Back to Journals » Clinical Pharmacology: Advances and Applications » Volume 10
A Phase I, open-label, randomized, crossover study in healthy subjects to evaluate the bioavailability of, and the food effect on, a pomalidomide oral liquid suspension
Authors Li Y, Liu L, Huang L, Wang X ![]() , Hoffmann M, Reyes J, Palmisano M, Zhou S
, Hoffmann M, Reyes J, Palmisano M, Zhou S
Received 21 April 2018
Accepted for publication 31 May 2018
Published 19 July 2018 Volume 2018:10 Pages 89—99
DOI https://doi.org/10.2147/CPAA.S171735
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Arthur E. Frankel
Yan Li,1 Liangang Liu,2 Lian Huang,3 Xiaomin Wang,4 Matthew Hoffmann,4 Josephine Reyes,1 Maria Palmisano,1 Simon Zhou1
1Translational Development and Clinical Pharmacology, 2Biometrics and Data Operations, 3Pharmaceutical Science Drug Product Development, 4Non-Clinical Development, Celgene Corporation, Summit, NJ, USA
Objective: The aim of this study was to evaluate the bioavailability of a pomalidomide oral liquid suspension relative to the commercial capsule formulation and to assess the food effect on the pomalidomide oral liquid suspension when administered as a single 4 mg dose.
Methods: This was an open-label, randomized, three-period, two-sequence crossover study in healthy subjects consisting of a screening phase, a baseline assessment phase, a treatment phase with three periods, and a follow-up phone call phase. Blood samples for pharmacokinetics (PK) assessment were collected up to 48 h postdose during each treatment period. Safety was evaluated throughout the study.
Results: Pomalidomide exposures were comparable in healthy subjects administered with a single oral 4 mg dose as the reference capsule or as the test liquid suspension formulations, demonstrated as the 90% confidence intervals of the geometric mean ratios for area under the plasma concentration–time curve calculated from time 0 to the last measurable concentration at time t (AUC0–t), area under the plasma concentration–time curve from time 0 to infinity (AUC0–∞), and peak (maximum) plasma drug concentration (Cmax) were completely contained within the bioequivalence range of 80–125%. Administration of the pomalidomide liquid suspension with a high fat meal resulted in a 3.0 h delay in pomalidomide time to Cmax (tmax) and an ~ 34.5% reduction in Cmax. However, the AUCs were comparable after dose administration with and without food.
Conclusion: A single oral dose of 4 mg of liquid suspension was bioequivalent to a single oral dose of 4 mg of capsule formulation. There was no clinically relevant impact of food on pomalidomide liquid suspension. Single oral doses of 4 mg pomalidomide were safe and well tolerated when administered as a liquid suspension under fed and fasted conditions or as a capsule under fasted conditions.
Keywords: pomalidomide, liquid suspension, bioavailability, bioequivalence, food effect
Introduction
Pomalidomide, an analog structurally similar to thalidomide,1,2 is an immunomodulatory agent with antineoplastic activity.3,4 In in vitro cellular assays, pomalidomide inhibited proliferation and induced the apoptosis of hematopoietic tumor cells5–9 and showed immunomodulatory activity.10–12 Pomalidomide has been studied for the treatment of various hematologic and nonneoplastic hematologic disorders,1,13–15 and the dosage of 4 mg per day taken orally on Days 1–21 of repeated 28-day cycles was approved (in combination with dexamethasone) in the European Union and the United States for the treatment of patients with multiple myeloma (MM) who have received ≥2 prior therapies, including lenalidomide and bortezomib (in the European Union) and a proteasome inhibitor (in the United States) and who have progressed on or within 60 days of completion of the last therapy or have disease progression on the last therapy.1,13,15 This combination (pomalidomide with low-dose dexamethasone) increased progression-free survival and overall survival compared with high-dose dexamethasone.1,13 Thrombocytopenia, neutropenia, and anemia were the most common grade 3/4 adverse events (AEs).13
Pomalidomide pharmacokinetics (PK) has been well characterized both in healthy subjects and in subjects with relapsed and refractory MM.2,16–18 Pomalidomide commercial capsules were well absorbed, and the systemic exposures increased in an approximately dose-proportional manner over the dose range of 1–50 mg.19,20 Pomalidomide reached steady-state by Day 3 with minimal accumulation (<14.0% for both AUC and peak [maximum] plasma drug concentration [Cmax]). Coadministration of pomalidomide commercial capsule formulation with a high-fat meal decreased the rate but not the extent of absorption; therefore, pomalidomide capsules can be administered without regard to food intake.19,20 The [14C]-pomalidomide study in healthy adult males showed that pomalidomide was extensively metabolized in vivo via multiple metabolic pathways, including oxidation, hydroxylation, and hydrolysis.18 Pomalidomide-related material was eliminated predominantly through renal excretion (~73.0% of the administered dose), with a low fraction of the dose excreted in urine as unchanged drug (overall <5.0% of the administered dose).18 The mean apparent terminal half-life (t1/2) of pomalidomide was ~7.5 h and the apparent total plasma clearance (CL/F) ranged from 6.5 to 10.8 L/h.19,20
Following US Food and Drug Administration (FDA) approval of pomalidomide for the treatment of adult patients with relapsed and refractory MM, the only available commercial formulation is capsule with the following four strengths: 1 mg (size 4 dark blue cap and yellow body with black and white ink), 2 mg (size 2 dark blue cap and orange body with white ink), 3 mg (size 2 dark blue cap and green body with white ink), and 4 mg (size 2 dark blue cap and blue body with white ink) containing mannitol, pregelatinized starch, and sodium stearyl fumarate;20 there is no alternative formulation option for adult patients with dysphagia who are unable to swallow an intact pomalidomide capsule. In addition, pomalidomide is currently under investigation for the treatment of solid tumors in the pediatric population and it is challenging to dose pediatric subjects, especially for pediatrics younger than 2 years who are not willing or not able to swallow an intact pomalidomide capsule. Therefore, the development of a pomalidomide liquid formulation is necessary to provide additional formulation options for adult patients with dysphagia and to support the clinical development of pomalidomide for pediatric indications.
Previously, a food effect study has been conducted on pomalidomide commercial capsule formulation. It was concluded that there was no clinically relevant impact of food intake on the absorption of pomalidomide, ie, pomalidomide capsules may be taken with or without food in the prescribing labels.19,20 Since liquid formulations such as suspension do not require disintegration of the formulation and thus are generally absorbed more readily than solid formulations and food may change or influence the bioavailability (BA) of liquid suspension formulations differently as that of solid capsule formulations, a food effect study on the liquid suspension is necessary and the results may help designing future clinical safety and efficacy studies using liquid suspension formulation in pediatric populations.
