Back to Journals » Clinical Ophthalmology » Volume 13
A phase 1, open-label, single-arm study evaluating the ocular safety of OTX-101 and systemic absorption of cyclosporine in healthy human volunteers
Authors Karpecki PM ![]() , Weiss SL
, Weiss SL ![]() , Kramer WG, O'Connor P, Evans D, Johnston J, Jasper AL, Justice A
, Kramer WG, O'Connor P, Evans D, Johnston J, Jasper AL, Justice A ![]() , Ogundele AB, Devries D
, Ogundele AB, Devries D
Received 18 September 2018
Accepted for publication 29 January 2019
Published 5 April 2019 Volume 2019:13 Pages 591—596
DOI https://doi.org/10.2147/OPTH.S187945
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Scott Fraser
Paul M Karpecki,1,2 Sidney L Weiss,3 William G Kramer,4 Patrick O’Connor,5 David Evans,6 Josh Johnston,7 April L Jasper,8 Angela Justice,9 Abayomi B Ogundele,9 Doug Devries10
1Kentucky Eye Institute, Lexington, KY, USA; 2Gaddie Eye Centers, Louisville, KY, USA; 3i-novion, Inc., Randolph, NJ, USA; 4Kramer Consulting, LLC, North Potomac, MD, USA; 5Auven Therapeutics, Delray Beach, FL, USA; 6Total Eye Care, Memphis, TN, USA; 7Georgia Eye Partners, Atlanta, GA, USA; 8Advanced Eyecare Specialists, West Palm Beach, FL, USA; 9Sun Ophthalmics, Sun Pharmaceutical Industries, Ltd., Princeton, NJ, USA; 10Eye Care Associates of Nevada, Sparks, NV, USA
Purpose: To evaluate the ocular safety of OTX-101 0.09% – a novel, nanomicellar, clear, aqueous solution of cyclosporine (CsA) – and to determine the systemic exposure to CsA following ophthalmic administration.
Patients and methods: Healthy volunteers ≥18 years of age were recruited for participation in this phase 1, open-label, single-center, single-arm, study. Subjects received one drop of OTX-101 0.09% in each eye every 12 hours for 7 days, and once on day 8. Blood samples were collected predose, and 0.25, 0.5, 1, 2, 4, 8, and 12 hours post-first dose on day 1 and day 8. CsA levels in whole blood samples were analyzed using liquid chromatography-tandem mass spectrometry. Pharmacokinetic parameters (maximal whole blood concentration [Cmax, ng/mL], time to Cmax [Tmax, hours]), and area under the concentration-time curve from 0 to the last measurement [AUC(0–t), h·ng/mL]) were calculated using noncompartmental analysis. Safety assessments included subject-reported adverse events (AEs), vital signs, visual acuity, intraocular pressure measurement, biomicroscopy, and direct ophthalmoscopy.
Results: A total of 16 subjects were enrolled; 15 subjects completed the study. Blood sample analysis indicated limited systemic exposure to CsA; three subjects had a CsA concentration greater than or equal to the lower limit of quantitation (LLOQ) on day 1; only four subjects had three consecutive CsA concentration measurements ≥LLOQ on day 8; the mean±SD for Cmax was 0.17±0.02 ng/mL, Tmax was 1.5±0.58 hours, and AUC(0–t) was 0.53±0.06 h·ng/mL. Three subjects reported three AEs (eye pain, eye pruritis, and eye irritation) during the study. No clinically significant changes in the safety assessments were noted.
Conclusion: The OTX-101 formulation was well tolerated. Systemic exposure to CsA was negligible in healthy volunteers after twice-daily ocular administration. No evidence for systemic accumulation of CsA was observed.
