Back to Journals » Drug Design, Development and Therapy » Volume 9
A phase 1 randomized study evaluating the effect of omeprazole on the pharmacokinetics of a novel 5-hydroxytryptamine receptor 4 agonist, revexepride (SSP-002358), in healthy adults
Authors Pierce D, Corcoran M, Velinova M, Hossack S, Hoppenbrouwers M, Martin P
Received 22 March 2014
Accepted for publication 14 May 2014
Published 27 February 2015 Volume 2015:9 Pages 1257—1268
DOI https://doi.org/10.2147/DDDT.S64621
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
David Pierce,1 Mary Corcoran,2 Maria Velinova,3 Stuart Hossack,4 Mieke Hoppenbrouwers,5 Patrick Martin,2
1Shire, Basingstoke, UK; 2Shire, Wayne, PA, USA; 3PRA International, Zuidlaren, the Netherlands; 4Covance, Leeds, UK; 5Shire-Movetis NV, Turnhout, Belgium
Background: About 30% of patients with gastroesophageal reflux disease continue to experience symptoms despite treatment with proton pump inhibitors. The 5-hydroxytryptamine 4 receptor agonist revexepride (SSP-002358) is a novel prokinetic that stimulates gastrointestinal motility, which has been suggested as a continued cause of symptoms in these patients. The aim of this study was to assess whether revexepride pharmacokinetics were affected by co-administration of omeprazole, in preparation for a proof-of-concept evaluation of revexepride added to proton pump inhibitor treatment.
Methods: In this phase 1, open-label, randomized, two-period crossover study, healthy adults aged 18–55 years were given a single dose of revexepride 1 mg or revexepride 1 mg + omeprazole 40 mg. Pharmacokinetic parameters were assessed for up to 48 hours after administration of the investigational product. Adverse events, clinical chemistry and hematology parameters, electrocardiograms, and vital signs were monitored.
Results: In total, 42 participants were enrolled and 40 completed the study. The median age was 24 years (18–54 years), 55% were women and 93% were white. The pharmacokinetic parameters of revexepride were similar without or with omeprazole co-administration. The mean area under the plasma concentration–time curve from time 0 to infinity (AUC0–∞) was 23.3 ng · h/mL (standard deviation [SD]: 6.33 ng · h/mL) versus 24.6 ng · h/mL (SD: 6.31 ng · h/mL), and maximum plasma concentrations (Cmax) were 3.89 ng/mL (SD: 1.30 ng/mL) and 4.12 ng/mL (SD: 1.29 ng/mL) in participants without and with omeprazole, respectively. For AUC0–∞ and Cmax, the 90% confidence intervals for the ratios of geometric least-squares means (with:without omeprazole) were fully contained within the pre-defined equivalence limits of 0.80–1.25. Mean apparent terminal phase half-life was 9.95 hours (SD: 2.06 hours) without omeprazole, and 11.0 hours (SD: 3.25 hours) with omeprazole.
Conclusion: Co-administration of the 5-hydroxytryptamine receptor 4 agonist revexepride with omeprazole did not affect the pharmacokinetics of revexepride in healthy adults.
Keywords: revexepride, omeprazole, pharmacokinetics, gastroesophageal reflux disease
Introduction
Gastroesophageal reflux disease (GERD) is a chronic condition characterized by the symptoms of heartburn and regurgitation, which are caused by gastroesophageal reflux.1 It has been reported to affect 17%–28% of patients in primary care2 and is associated with lower health-related quality of life than that found in the general population.3
Treatments for GERD include over-the-counter antacids, as well as prescription drugs that reduce gastric acid secretion. The latter group includes proton pump inhibitors (PPIs) such as omeprazole, esomeprazole, and rabeprazole, and histamine type 2 receptor antagonists.4 PPIs are the most effective and widely used prescription treatments for GERD, achieving marked improvement in symptoms in most patients; however, about 30% of individuals with GERD continue to experience symptoms despite PPI treatment.4
In some of these patients, symptoms persist even when acid secretion is effectively suppressed.5 Dysmotility of the gastrointestinal tract has been suggested to be a cause of continued symptoms, and prokinetic agents may therefore be of benefit.6 Revexepride (SSP-002358) is one of a new class of 5-hydroxytryptamine 4 receptor (5-HT4) agonists that has been developed with the aim of stimulating gastrointestinal motility, accelerating gastric emptying, and increasing lower esophageal sphincter pressure. The chemical structure of the compound is shown in Figure 1. It is a highly potent and specific 5-HT4 agonist, which enhances the physiological release of acetylcholine at the myenteric plexus.
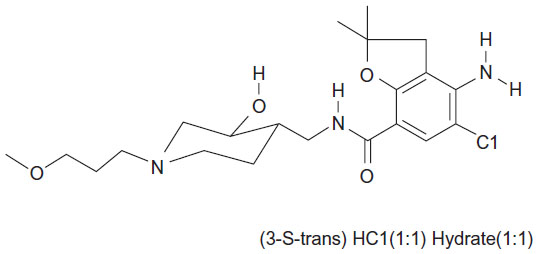 | Figure 1 Structural formula of revexepride (SSP-002358). |
As a potential therapy for GERD, revexepride would be used in combination with or directly following treatment with PPIs, which have a lasting inhibitory effect on gastric acid secretion.7 Changes in gastric pH affect the absorption of some drugs. Revexepride has high solubility across the gastric pH range, so an effect on its pharmacokinetics due to a change in gastric pH is unlikely. There is, however, potential for a drug–drug interaction (DDI) between revexepride and PPIs through the enzyme cytochrome P450 (CYP) 3A4/5. An in vitro study demonstrated that CYP3A4/5 is involved in the metabolism of revexepride and the formation of the associated normetabolite (Supplementary material), and CYP3A4 is also involved in the metabolism of PPIs.8 Furthermore, when ketoconazole, a known CYP3A4 inhibitor, was co-administered with revexepride, there was a two to three fold increase in the systemic exposure to revexepride-base (Supplementary material).