This was an open-label, randomized, three-period, two-sequence crossover study in healthy subjects. The primary objective of the study was to evaluate the BA of a pomalidomide oral liquid suspension (2 mg/mL, test formulation) relative to the commercial capsule formulation (4 mg capsule strength, reference formulation) when administered as a single 4 mg dose, and the secondary objectives were to assess the effect of food on the BA of the pomalidomide oral liquid suspension and to assess the safety and tolerability of the pomalidomide oral liquid suspension after a single dose. Blood samples for PK assessment were collected up to 48 h postdose during all three treatment periods, and safety was evaluated throughout the study.
Methods
This study was conducted and monitored in accordance with Celgene procedures and the study protocol. These procedures complied with the ethical principles of the International Conference on Harmonisation (ICH) harmonized tripartite guideline E6 (R1): Good Clinical Practice (GCP), as required by the major regulatory authorities. The conduct also complied with the Declaration of Helsinki, Title 21 of the US Code of Federal Regulations, Parts 50 and 56 concerning informed consent and institutional review board regulations and applicable national, state, and local laws or regulations. This study was conducted by Covance Clinical Research Unit Inc. (Daytona Beach, FL, USA) and was approved by the Salus Independent Review Board (Austin, TX, USA). The investigator obtained written informed consent from the subjects prior to any study-related procedures.
Study design
This was an open-label, randomized, three-period, two-sequence crossover study in healthy subjects. The study consisted of a screening phase (Days –21 to –2), a baseline assessment phase (Day –1), a treatment phase with three periods, and a follow-up phone call phase. Each treatment period was 3 days in duration for dosing and PK sample collection. The washout period between doses was 4 days. The eligibility of the subjects was assessed during the screening period. Eligible subjects were admitted to the study center on Day –1 for baseline assessments. On Day 1 of Treatment Period 1, subjects were assigned randomly to one of the two crossover sequences (sequence ABC or sequence BAC; 14 subjects per treatment sequence). On Day 1 of each treatment period, subjects received a single 4 mg oral dose of pomalidomide, according to the assigned sequence:
- Treatment A: single oral dose of 4 mg of pomalidomide (reference formulation, 1×4 mg of capsule) under fasted condition.
- Treatment B: single oral dose of 4 mg of pomalidomide (test formulation, 2.0 mL of oral liquid suspension) under fasted condition.
- Treatment C: single oral dose of 4 mg of pomalidomide (test formulation, 2.0 mL of oral liquid suspension) under fed condition.
The balanced crossover design removed the inter-subject variability from the comparison between formulations, while randomly assigning the subjects to one of the sequences-controlled bias that might otherwise have influenced the comparison. This portion of the study (Treatment Periods 1 and 2) was powered appropriately for the assessment of the bioequivalence (BE) between the two formulations. Pomalidomide was given under fasting conditions for this evaluation to avoid the effect of food on the oral absorption of pomalidomide. Food is known to moderately reduce the extent and rate of pomalidomide absorption when given as a capsule. Thus, an effect of food on the oral liquid suspension was anticipated and was assessed in this study. Since the present study was not intended to claim a no food effect, a fixed-sequence design (fasted followed by fed) was used for the evaluation of food effect.
In Treatment Period 3, following the 10 h overnight fast, subjects were served a high-fat and high-calorie breakfast ~30 min prior to the planned dose. The meal consisted of ~150, 250, and 500–600 calories derived from protein, carbohydrates, and fat, respectively. The subjects were instructed to eat the entire meal within 30 min of serving. Dosing must occur 30 min (±5 min) after the start of the meal.
The liquid suspension consisted of single-use vials each containing 20 mg of pomalidomide as the active ingredient with the same particle size as that in capsule formulation and the following components as the inactive ingredients: mannitol, pregelatinized starch, and sodium stearyl fumarate. The blend in single-use vials was constituted with 4 mL of water and 5 mL of Versa Plus® suspension vehicle to make the administration ready yellow opaque suspension with the final volume of 10 mL. The taste of the oral liquid suspension was close to the taste of diluent based on the results of the ASTREE Electronic Tongue instrument (Alpha MOS, Toulouse, France).
Subjects were confined to the study center from Day –1 (baseline) until Day 3 of Treatment Period 3, including the 4 days between dose washouts. Subjects were discharged from the study center on Day 3 of Treatment Period 3 following the completion of all required study procedures. A follow-up phone call to each subject was made ~4 days (±2 days) after discharge from the study center.
A total of 28 healthy adult male or female subjects were enrolled to obtain ~24 subjects who completed sufficient evaluable PK time points during both Treatment Periods 1 and 2 and ~16 subjects who completed sufficient evaluable PK time points during Treatment Period 3. The study design is presented in Figure 1.
 | Figure 1 Study design. Note: *Washout period was 4 days between doses. Abbreviation: PK, pharmacokinetic. |
Blood collection for PK analysis
During each treatment period, serial blood samples were collected predose and up to 48 h postdose at the following time points for the determination of pomalidomide plasma concentrations: 0 h predose and 0.25, 0.5, 1, 1.5, 2, 2.5, 3, 6, 8, 12, 24, 36, and 48 h postdose.
Safety assessment
Safety was evaluated throughout the study by the monitoring of AEs, electrocardiograms (ECGs), physical examinations (PEs), clinical laboratory tests, and pregnancy tests for female subjects, vital signs, and recording of concomitant medications and procedures. All AEs were monitored and recorded throughout the study from the time the informed consent form (ICF) was signed until study completion and when made known to the investigator within 28 days after the last dose of pomalidomide (and those serious AEs [SAEs] made known to the investigator at any time thereafter that are suspected of being related to pomalidomide). All concomitant medications and procedures were reviewed and recorded from the time the subject signs the ICF until study completion.
Bioanalytical methodology
Validated liquid chromatography–tandem mass spectrometry (LC–MS/MS) assays were used to assess pomalidomide concentrations in the plasma samples.18 Plasma samples were spiked with stable 13C5-labeled pomalidomide as internal standard. Pomalidomide and 13C5-labeled pomalidomide were extracted using liquid–liquid extraction from 0.2 mL of acidified human plasma samples (acidified with citric acid, pH =2.5). After transfer to a new tube, the solvent was evaporated, and the samples were reconstituted and injected for LC–MS/MS analysis using an analytical column (Luna C18 (2), 5 μm, 50 × 2.0 mm column; Phenomenex, Torrance, CA, USA). Positive ions were measured in the multiple reactions’ monitoring mode (m/z 274.1/201.1 for pomalidomide and 279.1/204.0 for the internal standard) using a SciexAPI-5000 tandem mass spectrometer (Sciex, Framingham, MA, USA) equipped with an Ion Spray source. The pomalidomide standard curves were linear (r2>0.99), from 0.25 to 100 ng/mL. The lower limit of quantitation was 0.25 ng/mL. Plasma quality control samples at three concentrations (0.75, 40, and 80 ng/mL) were used to determine assay accuracy and precision. Inter-day assay accuracy ranged from -0.37 to 2.13 deviating from the nominal concentration for plasma sample analysis. Inter-day precision did not exceed 5.48% for plasma assays.
