Back to Journals » Clinical Interventions in Aging » Volume 13
A pathogenic PSEN2 p.His169Asn mutation associated with early-onset Alzheimer’s disease
Authors Giau VV ![]() , Pyun JM, Bagyinszky E, An SSA
, Pyun JM, Bagyinszky E, An SSA ![]() , Kim SY
, Kim SY ![]()
Received 6 April 2018
Accepted for publication 24 May 2018
Published 31 July 2018 Volume 2018:13 Pages 1321—1329
DOI https://doi.org/10.2147/CIA.S170374
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Richard Walker
Vo Van Giau,1,* Jung-Min Pyun,2,* Eva Bagyinszky,2 Seong Soo A An,1 SangYun Kim2
1Department of BioNano Technology, Gachon Medical Research Institute, Gachon University, Seongnam, South Korea; 2Department of Neurology, Seoul National University College of Medicine & Neurocognitive Behavior Center, Seoul National University Bundang Hospital, Seongnam, South Korea
*These authors contributed equally to this work
Background: Autosomal dominant early-onset Alzheimer’s disease (EOAD) is genetically heterogeneous and has been associated with mutations in 3 different genes, coding for amyloid precursor protein (APP), presenilin 1 (PSEN1), and presenilin 2 (PSEN2). Most frequent cases are associated with mutations in the PSEN1 gene, whereas mutations in the APP and PSEN2 genes are rare.
Methods: Patient who presented progressive memory decline in her 50s was enrolled in this study. A broad battery of neuropsychological tests and neuroimaging was applied to make the diagnosis. Genetic tests were performed in the patient to evaluate possible mutations using next-generation sequencing (NGS). The pathogenic nature of missense mutation and its 3D protein structure prediction were performed by in silico prediction programs.
Results: A pathogenic mutation in the PSEN2 gene in a Korean patient associated with EOAD was identified. Targeted Next-generation sequencing and Sanger sequencing revealed a heterozygous C to A transition at position 505 (c.505C>A), resulting in a probably missense mutation at codon 169 (p.His169Asn) in PSEN2. PolyPhen-2 and SIFT software analyses predicted this mutation to be a probable damaging variant. This hypothesis was supported by the results of 3D in silico modelling analyses that predicted the p.His169Asn may result in major helix torsion due to histidine to asparagine substitution. Mutation may cause additional stresses with hydrophobic residues on the surface that interact inside the transmembrane domain III, which is a conserved domain in PSEN2 His169.
Conclusion: These findings revealed that the p.His169Asn might be an important residue in PSEN2, which may alter the functions of PSEN2, suggesting its potential involvement with AD phenotype. Future functional studies are needed to evaluate the role of PSEN2 p.His169Asn mutation in AD disease progression.
Keywords: Alzheimer’s disease, p.His169Asn mutation, presenilin-2, next-generation sequencing
Introduction
Alzheimer’s disease (AD) is a devastating neurodegenerative disease, which can be divided into 2 main categories: early-onset AD (EOAD) and late-onset AD (LOAD), with a dividing age of 65 years. Mutations in 3 genes have been associated with the etiology of EOAD form of AD, the amyloid precursor protein (APP), presenilin 1 (PSEN1), and presenilin 2 (PSEN2) genes. Approximately half of early-onset can be attributed to mutation in any of three genes, all of which affect amyloid β (Aβ) deposition.1,2 Remarkably, PSEN1 is the most commonly involved gene, with 247 mutations reported as pathogenic in the Alzforum database (www.alzforum.org/mutations). The second most commonly involved gene is APP, with 58 pathogenic mutations described (www.alzforum.org/mutations), while at least 31 PSEN2 mutations have been identified in AD patients, 15 pathogenic in 24 probands and 16 with pathogenicity nature unclear.3 Although PSEN mutations are commonly inherited in an autosomal dominant manner, several de novo mutations in PSEN1 and PSEN2 have been reported EOAD.4–6 In Asia, a few PSEN2 mutations were previously reported, but emerging recent studies discovered several novel mutations in Korean and Chinese patients.6–10 It is known that phenotypes in AD patients with PSEN2 mutations are milder than those in with PSEN1 mutations, and PSEN2 mutation carriers have generally a wider onset age range from 40 to 70 years.11,12 However, several PSEN2 mutations may exhibit an incomplete penetrance and variable clinical expression, overlapping with LOAD.13,14 Interestingly, PSEN2 mutations appeared not only in AD patients but also in patients with other disorders, including frontotemporal dementia (FTD), dementia with Lewy bodies, breast cancer, dilated cardiomyopathy, and Parkinson’s disease with dementia.15
In this study, we described the detailed case report of the first Korean EOAD patient with a PSEN2 p.His169Asn mutation with “pathogenic nature unclear” in East Asia.
