Back to Journals » International Journal of Nanomedicine » Volume 12
A novel nanoemulsion-based method to produce ultrasmall, water-dispersible nanoparticles from chitosan, surface modified with cell-penetrating peptide for oral delivery of proteins and peptides
Authors Barbari GR, Dorkoosh FA, Amini M, Sharifzadeh M, Atyabi F, Balalaei S, Rafiee Tehrani N ![]() , Rafiee Tehrani M
, Rafiee Tehrani M
Received 29 June 2016
Accepted for publication 8 December 2016
Published 2 May 2017 Volume 2017:12 Pages 3471—3483
DOI https://doi.org/10.2147/IJN.S116063
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Thomas Webster
This paper has been retracted.
Ghullam Reza Barbari,1 Farid Abedin Dorkoosh,1 Mohsen Amini,2 Mohammad Sharifzadeh,3 Fateme Atyabi,1 Saeed Balalaie,4 Niyousha Rafiee Tehrani,5 Morteza Rafiee Tehrani1
1Department of Pharmaceutics, 2Department of Medicinal Chemistry, 3Department of Pharmacology, School of Pharmacy, Tehran University of Medical Sciences, 4Department of Chemistry, Khaje Nasiroddin University, 5Department of Biochemistry, School of Medicine, Tehran University of Medical Sciences, Tehran, Iran
Abstract: A simple and reproducible water-in-oil (W/O) nanoemulsion technique for making ultrasmall (,15 nm), monodispersed and water-dispersible nanoparticles (NPs) from chitosan (CS) is reported. The nano-sized (50 nm) water pools of the W/O nanoemulsion serve as “nanocontainers and nano-reactors”. The entrapped polymer chains of CS inside these “nano-reactors” are covalently cross-linked with the chains of polyethylene glycol (PEG), leading to rigidification and formation of NPs. These NPs possess excessive swelling properties in aqueous medium and preserve integrity in all pH ranges due to chemical cross-linking with PEG. A potent and newly developed cell-penetrating peptide (CPP) is further chemically conjugated to the surface of the NPs, leading to development of a novel peptide-conjugated derivative of CS with profound tight-junction opening properties. The CPP-conjugated NPs can easily be loaded with almost all kinds of proteins, peptides and nucleotides for oral delivery applications. Feasibility of this nanoparticulate system for oral delivery of a model peptide (insulin) is investigated in Caco-2 cell line. The cell culture results for translocation of insulin across the cell monolayer are very promising (15%–19% increase), and animal studies are actively under progress and will be published separately.
Keywords: ultrasmall, cell-penetrating peptide, chitosan, oral insulin, nanoemulsion, Caco-2 cell
Introduction
Oral delivery of proteins and peptides has attracted great attention in recent years,1 and new breakthroughs in the field of nanotechnology and biology have opened a new avenue to meet this goal. Among the large number of peptides and hydrophilic compounds, insulin is the main peptide whose oral delivery has been the dream of many accredited universities and pharmaceutical companies around the world.2,3 Oral delivery of insulin has some crucial advantages, including high patient compliance, ease of administration and more patient adherence to the therapy, and, more importantly, it is the sole route of administration that potentially mimics the physiological insulin secretion seen in nondiabetic individuals,4 thus relieving the patients from hyperinsulinemia and its consequent complications such as nephropathy and neuropathy.5 However, oral delivery of insulin has two main bottlenecks, the harsh and degrading enzymatic situation of the gastrointestinal tract that inactivates insulin very quickly and the mucosal barrier that limits insulin’s oral bioavailability.6,7 Numerous approaches have been applied to bypass the enzyme barrier, and some of them have achieved promising results.8 Yet, till now, there has been no outstanding breakthrough in resolving the mucosal barrier, which is the main reason for the failure of oral insulin formulations. To resolve the mucosal barrier, many wise and complicated approaches have been undertaken, including application of muco-adhesive nanoparticulate system, muco-adhesive composites,9 nanoemulsions, double emulsions,10 chemical modification of insulin molecules such as acylation and PEGylation to alter hydrophobic/hydrophilic balance of insulin11 and application of cell-penetrating peptides (CPPs) in both chemical4 and physical conjugations (co-administrations).12