PK analyses
Noncompartmental PK parameters, such as Cmax, time to peak (maximum) plasma drug concentration (tmax), area under the plasma concentration–time curve calculated from time 0 to the last measurable concentration at time t (AUC0–t), area under the plasma concentration–time curve from time 0 to infinity (AUC0–∞), t1/2, CL/F, apparent volume of distribution (Vz/F), and percentage of AUC0–∞ due to extrapolation from the time for the last quantifiable concentration to infinity (AUC%extrap), were calculated from the plasma concentration–time data with Phoenix™ WinNonlin® Professional Version 6.3 (Pharsight®; Certara, St Louis, MO, USA). Actual sampling times were used in the calculations. Descriptive statistics (N, mean, standard deviation [SD], percentage of coefficient of variation [CV%], geometric mean, geometric CV%, median, minimum, and maximum) were provided for concentrations at each time point and for all PK parameters.
Statistical analyses
Formal sample size calculation for the BE between Treatments A and B was performed based on a 2´2 crossover design. A sample size of 24 subjects was selected to reach 89% power so that the 90% CIs for the ratio of the geometric mean values of Cmax and AUC fell within the 80–125% limits. This calculation was based on an intra-subject SD of 0.2 and the mean ratio of 0.95–1.05 between test and reference formulations. An additional four subjects were proposed to account for possible dropouts. Therefore, the total sample size for this study was 28 subjects.
To assess the BA of pomalidomide test formulation (Treatment B) and reference formulation (Treatment A) under fasted conditions, an analysis of variance (ANOVA) model, with treatment, sequence, and period as fixed effects and subject nested within sequence as a random effect, was performed on the natural log-transformed AUCs and Cmax from Treatment Periods 1 and 2. The geometric mean values and percentage ratios (Treatment B/Treatment A) of the geometric mean values between test and reference formulations were presented, along with the 90% CIs for the ratios. Since tmax is a discrete variable reflecting the time of highest plasma concentration based on the discrete series of prespecified sampling time, a nonparametric analysis was used to produce a median difference between treatments. The median, median difference (test reference), and 90% CI of the median difference was obtained from the Hodges–Lehmann estimate. The P-value was derived from the Wilcoxon signed-rank test. The two pomalidomide formulations were considered bioequivalent if the 90% CIs of the ratio of geometric mean values between the two formulations for Cmax and AUC0–t were completely contained within the range of 80–125%.
To assess the effect of food on the pomalidomide liquid formulation, an ANOVA model, with treatment as a fixed effect and subject as a random effect, was performed on the natural log-transformed AUCs and Cmax for Treatment C (test formulation, fed) and Treatment B (test formulation, fasted) from Treatment Periods 1, 2, and 3. The geometric mean values and percentage ratios (Treatment C/Treatment B) of the geometric mean values between Treatments C and B were presented, along with the 90% CIs for the ratios. For tmax, a nonparametric analysis was used to produce a median difference between treatments. The median, median difference (fed–fasted), and 90% CI of the median difference were obtained from the Hodges–Lehmann estimate. The P-value was derived from the Wilcoxon signed-rank test.
All safety assessments, including AEs, vital sign measurements, clinical laboratory information, concomitant medications, physical examinations, and 12-lead ECG interpretations, were tabulated and summarized as appropriate. AEs were recorded and classified using the Medical Dictionary for Drug Regulatory Activities (MedDRA) classification system. Treatment emergent AEs (TEAEs) were summarized by frequency, severity, and relatedness to study drug. The frequency (the number of TEAEs and the number of subjects experiencing a TEAE) of TEAEs was tabulated by system organ class and preferred term. In the per-subject analyses, a subject having the same event more than once was counted only once. Laboratory and vital sign data were summarized descriptively (sample size [N], mean, SD, minimum [minimum], median, and maximum [maximum]). There was no statistical comparison of safety parameters between treatments.
Results
Demographic and other baseline characteristics
A total of 28 subjects were randomized and completed the study. A summary of demographics and baseline characteristics is presented in Table 1. Demographic data were similar between the two treatment sequences. Of the subjects, 15 (53.6%) were Hispanic or Latino and 13 (46.4%) were not Hispanic or Latino. The majority of subjects were male (75.0%), white (85.7%), with age ranging between 18 and 63 years, body weight ranging between 54.5 and 113.4 kg, and the body mass index ranging between 21.9 and 31.6 kg/m2.

Assessment of BE
The relative BA of a pomalidomide oral liquid suspension (test formulation, Treatment B) to the commercial capsule formulation (reference formulation, Treatment A) was evaluated as the primary endpoint in the present study.
Mean (± SD) pomalidomide plasma concentration versus time profiles from Treatment A and Treatment B are summarized in Figure 2. Mean pomalidomide plasma concentration time profiles were well characterized over the 48 h postdose sampling interval, and the AUC%extrap from all subjects was <20% (ranging from 0.47 to 4.86%), suggesting an adequate PK sampling schedule for both treatments.