Materials and methods
Subjects
The study subject provided written informed consent allowing genetic and clinical data to be used for research purposes. A diagnosis of probable AD was made according to the criteria of the National Institute of Neurological and Communicative Disorders and Stroke Alzheimer’ Disease and Related Disorders Association.16 This study was conducted with approval from the Institutional Review Board of Seoul National University College of Medicine & Neurocognitive Behavior Center, Seoul National University Bundang Hospital (B-1302/192-006).
Molecular genetic analysis
White blood cells were isolated by centrifugation of blood samples at 800× g for 30 minutes. Genomic DNA was purified using a GeneAll blood kit (GeneAll Biotechnology Co. Ltd, Seoul, Republic of Korea), following the manufacturer’s instructions. All DNA samples were stored at −20°C for further analysis.
A complex genetic screen was performed on the proband, using a specifically designed gene panel of 50 causative and risk factor genes for various neurodegenerative diseases.17 The DNA sample was analyzed by next-generation sequencing (NGS) from the Theragen Etex Bio Institute (Seoul, Republic of Korea). Briefly, fragment libraries were constructed by DNA fragmentation, and 20 ng of DNA was used for multiplex PCR of a panel covering 50 genes, where causative or probably causative variants were previously reported17 (Ion AmpliSeq Customized Panel, Thermo Fisher Scientific, Waltham, MA, USA). Barcode and adaptor ligation and library amplification using the Ion DNA Barcoding kit (Thermo Fisher Scientific) were done according to the manufacturer’s instructions. The size distribution of the DNA fragments was analyzed on the Agilent Bioanalyzer using the High Sensitivity Kit (Agilent Technologies, Santa Clara, CA, USA). Template preparation, emulsion polymerase chain reaction (PCR), and Ion Sphere Particle (ISP) enrichment were performed using the Ion Xpress Template kit (Thermo Fisher Scientific), according to the manufacturer’s instructions. The ISPs were loaded onto a P1 chip and sequenced using an Ion P1 sequencing 200 kit (Thermo Fisher Scientific).
To confirm the presence of identified mutations, Sanger sequencing was also performed in both directions, using the same primer sets,17 and the assay was carried out by BioNeer Inc. (Dajeon, Republic of Korea). The novelty of identified mutations was analyzed by screening for their inclusion in the Korean Reference Genome Database (KRGDB, http://152.99.75.168/KRGDB/menuPages/intro.jsp), which performed whole-genome sequencing on 622 Korean individuals who were not affected with any kind of disorder. Mutations were also checked against the Alzforum (www.alzforum.org/mutations), the Exome Aggregation Consortium (ExAC, http://exac.broadinstitute.org/), and 1000 Genomes (http://www.1000genomes.org/) databases.
In silico analyses and protein structure prediction
Mutations were screened by PolyPhen-2 (http://genetics.bwh.harvard.edu/pph2/), SIFT (http://sift.jcvi.org/), and PROVEAN (http://provean.jcvi.org/index.php) software, which were used to determine the nature of mutations to be benign or possible damaging. In addition, prediction of mutational change on protein properties was also made by using PyMOL programs at ExPASy server (https://www.expasy.org/), in which different parameters have been analyzed, such as bulkiness, polarity, or hydrophobicity (Kyte and Doolittle) index. A 3D protein structure prediction model was determined with the online Raptor X software (http://raptorx.uchicago.edu/), and the PSEN2 variant was compared with the normal X-ray structure. Superimposed images of variant and normal proteins were processed using the Discovery Studio 3.5 Visualizer software, designed by Accelrys (San Diego, CA, USA).18
Cytoscape (Cytoscape Consortium, New York, NY, USA) is a powerful software program used to visualize the relationship between proteins or to assess genetic interaction. The Cytoscape plugin ClueGO was used for analyzing and biological processes in concert with other interacting proteins.19 Gene Ontology categories were used to capture biological information. κ statistics is supported to create networks of genetic or protein interactions. After starting functional analysis, ClueGO displayed the visualized network interactions, an information table for the associated protein, a significance histogram of each group, as well as a chart overview of the functional groups. Each biological process was represented with nodes which are connected with edges to indicate interactions. Since we performed a complex genetic screening, association may be possible between the different mutation-carrier genes. In order to analyze the biological network of 15 genes with identified mutations in this study, the ClueGO software was used. This algorithm was designed to analyze the biological network of genes and to identify which pathway could be responsible for different disorders.