To evaluate a potential DDI between revexepride and PPIs, the pharmacokinetics of revexepride were compared in the presence or absence of omeprazole, a commonly used PPI that has been shown to be metabolized by CYP3A4.8 The study did not aim to address the impact of revexepride on the pharmacokinetics of omeprazole. Revexepride was not expected to alter the pharmacokinetics of co-administered omeprazole, because an in vitro study had previously demonstrated that revexepride had minimal effects on the activities of the major CYP groups (Supplementary material). Moreover, the typical dose of omeprazole in clinical practice (40 mg once daily) is at least 20 fold higher than the proposed doses of revexepride in the present proof-of-concept study (≤2 mg).
Materials and methods
Objectives
The primary objective of this study was to examine the effect of co-administration of omeprazole on the pharmacokinetics of revexepride. The secondary objective was to provide additional safety information on revexepride when administered alone or in combination with omeprazole.
Study design
This was a phase 1, open-label, randomized, two-period crossover, drug interaction study investigating the pharmacokinetic profile of revexepride when administered alone and in combination with omeprazole (Figure 2; ClinicalTrials.gov identifier: NCT01415349). Participants were randomized in a 1:1 ratio on Day 1 of Treatment Period 1 to receive one of two treatment sequences, A followed by B or B followed by A, in which the treatments were: A) a single oral dose of revexepride 1 mg (expressed as base-equivalents); B) a single oral dose of revexepride 1 mg + a single oral dose of omeprazole 40 mg. The alternative treatment was administered on Day 1 of Treatment Period 2 following a 4 day washout period and overnight fasting. Participants received a follow-up telephone call 7 days after the last dose of investigational product. A 1 mg dose was used because it was in the potential clinical range and would allow a multifold increase in revexepride plasma concentrations without compromising safety and tolerability.
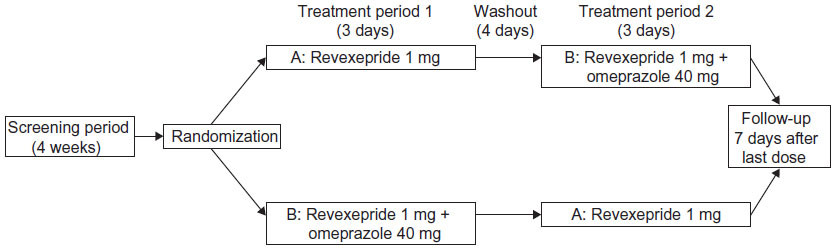 | Figure 2 Study design flow chart. |
Clinical data collection and safety analyses were carried out at PRA International, Zuidlaren, the Netherlands, and PRA International, Raleigh, NC, USA, respectively. The study took place in August–September 2011 and was carried out in accordance with the International Conference on Harmonisation of Good Clinical Practice, the principles of the Declaration of Helsinki, and applicable local ethical and legal requirements.9,10 Before entering the study, all participants provided written informed consent, which was reviewed, along with the study protocol, by the institutional review board of the clinical research center and an independent ethics committee (Medische Ethische Toetsings Commisssie [METC] van de Stichting Beoordeling Ethiek Biomedisch Onderzoek [BEBO], Assen, the Netherlands).
Study participants
Healthy men and healthy non-pregnant women aged 18–55 years were included in the study. Study participants had to have a body mass index in the range 18.5–30 kg/m2 at the first visit and no current or recurrent disease that could affect the pharmacokinetics or safety assessments of the investigational product. They were also required to: have no known intolerance or hypersensitivity to the drugs used in the study; not be taking any medication apart from hormone replacement therapy or hormonal contraceptives in the 14 days before receiving the first dose of investigational product; and to not have taken either drug used in the study in the 30 days before receiving their first dose.
Participants were excluded if they had: donated blood in the 60 days before receiving the first dose of investigational product; a history of drug or alcohol abuse in the past year; consumed more than 300 mg of caffeine a day; smoked or used nicotine-containing products; drunk more than 21 (men) or 14 (women) units of alcohol per week; or eaten any product containing grapefruit or Seville oranges (which inhibit CYP activity)11 in the 7 days before receiving the first dose of investigational product.
Treatments
The investigational product was administered on Day 1 of each treatment period. Study participants were required to fast overnight before drug administration and until 4 hours post-dose. Participants remained in the study center for assessments from Day 1 until the completion of all study assessments on Day 3 for each treatment period.
Revexepride was administered as 2×0.5 mg (expressed as base-equivalents) monohydrochloride monohydrate salt tablets (batch number AF110101). Omeprazole was administered as a 40 mg capsule (Losec®; AstraZeneca, London, UK; batch number AF1101010). Both revexepride tablets and omeprazole capsules were swallowed whole with approximately 240 mL water.
Pharmacokinetic assessments
Bioanalysis and sample storage took place at Analytical Biochemical Laboratory BV, Assen, the Netherlands. Serial 2 mL blood samples were collected before administration of the investigational product and at the following times after drug administration in each treatment period: 0.5, 1, 1.5, 2, 3, 4, 6, 8, 10, 12, 24, 36, and 48 hours. Plasma revexepride-base concentrations were determined using a validated high-performance liquid chromatography with tandem mass spectrometry assay, which was linear over the range 2.08–1,040 pg/mL, with a lower limit of quantification of 2.08 pg/mL. Where applicable, samples were diluted ten fold in blank human heparin to bring the drug concentration within the quantitation limits.
Quality control and calibration standard data were collected in accordance with the US Food and Drug Administration Guidance for Industry: Bioanalytical Method Validation.12 Quality control samples were prepared in control human plasma at three concentration levels (6.23, 83.1, and 831 pg/mL), stored with the study samples, and analyzed with each batch against freshly prepared calibration standards. Mean estimates of accuracy ranged from −2.7% to −7.1% and mean estimates of precision ranged from 3.2% to 8.7% across the three concentration levels. Although plasma concentrations of revexepride were determined in pg/mL during bioanalysis, they were converted to ng/mL before pharmacokinetic analysis for consistency with previous data.
