Back to Journals » Clinical Ophthalmology » Volume 12
A Phase II/III, randomized, double-masked, vehicle-controlled, dose-ranging study of the safety and efficacy of OTX-101 in the treatment of dry eye disease
Authors Tauber J ![]() , Schechter BA, Bacharach J, Toyos MM
, Schechter BA, Bacharach J, Toyos MM ![]() , Smyth-Medina R, Weiss SL
, Smyth-Medina R, Weiss SL ![]() , Luchs JI
, Luchs JI ![]()
Received 23 May 2018
Accepted for publication 18 August 2018
Published 2 October 2018 Volume 2018:12 Pages 1921—1929
DOI https://doi.org/10.2147/OPTH.S175065
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 5
Editor who approved publication: Dr Scott Fraser
Joseph Tauber,1 Barry A Schechter,2 Jason Bacharach,3 Melissa M Toyos,4 Robert Smyth-Medina,5 Sidney L Weiss,6 Jodi I Luchs7
1Tauber Eye Center, Kansas City, MO, USA; 2Florida Eye Microsurgical Institute, Boynton Beach, FL, USA; 3North Bay Eye Associates, Petaluma, CA, USA; 4Toyos Clinic, Nashville, TN, USA; 5North Valley Eye Medical Group, Mission Hills, CA, USA; 6i-Novion, Inc, Randolph, NJ, USA; 7South Shore Eye Care, Wantagh, NY, USA
Purpose: The aim of this study was to evaluate the safety and efficacy of OTX-101, a clear nanomicellar aqueous solution of cyclosporine, in the treatment of dry eye disease (DED).
Patients and methods: This was a 12-week multicenter, randomized, prospective, double-masked, vehicle-controlled, dose-ranging clinical trial. Subjects were adults aged ≥18 years, with a total conjunctival staining score of ≥3 and ≤9, and global DED symptom score ≥40 (0–100 visual analogue scale). Following a 14-day vehicle run-in, subjects were randomized in a 1:1:1 ratio to twice daily treatment with OTX-101 0.09%, OTX-101 0.05%, or vehicle for 84 days. Co-primary efficacy end points were changes, from baseline to Day 84, in the total lissamine green conjunctival staining score in the designated study eye and in the global symptom score (both eyes). Secondary end points included total corneal fluorescein staining score, tear breakup time, and Schirmer’s test score.
Results: In total, 455 subjects were randomized. Subjects treated with active drug experienced greater improvement in conjunctival staining than vehicle-treated patients (P<0.01 for both concentrations). All groups demonstrated improvements in global symptom score, but there were no differences among groups. Nominally significant differences were found between the active drug arms and vehicle for corneal staining scores and Schirmer’s test scores. Most treatment-emergent adverse events were mild in severity; no serious ocular adverse events were reported.
Conclusions: Both concentrations of OTX-101 met the co-primary sign end point (conjunctival staining) but not the co-primary symptom end point. OTX-101 0.09% demonstrated a notable impact on multiple signs of DED relative to vehicle and was well-tolerated.
Keywords: dry eye disease, cyclosporine, inflammation, Schirmer’s test
Corrigendum for this paper has been published
Further corrigendum has been published
Introduction
Dry eye disease (DED), or keratoconjunctivitis sicca (KCS), is a common chronic condition affecting the ocular surface, characterized by the loss of homeostasis of the tear film. It is associated with a range of ocular symptoms, including ocular dryness and discomfort, grittiness, foreign body sensation, and blurred or fluctuating vision.1,2 Tear film instability and hyperosmolarity, ocular surface inflammation and damage, as well as neurosensory abnormalities are recognized as potential contributing factors in the development of DED.3
The etiology of DED is complex, with a range of potential triggers and conditions that result in a reduction in the quantity of the components of the tear film that is produced or a tear film instability that leads to evaporative forms of dry eye.1 Loss of homeostasis of the tear film results in desiccation of the ocular surface tissue and the production of inflammatory cytokines. Increases in tear film osmolarity, combined with the presence of inflammatory cells, lead to epithelial and goblet cell death. Damage and loss of cells on the ocular surface as well as alterations in the normal functioning of the lacrimal and meibomian glands, further perpetuates the chronic cycle of inflammation that is characteristic of DED.1,2
DED is most prevalent in women and patients aged >50 years.4 Within the USA, DED has been estimated to affect up to 40 million people, and the number is expected to continue to increase.5–7
Treatment options for DED are often staged according to the nature and severity of a patient’s condition. Patient education and ocular lubricants may be sufficient for patients with mild forms of DED, while tear conservation methods and topically applied medications are recommended for patients who require additional treatment. Topical anti-inflammatory agents used to treat DED include cyclosporine A, lymphocyte function-associated antigen 1 (LFA-1) antagonists, and corticosteroids.8
Cyclosporine is an immunomodulatory agent with anti-inflammatory properties. Cyclosporine inhibits calcineurin, inhibiting the activation of T lymphocytes and subsequent release of proinflammatory mediators.9,10 Treatment of patients with DED using topical ophthalmic application of cyclosporine has been shown to reduce the clinical signs (reduction in corneal staining, increase in Schirmer’s test scores) of the condition.11–13 Other studies have demonstrated an increase in conjunctival goblet cell density and a decrease in inflammatory cell activation markers following treatment with cyclosporine.12–15
Restasis™ (Allergan, Irvine, CA, USA), the only topical ophthalmic formulation of cyclosporine currently marketed in the USA, is a turbid oil-in-water emulsion; as an emulsion, shaking is required prior to instillation.16 OTX-101 is a novel, nanomicellar, clear aqueous solution of cyclosporine (Sun Pharmaceutical Industries, Cranbury, NJ, USA). The smaller particle size of nanomicellar technology provides improved bioavailability and physicochemical stability, potentially resulting in better long-term ocular tolerance and efficacy. The purpose of this study was to evaluate the safety and efficacy of 2 concentrations of OTX-101 relative to its vehicle in the treatment of subjects with DED.