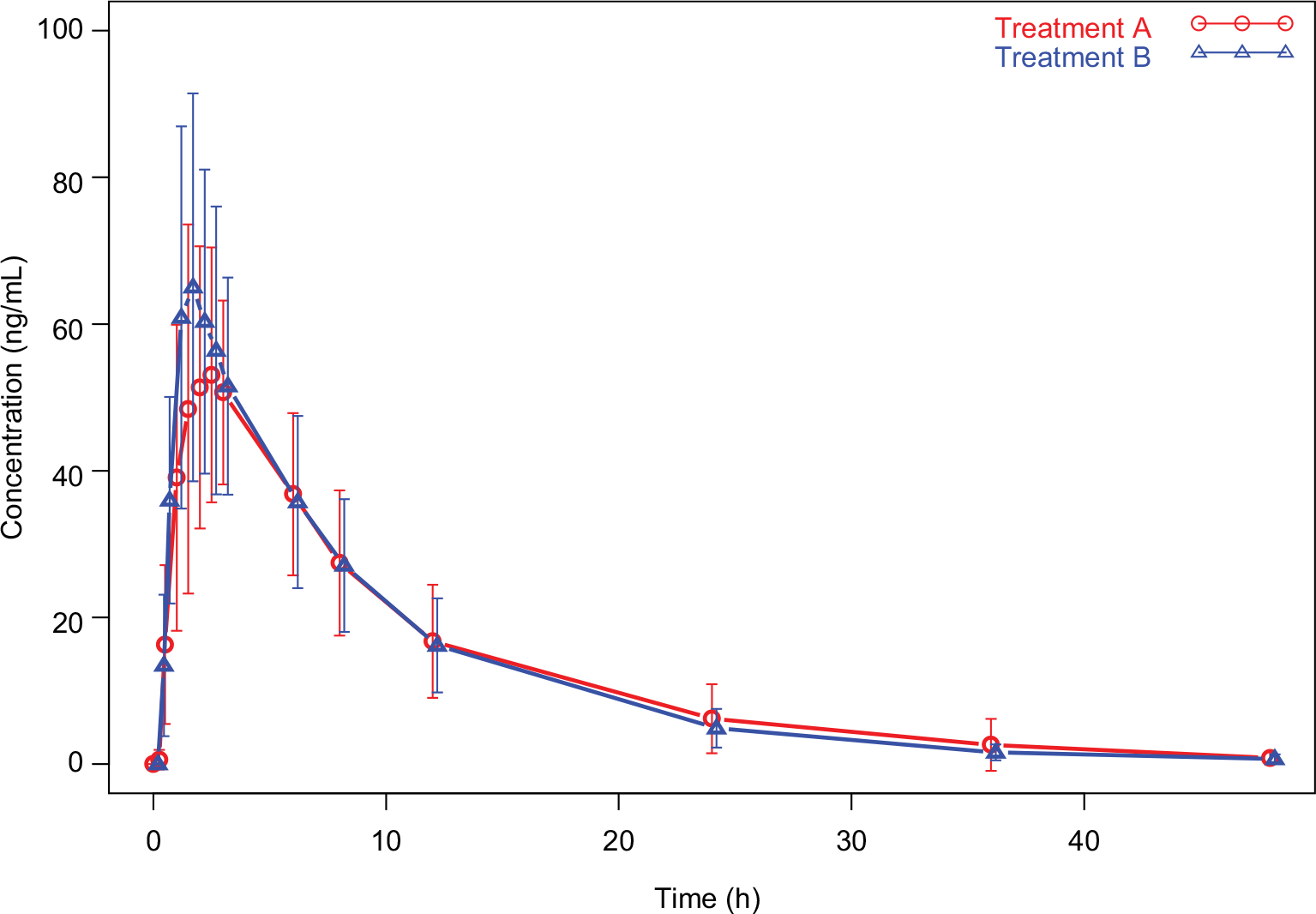 | Figure 2 Pomalidomide plasma concentration–time profiles: bioavailability assessment between oral liquid suspension and capsule (mean ± SD). Notes: Red lines and symbols represent data from Treatment A, and blue lines and symbols represent data from Treatment B. Treatment A: 4 mg of oral pomalidomide capsule, reference formulation. Treatment B: 4 mg of oral pomalidomide liquid suspension, test formulation. Abbreviation: SD, standard deviation. |
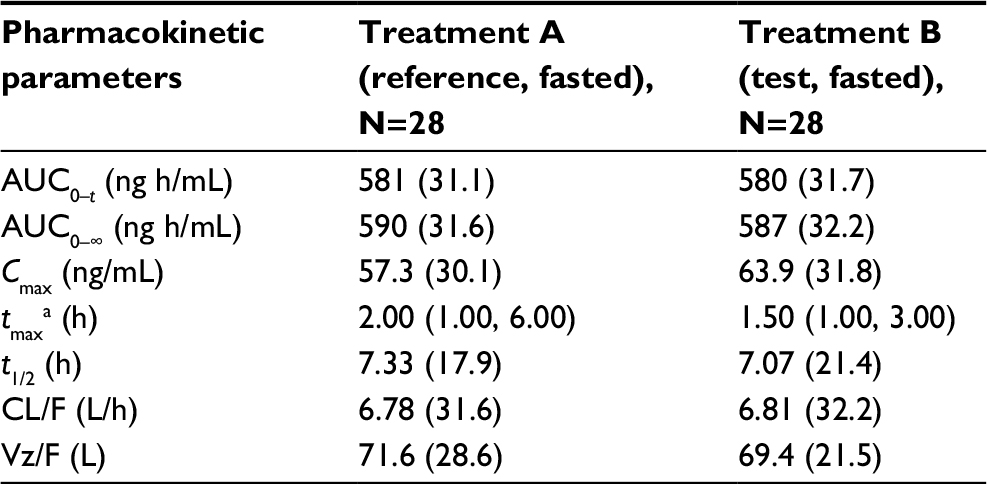
The plasma PK parameters of pomalidomide by treatment are summarized in Table 2. The rate of absorption, as reflected by tmax and Cmax, was slightly faster after the administration of Treatment B compared with Treatment A. As detailed in Table 2, the median tmax after the administration of Treatment B (1.5 h) was 0.5 h shorter than that observed after the administration of Treatment A (2.0 h). Additionally, the geometric mean pomalidomide Cmax values after the administration of Treatment B was slightly higher than that observed after the administration of Treatment A (63.9 and 57.3 ng/mL for Treatments B and A, respectively). Metrics of overall systemic exposure, AUC0–∞, and AUC0–t, was comparable across treatments (Table 2), suggesting that differences in formulation did not affect the extent of absorption. Similarly, pomalidomide t1/2, CL/F, and Vz/F were similar after the administration of Treatments A and B (Table 2). Hence, only the rate of pomalidomide absorption appeared to be slightly affected by the formulation differences.
Statistical analyses of the systemic exposure parameters, such as Cmax, AUC0–∞, and AUC0–t, were conducted to assess the BE of test and reference formulations. As shown in Table 3, the 90% CIs around the geometric mean ratios for Cmax, AUC0–∞, and AUC0–t fell within the 80–125% limits. BE between Treatment A and Treatment B was demonstrated.