Results
Clinical findings
The proband was a 58-year-old woman who presented progressive memory decline in her 50s. She exhibited forgetfulness, such as forgetting to attend important meetings or forgetting the password number of her bank account, from the age of 56. From the age of 58, she had difficulty in performing routine household chores, wore clothes in layers inappropriately, wandered about, and lost her way. She was unable to speak fluently, became apathic, and showed aggressiveness toward people. Her past medical history was unremarkable. There was no medical history of dementia in her family. Her score on the Korean version of the Mini-Mental Status Examination was 21/30, and global deterioration scale score was 4 at 1 year after symptom onset. In follow-up tests after 1 year, Korean version of the Mini-Mental Status Examination score was 5 and global deterioration scale score was 7. Her brain magnetic resonance imaging at 1 year after symptom onset revealed atrophy of left-dominant bilateral temporal lobe (Figure 1A). 18 F-fludeoxyglucose positron emission tomography (FDG-PET) at 2 years after symptom onset showed hypometabolism in the left temporal lobe, right anterior temporal lobe, and bilateral frontal cortex (Figure 1B). Patient had an APOE ε 3/3 polymorphism. Family history of the proband’s generations was negative for any neurological disease, suggesting it as a de novo case of AD. All living family members declined genetic testing (Figure 1C).
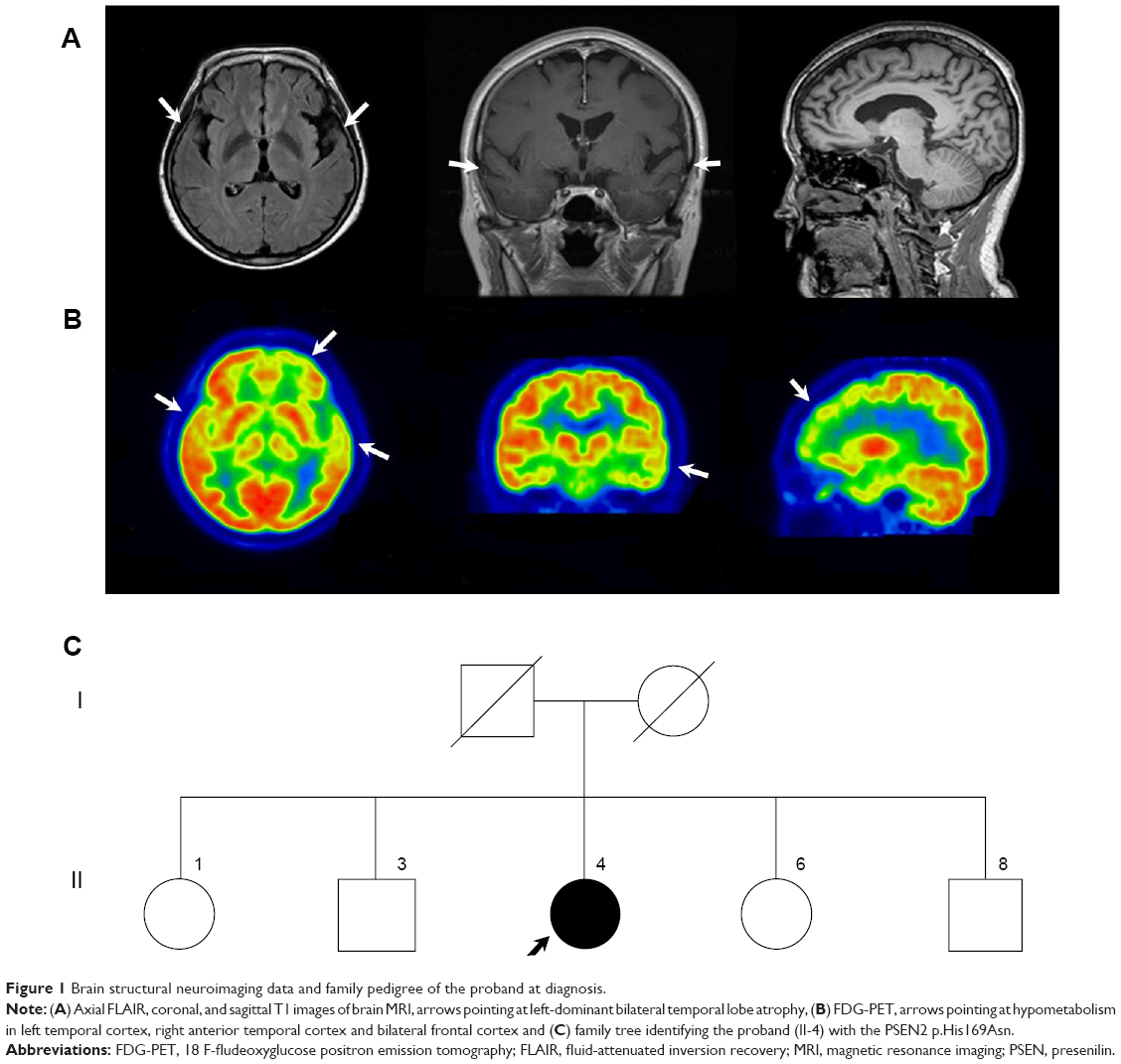 | Figure 1 Brain structural neuroimaging data and family pedigree of the proband at diagnosis. |
Genetic analyses
A heterozygous C > A substitution was identified and was confirmed to occur in the PSEN2 coding region using both NGS and standard sequencing (Figure 2A). The mutation consisted of a single nucleotide substitution at codon 169 (nucleotide c.505C>A, in heterozygous state), located at exon 6 of PSEN2 gene, which changed the correspondent amino acid (His to Asn) and in transmembrane (TM) domain III of the PSEN2 protein. Interestingly, this mutation PSEN2 p.His169Asn was previously identified in 1 patient with familial LOAD and in 1 patient with sporadic FTD from People’s Republic of China,9 but its pathogenic nature was not clarified yet.
 | Figure 2 Identification and in silico analysis of function the mutation in the patient. |