Keywords: dry eye disease, cyclosporine, pharmacokinetic, systemic exposure
Introduction
Cyclosporine (CsA) is a fungal-derived cyclic polypeptide with immunosuppressant activity. Cyclosporine modulates the immune response through the inhibition of calcineurin, preventing the activation of T lymphocytes and the production of inflammatory cytokines.1,2 The specific action of CsA on T-cell activation facilitated the use of the agent in the treatment of autoimmune and immune-mediated conditions. Cyclosporine has been administered systemically in the treatment of organ recipients, limiting immune-mediated rejection of transplanted tissue.3
The immunomodulatory properties of CsA are utilized in the treatment of dry eye disease (DED) via topical ophthalmic administration. Application of topical CsA ophthalmic emulsion 0.05% (Restasis; Allergan, Irvine, CA, USA) in patients with DED is indicated in the USA to increase tear production (twice daily administration, ~12 hours apart).4,5 Increased tear production and a reduction in clinical signs and symptoms have been observed following topical administration of cyclosporine ophthalmic emulsion 0.05% in patients with DED.6
The hydrophobic nature of CsA is responsible for a low aqueous solubility of the molecule, necessitating the development of alternative formulation approaches for topical delivery.7 OTX-101 is a novel, nanomicellar, clear aqueous solution of CsA recently approved by the US Food and Drug Administration (Cequa™; Sun Pharmaceutical Industries, Cranbury, NJ, USA) for the increase of tear production in patients with keratoconjunctivitis sicca (DED). The OTX-101 formulation solubilizes CsA in water by forming nanosized particles in which CsA is encapsulated in hydrophilic shells. The objectives of this clinical study were to evaluate the ocular and systemic safety of the OTX-101 0.09% formulation and to determine the systemic exposure to CsA following ophthalmic administration.
Materials and methods
Study design
This was a phase 1, open-label, single-arm study. Subjects were enrolled at a single center in the USA. Institutional Review Board (Optimum Clinical Research, Inc., Oshawa, ON, Canada) approval was obtained for the protocol and consent forms. Written informed consent was obtained from all subjects at the screening visit, prior to the conduct of any study-specific procedures. The study was conducted in accordance with the principles of the Declaration of Helsinki and all applicable legal and regulatory requirements.
Total study participation for subjects ranged from 14 to 16 days. The study duration included a screening visit and 8 days of treatment with OTX-101 0.09%. Subjects received instillations of OTX-101 0.09% in each eye twice daily, starting the morning of the baseline visit (day 1) through day 7, and a single instillation of OTX-101 0.09% on the morning of day 8. The schematic for the study design is presented in Figure 1.
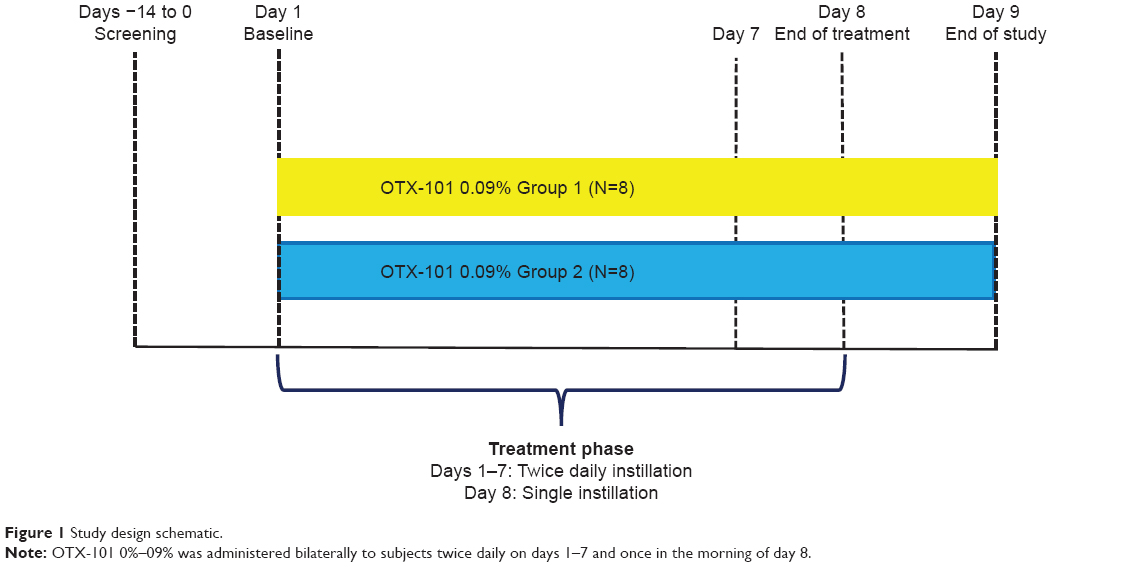 | Figure 1 Study design schematic. |
Study protocol
Screening and subject eligibility
The screening visit was conducted within 14 days prior to the initiation of treatment at the baseline visit (day 1). Subjects were evaluated for eligibility based on the inclusion/exclusion criteria defined in the study protocol. Demographic information, including age, gender, ethnicity, race, and iris color was collected. Subject information on prior medical/surgical histories was also collected, along with any concomitant medication usage. A physical examination was conducted to determine the subject’s height and weight, and vital signs (heart rate, blood pressure, and temperature) were obtained. An electrocardiogram (ECG) was performed. Blood samples were collected and a urine pregnancy test was performed, as appropriate (female subjects of child-bearing potential only). Ocular examinations included corrected visual acuity assessment (via a Snellen chart), intraocular pressure (IOP), biomicroscopy, and direct ophthalmoscopy.