Covance Clinical Research Unit, Leeds, UK, carried out pharmacokinetic and related statistical analysis and reporting of these data. The following pharmacokinetic parameters were analyzed from the concentration–time data using WinNonlin® version 5.2 (Pharsight Corporation, Mountain View, CA, USA):maximum plasma concentration (Cmax); time to Cmax (tmax); time of last quantifiable plasma concentration (tlast); apparent terminal phase half-life (t½); apparent terminal phase rate constant (λz); area under the plasma concentration–time curve from time 0 to tlast (AUC0–t); area under the plasma concentration–time curve from time 0 to infinity (AUC0–∞), calculated as “AUC0–t + (last measurable plasma concentration [Ct]/λz)”; proportion of AUC that is due to extrapolation from tlast to infinity (%extrap).
Safety assessments
Participant health was assessed at screening, and the day before and 3 days after administration of the investigational product. Assessment included physical examination, electrocardiography, and clinical laboratory tests. Information on adverse events (AEs) was recorded from the time of informed consent until the end of follow-up or until the AE resolved.
Statistical analysis
Endpoints
The primary endpoints were Cmax and AUC0–∞ of revexepride-base following treatment with revexepride alone or revexepride + omeprazole. Secondary endpoints were AUC0–t and tmax for revexepride-base.
Analysis populations
The Safety Analysis Set included all study participants who took at least one dose of investigational product and had at least one post-dose safety assessment. Participants in the Safety Analysis Set whose primary pharmacokinetic data were considered sufficient and interpretable were included in the Pharmacokinetic Analysis Set. Participants were excluded from the pharmacokinetic descriptive statistics and statistical analysis for the treatment period if they vomited or experienced significant diarrhea that crossed two or more sampling times in the 10 hours after dosing.
Estimation of sample size
The sample size was estimated based on the probability of achieving equivalence for each of the primary endpoints (revexepride-base Cmax and AUC0–∞). Based on the findings of a previous open-label, randomized, three-way crossover, single-dose pharmacokinetic study in healthy adults receiving revexepride 1 mg capsules (Supplementary material), a coefficient of variation of 25% for both Cmax and AUC0–∞ was assumed. To demonstrate equivalence, allowing for a 5% difference in true means (ratio of 1.05) and true within-subject coefficient of variation of 25%, 36 participants were required to achieve 90% power. To ensure that 36 participants completed the study, it was decided to enroll a total of 42 individuals.
Analysis of pharmacokinetic parameters
Following logarithmic transformation, the primary endpoints, Cmax and AUC0–∞, for participants in the Pharmacokinetic Analysis Set were analyzed using a mixed-effect linear model with sequence group (AB or BA), treatment period (1 or 2), and treatment (revexepride, or revexepride + omeprazole) as fixed effects. Subject-within-sequence was included as a random effect. For each endpoint, the least-squares (LS) mean and associated standard error were determined for both treatments. The LS mean and standard error were also calculated for the difference between treatments, and used to evaluate a 90% confidence interval (CI) for the difference between the logs of the two treatments. To return to the original scale, an exponential transformation was applied to the lower and upper limits of the CI. This created a 90% CI for the ratio of revexepride + omeprazole to revexepride alone. Point estimates and 90% CIs for the primary endpoints, Cmax and AUC0–∞, were assessed against the accepted bioequivalence criteria for log-transformed data (0.80, 1.25).
The main secondary pharmacokinetic endpoint, AUC0–t, was analyzed by the same methods as the primary endpoints but was not assessed against equivalence criteria. The analysis of the other secondary endpoint, tmax, was conducted without transformation or modeling. Each value from participants treated with revexepride alone was subtracted from each value from participants in the revexepride + omeprazole group to create a set of all differences in observations between the two treatment regimens. The median difference and 90% CI were then calculated based on Hodges–Lehmann estimate for Wilcoxon’s signed-rank test.
Results
Participant disposition and demographics
Forty-two participants were enrolled and included in the Safety Analysis Set, of whom 40 completed the study (Figure 3). Two participants were withdrawn from the study during Treatment Period 1: one participant withdrew informed consent and the other participant discontinued owing to an AE (cystitis). Both withdrawals occurred on day 3 of Treatment Period 1 after receiving revexepride + omeprazole. These two participants were included in the Pharmacokinetic Analysis Set. One participant was excluded from the Pharmacokinetic Analysis Set owing to vomiting in Treatment Period 1 and significant diarrhea in both treatment periods.
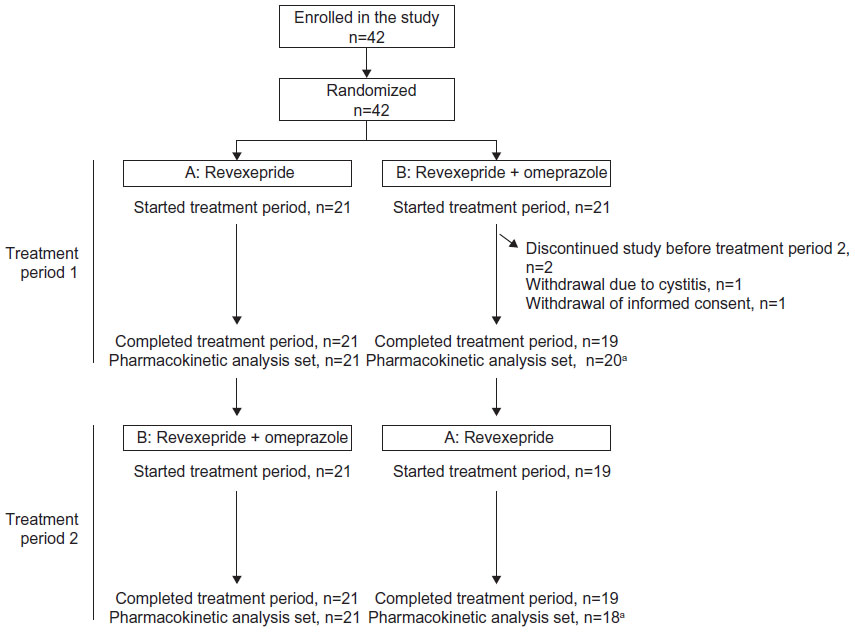 | Figure 3 Overview of disposition of participants in the study. |
In total, 23 (54.8%) participants in the Safety Analysis Set were women, and the median age was 24 years (range, 18–54 years; Table 1). Most participants (92.9%) were white. Patient demographics were similar between treatment groups.
