JF. Predictors of treatment initiation with tumor necrosis factor-alpha inhibitors in patients with rheumatoid arthritis. J Manag Care Pharm. 2014;20(11):1110–1120. | ||
Biogen [webpage on the Internet]. FLIXABI®, Biogen’s infliximab biosimilar referencing Remicade®, approved in the European Union; c2016. Available from: http://media.biogen.com/press-release/biosimilars/flixabi-biogens-infliximab-biosimilar-referencing-remicade-approved-Europe. Accessed June 17, 2016. | ||
Biogen, Samsung BioLogics [webpage on the Internet]. BENEPALI®, the first etanercept biosimilar referencing Enbrel®, approved in the European Union. Zug, Switzerland: Business Wire; c2016. Available from: http://www.businesswire.com/news/home/20160116005011/en/BENEPALI%C2%AE-Etanercept-Biosimilar-Referencing-Enbrel%C2%AE-Approved-European. Accessed June 28, 2016. | ||
European Medicines Agency [webpage on the Internet]. European Medicines Agency recommends approval of first two monoclonal-antibody biosimilars [press release]; c2013 [updated June 28, 2013]. Available from: http://www.ema.europa.eu/ema/index.jsp?curl=pages/news_and_events/news/2013/06/news_detail_001837.jsp&mid=WC0b01ac058004d5c1. Accessed July 31, 2015. | ||
Generics and Biosimilars Initiative (GaBI) [webpage on the Internet]. Biosimilar monoclonal antibody approved in Korea. Mol, Belgium: GaBI Online; c2012 [updated March 8, 2012]. Available from: http://www.gabionline.net/Biosimilars/News/Biosimilar-monoclonal-antibody-approved-in-Korea. Accessed May 10, 2016. | ||
Generics and Biosimilars Initiative (GaBI) [webpage on the Internet]. Biosimilar infliximab launched in Japan [media release]. Mol, Belgium: Pro PharmaCommunications International; c2014 [updated December 5, 2014]. Available from: http://www.gabionline.net/Biosimilars/News/Biosimilar-infliximab-launched-in-Japan. Accessed May 10, 2016. | ||
United States Food and Drug Administration [webpage on the Internet]. FDA approves Inflectra, a biosimilar to Remicade; c2016. Available from: http://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm494227.htm. Accessed June 17, 2016. | ||
US Food and Drug Administration. Scientific Considerations in Demonstrating Biosimilarity to a Reference Product. Guidance for Industry. Silver Spring, MD: US Department of Health and Human Services, Food and Drug Administration, Center for Drug Evaluation and Research (CDER), Center for Biologics Evaluation and Research (CBER); c2015. Available from: http://www.fda.gov/downloads/drugs/guidancecomplianceregulatoryinformation/guidances/ucm291128.pdf. Accessed May 20, 2015. | ||
Jeske W, Walenga JM, Hoppensteadt D, Fareed J. Update on the safety and bioequivalence of biosimilars – focus on enoxaparin. Drug Healthc Patient Saf. 2013;5:133–141. | ||
Mysler E, Pineda C, Horiuchi T, et al. Clinical and regulatory perspectives on biosimilar therapies and intended copies of biologics in rheumatology. Rheumatol Int. 2016;36(5):613–625. | ||
European Medicines Agency. Guideline on Similar Biological Medicinal Products Containing Biotechnology-Derived Proteins as Active Substance: Non-Clinical and Clinical Issues. London: European Medicines Agency; c2014. Available from: http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2015/01/WC500180219.pdf. Accessed July 27, 2015. | ||
World Health Organization. Guidelines on Evaluation of Similar Biotherapeutic Products (SBPs). Geneva: Expert Committee on Biological Standardization; c2009. Available from: http://www.who.int/biologicals/areas/biological_therapeutics/BIOTHERAPEUTICS_FOR_WEB_22APRIL2010.pdf. Accessed March 17, 2016. | ||