The inclusion criteria for the study were male or female subjects aged 18 or older, body mass index (BMI) of 18–32 kg/m2, medically healthy subjects with clinically insignificant screening results (laboratory profiles, medical history, ECG results, and physical profile), current non-tobacco/nicotine use (6 months prior to the study initiation), evaluation of two normal (non-diseased) eyes, IOP measurements between 10 and 21 mm Hg in each eye at screening, corrected visual acuity of 20/40 (Snellen) or better in each eye at screening, willingness, and ability to provide informed consent and to comply with study requirements.
The ophthalmic exclusion criteria for the study were history of glaucoma, presence of retinopathies or maculopathies in either eye, known hypersensitivity to any component of the study formulation or topical anesthetics or mydriatic agents, unwillingness to discontinue use of contact lenses during the study period, any history of surgery or trauma to either eye, presence of any other clinically relevant ocular condition as determined by the Investigator, use of any other ocular medication or artificial tears within 30 days of the study initiation, or use of any other ocular medication during the study. Systemic exclusion criteria included report of a blood donation totaling between 100 and 499 mL of blood within 30 days prior to the initiation of treatment (day 1) or donation of >499 mL of blood within 56 days prior to treatment initiation; use of any topical or systemic medications throughout the study period; blood dyscrasia or use of anticoagulation treatment; clinically significant abnormalities observed in laboratory tests at screening; clinically significant systemic disease that was considered to potentially interfere with the study; participation in any other investigational study within 30 days of study initiation; presence of allergic rhinitis (seasonal or perennial); women of childbearing potential who were pregnant, nursing or planning a pregnancy, or not using a medically acceptable form of birth control; and demonstration of veins that were considered unsuitable for venipuncture.
Baseline and treatment visits
Subjects who were considered eligible for study participation at the screening visit were instructed to return to the clinic for the baseline visit (day 1) between 7 and 9 am. Prior to instillation of study medication, vital signs were measured and blood samples were obtained, concomitant medications were recorded, and ocular examinations (visual acuity, IOP, and biomicroscopy) were conducted to confirm study eligibility. The first dose of study drug was administered to each of the subject’s eyes by clinical personnel. Vital signs were monitored 30 minutes (±5 minutes) prior to and 30 minutes (±5 minutes) and 12 hours (±30 minutes) following study drug administration. Blood samples for pharmacokinetic analysis were collected at −30 minutes, +15 minutes, +30 minutes, +1 hour, +2 hours, +4 hours, +8 hours, and +12 hours time points relative to study drug administration. All samples were to be collected no later than 1 minute after the scheduled time. The evening dose of study drug was administered by clinic personnel at +12 hours (±30 minutes) relative to the initial administration, after the measurement of vital signs and blood sample collection. Subjects remained at the clinical site for 30 minutes following the evening administration of study drug. Adverse events (AEs) were monitored and recorded throughout the study period.
On days 2–7, subjects returned to the clinic twice daily for the morning and evening administrations of study drug by clinic personnel and for the recording of any spontaneously reported AEs. The end-of-treatment visit on day 8 was scheduled between 7 and 9 am. Ophthalmic evaluations (visual acuity, IOP, and biomicroscopy) were conducted, and vital signs were obtained (heart rate and blood pressure). Blood samples were collected at the following time points relative to study drug administration: −30 minutes, +15 minutes, +30 minutes, +1 hour, +2 hours, +4 hours, +8 hours, and +12 hours. All samples were to be collected no later than 1 minute after the scheduled time. Subjects returned to the clinic on day 9 for the end-of-study visit. Final ophthalmic examinations (visual acuity, IOP, biomicroscopy, and direct ophthalmoscopy) were conducted, and vital signs (heart rate and blood pressure) were recorded. A urine pregnancy test was performed, as appropriate.