Materials and methods
Study design
This was a Phase II/III randomized, multicenter, double-masked, vehicle-controlled, dose-ranging study. Subjects were enrolled at 29 sites in the USA. All study-related documents were submitted for review and approved by an institutional review board (Alpha Institutional Review Board, San Clemente, CA, USA). Written informed consent was obtained from all subjects prior to study enrollment and any study-related procedures were performed. The study was conducted in accordance with the principles of the Declaration of Helsinki and all applicable legal and regulatory requirements. The study was registered through ClinicalTrials.gov: NCT02254265 (https://ClinicalTrials.gov/ct2/show/NCT02254265).
Subject participation in the study was approximately 100 days in duration, including an 84-day treatment period (Days 1–84). Subjects were required to attend a total of 7 study visits during the study for safety and efficacy evaluations. Subjects were randomized to one of OTX-101 0.09%, OTX-101 0.05%, or vehicle groups in a 1:1:1 ratio at the baseline visit (Figure 1). Subjects were instructed to instill 1 drop of the ophthalmic formulation provided in their assigned study kit in each eye twice a day, approximately 12 hours apart.
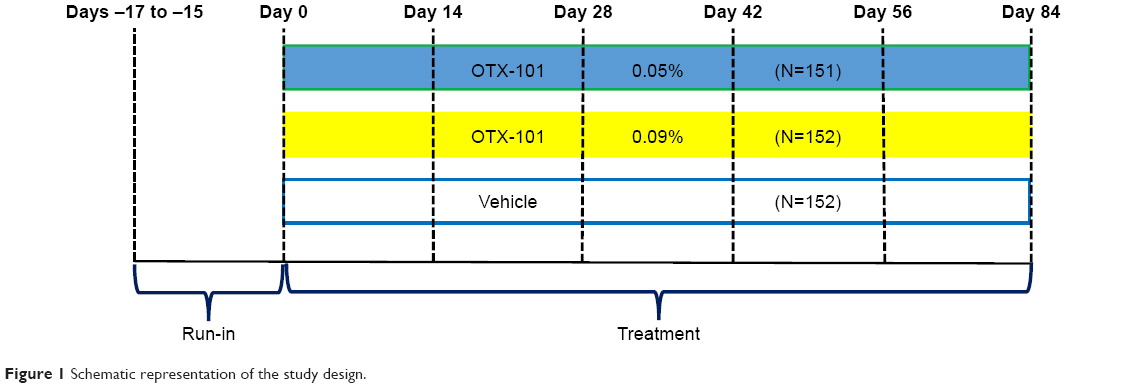 | Figure 1 Schematic representation of the study design. |
Study protocol
Screening and subject eligibility
Screening was conducted at Visit 1 (Day −17 to −15). After written informed consent had been collected, subjects were evaluated for eligibility according to the protocol-defined inclusion/exclusion criteria. Subject demographic information and medical/ocular/concomitant medication histories were collected. Comprehensive ophthalmic examinations were conducted as part of the screening process, including the evaluation of selected signs and symptoms of DED to confirm the presence of the disease.
Subjects were required to meet all inclusion criteria to be considered for enrollment. The main inclusion criteria were as follows: 18 years of age or older, with a patient-reported history of DED for a period of at least 6 months supported by a clinical diagnosis of bilateral DED; Snellen visual acuity (VA) of 20/200 or better in both eyes; lissamine green conjunctival staining sum score of ≥3 to ≤9 out of a total possible score of 12 in the same eye; and a global symptom score (symptoms of dryness and/or irritation) of ≥40 rated by the subject on a 0–100 visual analogue scale (VAS) at both the screening and the baseline visits. Subjects were also required to discontinue the use of any current therapy for DED throughout the duration of the study, beginning at the screening visit.
Potential subjects were excluded from participation in the study based on the following criteria: use of cyclosporine ophthalmic emulsion 0.05% (Restasis) within 3 months prior to screening or a report of a previous treatment failure (lack of efficacy) on topical cyclosporine; a diagnosis of Sjögren’s Syndrome of >5 years’ duration; presence or history of seasonal/perennial allergic conjunctivitis; or another current active eye disease other than DED. Patients were considered ineligible for study participation if the following medications were used within 7 days prior to screening or throughout the study period: cholinergics, antimuscarinics, antihistamines, antidepressants, phenothiazines, retinoids, or corticosteroids. The continued use of immunomodulators and omega-3 fatty acid supplements was permitted if the subject’s dose was stable for 3 months prior to screening. No other ophthalmic medications or over-the-counter (OTC) artificial tears were permitted during the study period. Subjects were also excluded from study participation if they had a history of any ocular surgery within 3 months of screening or were unwilling to discontinue use of contact lenses for the duration of the study; pregnant or breastfeeding women were also excluded.