The mutation was missing in the KRGDB. His169Arg was also checked in the ExAC, which shows all variants from 60,706 unrelated individuals. ExAC is a useful reference database for disease-associated genetic analyses. Frequency of PSEN2 p.His169Asn was 0.0001648 with heterozygosity only. While no other additional mutation was found in the APP, PSEN1, and PSEN2, an additional 25 missense variants from AD and other disorders risk factor genes were discovered in this proband, as indicated in the Table S1.
In silico predictions
PolyPhen-2 suggested PSEN2 p.His169Asn as a probably damaging variant with HumDiv and HumVar scores of 0.985 and 0.925, respectively. SIFT also revealed categorizing PSEN2 H169N as a damaging variant with a score of 0.005. PROVEAN also revealed H169N as damaging, with a score of −5.95. ExPASy tools revealed that the mutation could affect significantly the presenilin structure through different parameters, such as bulkiness, polarity, and hydrophobicity. For example, scores of bulkiness were reduced slightly due to H169N (17.292 Dal) in comparison to normal PSEN2 (17.389 Dal), and several amino acids near the mutation site were also affected (Figure 2B).
Three-dimensional structure modeling revealed that PSEN2 139–169 TM-III domain had an N-terminal loop structure disturb to residue 169 (Figure 2C), suggesting divergent structures between normal PSEN2 and its mutant p.His169Asn. The mutation may not have a strongly defined structure, but reveals dynamic motion in the loop structure. The highly distinct properties of His and Asn could have a dramatic impact on the structure and functions of PSEN2. Histidine is hydrophilic and has a positive charge, similar to arginine, but has a smaller size. Asparagine is hydrophilic and polar and can form hydrogen bonds via hydroxyl and amide groups; consequently, the mutation can change the coil conformation. In the 3D model, significant changes could be seen in the TM-III helix conformation. This mutation is located at TM-III region which tends to have hydrophobic residues on the surface that interact with the membrane.
ClueGO Ontology analyses (Figure 2D) revealed 2 important pathways in which the genes with missense variants could be involved: amyloid metabolism and regulation of mitochondrial fission. PSEN2 would play a central role in both metabolic pathways. PSEN2 p.His169Asn may be involved in disease phenotype, but the disease-associated mechanism may be unclear. As an be perceived from the ClueGo network analysis, PSEN2 may affect the Aβ network together SORL1, BACE1, and ABCA7 molecules. It is still unclear that how the other risk factor mutations could affect the disease phenotypes, as listed in the Table S1.