Outcome measures
Safety and tolerability
AEs, observed and spontaneously reported by subjects, were recorded throughout the study period. Reported AEs were followed until the event(s) were resolved to the satisfaction of the investigator.
Corrected visual acuity was evaluated and monitored during the study. The subject was required to wear the same glasses at each visit, as necessary. If the subject’s glasses were not available at the study visit, the necessary prescription lenses were placed into a trial frame for evaluation of corrected visual acuity.
Slit lamp biomicroscopy was conducted to evaluate the anterior segment. The lids, tear film, conjunctiva, cornea, sclera, iris, lens, and anterior/posterior chambers were evaluated for any abnormalities.
The IOP assessment was conducted using Goldmann applanation tonometry. The IOP measurements for each patient were obtained using the same tonometer and observer throughout the study, if possible.
The direct ophthalmoscopy evaluation with dilation was conducted to evaluate the posterior segment. The optic nerve head was assessed for pallor and cupping. The vitreous, optic nerve, macula, and peripheral retina were evaluated as part of the fundus examination, with any abnormalities recorded.
In addition to the collection of whole blood sample for the pharmacokinetic analysis, blood samples from subjects were evaluated through the use of a complete blood count with differential, clinical chemistry, and serology screening. Urine samples were utilized for a urinalysis, drug screening, and pregnancy testing.
Efficacy
No efficacy assessments were conducted as part of this study.
Bioanalytical and pharmacokinetic analyses
Blood samples for bioanalytical analysis were collected in 6 mL dipotassium ethylenediaminetetraacetic acid (K2EDTA) anticoagulant tubes. A total of 16 samples were collected for each subject; 8 samples on day 1, and 8 samples on day 8. Sample tubes were labeled with the study number, subject number, dosing day, date, and collection time (study hour). Following collection, the blood samples were split into 3-mL portions and stored at −20°C until shipment on dry ice to the bioanalytical laboratory (Intertek Pharmaceutical Services, San Diego, CA, USA).
Cyclosporine was extracted from human whole blood samples by liquid–liquid extraction using methyl-t-butyl ether after acidification with hydrochloric acid (0.1 n). The organic extract was washed with an alkaline (sodium hydroxide; 0.1 n) solution. Samples were evaporated to dryness and reconstituted in 50:50 acetonitrile:5 mM ammonium acetate for analysis. Concentrations of CsA were measured using a validated liquid chromatography with tandem mass spectrometry method (SCIEX API 4000; Applied Biosystems/MDS SCIEX, Concord, ON L4K 4V8, Canada). The validated concentration range for CsA in whole blood was from 0.1 ng/mL to 100 ng/mL. The precision of the method ranged from 0.400% to 16.5% and the within-run coefficients of variation ranged from 1.86% to 7.44%.
All pharmacokinetic parameters were calculated using non-compartmental analysis. Only CsA concentrations that were greater than the validated lower limit of quantitation (LLOQ) of 0.1 ng/mL were included in the analysis. Actual sampling times were used for the pharmacokinetic analysis. The maximum whole blood concentration of CsA (Cmax) and time to Cmax (Tmax) were determined directly from the CsA measurement data. Area under the whole blood concentration of CsA vs time curve from 0 to the final sample with a concentration > LLOQ (AUC(0–t)) was calculated using the linear trapezoidal method.
Preparation of descriptive statistics for demographic information and pharmacokinetic parameter calculations were performed using SAS for Windows, version 9.4 (SAS Institute Inc., Cary, NC, USA).