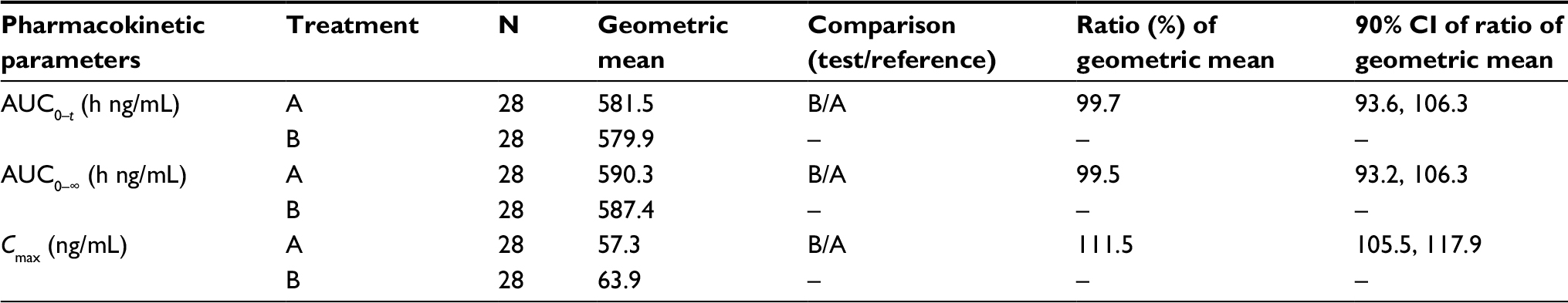 | Table 3 Statistical comparison of pomalidomide plasma pharmacokinetic parameters for relative bioavailability from oral liquid suspension to capsule: AUC and Cmax Notes: Treatment A: 4 mg of oral pomalidomide capsule, reference formulation, fasted. Treatment B: 4 mg of oral pomalidomide liquid suspension, test formulation, fasted. Abbreviations: CI, confidence interval; AUC0–∞, area under the plasma concentration–time curve from time 0 to infinity; AUC0–t, area under the plasma concentration–time curve calculated from time 0 to the last measurable concentration at time t; Cmax, peak (maximum) plasma drug concentration; N, number of subjects. |
Assessment of food effect on the pomalidomide oral liquid suspension
Mean (± SD) pomalidomide plasma concentration versus time profiles from Treatment B (pomalidomide liquid suspension administered under fasted conditions) and Treatment C (pomalidomide liquid suspension administered under fed conditions) are summarized in Figure 3. Mean pomalidomide plasma concentration time profiles are well characterized over the 48 h postdose sampling interval. The AUC%extrap from all subjects was <20% (ranging from 0.47 to 3.52%), suggesting an adequate PK sampling schedule for both treatments.
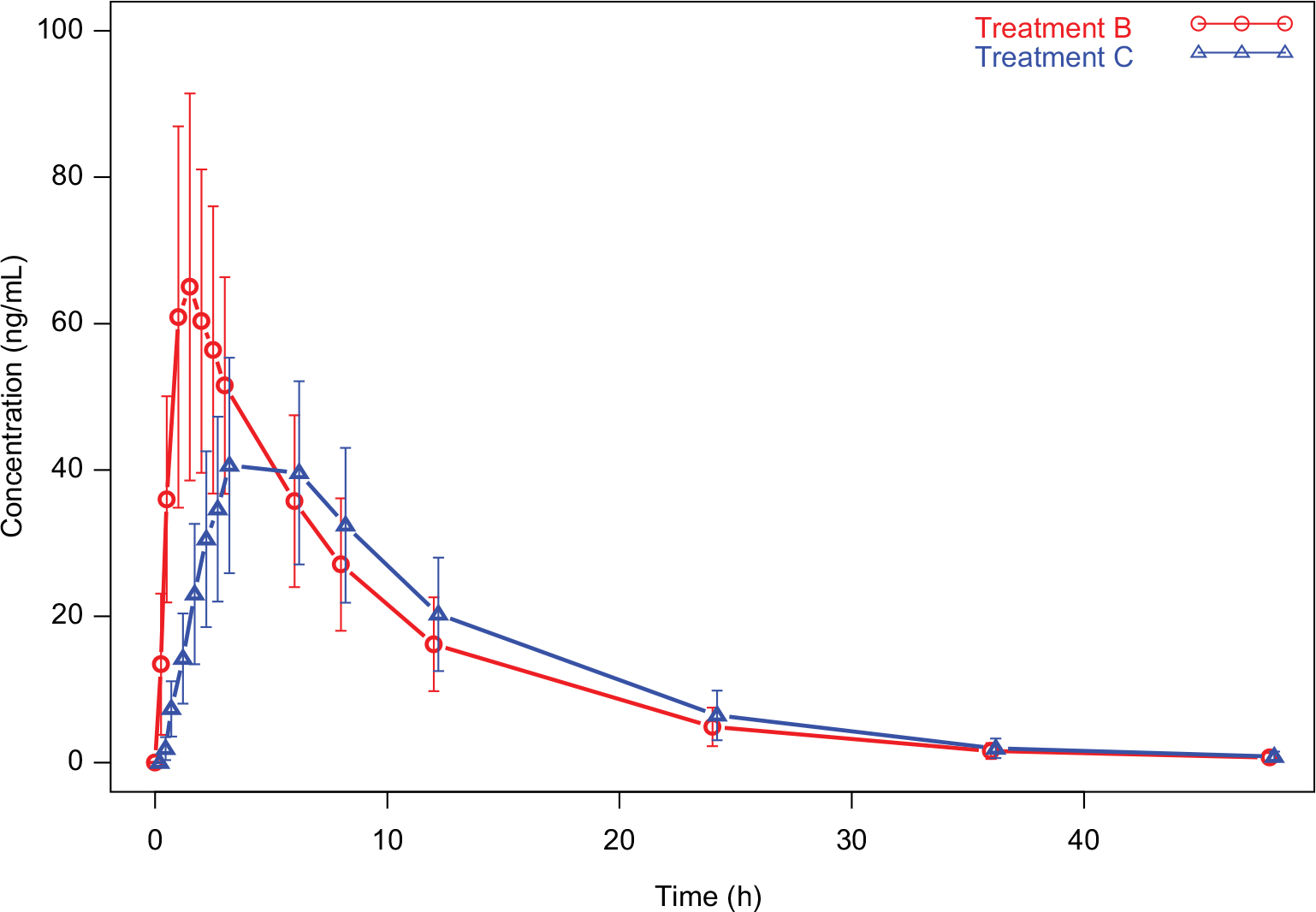 | Figure 3 Pomalidomide plasma concentration–time profiles: food effect on oral liquid suspension (mean ± SD). Notes: Red lines and symbols represent data from Treatment B, and blue lines and symbols represent data from Treatment C. Treatment B: 4 mg of oral pomalidomide liquid suspension, test formulation, fasted. Treatment C: 4 mg of oral pomalidomide liquid suspension, test formulation, fed. Abbreviation: SD, standard deviation. |
The plasma PK parameters of pomalidomide by treatment are summarized in Table 4. The mean pomalidomide plasma concentration–time profiles illustrated in Figure 3 reflect a significant (P<0.0001, Table 5) delay in absorption under fed conditions. Accordingly, the median tmax after Treatment C (4.5 h) was 3.0 h slower than that observed after Treatment B (1.5 h) as detailed in Table 5. There is a corresponding 34.5% decrease in geometric mean pomalidomide Cmax after the coadministration of pomalidomide test formulation with a high-fat meal (41.8 ng/mL) versus administration under fasted conditions (63.9 ng/mL) (Tables 4 and 5). However, there was only a ~3% decrease in AUC0–∞ and AUC0–t under fed conditions (Tables 4 and 5). Statistical analyses of pomalidomide systemic exposure parameters (Table 5) under fed and fasted conditions indicate that the 90% CI of the geometric mean ratios for AUC0–t and AUC0–∞ fell within the range of 80–125%, but that for Cmax was less than the lower BE boundary (a geometric mean ratio of 65.5% with a 90% CI of 61.9–69.3%).