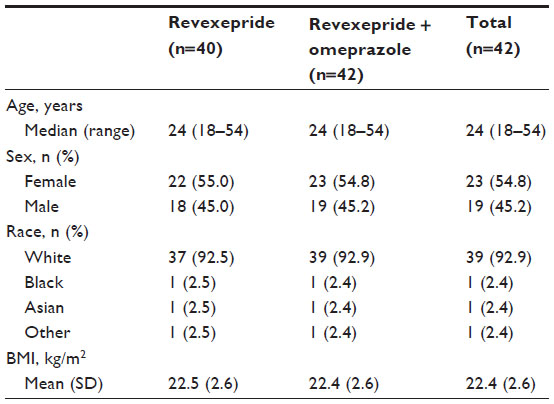 | Table 1 Baseline demographics of the Safety Analysis Set |
Pharmacokinetics
Six participants in the Pharmacokinetic Analysis Set experienced significant diarrhea between dosing and 10 hours post-dose in Treatment Period 1. The plasma concentration data and the pharmacokinetic data for these six participants were excluded from the descriptive statistics and statistical analyses for Treatment Period 1 (three received revexepride and three received revexepride + omeprazole).
The pharmacokinetic profile of revexepride in the presence or absence of co-administration with omeprazole is shown in Figure 4. Mean plasma concentrations of revexepride peaked 1 hour after administration of investigational product to similar levels in each treatment group.
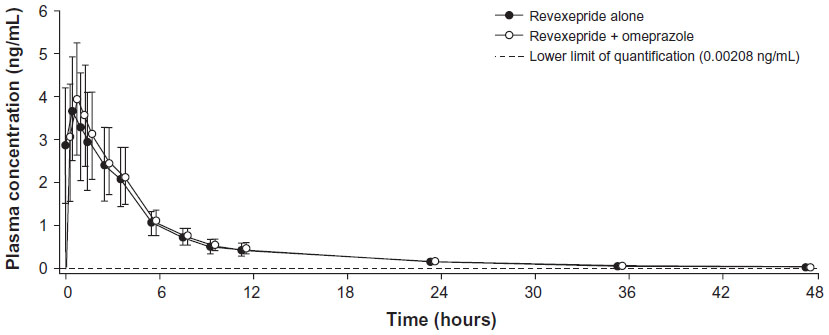 | Figure 4 Plasma concentration–time profiles for revexepride-base for the pharmacokinetic analysis set following a single dose of revexepride 1 mg alone or co-administered with omeprazole 40 mg. |
Pharmacokinetic parameters of revexepride following a single dose administered alone or in combination with omeprazole are presented in Table 2. The primary endpoints, AUC0–∞ and Cmax mean values, were similar in participants who had taken revexepride alone and in those who had received revexepride + omeprazole (Table 2). The mean AUC0–t was also similar between treatment groups. The median value of tmax was 1.00 hour for both treatment groups, with ranges of 0.50–4.00 hours with revexepride alone and 0.50–1.50 hours with revexepride + omeprazole (Table 2). Mean t½ values were similar for the two treatments, at 9.95 hours (standard deviation [SD]: 2.06 hours) for revexepride alone and 11.0 hours (SD: 3.25 hours) for revexepride + omeprazole.
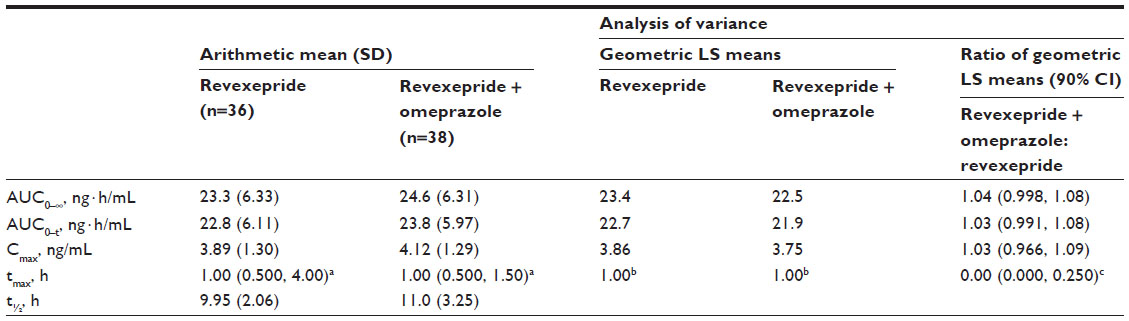 | Table 2 Arithmetic mean values and analysis of variance of plasma pharmacokinetic parameters of revexepride-base following a single dose of revexepride 1 mg administered alone or in combination with omeprazole 40 mg |
Analysis of variance demonstrated that there was no effect of co-administration with omeprazole on systemic exposure to revexepride (Table 2). The 90% CIs for the geometric LS mean ratios revexepride + omeprazole to revexepride alone of the primary endpoints (AUC0–∞: 0.998, 1.08; Cmax: 0.966, 1.09) fell within the predefined equivalence limits of 0.80 and 1.25.
Safety and tolerability
All but one of the participants experienced at least one treatment-emergent adverse event (TEAE) during the study. However, no TEAEs were considered serious, and only one TEAE led to early withdrawal. One participant experienced a severe TEAE that was considered related to the investigational product (vomiting following administration of revexepride + omeprazole). One participant withdrew from the study following a case of cystitis that was considered by the investigator not to be related to the investigational product.
The incidence of TEAEs was similar between treatment groups, with TEAEs affecting 93% and 91% of participants treated with revexepride and revexepride + omeprazole, respectively (Table 3). The most common TEAEs were gastrointestinal disorders (revexepride: 75%, revexepride + omeprazole: 79%), particularly diarrhea (revexepride: 53%, revexepride + omeprazole: 62%). Headache was also common, affecting 30 participants (75%) in the revexepride group and 33 participants (79%) in the revexepride + omeprazole group.