Once a subject was evaluated according to the inclusion/exclusion criteria, eligible subjects entered a run-in period (14–17 days). During the run-in period, all subjects were instructed to instill 1 drop of vehicle (placebo) into each eye twice daily. Subjects were evaluated for eligibility based on the inclusion/exclusion criteria at both the screening and the baseline visits.
Study masking
Subjects, investigators, other clinical site staff, and all personnel responsible for monitoring and medical evaluation of the data remained masked to the subject’s randomized treatment assignment throughout the study. Double-masked study treatment kits and the single-use vials contained therein were identical in appearance for all treatments.
Outcome measures
Efficacy
Efficacy was assessed through the evaluation of the signs and symptoms of DED, collected at all study visits. The clinical signs included corneal fluorescein staining score, lissamine green staining score of the conjunctiva, tear breakup time (TBUT), and Schirmer’s test score (baseline and Day 84 visits only). Patient-reported symptoms were evaluated for the frequency and severity of dryness and/or irritation using a modified “symptom assessment in dry eye” (SANDE) questionnaire.17
The Expanded National Eye Institute (NEI)/Industry Workshop scales18 for corneal and conjunctival staining were used to grade each area of the cornea and conjunctiva. Lissamine green staining was assessed for each of the 6 regions of the conjunctiva.18 The total lissamine green staining score was calculated for each subject at each visit by adding the scores of the individual regions, excluding the 2 superior regions (0–12 range); the superior zones were excluded in an effort to reduce variability across the treatment groups. Corneal fluorescein staining was assessed for each of the 5 regions of the cornea,18 including 0.5 increments on an individual basis and summed for a total corneal staining score (0–20 range). TBUT was recorded in seconds following instillation of fluorescein stain. A Schirmer’s test (unanesthetized) was also performed.
A modified SANDE questionnaire was used to monitor subject-reported symptoms. Subjects were asked to report on VASs (range of 0–100) both the frequency and the severity of their symptoms of ocular dryness/irritation. A global symptom score was calculated based on the subject’s responses at each visit using the square root of the product of the modified SANDE frequency and VAS severity scores.
The co-primary efficacy end points for this study were mean changes from baseline at Day 84 for the total conjunctival lissamine green staining score in the designated study eye and the global symptom score. The study eye was determined as the eye that had the higher total lissamine green staining score at baseline. If both eyes were equal, the right eye was designated as the study eye. The secondary efficacy end points included mean changes from baseline in TBUT, total corneal fluorescein staining score, and Schirmer’s test score (average of both eyes).
Safety and tolerability
At all study visits, slit lamp examinations were conducted, Snellen VA was measured, and adverse events (AEs) were recorded. Other safety assessments, including ophthalmoscopy/dilated fundoscopy and tonometry, were conducted at specified study visits. The comfort/tolerability of the study medication was evaluated at baseline (Day 0) and Day 84 of the treatment period. Subjects were asked to grade study medication comfort on a 0–3 scale (0= no discomfort, 3= severe discomfort), 3 and 10 minutes postinstillation.
Statistical methods
The sample size for the study was determined based on the following calculations. A sample size of 140 in each group was determined to have 80% power to detect a mean difference of 0.68 in the total lissamine green staining score, assuming that the common SD was 2.0, using a 2-group t-test with a 2-sided significance level of 0.05. A sample size of 140 in each group was determined to have 80% power to detect a difference of 8.5 in the mean global symptom score, assuming that the common SD was 25, using a 2-group t-test with a 2-sided significance level of 0.05.
The intent-to-treat (ITT) population was defined as all randomized subjects. The safety population included all subjects who received at least 1 dose of study medication. The ITT population was used for the primary efficacy analysis.
An analysis of covariance (ANCOVA) was used for the co-primary end points, with treatment and clinical site as fixed factors and baseline value as a covariate, to compare the mean change from baseline at each postbaseline time point for each efficacy end point. Categorical variables were analyzed using the chi-squared test, Fisher’s exact test, or the Cochran–Mantel–Haenszel test (adjusted for clinical site), as appropriate. The analysis of the co-primary end points was conducted without adjustment for multiplicity; however, a closed testing procedure was conducted with respect to testing for multiple doses. The higher concentration of OTX-101 was tested against vehicle first, and if the P≤0.05, then the lower concentration of OTX-101 was tested against vehicle. A 2-sided significance level of 0.05 was adopted.
Continuous variables were summarized with descriptive statistics (n, mean, median, SD, standard error, minimum, and maximum), and categorical variables were summarized with counts and percentages.
Results
A total of 585 subjects were evaluated for eligibility at the screening and baseline visits. There were 130 subjects who failed screening, leading to 455 subjects who were randomized to 1 of the 3 treatment groups (OTX-101 0.05%, OTX-101 0.09%, or vehicle). A total of 426 subjects completed the study through Day 84. The disposition of the subjects is presented in Figure 2.