CPPs are small peptides (usually <30 amino acids), also known as protein transduction domains,13 and have attracted much attention in the recent two decades, and their application has drastically been increasing in the field of noninvasive peptide or nucleotide delivery.14 They have provided a promising and powerful tool for drug delivery into the cells as well as trafficking inside the cells and finally resolving the cell barrier. Both cell culture and animal studies have shown that covalently,15 and even physically, linking a CPP (eg, trans-activated-transcriptions [TAT]) to almost any type of drugs, including hydrophilic compounds and large proteins (molecular weight [Mw] >150 kDa), facilitates translocation of the attached species into the cells of all organ types, including the brain.16 In this innovative study, a wise combination of the most successful and modern solutions to circumvent both the enzymatic situation and the mucosal barrier is reported. To overcome the enzyme barrier, insulin was encapsulated and protected within the polymeric chains of the biodegradable, biocompatible and muco-adhesive polymer chitosan (CS), and the cell membrane barrier was overridden through both tight-junction-opening capabilities of CS and, more importantly, direct penetration and transcytosis properties of a very potent and newly developed CPP, PenetraMax, where the CPP moiety was tagged on the surface of the nanoparticles (NPs) via covalent conjugation.
Materials and methods
Materials
Medium Mw CS (75 kDa), 90% deacetylated, was obtained from Primex (Siglufjörður, Iceland). Triton X-100, N-hydroxysuccinimide (NHS), N-ethyl-N′-(3-dimethylaminopropyl)carbodiimide (EDC) and dicarboxylic acid polyethylene glycol (PEG; average Mn, 500 Da) were purchased from Merck KGaA (Darmstadt, Germany). Dialysis bag (molecular weight cut-off, 12,000 Da) was obtained from Sigma-Aldrich (St Louis, MO, USA). Regular human insulin USP (recombinant; regular human insulin, 28.9 IU/mg) was a gift from Ronak Ltd (Tehran, Iran), and aspart insulin was procured from Novo Nordisk (Bagsvaerd, Denmark). Caco-2 cell line with passage number 30–40 was provided by Pasteur Institute (Tehran, Iran). Human Insulin & Insulin Analogs (lispro, aspart) enzyme-linked immunosorbent assay (ELISA) kit was procured from Alpha Diagnostic International (San Antonio, TX, USA).
Methods
Synthesizing the CPP and the rationale behind the selection of its amino acid sequence
The CPP that we were enthusiastically interested to study its cell permeation properties and its potential to translocate the covalently conjugated cargos is a newly developed and different analog of the famous and well-established CPP “penetratin”. As we know, cell penetration and cargo delivery properties of penetratin are well-known facts to scientists involved in the fields of cell permeation and peptide delivery.12–17 According to the promising results reported for a special analog of penetratin in an intranasal insulin delivery (a physical mixture formulation),18 this 16-amino-acid peptide sequence was synthesized in our peptide laboratory in Khaje Nasir University and purified to more than 95% purity. Table 1 provides the amino acid sequence of penetratin and the newly developed analog of penetratin (PenetraMax) investigated in this study. To facilitate the conjugation of the CPP sequence with the free amine groups at the surface of CS NPs and to preserve the CPP configuration after conjugation, a very short-chain (nine carbon atoms) PEG with a free carboxylic acid at one side was attached to the C-terminus of the CPP sequence.