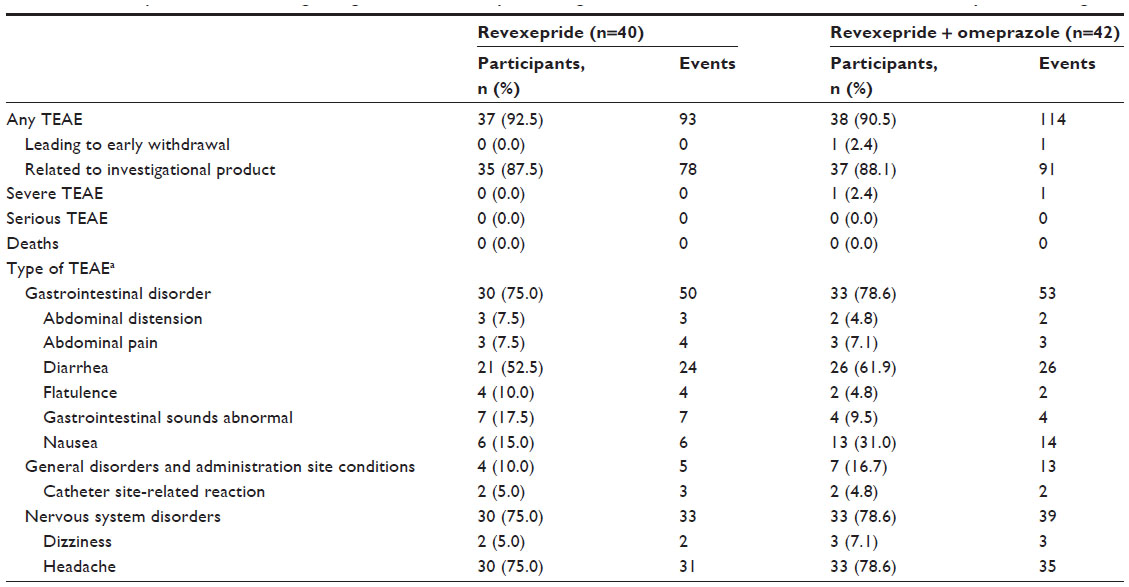 | Table 3 Summary of TEAEs following a single dose of revexepride 1 mg administered alone or in combination with omeprazole 40 mg |
No cardiac TEAEs were observed during the study. No clinically relevant changes compared with baseline were observed in electrocardiogram parameters in any of the study participants. There were no clinically significant changes in pulse rate or blood pressure compared with baseline in either treatment group, and no QT prolongation or other evidence of effects on cardiac repolarization were noted (data not shown).
Discussion
The results of the present study demonstrate that co-administration of omeprazole did not have an effect on the pharmacokinetics of revexepride.
The concentration–time profiles and rates and extents of absorption of revexepride-base were similar in the presence and absence of omeprazole. These observations are consistent with the fact that revexepride is highly soluble across a range of acidities (pH 1–8), and confirm that alteration of gastrointestinal pH by omeprazole therefore does not affect revexepride absorption.
In vitro data have shown CYP3A4/5 to be involved in the metabolism of revexepride-base (Supplementary material). In addition, a DDI study demonstrated a two to three fold increase in systemic exposure to revexepride when revexepride 1 mg was co-administered with the potent CYP3A inhibitor ketoconazole (Supplementary material). Together, these data indicate that revexepride is metabolized predominantly by CYP3A4/5. Omeprazole is also metabolized to some extent by CYP3A4/5, suggesting that co-administration might interfere with revexepride metabolism.8,13 The lack of such interference in the present study can be explained because omeprazole is also metabolized by CYP2C19, for which it has an approximately ten fold higher affinity.13
The most commonly observed TEAEs in the present study were gastrointestinal disorders and headache. These are consistent with known AEs for omeprazole and AEs expected of a prokinetic drug, as well as with data from in-house pharmacokinetic studies in healthy participants (Supplementary material), and likely reflect the enterokinetic (gastrointestinal disorders) and serotonergic (headache) properties of the drug. There have been concerns over the potential for 5-HT4 receptor agonists to cause cardiovascular side effects, which led to the withdrawal of cisapride, the first compound in this class;11 however, this compound exhibited additional pharmacology as a potent blocker of the human ether-à-go-go-related gene channel. This channel is responsible for the repolarization phase of the cardiac action potential, which is believed to be the cause of these cardiovascular AEs.11 No cardiac TEAEs were seen in the present study in which participants were treated with the more selective 5-HT4 agonist revexepride, and there were no clinically significant changes in cardiovascular parameters or vital signs.
A possible limitation of the study is that the pharmacokinetics of omeprazole were not assessed. As discussed above, however, previous evidence indicates that revexepride has minimal effects on the activities of any of the major CYP enzyme groups (Supplementary material). Moreover, because the dose of revexepride administered was much lower than that of omeprazole, concentrations of revexepride reaching the shared drug metabolizing enzymes CYP3A4/5 would also have been substantially lower than those of omeprazole. Therefore, the pharmacokinetics of omeprazole in these participants were not expected to be affected by revexepride at the relative doses of the two drugs used in this study.
Additional treatment options are needed for patients with GERD whose symptoms are not relieved by PPIs. Promotility drugs could be useful for increasing gastrointestinal motility and alleviating symptoms in these patients.4 The data presented here suggest that, from a pharmacokinetics perspective, revexepride could be used in addition to PPIs. Further studies are warranted to evaluate the efficacy and safety of revexepride in patients with GERD.
Conclusion
There was no effect of co-administration of omeprazole 40 mg with the novel 5-HT4 receptor agonist revexepride 1 mg on the pharmacokinetics of revexepride, compared with revexepride 1 mg alone. The safety and tolerability profiles of revexepride were similar when taken alone or in combination with omeprazole 40 mg, and no serious TEAEs were reported.