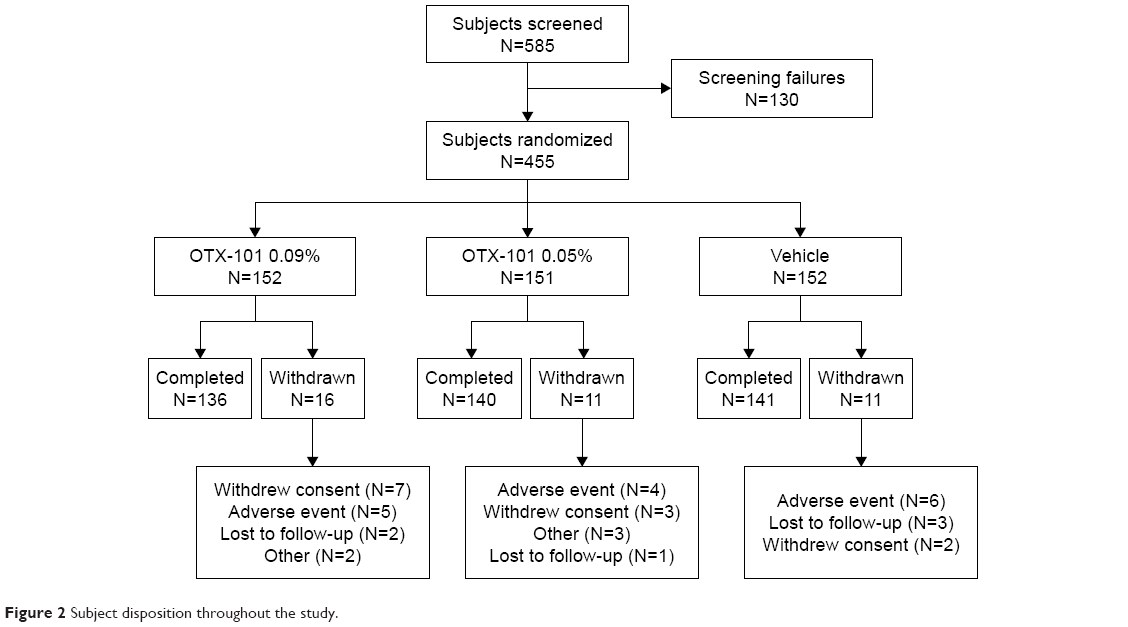 | Figure 2 Subject disposition throughout the study. |
A summary of the subject demographic information by treatment group and for the overall study population is presented in Table 1. The demographic characteristics were generally similar between the treatment groups. The majority of subjects who participated in the study were female, were white, and identified themselves as neither Hispanic nor Latino.
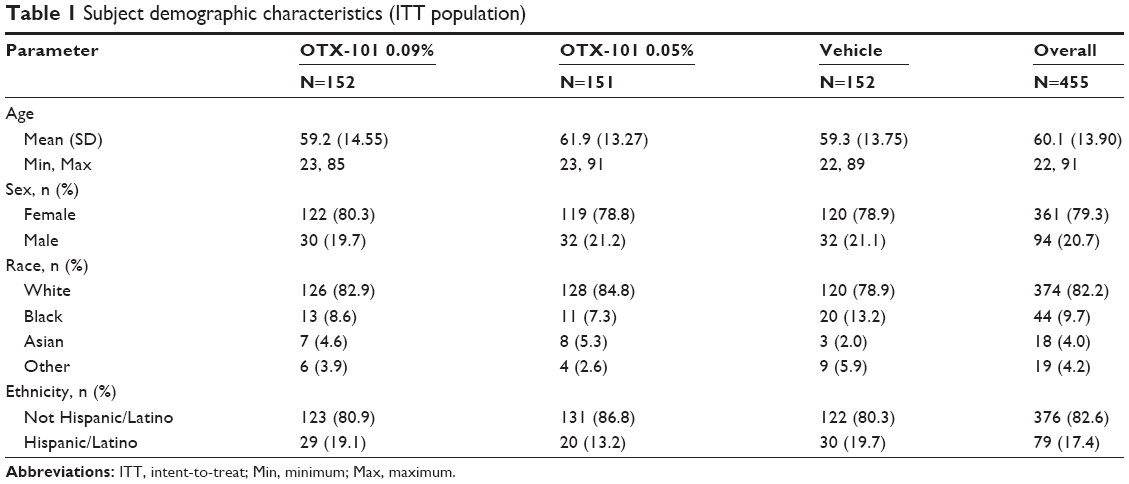 | Table 1 Subject demographic characteristics (ITT population) |
Efficacy end points
Conjunctival staining
The mean (SD) values at baseline for the total conjunctival lissamine green staining scores (excluding the superior zones) in the OTX-101 0.09%, OTX-101 0.05%, and vehicle groups were 5.8 (1.8), 6.2 (1.7), and 6.0 (1.7), respectively. Statistically significant reductions in the mean change from baseline in the study eye at Day 84 relative to vehicle were observed for both OTX-101 concentrations (P<0.01). The results by study visit are depicted in Figure 3.