Discussion
Here, we reported PSEN2 p.His169Asn mutation in a 58-year-old AD patient from Korea. Mutation was initially described in 1 familial LOAD patient and 1 sporadic early-onset FTD patient from People’s Republic of China. Several de novo cases of dementia associated with familial LOAD and patients with FTD, progressive nonfluent aphasia were previously reported.9 The age of onset was between 62 and 68 years, with disease duration of 3–4 years. These patients presented 2 atypical phenotypes: frontal variant of AD and FTD. In this study, the proband patient presented typical AD symptoms with memory decline at the beginning. After 2 years, behavioral changes reflecting frontal lobe dysfunction and language problems developed, which was consistent with left-side dominant frontotemporal lobe hypometabolism in the FDG-PET image. Her phenotype was a rapidly progressing AD with components of FTD. Compared with the 2 Chinese patients, our proband showed a similar clinical manifestation with the proband with frontal variant AD; however, brain imaging showed that the proband had FTD. This might imply that the PSEN2 p.His169Asn mutation could lead to a pathology affecting frontal and temporal lobes. Although there is no additional mutation reported at residue 169 of the PSEN2 protein, the p.His169Asn mutation was also located at the conserved TM-III region of PSEN2, containing the pathogenic variants (M174V and S175C) (Table 1), based on the algorithms to predict the pathogenicity of the mutations described by Guerreiro et al.20
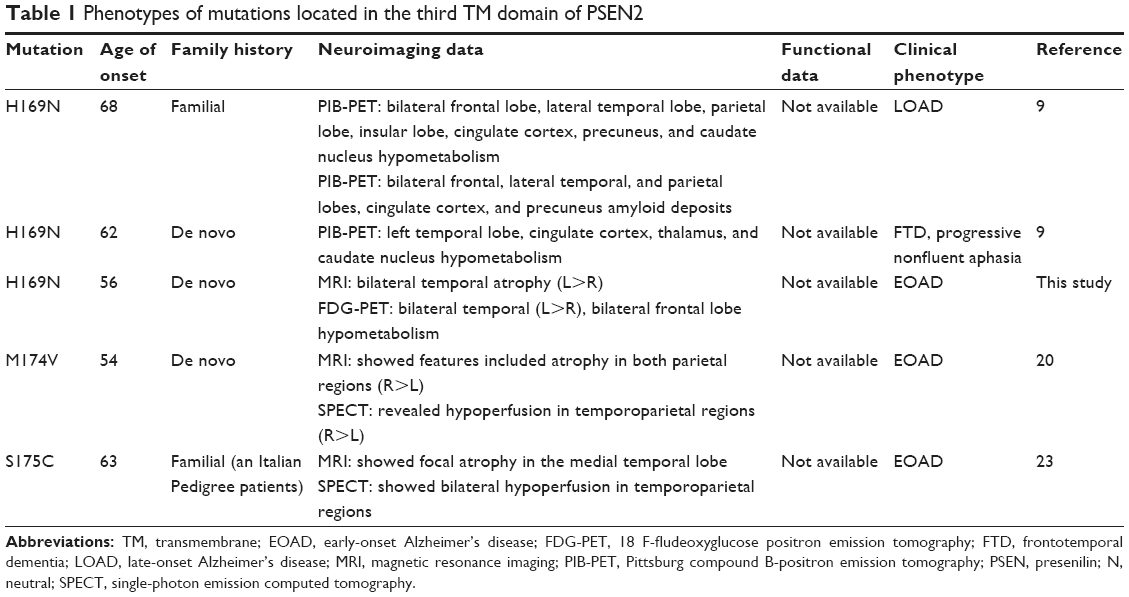 | Table 1 Phenotypes of mutations located in the third TM domain of PSEN2 |
Since the functional or cerebrospinal fluid triple biomarker studies have not yet been performed yet for this mutation, several bioinformatics methods were used to assess the significant role of PSEN2 p.His169Asn in the disease progression, especially since the age of onset of disease was 56 years. The pathologic nature of His169Asn mutation was supported by the bioinformatics analysis demonstrating the conservation of the residue. PolyPhen-2, SIFT, and PROVEAN tools revealed the mutation as a damaging variant. ExPASy tool showed that PSEN2 p.His169Asn could affect the protein structure and function through changes in bulkiness. From the protein structure prediction, this mutation could disturb the protein structure; since the asparagine is hydrophilic and polar and can form hydrogen bonds via hydroxyl and amide groups, it can consequently change the coil conformation. Hence, the change in protein structure and hydrophobic interactions would have higher significance in PSEN2 p.His169Asn. To date, only 3 mutations have been reported in PSEN2 TM-III (H169N, M174V, and S175C) with unclear pathogenic nature (Table 1). Following the American College of Medical Genetics and Genomics variant interpretation guideline,21,22 this variant was properly validated by PolyPhen-2, SIFT, PROVEAN, ExPASy, as well as 3D in silico modeling analyses; it was predicted to have deleterious effect. Taken together, these results indicate that the mutation was probably the cause of AD in this patient.