 | Table 1 Amino acid sequence of penetratin and its newly developed analog (PenetraMax) |
CS NPs are usually developed by an ionic gelation method using sodium tripolyphosphate ionic cross-linker or polyelectrolyte complexation,19,20 producing particles that are >100 nm in size and are not water dispersible in neutral and alkaline pHs. Since these particles are stabilized via ionic interactions, acidic pH environment could easily compromise its integrity, and in neutral or alkaline pH NPs are not water dispersible and cannot release the encapsulated ingredients. Therefore, covalent cross-linking is more attractive than the ionic gelation or electrostatic complexation techniques. Bodnar et al21 reported the synthesis of CS NPs covalently cross-linked with tartaric acid that resulted in particles of 60–280 nm size range. Only a few reports have demonstrated water-in-oil (W/O) microemulsion synthesis of covalently cross-linked CS NPs (using glutaraldehyde as the cross-linker) in the size range between 30 and 150 nm.22 Other scientists reported the synthesis of tartaric acid cross-linked, ultrasmall fluorescent carbon NPs via a W/O microemulsion technique in which the resulted NPs were highly water dispersible.23
Fabrication of the cross-linked NPs
The NPs were produced using a homogeneous W/O nanoemulsion technique based on a published protocol,24 but with certain modifications and optimizations and application of design of expert (DoE) methodology to find the minimum particle size, minimum polydispersity, optimum zeta potential and maximum swelling ratio. The NP fabrication technique consisted of preparation of two separate homogeneous and robust W/O nanoemulsions: nanoemulsion I and nanoemulsion II. The nanoemulsion medium consists of cyclohexane (outer continuous phase), aqueous solution containing CS polymer or the cross-linker PEG (inner dispersed phase), n-hexanol (cosurfactant) and Triton X-100 (surfactant). We found that very stable and reproducible W/O nanoemulsions with the minimum size water pools can be formed using the following ratios of cyclohexane, water, cosurfactant and surfactant, 11:8:6:5, respectively. Comparing the reference protocol, the ratio of aqueous inner phase was increased by more than two times, resulting in 100% increase in process throughput and less consumption of hazardous cyclohexane solvent while the harvested NPs were considerably smaller and polydispersity was obviously decreased. To prepare the CS solution, 0.75 g of medium Mw CS (Mv ~75 kDa, degree of deacetylation =90%) was dissolved in 99 mL of 1% (w/w) acetic acid solution to achieve the optimized polymer solution viscosity. To prepare the nanoemulsion I, 6 mL of n-hexanol and 5 mL of Triton X-100 were added to 11 mL of cyclohexane and magnetically stirred at 4°C till the solution became completely clear and transparent. Then, 8 mL of CS solution was added dropwise to the mixture of cyclohexane, surfactant and cosurfactant and stirred until the emulsion became perfectly clear and transparent, indicating the formation of a stable nanoemulsion. Nanoemulsion II was prepared using the same protocol, and the aqueous phase of nanoemulsion II consisted of cross-linker dicarboxylic acid PEG and water-soluble EDC. NPs were produced by dropwise addition of nanoemulsion II to nanoemulsion I and mild magnetic stirring for 24 h at room temperature (RT) in a capped container. The overnight stirring provided the chance for the water pools of nanoemulsion I and II to collide with each other, and the CS polymer inside the water pools of nanoemulsion I was cross-linked by the cross-linker PEG inside the water pools of nanoemulsion II. By adding ethanol, the nanoemulsion was disrupted and the formed and cross-linked NPs were precipitated by centrifuging the water phase at 14,000 rpm for 20 min. The harvested NPs were thoroughly washed with acetone and dialyzed (Mw cutoff point: 12,000 Da) against distilled water for 2 days to wash away the unreacted PEG, EDC and surfactant or cosurfactant.
Optimization with DoE
To find the minimum particle size, minimum polydispersity, optimum zeta potential and maximum swelling ratio, Box–Behnken response surface methodology was applied using Design-Expert® software (V7.0.0; Stat-Ease, Inc., Minneapolis, MN, USA). In these study responses, particle size, polydispersity, zeta potential and swelling ratio were functions of the proportions of the independent factors including the concentration of CS (mg/100 mL), concentration of PEG (mg/100 mL), volume ratio of Triton X-100/cyclohexane (%) and volume ratio of water/cyclohexane (%). Based on preliminary studies (Table 2), the lower and upper levels of the independent variable were determined (Table 3). According to statistical calculations, 25 runs of experiment were carried out to develop an appropriate model to give the optimized responses (data not shown).