Acknowledgments
Clinical research was funded by Shire International GmBH, Eysins, Switzerland. We would like to thank Richard Abbott and Analytical Biochemical Laboratory BV, Assen, the Netherlands for sample storage and excellent bioanalysis. Clinical data collection, safety analyses, and data management were carried out at PRA International, Zuidlaren, the Netherlands, PRA International, Raleigh, NC, USA, and PRA International, Swansea, UK, respectively. Illingworth Research Ltd, Macclesfield, UK was responsible for clinical monitoring. Covance Clinical Research Unit, Leeds, UK, carried out pharmacokinetic and related statistical analysis and reporting of these data. Under the direction of the authors, Rosalind Morley, PhD, from PharmaGenesis™ London, London, UK, provided medical writing assistance for this manuscript, funded by Shire.
Disclosure
The study was funded by Shire International GmbH, Eysins, Switzerland. MC and PM are employees of Shire and own stock in Shire. When the study was conducted, DP and MH were employees of Shire and hold stock and/or stock options in Shire. MV is an employee of PRA International, which received funding from Shire for the clinical conduct of the study. SH is an employee of Covance, which received funding from Shire to carry out pharmacokinetic and statistical analysis and reporting of the study. PharmaGenesis™ London, London, UK, was funded by Shire to provide medical writing assistance in the preparation of the manuscript.
References
Vakil N, van Zanten SV, Kahrilas P, Dent J, Jones R, Global Consensus Group. The Montreal definition and classification of gastroesophageal reflux disease: a global evidence-based consensus. Am J Gastroenterol. 2006;101(8):1900–1920. | |
El-Serag H, Becher A, Jones R. Systematic review: persistent reflux symptoms on proton pump inhibitor therapy in primary care and community studies. Aliment Pharmacol Ther. 2010;32:720–737. | |
Ponce J, Beltran B, Ponce M, et al. Impact of gastroesophageal reflux disease on the quality of life of Spanish patients: the relevance of the biometric factors and the severity of symptoms. Eur J Gastroenterol Hepatol. 2009;21(6):620–629. | |
Bredenoord AJ. New therapies for gastroesophageal reflux disease. Minerva Gastroenterol Dietol. 2010;56(2):129–138. | |
Vela MF, Camacho-Lobato L, Srinivasan R, Tutuian R, Katz PO, Castell DO. Simultaneous intraesophageal impedance and pH measurement of acid and nonacid gastroesophageal reflux: effect of omeprazole. Gastroenterology. 2001;120(7):1599–1606. | |
Farre R, Sifrim D. Regulation of basal tone, relaxation and contraction of the lower oesophageal sphincter. Relevance to drug discovery for oesophageal disorders. Br J Pharmacol. 2008;153(5):858–869. | |
Vakil N. Treatment of gastroesophageal reflux disease: defining endpoints that are important to patients. Rev Gastroenterol Disord. 2004;4 Suppl 4:S3–S7. | |
Meyer UA. Interaction of proton pump inhibitors with cytochromes P450: consequences for drug interactions. Yale J Biol Med. 1996;69(3):203–209. | |
International Conference on Harmonisation of Technical Requirement for Registration of Pharmaceuticals for Human Use. Ich Harmonised Tripartite Guidelines for Good Clinical Practice, 1996. Available from: http://www.ich.org/fileadmin/Public_Web_Site/ICH_Products/Guidelines/Efficacy/E6_R1/Step4/E6_R1__Guideline.pdf. Accessed December 17, 2013. | |
World Medical Association. Declaration of Helsinki: Ethical Principles for Medical Research Involving Human Subjects. Available from: http://www.wma.net/en/30publications/10policies/b3/. Accessed June 3, 2013. | |
Quigley EM. Cisapride: what can we learn from the rise and fall of a prokinetic? J Dig Dis. 2011;12(3):147–156. | |
Center for Drug Evaluation and Research (CDER). Guidance for Industry: Bioanalytical Method Validation, 2001. Available from: http://www.fda.gov/downloads/Drugs/Guidances/ucm070107.pdf. Accessed March 11, 2014. | |
Ishizaki T, Horai Y. Review article: cytochrome P450 and the metabolism of proton pump inhibitors – emphasis on rabeprazole. Aliment Pharmacol Ther. 1999;13 Suppl 3:27–36. |
Supplementary materials
In vitro experiments showing that CYP3A4 was involved in the metabolism of revexepride-base
Cytochrome P450 isoenzymes involved in metabolism
Based on in vitro inhibitor experiments and in vitro metabolism experiments with heterologous expression systems, the human CYP isoenzymes involved in the metabolism of revexepride were identified and quantified. Revexepride is mainly metabolized in vitro in humans by CYP3A4 (99.9%) with a minor contribution of CYP2D6 (0.1%). However, the intrinsic clearance in human hepatic microsomes is low, and when scaled to in vivo liver metabolic clearance in humans it was also low, which indicates that metabolism may not be a major clearance route in humans; thus CYP mediated drug–drug interactions where revexepride is the victim are unlikely. Revexepride was not a substrate for human CYP3A7 (a CYP enzyme expressed in newborns and infants).
Cytochrome P450 inhibition and induction
Revexepride was not a direct or metabolism-dependent inhibitor of human CYP1A2, CYP2A6, CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6 and CYP2E1 in vitro. Half maximal inhibitory concentration (IC50) direct inhibition values were higher than 100 μM, the highest revexepride concentration tested. Revexepride showed direct inhibition of human CYP3A4 in vitro with IC50 values of 16–49 μM. Revexepride showed metabolism-dependent inhibition of CYP3A4 in vitro with a decrease in IC50, following a 30 minute pre-incubation with nicotinamide adenine dinucleotide phosphate-oxidase of 5.6–14 fold. Metabolism-dependent inhibition of CYP3A4 was confirmed in vitro with maximal rate constants of inactivation of 0.0434 min−1, and the concentration of revexepride required to achieve half-maximal inactivation was 8.09 μM. Data suggest that revexepride is not a reversible CYP3A4 inhibitor, but it is not possible to distinguish between a quasi-irreversible or irreversible inhibition mechanism. At the predicted maximal plasma concentration after a 2 mg three times daily dose, this inhibition of CYP3A4 could be clinically significant. SSP-002833 (a metabolite of revexepride) showed inhibition of CYP3A4 in vitro with a calculated IC50 value of 85 μM, while the IC50 values of SSP-002834, SSP-002835, SSP-002836, and SSP-002837 (other metabolites of revexepride) were higher than 100 μM. Revexepride did not induce human CYP1A2 in vitro (based on both catalytic and gene expression measurements). Revexepride seems to be a slight inducer (at least in some hepatocyte donors used in the study) of the CYP2B6 enzyme in vitro, but the observed increase was significantly lower than that observed after treatment with the known inducer rifampicin. Revexepride is a potential inducer of the CYP3A4 enzyme (based on gene expression evaluation), and its in vitro induction potential is, at least in some donors, comparable to that of the known inducer rifampicin.