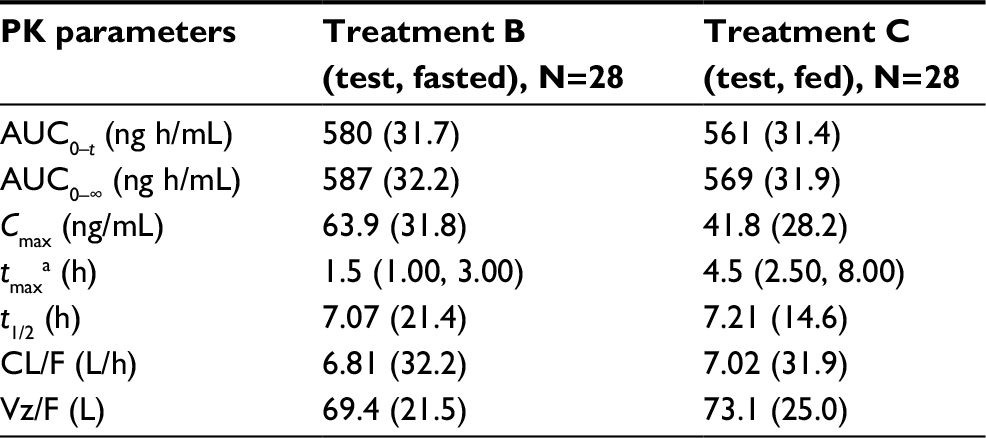 | Table 4 Summary of pomalidomide plasma PK parameters by treatment: food effect on oral liquid suspension Notes: Treatment B: 4 mg of oral pomalidomide liquid suspension, test formulation, fasted. Treatment C: 4 mg of oral pomalidomide liquid suspension, test formulation, fed. Geometric mean (geometric CV%) data are presented. aMedian (minimum, maximum) data are presented. Abbreviations: AUC0–∞, area under the plasma concentration–time curve from time 0 to infinity; AUC0–t, area under the plasma concentration–time curve calculated from time 0 to the last measurable concentration at time t; CL/F, apparent plasma clearance; Cmax, peak (maximum) plasma drug concentration; CV%, percentage of coefficient of variation; N, number of subjects; PK, pharmacokinetic; t1/2, terminal elimination half-life; tmax, time to peak (maximum) plasma drug concentration (Cmax); Vz/F, apparent volume of distribution. |
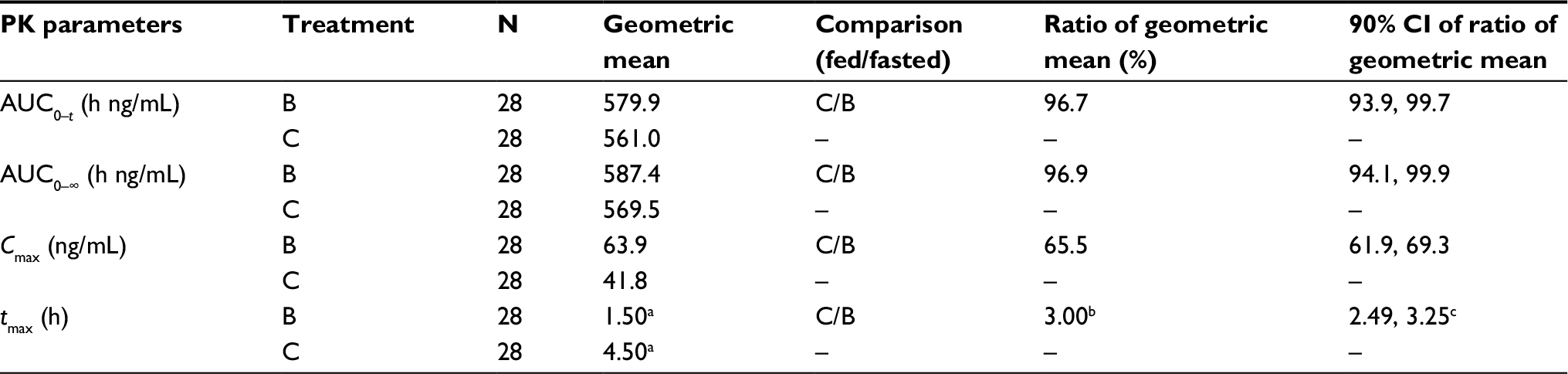 | Table 5 Statistical comparison of pomalidomide plasma pharmacokinetic parameters for food effect on oral liquid suspension: AUCs, Cmax, and tmax Notes: Treatment B: 4 mg of oral pomalidomide liquid suspension, test formulation, fasted. Treatment C: 4 mg of oral pomalidomide liquid suspension, test formulation, fed. aMedian. bMedian difference (P<0.0001). c90% CI of median difference. Abbreviations: AUC0–∞, area under the plasma concentration–time curve from time 0 to infinity; AUC0–t, area under the plasma concentration–time curve calculated from time 0 to the last measurable concentration at time t; CI, confidence interval; Cmax, peak (maximum) plasma drug concentration; N, number of subjects; tmax, time to peak (maximum) plasma drug concentration. |
Collectively, these data indicate that ingestion of food decreased the rate of oral absorption of liquid suspension as evidenced by the longer tmax and 34.5% decrease in Cmax under fed conditions. However, there appeared no significant impact of food on overall extent of absorption of liquid suspension as reflected by the 3% decrease in AUCs.
Safety
A single dose of 4 mg of pomalidomide was well tolerated when given as an oral liquid suspension (test formulation) under fasted or fed conditions and as a capsule (reference formulation) under fasted conditions. The incidence of TEAEs was low, and no TEAEs were reported following administration of the reference formulation. Overall, six of the 28 (21.4%) subjects reported eight TEAEs in this study. Seven of the eight TEAEs reported were judged by the investigator as suspected of being related to the study drug. No SAEs or severe TEAEs were reported, and no TEAE led to discontinuation of a subject from the study. All TEAEs were mild in severity and were reported by single subject only, except papular rash, which was reported by a total of two subjects. Except headache, all reported TEAEs were judged by the investigator as suspected of being related to study drug. All TEAEs resolved without treatment by the end of the study.
There were no apparent differences in mean clinical laboratory measurements prior to the first dose of study drug and at the end of the study. Sporadic out-of-range values occurred in some subjects for various clinical laboratory measurements. Prior to first dose administration, two laboratory measurements were recorded as mild AEs of hematuria and platelet count increased. One clinical laboratory value was reported as a TEAE by the investigator. One subject had a baseline (Day –1) alanine aminotransferase (ALT) value of 41 IU/L (reference range: 0–32 IU/L). At the next assessment on Day 3 of Treatment Period 3, the ALT value had increased to 122 IU/L and this was recorded as a mild TEAE with a suspected relationship to study drug. The ALT value had returned to within the normal range (31 IU/L) upon follow-up 10 days later.