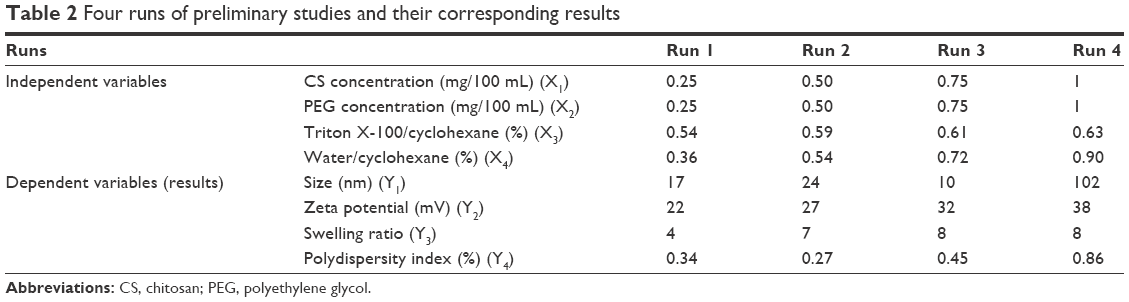 | Table 2 Four runs of preliminary studies and their corresponding results |
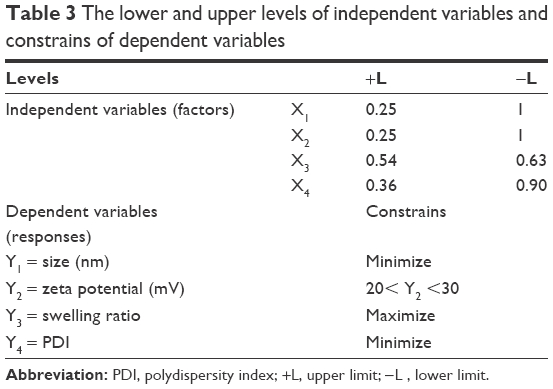 | Table 3 The lower and upper levels of independent variables and constrains of dependent variables |
Covalent linkage of CPP sequence at the surface of preformed NPs to form CPP–NP conjugates
To prevent the possible amide reaction between the final carboxyl group of CPP and the amine groups of lysine amino acids in the structure of CPP sequence upon activation, the amine groups in the CPP sequence were fluorenylmethoxycarbonyl (Fmoc) protected by a very environmentally friendly and expeditious method,25 prior to the exposure of CPP carboxyl group to primary amine groups of CS. After the protection of amine groups, the CPP sequence was separated from the unreacted 9-Fmoc chloride and purified via a semi-preparative reverse-phase high-performance liquid chromatography (HPLC) method.26
The Fmoc-protected CPP sequence was covalently conjugated to the surface amine groups of the preformed NP using carbodiimide chemistry.27 Briefly, Fmoc–CPP sequence was solubilized in distilled water (20 mg/mL) and completely mixed with NHS (40% w/w) and ethylenediaminetetraacetic acid (EDTA) (60% w/w) and stirred for 2 h at RT. Aqueous dispersion of preformed CS NPs (200 mg/mL) was added to the reaction mixture and stirred at RT for 20 h. The CPP–NP conjugates were dialyzed extensively against distilled water for 2 days in dialysis bags with Mw cutoff point of 12,000 Da and lyophilized. Figure 1 shows the schematic