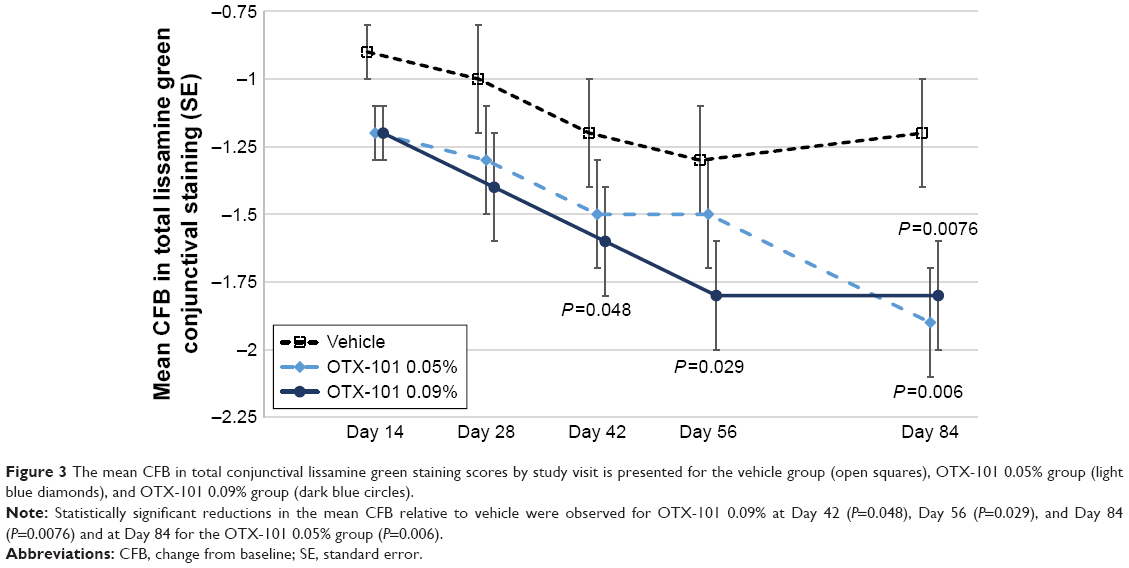 | Figure 3 The mean CFB in total conjunctival lissamine green staining scores by study visit is presented for the vehicle group (open squares), OTX-101 0.05% group (light blue diamonds), and OTX-101 0.09% group (dark blue circles). |
Global symptom score
The mean (SD) values for the global symptom score at baseline for the OTX-101 0.09%, OTX-101 0.05%, and vehicle groups were 62.6 (14.6), 61.8 (15.3), and 61.5 (14.3), respectively. The global symptom score decreased from baseline throughout the 84 days of treatment by approximately 30% in all treatment arms. Neither of the OTX-101 concentrations differed significantly from the vehicle with respect to mean change from baseline at Day 84.
Corneal staining
Baseline mean (SD) values for the total corneal fluorescein staining assessment scores in the study eye for the OTX-101 0.09%, OTX-101 0.05%, and vehicle groups were 4.5 (3.1), 5.1 (3.2), and 4.6 (2.8), respectively. Significant improvements in total corneal staining at Day 84 were observed in both OTX-101 treatment groups (P=0.0003 for the OTX 0.09% group, and P=0.024 for the OTX 0.05% group). The mean changes from baseline in the total corneal fluorescein staining scores are presented by study visit in Figure 4.
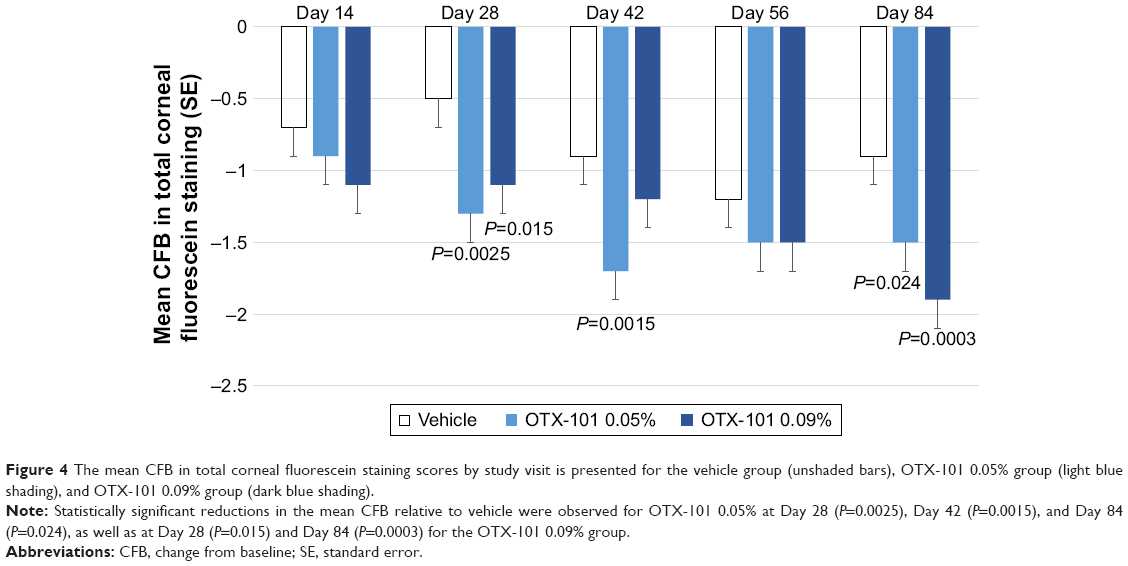 | Figure 4 The mean CFB in total corneal fluorescein staining scores by study visit is presented for the vehicle group (unshaded bars), OTX-101 0.05% group (light blue shading), and OTX-101 0.09% group (dark blue shading). |
Schirmer’s test scores
The mean (SD) baseline Schirmer’s test values (average of both eyes) for the OTX-101 0.09%, OTX-101 0.05%, and vehicle groups were 12.3 (8.67), 10.7 (7.57), and 12.2 (8.25) mm, respectively. Both OTX-101 treatment arms were superior to the vehicle arm in terms of mean change from baseline. Using an ANCOVA model, mean improvements from baseline were 3.5 and 2.6 mm for the OTX-101 0.09% and 0.05% arms vs 0 mm for the vehicle arm (nominal P-values: P=0.0003 and P=0.0076 vs vehicle, respectively).
A post hoc responder analysis was conducted to evaluate the proportion of subjects in which an increase of ≥10 mm in Schirmer’s test scores from baseline to Day 84 was observed. A higher proportion of subjects in the OTX-101 0.09% treatment group (17.9% vs 7.6%; P=0.007) increased ≥10 mm in the Schirmer’s tear test scores as compared to the subjects in the vehicle group (Figure 5).