Many previous studies suggested that the age of onset of disease associated with mutations in PSEN2 varied widely from 39 to 85 years,10,20,23,24 suggesting the role of other genetic or environmental factors in modifying the clinical phenotype. In our patient, sporadic AD was associated with young age of onset – 56 years. It is unclear why mutations in PSEN2 are rarely discovered in AD patients. To our knowledge, only 5 mutations in PSEN2 gene have been reported in People’s Republic of China8–10 and 2 mutations in Korea.6–7 Comparing these patients with the reported mutations, we noted that our proband also presented with memory loss, cognitive decline, and behavioral disturbance, followed by abnormal neurological imaging data. The reason for phenotypic heterogeneity of PSEN2 mutant is still unclear. However, the pathological dysfunction of PSEN2 could be due to a variable Aβ42/Aβ40 ratio and the proximal of the region to the C-terminus including the His169Asn residue.9,25,26
Despite the functional consequence of PSEN2 p.His169Asn, the effect of the mutation in AD pathogenesis remains unclear. On the other hand, several variants in risk factor genes, such as ABCA7, CTNNA3, and CR1, were also discovered. The Ontology program, ClueGO, for the interaction prediction of the variant carrier proteins, could also help to understand the potential roles of PSEN2 in 2 major processes: amyloid-associated mechanisms and receptor-associated processes. Variants in BACE1, ABCA7, and SORL1 could affect their levels and functions in the amyloid-associated mechanisms. Since SORL1 would be involved in amyloid trafficking in AD, SORL1 and ABCA7 were suggested as risk factors for LOAD. The decreased SORL1 expression would increase the amyloid deposition.27 ABCA7 loss-of-function variants may also be involved in increased β secretase cleavage and higher amyloid depositions.28 Therefore, we suggest that the histidine substitution of asparagine could affect the role of PSEN2 protein by altering the posttranslational modification, spatial structure, or interaction with other proteins. Limitations of the current study included a failure to conduct a segregation analysis of the PSEN2 p.His169Asn mutation, because the parents and siblings of the proband were all deceased, and all living family members refused the genetic test. In addition, we could not conduct in vitro studies to confirm how PSEN2 p.His169Asn could be involved in disease progression.
Conclusion
We discovered a PSEN2 p.H169N in a female Korean patient with EOAD for the first time in East Asia. From the analyses against major AD databases, AD and FTD mutation databases, and the Alzgene databases, PSEN2 p.His169Asn was verified as a rare mutation with unclear pathogenic nature. However, p.His169Asn might be involved in pathogenicity, since in silico analyses from PolyPhen-2, SIFT, and 3-dimensional modeling suggested pathogenic effects without clear mechanisms. Our finding supports recent studies, suggesting that a rare coding variability in PSEN2 contributes to susceptibility for apparently sporadic EOAD due to the fact that rare coding variants in other genes were not major players in the development of the disease. Future functional studies are needed to elucidate the underlying mechanisms by which this mutation contributes to AD pathogenesis.
Acknowledgment
This research was supported by a National Research Foundation of Korea (NRF) Grants awarded by the Korean government (MEST, No 2017R1A2B4012636 & 2017R1C1B5017807).
Disclosure
The authors report no conflicts of interest in this work.