representation of cross-linking and subsequent conjugation of CPP with the cross-linked NPs. The collected dialyzed sample was used for estimation of unbound CPP sequence using HPLC method and respective linear fitting curve established for the CPP sequence at ƛmax =214 nm. After the chemical conjugation of Fmoc-protected CPP sequences with the preformed CS NPs, the Fmoc-protected amine groups of CPP (conjugated to NPs) were de-protected using a very mild method according to a published protocol.28
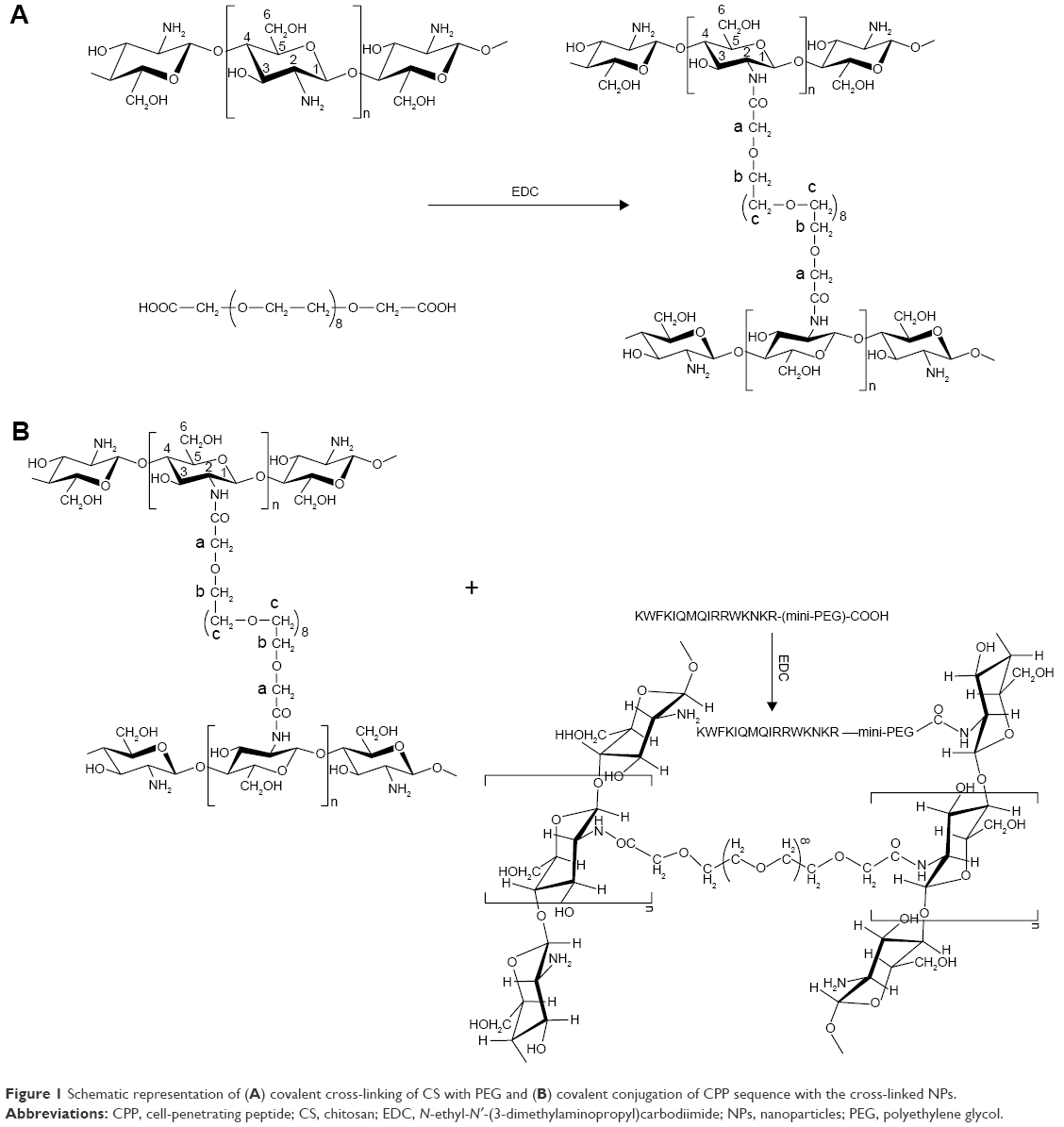 | Figure 1 Schematic representation of (A) covalent cross-linking of CS with PEG and (B) covalent conjugation of CPP sequence with the cross-linked NPs. |
Characterization of CPP–NP conjugates
Analyzing particle size and morphology of NPs before conjugation with CPP
To investigate the characteristics of the NPs produced by the optimized W/O nanoemulsion technique, particles size and morphology were studied both before and after conjugation of CPP with the NPs. As far as these NPs are cross-linked with the chains of hydrophilic PEG at the time of fabrication, they should naturally have good swelling properties in aqueous medium. Therefore, these NPs were studied in both shrank (dry) and swollen states. For particle size studies in swollen state, NPs were dispersed in deionized (DI) water and size distribution was studied by dynamic light scattering technique (DLS; Malvern Zetasizer). To investigate the size and morphology of NPs in shrank (dry) state, samples were prepared and studied by scanning electron microscopy (SEM).