Discussion
The literature suggests that dysphagia (swallowing dysfunction) is a debilitating, depressing, and potentially life-threatening complication in cancer patients that is likely underreported.21–24 Dysphagia is related to many factors such as direct impact of the tumor, cancer resection, chemotherapy, and radiotherapy and to newer therapies such as epidermal growth factor receptor inhibitors. Concomitant solid oral anticancer medications may exacerbate subjective dysphagia.21–24 Most literatures focused on head and neck cancer, but dysphagia is also common in other types of cancer such as MM. Lindgren and Janzon showed that swallowing difficulties and other esophageal symptoms such as heartburn and globus sensation are common complaints among patients who seek medical care.25 Pomalidomide capsules (1, 2, 3, and 4 mg) are the only available commercial formulations indicated, in combination with dexamethasone, for adult patients with MM and there is no alternative formulation option for adult patients with dysphagia who are unable to swallow an intact pomalidomide capsule. The intended patient population for pomalidomide is generally elderly, and dysphagia is a major concern in this elderly population and may result in reduced patient adherence to medications. In addition, pomalidomide is currently under investigation in the pediatric population for solid tumors. Therefore, the development of a pomalidomide liquid formulation is necessary to provide additional formulation options for adult patients with dysphagia and to support the clinical development of pomalidomide for pediatric indications. However, liquid suspension may alter the BA especially the absorption properties of pomalidomide. Consequently, in vivo assessment of alternative liquid suspension formulation versus commercial capsule formulation, with respect to BA and PK properties, is necessary.
The global regulatory requirement for the equivalent BA, or the BE, of medications with different formulations has become crucial in medicine and pharmaceutical development.26 FDA guidance of BA and BE studies for orally administered drug products suggests that studies to measure BA and/or establish BE of a product are important elements in support of investigational new drugs (INDs), new drug applications (NDAs), abbreviated new drug applications (ANDAs), and their supplements. BA studies focus on determining the process by which a drug is released from the oral dosage form and moves to the site of action.27 BA data provide an estimate of the fraction of the drug absorbed, as well as its subsequent distribution and elimination, and studies to establish BE between two products are important for certain changes before approval for a pioneer product in NDA and ANDA submissions and in the presence of certain postapproval changes in NDAs and ANDAs. Consistently, the primary objective of the present study was to evaluate the BA of a pomalidomide oral liquid suspension (2 mg/mL, test formulation) relative to the commercial capsule formulation (4 mg capsule strength, reference formulation) when administered as a single 4 mg dose. The present study showed that exposures to pomalidomide were similar following single oral dose administration of test (Treatment B) or reference (Treatment A) formulation in the fasted state. BE between Treatment A and Treatment B was demonstrated by statistical analysis, which showed that the 90% CIs of geometric mean ratios (test versus reference) for AUC0–t and AUC0–∞ and Cmax were within the acceptable range of BE (80–125%). The study also reported the comparison tmax. The median (range) values of tmax of pomalidomide were 1.5 (1.0, 3.0) and 2.0 (1.0, 6.0) h for the liquid suspension and commercial capsule formulations, respectively, which is consistent with the fact that liquid formulations do not require disintegration of the formulation and thus are generally absorbed more readily than solid formulations. Wilcoxon matched-pairs test showed the difference in tmax between the two formulations was statistically significant (data not shown). Clinically, however, the difference was not meaningful since both the test and reference formulations reached the tmax quickly and showed nonsignificant impact on the maximum drug exposures.
FDA guidance of food effect BA and fed BE studies suggests that administration of a drug product with food may change the BA by affecting either the drug substance or the drug product and recommends that a food effect BA study be conducted for all new chemical entities (NCEs) during the IND period and for the to-be-marketed commercial formulations for NDA.28 A previous food effect BA study has been conducted on pomalidomide capsule formulation, and it showed that coadministration of pomalidomide with a high-fat meal (~50% of the total caloric content) and a high-calorie meal (~800–1000 calories) (the meal contained ~150, 250, and 500–600 calories from protein, carbohydrates, and fat, respectively) delayed the tmax by 2.5 h, decreased mean plasma Cmax and AUC in healthy volunteers by ~27 and 8%, respectively. Based on these findings, it was concluded that pomalidomide may be taken with or without food in the prescribing label. Considering that food is known to moderately reduce both the rate and extent of pomalidomide absorption when given as a capsule and the potential different in vivo absorption properties between solid formulations such as capsules and liquid suspension formulations such as suspension, a food effect study on the liquid suspension is necessary and the results may help to design future clinical safety and efficacy studies using liquid suspension in pediatric populations. Accordingly, the secondary objective of the present study was to assess the effect of food on the BA of the pomalidomide oral liquid suspension after a single 4 mg dose. Fairly consistent with those from the previous food effect assessment on the commercial capsule formulations, the ingestion of a high-fat meal within 30 min prior to the administration of pomalidomide liquid suspension resulted in a reduction in Cmax (34.5%) and AUC (3%) relative to the test liquid suspension formulation administered in the fasted state. Food delayed tmax with the median tmax values of 1.5 and 4.5 h observed for the fasted and fed states, respectively. Our previous exposure versus response analysis of data collected from a Phase III trial suggested no obvious exposure–efficacy relationship for pomalidomide within a wide range of pomalidomide plasma exposures under the clinical dose of 4 mg; however, dose interruption within Cycle 1 was statistically significantly associated with higher drug exposure (data on file). Therefore, it can be concluded from this analysis that the mild-to-moderate difference in AUC and Cmax observed when pomalidomide liquid suspension was administered with food would not appear to influence pomalidomide’s efficacy and safety profiles.
Regarding the safety, a single dose of 4 mg pomalidomide was safe and well tolerated when given as a capsule under fasted conditions and when given as an oral liquid suspension under fasted or fed conditions. No SAEs or severe TEAEs were reported during the study, and no subjects were discontinued from the study due to a TEAE. Except for a laboratory value that was reported as a TEAE, there were no clinically-significant findings in clinical laboratory evaluations, vital sign measurements, or ECGs during this study.
Conclusion
Our study showed that a single oral dose of 4 mg test liquid suspension was bioequivalent to a single oral dose of 4 mg reference commercial capsule formulation. Consistent with the results from the previous food effect study on the pomalidomide commercial capsule formulation, it was demonstrated a mild to modest but clinically insignificant effect of food on pomalidomide liquid suspension and it was recommended that pomalidomide liquid suspension may be taken regardless of food intake. Finally, single oral dose of 4 mg pomalidomide was safe and well tolerated by healthy subjects when administered as an oral liquid suspension under fed and fasted conditions or as a capsule under fasted conditions.