References
Rosenberg RN. The molecular and genetic basis of AD: the end of the beginning. Neurology. 2000;54:2045–2054. | ||
Brouwers N, Sleegers K, Van Broeckhoven C. Molecular genetics of Alzheimer’s disease: an update. Ann Med. 2008;40(8):562–583. | ||
Cacace R, Sleegers K, Van Broeckhoven C. Molecular genetics of early-onset Alzheimer’s disease revisited. Alzheimers Dement. 2016;12(6):733–748. | ||
Portet F, Dauvilliers YD, Campion G, et al. Very early onset AD with a de novo mutation in the presenilin 1 gene (Met 233 Leu). Neurology. 2003;61(8):1136–1137. | ||
Golan MP, Styczynska M, Jozwiak K, et al. Early-onset Alzheimer’s disease with a de novo mutation in the presenilin 1 gene. Exp Neurol. 2007;208(2):264–268. | ||
Park KW, An SS, Bagyinszky E, Kim S. A case of possibly pathogenic PSEN2 R62C mutation in a patient with probable early-onset Alzheimer’s dementia supported by structure prediction. Clin Interv Aging. 2017;12:367–375. | ||
Youn YC, Bagyinszky E, Kim H, Choi BO, An SS, Kim S. Probable novel PSEN2 Val214Leu mutation in Alzheimer’s disease supported by structural prediction. BMC Neurol. 2014;14:105. | ||
Niu F, Yu S, Zhang Z, et al. A novel mutation in the PSEN2 gene (N141Y) associated with early-onset autosomal dominant Alzheimer’s disease in a Chinese Han family. Neurobiol Aging. 2014;35(10):2420.e1–2420.e5. | ||
Shi Z, Wang Y, Liu S, et al. Clinical and neuroimaging characterization of Chinese dementia patients with PSEN1 and PSEN2 mutations. Dement Geriatr Cogn Disord. 2015;39(1–2):32–40. | ||
Xia M, Chen S, Shi Y, et al. Probable novel PSEN2 Pro123Leu mutation in a Chinese Han family of Alzheimer’s disease. Neurobiol Aging. 2015;36(12):3334.e13–3334.e18. | ||
Ryan NS, Rossor MN. Correlating familial Alzheimer’s disease gene mutations with clinical phenotype. Biomark Med. 2010;4(1):99–112. | ||
Pilotto A, Padovani A, Borroni B. Clinical, biological, and imaging features of monogenic Alzheimer’s disease. Biomed Res Int. 2013;2013:689591. | ||
Finckh U, Alberici A, Antoniazzi M, et al. Variable expression of familial Alzheimer disease associated with presenilin 2 mutation M239I. Neurology. 2000;54(10):2006–2008. | ||
Bird TD, Levy-Lahad E, Pookaj P, et al. Wide range in age of onset for chromosome-1 related familial Alzheimer’s disease. Ann Neurol. 1996;40(6):932–936. | ||
Cai Y, An SS, Kim S. Mutations in presenilin 2 and its implications in Alzheimer’s disease and other dementia-associated disorders. Clin Interv Aging. 2015;10:1163–1172. | ||