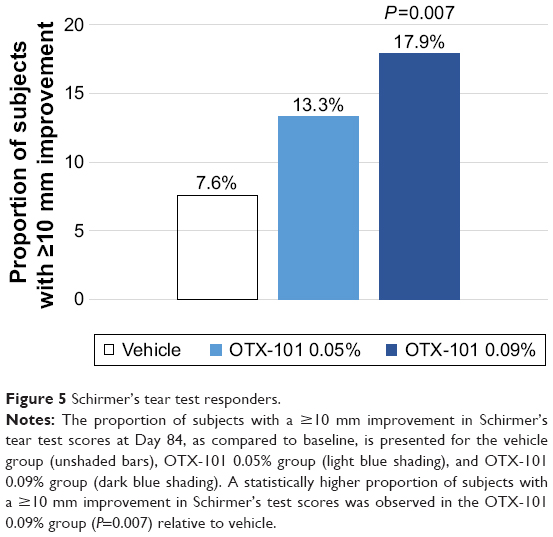 | Figure 5 Schirmer’s tear test responders. |
TBUT assessment
The mean values at baseline for the TBUT measurements of the study eye for the OTX-101 0.09%, OTX-101 0.05%, and vehicle groups were 4.8, 4.5, and 4.3 seconds, respectively. No significant differences among treatment groups in terms of changes from baseline in the TBUT were observed at any time point.
Safety and tolerability assessments
The safety population consisted of 455 subjects who received at least 1 dose of the assigned study medication. A total of 347 AEs associated with 147 subjects were reported during the study: 118 AEs from 52 subjects (34.2%) in the OTX-101 0.09% group, 124 AEs from 44 subjects (29.1%) in the OTX-101 0.05% treatment group, and 105 AEs in 51 subjects (33.6%) in the vehicle group. The majority of ocular AEs were considered mild or moderate in severity. A summary of the most common ocular and non-ocular treatment-emergent AEs is presented in Table 2.
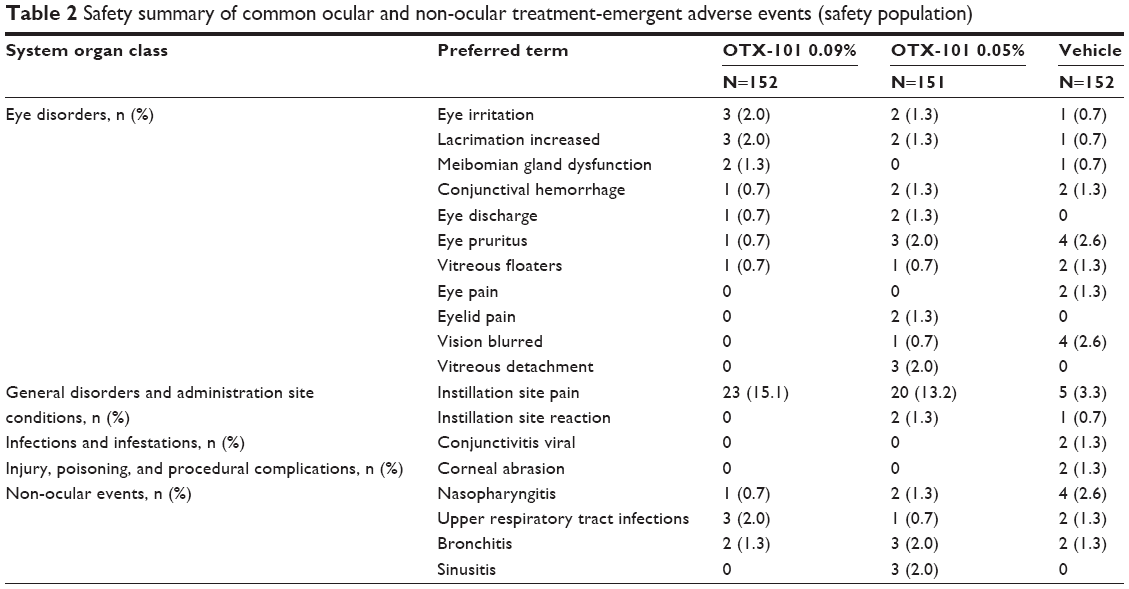 | Table 2 Safety summary of common ocular and non-ocular treatment-emergent adverse events (safety population) |
No deaths occurred during the study. A total of 5 subjects experienced serious adverse events (SAEs): 1 subject (0.7%) in the OTX-101 0.05% group, and 4 subjects (2.6%) in the vehicle group. None of the SAEs recorded during the study were considered to be related to study treatment.
Fifteen subjects exited the study due to AEs: 5 subjects (3.3%) in the OTX-101 0.09% group, 4 subjects (2.6%) in the OTX-101 0.09% group, and 6 subjects (3.9%) in the vehicle group. As shown in Table 3, the majority of subjects across treatment groups reported no or mild discomfort at the 3- and 10-minute postinstillation assessments conducted at the baseline (Day 0) and Day 84 visits. At the Day 84 visit, the proportion of subjects reporting worse than mild discomfort at 3 minutes postinstillation was slightly higher in the OTX-101 0.09% group than the vehicle group, but, at 10 minutes, the comfort rating was comparable across all 3 treatment groups.