Results
A total of 16 subjects were enrolled; 15 completed the study. Following screening, subjects were split into two groups (Group 1, subjects 1–8; Group 2, subjects 9–16). The study groups were staggered with respect to dosing time to facilitate evaluation and sample processing. One subject from Group two was removed from the study after day 2 due to non-compliance. A summary of the demographic information (age, gender, height, weight, BMI, race, and ethnicity) is presented in Table 1.
 | Table 1 Demographic characteristics of the full study population |
Bioanalytical and pharmacokinetic analyses
Following bioanalytical assay of the samples collected, only samples from 15 subjects were included in the pharmacokinetic analysis. There were 6 samples in 3 subjects on day 1, and 24 samples in 10 subjects on day 8 that had CsA concentrations ≥ LLOQ. On day 8, four subjects had ≥3 consecutive whole blood CsA concentrations ≥ LLOQ. The descriptive statistics for the pharmacokinetic parameters Cmax, Tmax, and AUC(0–t) calculated on day 8 for CsA measured in whole blood are presented in Table 2. No evidence for systemic accumulation of CsA was observed.
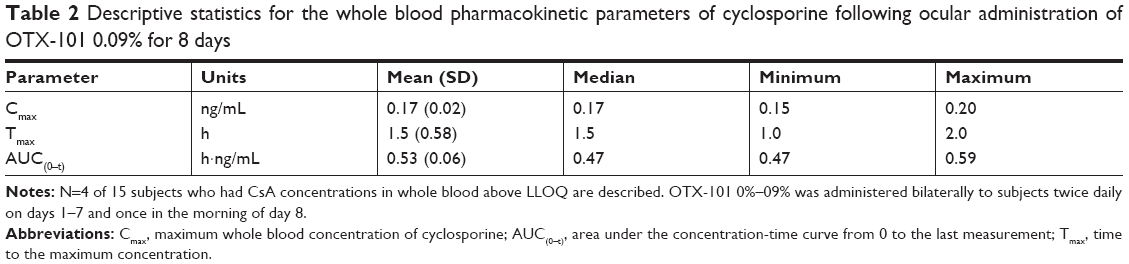 | Table 2 Descriptive statistics for the whole blood pharmacokinetic parameters of cyclosporine following ocular administration of OTX-101 0.09% for 8 days |
Safety and tolerability
A total of three AEs were reported by three subjects during the study period. All AEs were ocular in nature: eye pain, eye pruritis, and eye irritation. The AEs were considered to be mild in severity, and were suspected to be related to treatment. All AEs resolved without the need for additional treatment. No subjects were discontinued or withdrew due to an AE. No serious AEs were reported.
No clinically significant changes in the laboratory tests were observed during the study. Average subject vital signs did not exceed the recorded baseline measurements or specification ranges. All vital sign measurements were well within the maximum specification ranges.
The average IOP measurements at screening for the left and right eyes were 16.8 mm Hg and 17.1 mm Hg, respectively. The mean change in IOP from baseline (day 1) to the end of study (day 9) was 0.1 mm Hg for the left eye and 0.3 mm Hg for the right eye. The average IOP reading did not exceed 21 mm Hg during the course of the study. No abnormal IOP values for individual subjects were observed during the study; all values were within the normal range (10–21 mm Hg). No clinically significant changes in visual acuity were observed. Slit lamp biomicroscopy evaluations were conducted to evaluate any abnormalities in the anterior segment. Abnormalities were found for a total of four (25%) subjects on days 8 and 9. The findings were normal for all 16 subjects with regard to the direct ophthalmoscopy/dilated fundus examinations.
Discussion
This study reports the pharmacokinetics of CsA following topical ophthalmic administration of OTX-101 in humans. Systemic exposure to CsA was negligible during 7 days of twice-daily administration and a single administration of OTX-101 0.09% on day 8. Although the distribution of CsA in ocular tissues was not evaluated in this clinical study, the pharmacokinetic parameters of CsA in ocular tissues and fluids has been evaluated in New Zealand White rabbits.8 Following single and repeated topical ophthalmic instillations, higher concentrations of CsA were measured following OTX-101 administration compared with cyclosporine ophthalmic emulsion (Restasis; Allergan, Irvine, CA, USA) in relevant ocular tissues, including the cornea and conjunctiva.