BEL-14 – co-administration of ketoconazole leads to an increase in revexepride-base
Revexepride–ketoconazole interaction
Inhibition experiments in human liver microsomes revealed that CYP3A4 was a major CYP form involved in the metabolism of revexepride-base. Other CYP isoenzymes contributed to revexepride-base metabolism to a minor extent. The urine excretion results obtained in study BEL-1 showed that the percentage of the dose excreted in the urine as unchanged revexepride-base was approximately 12% after a single revexepride-HBr dose.
The double-blind, placebo-controlled, randomized, 2 period crossover study, BEL-14, was carried out from 18 May to 7 July 1999 at a single center in Belgium. On days 1–8 of each period, participants received either ketoconazole 200 mg or placebo twice daily. On day 2 of each period, individuals received a single dose of 1 mg revexepride. On days 4–7 of each period, participants received 1 mg revexepride three times daily, followed by a single morning dose on day 8, together with either ketoconazole 200 mg tablets or corresponding placebo tablets twice daily. After the single dose of revexepride on day 2, the pharmacokinetic parameters of revexepride were assessed until 48 hours post-dosing. At steady state (day 8), the pharmacokinetic parameters of revexepride-base were assessed (until 72 hours post-dosing). A washout interval of at least 1 week was required before the participants were crossed over to the second period.
Fourteen healthy volunteers (eight men and six women) were enrolled in the study. Their mean age was 41.1±2.3 years (range 23–55 years). All participants were fully compliant and took all doses of the investigational product(s) as specified in the protocol, with the exception of one individual who forgot to take one placebo tablet in the evening of day 7 in period 2. The pharmacokinetic parameters for revexepride-base in this study are listed in Table S1.
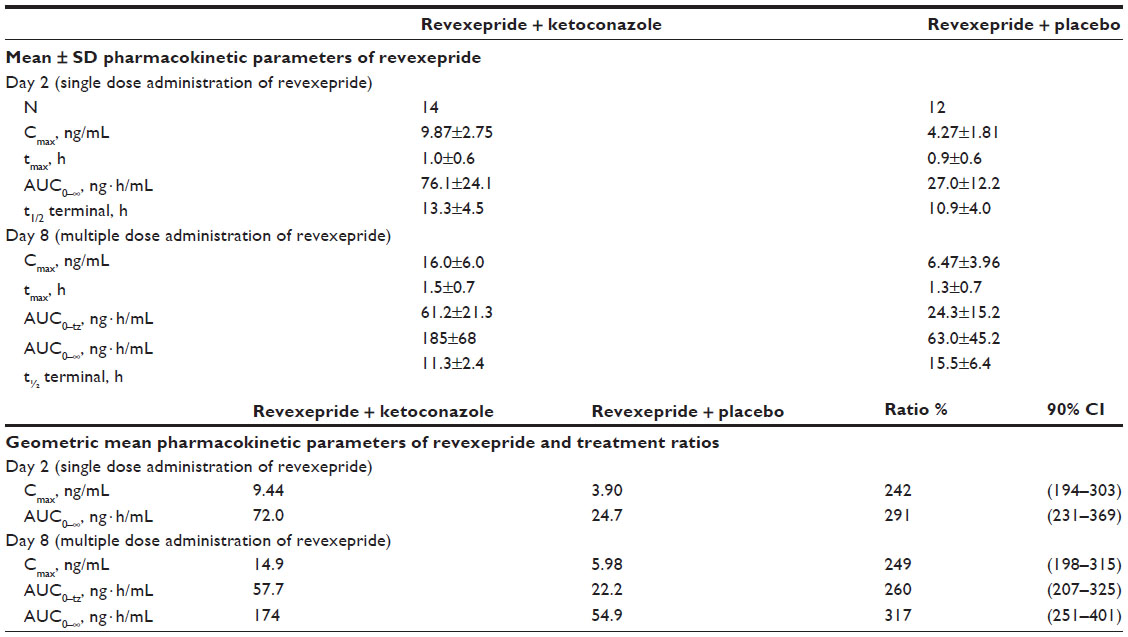 | Table S1 Overview of the mean pharmacokinetic parameters of revexepride-base after a single 1 mg dose and after a 1 mg three times daily dose regimen, with co-administration of ketoconazole 200 mg twice daily (or placebo) in study BEL-14 |
Co-administration of revexepride with ketoconazole triggered a clinically significant pharmacokinetic drug–drug interaction, which resulted in a two and three fold increase in the plasma concentrations of revexepride-base for maximum plasma concentration (Cmax) and area under the plasma concentration–time curve from time 0 to infinity, respectively, after either single or three times daily dosing. Whereas administration of 1 mg revexepride was well-tolerated, the combined administration of revexepride with ketoconazole was only moderately tolerated. Following intake of a single dose of revexepride, there was a similar number of adverse events (AEs) during co-treatment with ketoconazole (78.6% of participants) compared with placebo (75.0%). However, a higher level of abdominal pain (42.9% versus 0%) and diarrhea (71.4% versus 66.7%) was seen with ketoconazole compared with placebo. The incidence of AEs decreased during multiple dosing with revexepride, and were resolved before the end of the study (5 day multiple dose). Two participants discontinued co-administration of revexepride with ketoconazole due to AEs (vomiting and severe abdominal pain, both known side effects of ketoconazole). These results indicate that combined use of revexepride and strong CYP3A4-inhibitors should be met with caution.