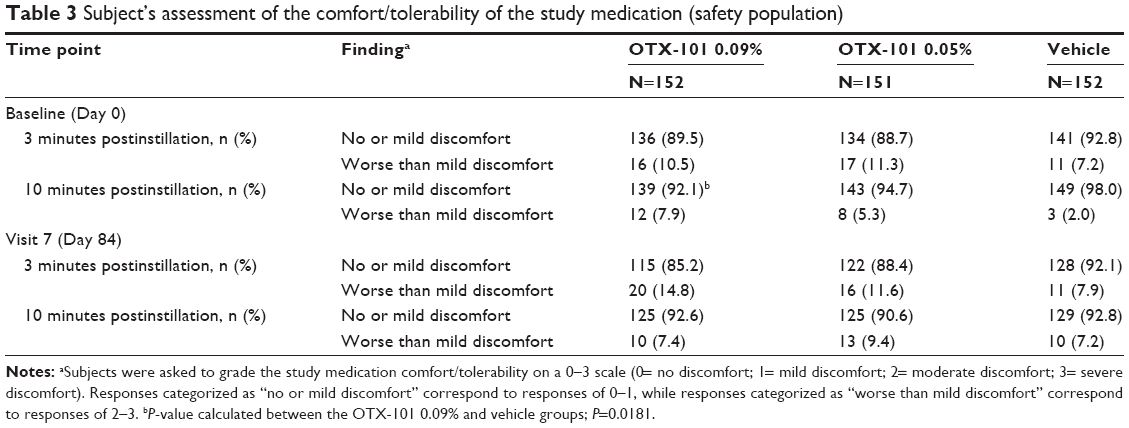 | Table 3 Subject’s assessment of the comfort/tolerability of the study medication (safety population) |
Discussion
This was the first clinical trial of OTX-101, an aqueous, nanomicellar ophthalmic formulation of cyclosporine, in the treatment of DED. Subjects were treated with OTX-101 0.05%, OTX-101 0.09%, or vehicle twice daily for 12 weeks. Both concentrations of cyclosporine demonstrated superiority to vehicle in terms of the co-primary end point, total conjunctival staining, as well as in the secondary signs of total corneal staining and Schirmer’s test. The co-primary end point related to subject-reported symptoms was not met. An approximate 30% mean improvement from baseline in the modified SANDE instrument was observed, with no differences seen among the 3 arms.
OTX-101 appears to be the first DED product candidate to have demonstrated statistically significant improvement relative to vehicle for both conjunctival staining and unanesthetized Schirmer’s test, in addition to reduction in corneal staining. A post hoc analysis also produced significant results for both OTX-101 concentrations vs vehicle for staining of the temporal conjunctiva, which may be a marker of more advanced DED.19 Post hoc analyses of corneal staining by region demonstrated superiority to vehicle for both OTX-101 concentrations in the inferior zone and for OTX-101 0.09% in the central, medial, and lateral zones.
The inability of this study to demonstrate a significant treatment effect with respect to patient-reported symptoms is not entirely unexpected as it has been repeatedly shown that clinical tests measuring tear deficiency and ocular surface damage are, in general, only weakly correlated with patient symptoms.20 However, it is possible that the scoring of individual symptoms of DED, rather than the general queries used in the modified SANDE instrument to evaluate the frequency and severity of symptoms, might have allowed for a separation in the active vs vehicle treatment groups. Additionally, the OTX-101 vehicle is a polymeric solution that may provide some degree of symptomatic relief and the 15- to 17-day run-in period used in the present study may not have been sufficient in length to identify vehicle responders.
Due to the poor aqueous solubility of cyclosporine, novel approaches have been used to deliver the drug to the ocular surface. Oil-in-water emulsions, such as cyclosporine ophthalmic emulsion 0.05% (Restasis; Allergan) in the USA, and ciclosporin 1 mg/mL emulsion (Ikervis; Santen SAS, Evry Cedex, France) in the European Union have been developed as topical agents to treat DED. Additionally, a formulation of cyclosporine (0.1%), using ethanol and surfactants to solubilize the active agent, has been marketed in Mexico and South America (Modusik-A Ofteno; Laboratorios Sophia, Zapopan, Jalisco, México) for the treatment of DED.21
Nanomicelle-based formulations involve the use of amphiphilic polymers that self-assemble in an aqueous solution to form organized supramolecular structures. The proprietary formulation technology used in OTX-101 allows for a >10-fold increase in aqueous concentrations of cyclosporine, from a typical 12 μg/mL to as high as 1,500 μg/mL, providing a higher distribution of the drug into ocular tissues.22
The safety and tolerability profiles of topical cyclosporine have been established via the postmarketing experience of cyclosporine ophthalmic emulsion 0.05% (Restasis; Allergan) in 2003. In the present study, no unexpected or serious ocular AEs attributed to the 0.05% or 0.09% cyclosporine concentration-containing formulations of OTX-101 were reported. Notably, the majority of ocular AEs were considered mild in severity and the most frequently reported AE coding term, instillation site pain, was similar between the 2 groups of OTX-101 treatment (15.1% for OTX-101 0.09%, and 13.2% for the OTX-101 0.05% group). The results of the evaluation of subject-rated study medication comfort indicated an acceptable tolerability profile, with most of the subjects in all 3 treatment groups reporting no or mild discomfort postinstillation.
Limitations of the present study include the lack of an active comparator, such as the currently marketed topical cyclosporine ophthalmic emulsion (0.05%), a treatment period of only 84 days (12 weeks), and a limited range of instruments used to evaluate and monitor subject-reported symptoms of DED.