McKhann G, Drachman D, Folstein M, Katzman R, Price D, Stadlan EM. Clinical diagnosis of Alzheimer’s disease: report of the NINCDS-ADRDA Work Group under the auspices of Department of Health and Human Services Task Force on Alzheimer’s disease. Neurology. 1984;34(7):939–944. | ||
Giau VV, An SS, Bagyinszky E, Kim SY. Gene panels and primers for next generation sequencing studies on neurodegenerative disorders. Mol Cell Toxicol. 2015;11(2):89–143. | ||
Källberg M, Wang H, Wang S, et al. Template-based protein structure modeling using the RaptorX web server. Nat Protoc. 2012;7(8):1511–1522. | ||
Bindea G, Mlecnik B, Hackl H, et al. ClueGO: a Cytoscape plug-in to decipher functionally grouped gene ontology and pathway annotation networks. Bioinformatics. 2009;25(8):1091–1093. | ||
Guerreiro RJ, Baquero M, Blesa R, et al. Genetic screening of Alzheimer’s disease genes in Iberian and African samples yields novel mutations in presenilins and APP. Neurobiol Aging. 2010;31(5):725–731. | ||
Richards S, Aziz N, Bale S, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17(5):405–424. | ||
Giau VV, Bagyinszky E, An SSA, Kim SY. Clinical genetic strategies for early onset neurodegenerative diseases. Mol Cell Toxicol. 2018;14(2):123–142. | ||
Piscopo P, Talarico G, Crestini A, et al. A novel mutation in the predicted TMIII domain of the PSEN2 gene in an Italian pedigree with atypical Alzheimer’s disease. J Alzheimers Dis. 2010;20(1):43–47. | ||
Jayadev S, Leverenz JB, Steinbart E, et al. Alzheimer’s disease phenotypes and genotypes associated with mutations in presenilin 2. Brain. 2010;133(Pt 4):1143–1154. | ||
Mastrangelo P, Mathews PM, Chishti MA, et al. Dissociated phenotypes in presenilin transgenic mice define functionally distinct gamma-secretases. Proc Natl Acad Sci U S A. 2005;102(25):8972–8977. | ||
Shirotani K, Takahashi K, Araki W, Maruyama K, Tabira T. Mutational analysis of intrinsic regions of presenilin 2 that determine its endoproteolytic cleavage and pathological function. J Biol Chem. 2000;275(5):3681–3686. | ||
Rogaeva E, Meng Y, Lee JH, et al. The neuronal sortilin-related receptor SORL1 is genetically associated with Alzheimer disease. Nat Genet. 2007;39(2):168–177. | ||
Satoh K, Abe-Dohmae S, Yokoyama S, St George-Hyslop P, Fraser PE. ATP-binding cassette transporter A7 (ABCA7) loss of function alters Alzheimer amyloid processing. J Biol Chem. 2015;290(40):24152–24165. |
Supplementary material
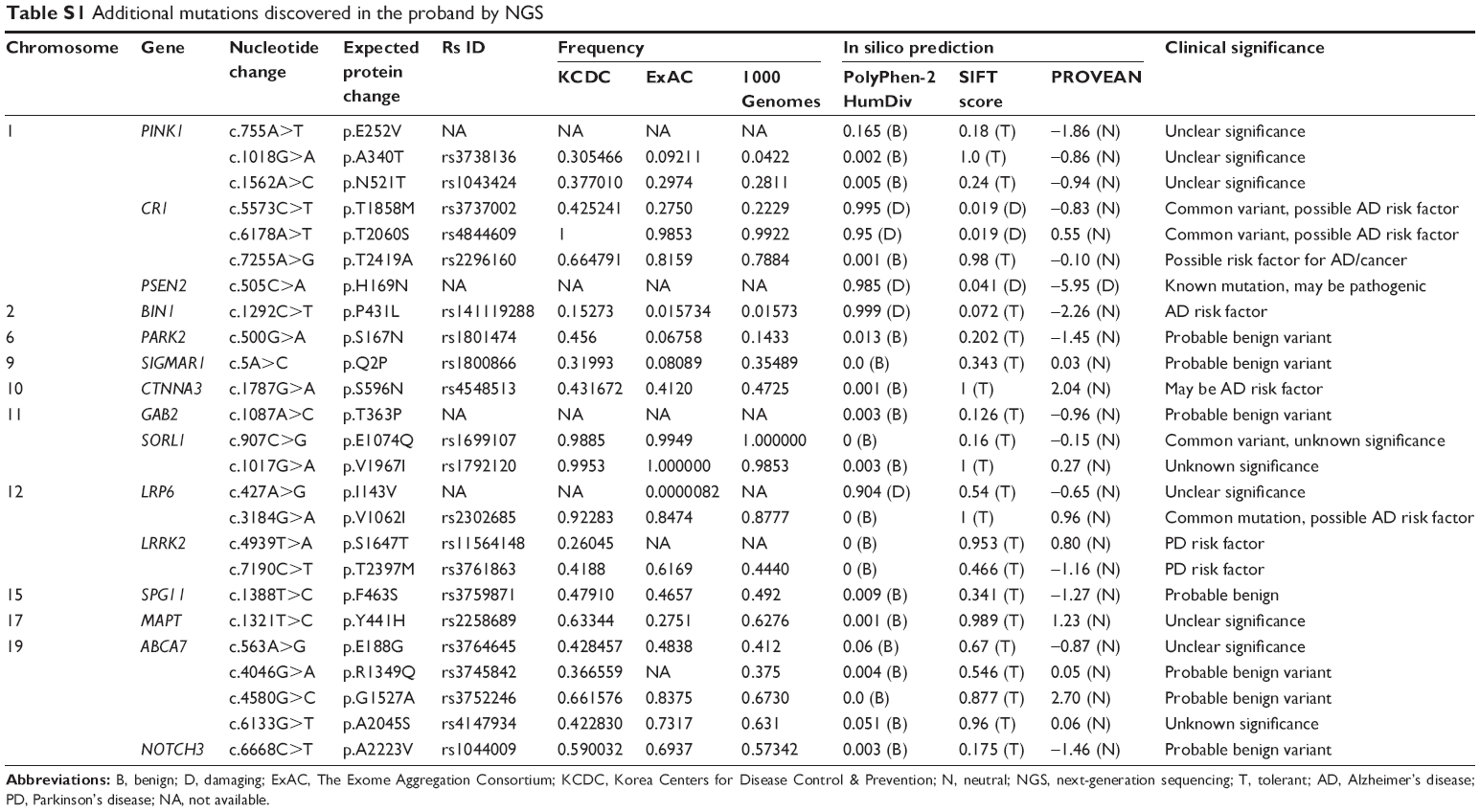 | Table S1 Additional mutations discovered in the proband by NGS |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.