European Medicines Agency. Guideline on Similar Biological Medicinal Products (CHMP/437/04 Rev 1). London: European Medicines Agency; c2014. Available from: http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2014/10/WC500176768.pdf. Accessed June 5, 2015. | ||
Scheinberg MA, Kay J. The advent of biosimilar therapies in rheumatology – “O brave new world”. Nat Rev Rheumatol. 2012;8(7):430–436. | ||
Christl L. FDA’s Overview of the Regulatory Guidance for the Development and Approval of Biosimilar Products in the US. Washington, DC: US Food and Drug Administration, Office of New Drugs (OND) Therapeutic Biologics and Biosimilars Team, Center for Drug Evaluation and Research. Available from: http://www.fda.gov/downloads/Drugs/DevelopmentApprovalProcess/HowDrugsareDevelopedandApproved/ApprovalApplications/TherapeuticBiologicApplications/Biosimilars/UCM428732.pdf. Accessed June 9, 2015. | ||
Declerck P, Farouk-Rezk M, Rudd PM. Biosimilarity versus manufacturing change: two distinct concepts. Pharm Res. 2016;33(2):261–268. | ||
Berkowitz SA, Engen JR, Mazzeo JR, Jones GB. Analytical tools for characterizing biopharmaceuticals and the implications for biosimilars. Nat Rev Drug Discov. 2012;11(7):527–540. | ||
European Medicines Agency. Guideline on Similar Biological Medicinal Products Containing Monoclonal Antibodies – Non-Clinical and Clinical Issues. European Medicines Agency; c2012. Available from: http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2012/06/WC500128686.pdf. Accessed July 27, 2015. | ||
European Medicines Agency. Guideline on Similar Biological Medicinal Products Containing Biotechnology-Derived Proteins as Active Substance: Quality Issues (Revision 1); c2012. Available from: http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2012/05/WC500127960.pdf. Accessed September 23, 2015. | ||
US Food and Drug Administration. Guidance for Industry: E9 Statistical Principles for Clinical Trials. Rockville, MD: US Department of Health and Human Services, Center for Drug Evaluation and Research (CDER), Center for Biologics Evaluation and Research (CBER); c1998 [updated September 1998]. Available from: http://www.fda.gov/downloads/drugs/guidancecomplianceregulatoryinformation/guidances/ucm073137.pdf. Accessed March 17, 2016. | ||
Isakov L, Jin B, Jacobs IA. Statistical primer on biosimilar clinical development. Am J Ther. 2016;23(6):e1903–e1910. | ||
Alten R, Cronstein BN. Clinical trial development for biosimilars. Semin Arthritis Rheum. 2015;44(6 Suppl):S2–S8. | ||
European Medicines Agency. European Public Assessment Report (EPAR) Annex I. Summary of Product Characteristics: Inflectra (infliximab). Warwickshire, UK: Hospira UK Limited; c2014 [updated N/A]. Available from: http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Product_Information/human/002778/WC500151489.pdf. Accessed July 29, 2015. | ||
European Medicines Agency. European Public Assessment Report (EPAR) Annex I. Summary of Product Characteristics: Remsima (infliximab). Budapest, Hungary: Celltrion Healthcare Hungary Kft; c2014 [updated N/A]. Available from: http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Product_Information/human/002576/WC500150871.pdf. Accessed July 29, 2015. | ||
Health Canada [webpage on the Internet]. Summary Basis of Decision (SBD) for Remsima. Ottawa, ON: Office of Regulatory Affairs, Biologics and Genetic Therapies Directorate. c2014. Available from: http://www.hc-sc.gc.ca/dhp-mps/prodpharma/sbd-smd/drug-med/sbd_smd_2014_remsima_160195-eng.php#sbd. Accessed July 29, 2015. | ||
Health Canada [webpage on the Internet]. Summary Basis of Decision (SBD) for Inflectra. Ottawa, ON: Office of Regulatory Affairs, Biologics and Genetic Therapies Directorate. c2015. Available from: http://www.hc-sc.gc.ca/dhp-mps/prodpharma/sbd-smd/drug-med/sbd_smd_2014_inflectra_159493-eng.php. Accessed July 29, 2015. | ||