Disclosure
YL, LL, LH, XW, MH, JR, MP, and SZ are employees of and hold equity ownership in Celgene Corporation. The authors report no other conflicts of interest in this work.
References
Gay F, Mina R, Troia R, Bringhen S. Pharmacokinetic evaluation of pomalidomide for the treatment of myeloma. Expert Opin Drug Metab Toxicol. 2013;9(11):1517–1527. | ||
Li Y, Zhou S, Hoffmann M, Kumar G, Palmisano M. Modeling and simulation to probe the pharmacokinetic disposition of pomalidomide R- and S-enantiomers. J Pharmacol Exp Ther. 2014;350(2):265–272. | ||
Mitsiades N, Mitsiades CS, Poulaki V, et al. Apoptotic signaling induced by immunomodulatory thalidomide analogs in human multiple myeloma cells: therapeutic implications. Blood. 2002;99(12):4525–4530. | ||
Zhu D, Corral LG, Fleming YW, Stein B. Immunomodulatory drugs Revlimid (lenalidomide) and CC-4047 induce apoptosis of both hematological and solid tumor cells through NK cell activation. Cancer Immunol Immunother. 2008;57(12):1849–1859. | ||
Gupta D, Treon SP, Shima Y, et al. Adherence of multiple myeloma cells to bone marrow stromal cells upregulates vascular endothelial growth factor secretion: therapeutic applications. Leukemia. 2001;15(12):1950–1961. | ||
Hideshima T, Chauhan D, Shima Y, et al. Thalidomide and its analogs overcome drug resistance of human multiple myeloma cells to conventional therapy. Blood. 2000;96(9):2943–2950. | ||
Lu L, Payvandi F, Wu L, et al. The anti-cancer drug lenalidomide inhibits angiogenesis and metastasis via multiple inhibitory effects on endothelial cell function in normoxic and hypoxic conditions. Microvasc Res. 2009;77(2):78–86. | ||
Reddy N, Hernandez-Ilizaliturri FJ, Deeb G, et al. Immunomodulatory drugs stimulate natural killer-cell function, alter cytokine production by dendritic cells, and inhibit angiogenesis enhancing the anti-tumour activity of rituximab in vivo. Br J Haematol. 2008;140(1):36–45. | ||
Verhelle D, Corral LG, Wong K, et al. Lenalidomide and CC-4047 inhibit the proliferation of malignant B cells while expanding normal CD34+ progenitor cells. Cancer Res. 2007;67(2):746–755. | ||
Corral LG, Haslett PA, Muller GW, et al. Differential cytokine modulation and T cell activation by two distinct classes of thalidomide analogues that are potent inhibitors of TNF-alpha. J Immunol. 1999;163(1):380–386. | ||
Hayashi T, Hideshima T, Akiyama M, et al. Molecular mechanisms whereby immunomodulatory drugs activate natural killer cells: clinical application. Br J Haematol. 2005;128(2):192–203. | ||
Teo SK, Chen Y, Muller GW, et al. Chiral inversion of the second generation IMiD CC-4047 (ACTIMID) in human plasma and phosphate-buffered saline. Chirality. 2003;15(4):348–351. | ||
San Miguel J, Weisel K, Moreau P, et al. Pomalidomide plus low-dose dexamethasone versus high-dose dexamethasone alone for patients with relapsed and refractory multiple myeloma (MM-003): a randomised, open-label, phase 3 trial. Lancet Oncol. 2013;14(11):1055–1066. | ||
Daver N, Shastri A, Kadia T, et al. Phase II study of pomalidomide in combination with prednisone in patients with myelofibrosis and significant anemia. Leuk Res. 2014;38(9):1126–1129. | ||
Richardson PG, Siegel DS, Vij R, et al. Pomalidomide alone or in combination with low-dose dexamethasone in relapsed and refractory multiple myeloma: a randomized phase 2 study. Blood. 2014;123(12):1826–1832. | ||
Li Y, Xu Y, Liu L, Wang X, Palmisano M, Zhou S. Population pharmacokinetics of pomalidomide. J Clin Pharmacol. 2015;55(5):563–572. | ||
Kasserra C, Assaf M, Hoffmann M, et al. Pomalidomide: evaluation of cytochrome P450 and transporter-mediated drug-drug interaction potential in vitro and in healthy subjects. J Clin Pharmacol. 2015;55(2):168–178. | ||
Hoffmann M, Kasserra C, Reyes J, et al. Absorption, metabolism and excretion of [14C]pomalidomide in humans following oral administration. Cancer Chemother Pharmacol. 2013;71(2):489–501. | ||
Pomalyst (pomalidomide) P. [package insert]. Summit, NJ: Celgene Corporation; 2016. | ||
Imnovid (pomalidomide) I. [Summary of Product Characteristics]. Uxbridge, UK: Celgene Europe; 2016. | ||
Russi EG, Corvo R, Merlotti A, et al. Swallowing dysfunction in head and neck cancer patients treated by radiotherapy: review and recommendations of the supportive task group of the Italian Association of Radiation Oncology. Cancer Treat Rev. 2012;38(8):1033–1049. | ||
Dysphagia Section, Oral Care Study Group, Multinational Association of Supportive Care in Cancer (MASCC)/International Society of Oral Oncology (ISOO), Raber-Durlacher JE, Brennan MT, et al. Swallowing dysfunction in cancer patients. Support Care Cancer. 2012;20(3):433–443. | ||
Tei K, Maekawa K, Kitada H, Ohiro Y, Yamazaki Y, Totsuka Y. Recovery from postsurgical swallowing dysfunction in patients with oral cancer. J Oral Maxillofac Surg. 2007;65(6):1077–1083. | ||
Cleary L. An occupational therapy program for patients with swallowing dysfunction following cancer treatment. Occup Ther Health Care. 1986;3(2):23–37. | ||
Lindgren S, Janzon L. Prevalence of swallowing complaints and clinical findings among 50–79-year old men and women in an Urvan population. Dysphagia. 1991;6(6):187–192. | ||
Tjandrawinata RR, Setiawati A, Nofiarny D, Susanto LW, Setiawati E. Pharmacokinetic equivalence study of nonsteroidal anti-inflammatory drug etoricoxib. Clin Pharmacol. 2018;10:43–51. | ||
FDA. Guidance for Industry: Bioavailability and Bioequivalence Studies for Orally Administered Drug Products – General Considerations. Silver Spring, MD: FDA; 2003. Available from: https://www.fda.gov/downloads/drugs/guidancecomplianceregulatoryinformation/guidances/ucm389370.pdf. Accessed April 1, 2018. | ||
FDA. Guidance for Industry: Food-Effect Bioavailability and Fed Bioequivalence Studies. Silver Spring, MD: FDA; 2002. Available from: https://www.fda.gov/downloads/drugs/guidancecomplianceregulatoryinformation/guidances/ucm070241.pdf. Accessed April 1, 2018. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.