Peak CsA concentrations detected in whole blood samples in this study following repeated topical ophthalmic administration were 3,459- to 5,906-fold less compared to the concentrations measured following oral CsA administration by patients for immunosuppressive therapy.9
The three ocular AEs reported by subjects in the study were likely related to ocular irritation or burning on instillation that is consistent with the most commonly reported safety/tolerability issues following the topical application of CsA; ocular burning was the most common adverse reaction (17%) reported by patients during the registration studies for CsA ophthalmic emulsion 0.05%.5 In the combined results of two Phase 3 studies evaluating topical ophthalmic CsA emulsion, the most common ocular AEs reported were ocular burning (14.7% for CsA 0.05% and 16.1% for CsA 1.0%, as compared to 6.5% for vehicle) and ocular stinging (3.4% for CsA 0.05%, and 4.5% for CsA 1.0% as compared to 1.4% for vehicle).6
OTX-101 – the novel, aqueous, nanomicellar formulation of CsA evaluated in this study – was well tolerated following repeated twice daily administration in healthy subjects. The findings for all subjects were within normal ranges for the safety assessments (vital signs, IOP, visual acuity, and direct ophthalmoscopy examinations).
Conclusion
The OTX-101 0.09% aqueous nanomicellar formulation of CsA demonstrated an acceptable profile in healthy volunteers. The negligible systemic exposure to CsA based on analysis of whole blood samples and the lack of any evidence of systemic accumulation along with the minimal safety findings observed in this study indicate that the OTX-101 0.09% formulation as compared with the other currently marketed topical ophthalmic formulation of CsA (cyclosporine ophthalmic emulsion 0.05%) is safe and suitable for the treatment of DED.
Acknowledgments
Writing and editorial assistance in preparation of the manuscript was provided by Kurt Brubaker of Bridge Over Brook, Inc., with funding provided by Sun Pharmaceutical Industries, Ltd.
Author contributions
All authors contributed to data analysis, drafting and revising the article, gave final approval of the version to be published and agree to be accountable for all aspects of the work.
Disclosure
This study was sponsored and funded by Ocular Technologies Sarl (now a wholly owned subsidiary of Sun Pharmaceutical Industries, Ltd.), which participated in the design of the study and oversaw its conduct, monitoring, and analysis. Dr Karpecki, and Dr Devries are consultants to Sun Pharmaceutical Industries, Ltd. Angela Justice and Abayomi Ogundele are employees of Sun Pharmaceutical Industries, Ltd. Sidney Weiss was an employee of Ocular Technologies Sarl. Dr Kramer, Dr O’Connor, Dr Evans, Dr Johnston, and Dr Jasper have no financial or proprietary interest in any product, material, or method mentioned. The authors report no other conflicts of interest on this work.
References
Pflugfelder SC. Antiinflammatory therapy for dry eye. Am J Ophthalmol. 2004;137(2):337–342. | ||
Fruman DA, Klee CB, Bierer BE, Burakoff SJ. Calcineurin phosphatase activity in T lymphocytes is inhibited by FK 506 and cyclosporin A. Proc Natl Acad Sci U S A. 1992;89(9):3686–3690. | ||
Moore CP, McHugh JB, Thorne JG, Te P, Phillips TE. Effect of cyclosporine on conjunctival mucin in a canine keratoconjunctivitis sicca model. Invest Ophthalmol Vis Sci. 2001;42:653–659. | ||
Bron AJ, de Paiva CS, Chauhan SK, et al. TFOS DEWS II pathophysiology report. Ocul Surf. 2017;15(3):438–510. | ||
Restasis® (package insert). Irvine, CA: Allergan; 2013. | ||
Sall K, Stevenson OD, Mundorf TK, Reis BL. Two multicenter, randomized studies of the efficacy and safety of cyclosporine ophthalmic emulsion in moderate to severe dry eye disease. Ophthalmol. 2000;107:631–639. | ||
Lallemand F, Schmitt M, Bourges JL, Gurny R, Benita S, Garrigue JS. Cyclosporine a delivery to the eye: a comprehensive review of Academic and industrial efforts. Eur J Pharm Biopharm. 2017;117:14–28. | ||
Weiss SL, Kramer W, Velagaleti P, Gilger BC. Ocular distribution of cyclosporine following topical administration of OTX-101 in New Zealand White rabbits. Poster (2677-A0404) presented at the Association for research and vision in ophthalmology; April 29–May 3; 2018; Honolulu, Hawaii. | ||
Aspeslet LJ, Legatt DF, Murphy G, Yatscoff RW. Effect of assay methodology on pharmacokinetic differences between cyclosporine Neoral and Sandimmune formulations. Clin Chem. 1997;43(1):104–108. |
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.