Conclusion
Both concentrations of the OTX-101 formulation tested in this study were observed to be efficacious in improving the multiple clinical signs of DED. The general similarity in the safety and tolerability profiles, the improved efficacy observed in the reduction of corneal staining, and the higher frequency of responders (≥10 mm increase) in the Schirmer’s tear test for subjects in the OTX-101 0.09% treatment group led to the decision to advance this concentration for further clinical development.
Acknowledgments
Writing and editorial assistance in preparation of the manuscript was provided by Kurt Brubaker of Bridge Over Brook, Inc, with funding provided by Sun Pharmaceutical Industries. This study was sponsored and funded by Ocular Technologies Sarl (now a wholly owned subsidiary of Sun Pharmaceutical Industries), which participated in the design of the study and oversaw its conduct, monitoring, and analysis.
Author contributions
All authors contributed to data analysis, drafting and revising the article, gave final approval of the version to be published, and agree to be accountable for all aspects of the work.
Disclosure
The authors report no conflicts of interest in this work.
References
Bron AJ, de Paiva CS, Chauhan SK, et al. TFOS DEWS II pathophysiology report. Ocul Surf. 2017;15(3):438–510. | ||
Stern ME, Schaumburg CS, Pflugfelder SC. Dry eye as a mucosal autoimmune disease. Int Rev Immunol. 2013;32(1):19–41. | ||
Craig JP, Nichols KK, Akpek EK, et al. TFOS DEWS II Definition and Classification Report. Ocul Surf. 2017;15(3):276–283. | ||
Stapleton F, Alves M, Bunya VY, et al. TFOS DEWS II Epidemiology Report. Ocul Surf. 2017;15(3):334–365. | ||
Schaumberg DA, Dana R, Buring JE, Sullivan DA. Prevalence of dry eye disease among US men: estimates from the Physicians’ Health Studies. Arch Ophthalmol. 2009;127(6):763–768. | ||
Schaumberg DA, Sullivan DA, Buring JE, Dana MR. Prevalence of dry eye syndrome among US women. Am J Ophthalmol. 2003;136(2):318–326. | ||
Ding J, Sullivan DA. Aging and dry eye disease. Exp Gerontol. 2012;47(7):483–490. | ||
Jones L, Downie LE, Korb D, et al. TFOS DEWS II Management and Therapy Report. Ocul Surf. 2017;15(3):575–628. | ||
Pflugfelder SC. Antiinflammatory therapy for dry eye. Am J Ophthalmol. 2004;137(2):337–342. | ||
Fruman DA, Klee CB, Bierer BE, Burakoff SJ. Calcineurin phosphatase activity in T lymphocytes is inhibited by FK 506 and cyclosporin A. Proc Natl Acad Sci U S A. 1992;89(9):3686–3690. | ||
Sall K, Stevenson OD, Mundorf TK, Reis BL. Two multicenter, randomized studies of the efficacy and safety of cyclosporine ophthalmic emulsion in moderate to severe dry eye disease. CsA Phase 3 Study Group. Ophthalmology. 2000;107(4):631–639. | ||
Baudouin C, Figueiredo FC, Messmer EM, et al. A randomized study of the efficacy and safety of 0.1% cyclosporine A cationic emulsion in treatment of moderate to severe dry eye. Eur J Ophthalmol. 2017;27(5):520–530. | ||
Leonardi A, van Setten G, Amrane M, et al. Efficacy and safety of 0.1% cyclosporine A cationic emulsion in the treatment of severe dry eye disease: a multicenter randomized trial. Eur J Ophthalmol. 2016;26(4):287–296. | ||
Kunert KS, Tisdale AS, Stern ME, Smith JA, Gipson IK. Analysis of topical cyclosporine treatment of patients with dry eye syndrome: effect on conjunctival lymphocytes. Arch Ophthalmol. 2000;118(11):1489–1496. | ||
Kunert KS, Tisdale AS, Gipson IK. Goblet cell numbers and epithelial proliferation in the conjunctiva of patients with dry eye syndrome treated with cyclosporine. Arch Ophthalmol. 2002;120(3):330–337. | ||
Restasis [package insert]. Irvine, CA: Allergan; 2013. | ||
Schaumberg DA, Gulati A, Mathers WD, et al. Development and validation of a short global dry eye symptom index. Ocul Surf. 2007;5(1):50–57. | ||
Lemp MA. Report of the National Eye Institute/Industry workshop on Clinical Trials in Dry Eyes. Clao J. 1995;21(4):221–232. | ||
Rolando M, Barabino S, Mingari C, et al. Distribution of conjunctival HLA-DR expression and the pathogenesis of damage in early dry eyes. Cornea. 2005;24(8):951–954. | ||
Begley CG, Chalmers RL, Abetz L, et al. The relationship between habitual patient-reported symptoms and clinical signs among patients with dry eye of varying severity. Invest Ophthalmol Vis Sci. 2003;44(11):4753–4761. | ||
Lallemand F, Schmitt M, Bourges JL, et al. Cyclosporine A delivery to the eye: A comprehensive review of academic and industrial efforts. Eur J Pharm Biopharm. 2017;117:14–28. | ||
Vaishya RD, Khurana V, Patel S, Mitra AK. Controlled ocular drug delivery with nanomicelles. Wiley Interdiscip Rev Nanomed Nanobiotechnol. 2014;6(5):422–437. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.