Pfizer Canada Inc [webpage on the Internet]. Health Canada approves Inflectra® (biosimilar infliximab) for three additional indications: Crohn’s disease, Fistulising Crohn’s disease and ulcerative colitis. Ottawa, ON. Available from: http://www.pfizer.ca/node/7526. Accessed June 27, 2016. | ||
Buer LC, Moum BA, Cvancarova M, Warren DJ, Medhus AW, Hoivik ML. Switching from Remicade(R) to Remsima(R) is safe and feasible: a prospective, open-label study. J Crohns Colitis. 2016. Epub 2016 Sep 22. | ||
Sieczkowska J, Jarzebicka D, Meglicka M, Oracz G, Kierkus J. Experience with biosimilar infliximab (CT-P13) in paediatric patients with inflammatory bowel diseases. Therap Adv Gastroenterol. 2016;9(5):729–735. | ||
Smits LJ, Derikx LA, de Jong DJ, et al. Clinical outcomes following a switch from remicade(R) to the biosimilar CT-P13 in inflammatory bowel disease patients: a prospective observational cohort study. J Crohns Colitis. 2016;10(11):1287–1293. | ||
Weise M, Bielsky MC, De Smet K, et al. Biosimilars: what clinicians should know. Blood. 2012;120(26):5111–5117. | ||
Park W, Hrycaj P, Jeka S, et al. A randomised, double-blind, multicentre, parallel-group, prospective study comparing the pharmacokinetics, safety, and efficacy of CT-P13 and innovator infliximab in patients with ankylosing spondylitis: the PLANETAS study. Ann Rheum Dis. 2013;72(10):1605–1612. | ||
Takeuchi T, Yamanaka H, Tanaka Y, et al. Evaluation of the pharmacokinetic equivalence and 54-week efficacy and safety of CT-P13 and innovator infliximab in Japanese patients with rheumatoid arthritis. Mod Rheumatol. 2015;25(6):817–824. | ||
Yoo DH, Hrycaj P, Miranda P, et al. A randomised, double-blind, parallel-group study to demonstrate equivalence in efficacy and safety of CT-P13 compared with innovator infliximab when coadministered with methotrexate in patients with active rheumatoid arthritis: the PLANETRA study. Ann Rheum Dis. 2013;72(10):1613–1620. | ||
Socinski MA, Curigliano G, Jacobs I, Gumbiner B, MacDonald J, Thomas D. Clinical considerations for the development of biosimilars in oncology. MAbs. 2015;7(2):286–293. | ||
Jung SK, Lee KH, Jeon JW, et al. Physicochemical characterization of Remsima. MAbs. 2014;6(5):1163–1177. | ||
McClellan JE, Udata C, Yin D, et al. Comparative structural, functional, nonclinical, and phase 1 similarity assessments of PF-06438179, a potential biosimilar to infliximab, and marketed reference products. Paper presented at: 10th Congress of European Crohn’s and Colitis Organisation (ECCO); February 18–21; 2015; Barcelona, Spain. | ||
Jung YS, Park DI, Kim YH, et al. Efficacy and safety of CT-P13, a biosimilar of infliximab, in patients with inflammatory bowel disease: a retrospective multicenter study. J Gastroenterol Hepatol. 2015;30(12):1705–1712. | ||
Farkas K, Rutka M, Balint A, et al. Efficacy of the new infliximab biosimilar CT-P13 induction therapy in Crohn’s disease and ulcerative colitis – experiences from a single center. Expert Opin Biol Ther. 2015;15(9):1257–1262. | ||
Udata C, Hua SY, Yin D, Salts S, Meng X, Rehman MI. A phase I pharmacokinetics trial comparing PF-06438179 (a potential biosimilar) and infliximab in healthy volunteers (REFLECTIONS B537-01). Ann Rheum Dis. 2014;73(Suppl 2):494. | ||
Udata C, Yin D, Cai CH, et al. Immunogenicity assessment of PF-06438179, a potential biosimilar to infliximab, in healthy volunteers [Abstr 1501]. Paper presented at: 2014 American College of Rheumatology (ACR)/Association of Rheumatology Health Professionals (ARHP); November 14–19; 2014; Boston, MA. |
 © 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.