NED-3 – single dose pharmacokinetic study, the results of which were used for sample size calculations
This open-label, randomized, three way crossover, Phase 1 study was conducted from 21 July 1999 to 27 August 1999. Participants were randomized to receive either a single revexepride 1 mg capsule (Treatment A, in the fasted state; Treatment C, after a high-fat, high-caloric breakfast), or 10 mL of a 0.1 mg/mL revexepride-HBr oral solution (Treatment B, in the fasted state). Treatments were separated by a washout period of 7 days. Blood samples were taken just before dosing on day 1 and at several time points thereafter, until 48 hours post-dosing.
Twelve men were enrolled in the study. Their mean age was 26 years (range: 20–47 years), and their mean body weight was 78 kg (range: 63–91 kg). No protocol deviations occurred during the study. Pharmacokinetic parameters are shown in Table S2. The ratio estimates for Cmax and area under the curve after intake of a 1 mg revexepride capsule in the fasted state, were 116% and 109%, respectively. The 90% confidence interval (CI)s for the ratio estimates were included within 80%–125% limits for area under the curve and within 70%–143% limits for Cmax. Peak plasma concentrations were attained 0.5–2 hours after dosing for both the revexepride capsule and the revexepride-HBr oral solution.
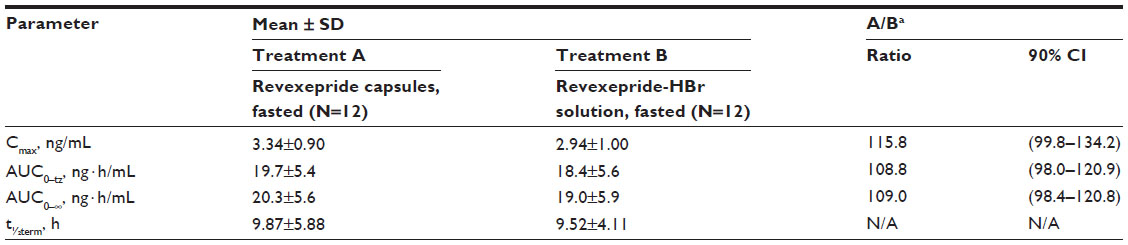 | Table S2 Summary of bioequivalence parameters and statistics after a single oral intake of 1 mg revexepride capsules or revexepride-HBr solution in the fasted state in study NED-3 |
The laboratory safety, cardiovascular safety and tolerability of capsules (fasted and fed) and oral solution were found to be comparable. There were no clinically relevant changes in any safety parameters. All three treatments were safe and well tolerated.
Treatment-emergent adverse events in healthy volunteers in previous phase 1 trials of revexepride
Safety data were pooled for eleven phase 1 studies (BEL-1, BEL-2, BEL-3, BEL-4, BEL-5, BEL-7, BEL-8, BEL-14, NED-2, NED-3, and SPD557-101). Overall, 93.5% of participants treated with revexepride had at least one treatment-emergent adverse event (TEAE), compared with 23.3% of those who received placebo. The most common TEAEs were headache, diarrhea, abnormal gastrointestinal sounds, and nausea. These TEAEs were mostly resolved by the end of the studies. Most TEAEs were reported on the day of intake and on the first day after the first administration of investigational product, and became less frequent during repeated dosing (ie, tolerability improved). Most TEAEs were mild to moderate in severity. A summary of the most common (≥5%) TEAEs by system organ class and preferred term are shown in Table S3.
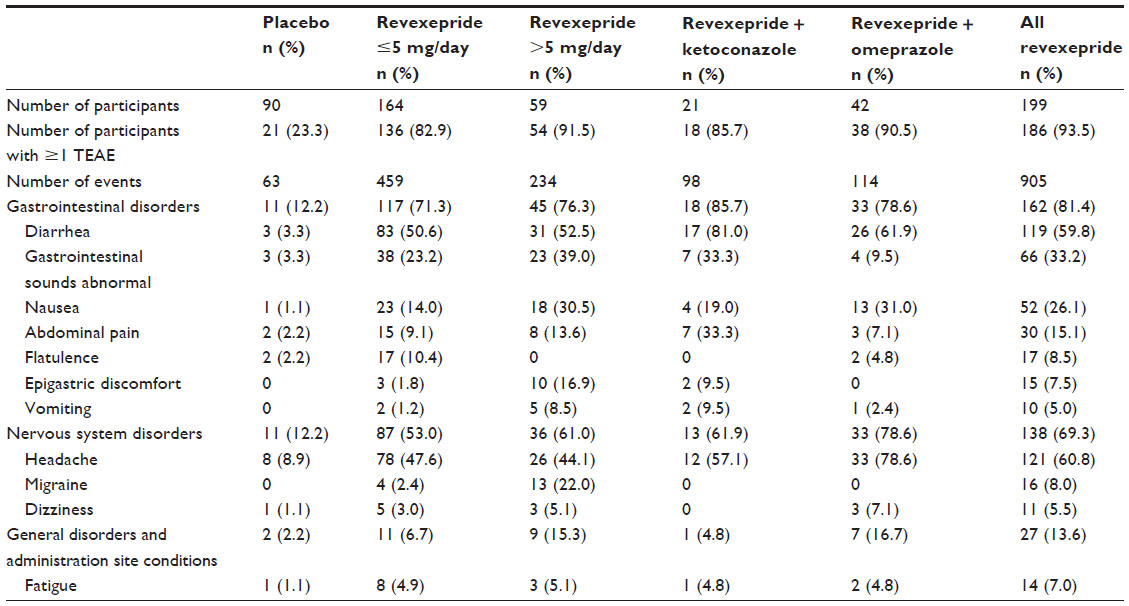 | Table S3 Summary of treatment-emergent adverse events observed for ≥5% of revexepride-treated participants in phase 1 studies (Pooled Safety Population) |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.