Analyzing particle size and morphology after conjugation with CPP
To evaluate the effect of conjugation of CPP sequences with the preformed NPs on the size and shape of the NPs, the CPP-conjugated NPs were also investigated for size distribution, surface morphology, zeta potential and swelling ratio, and their properties were compared to those of initial non-CPP-conjugated NPs.
Drug loading into the preformed CPP–NP conjugates
According to the results from preliminary studies and optimizations, only the optimized formulations with respect to the concentration of the polymer (CS) and cross-linking agent (PEG), at the time of NP fabrication, their particle size and swelling ratio were chosen for further investigations. Preliminary studies showed that aqueous medium containing CS with a concentration of 7.5 mg/mL and PEG with a concentration of 2.5 mg/mL leads to cross-linked NPs (in the novel W/O nanoemulsion technique) with desired swelling ratio and drug loading and drug release properties. To carry out the drug loading studies, a model peptide (insulin) was loaded in the optimized preformed CPP–CS NP conjugates by a post-loading method.29 An exact amount (50 mg) of blank CS-CPP NPs was dispersed in 2 mL of DI water, and the pH was adjusted to 5.5 using 0.01 N HCl solution. As we know, the isoelectric pH (pHi) of insulin is around 5.3;30 therefore, at pH >5.3, the total electric charge of insulin is negative.
However, CS has good positive charge in this pH because of protonation of its amine groups.31 In aqueous medium with this pH, the cross-linked NPs of CS was freely and extensively swelled because of hydrophilic nature of PEG in the structure of NPs as well as charge repulsion of protonated amine groups. The negatively charged peptide (insulin) was easily entrapped inside the positively charged mesh of CS, and the extra amount of insulin could be adsorbed at the surface of NPs due to the attraction between positive charge of free amine groups of CS and negative surface charge of insulin. In addition to the positive charge of free amine groups, the conjugated CPP sequence also introduced positive charge due to the presence of cationic amino acids such as arginine and lysine, thereby adsorbing more insulin at the surface of NPs. CPP–CS NP conjugates were incubated with insulin solution to be entrapped at four different concentrations, ie, 10, 20, 30, and 40% w/w of NPs. The resulting gel was kept incubated to saturate for 12 h, lyophilized and stored at 4°C. Figure 2 shows the flow diagram of step-by-step process of NP fabrication and drug loading.
 | Figure 2 Schematic representation of NP fabrication and drug loading process. |
Encapsulation efficiency (EE) and loading efficiency (LE) studies
To determine the EE% and LE%, an exact amount (30 mg) of drug-loaded NPs was dispersed in distilled water, the freshly prepared colloidal suspension was centrifuged at 14,000 rpm for 10 min at 4°C and the supernatant was analyzed for the determination of nonencapsulated insulin using HPLC method. The samples were injected to Agilent® 1260 infinity equipped with 1260 Quat pump VL, 1260 ALS auto sampler and 1260 DAD VL detector. The detector was set at 214 nm. C18 column was used for HPLC analysis of insulin using linearly regressed calibration curve. The mobile phase was a mixture of buffered aqueous phase and acetonitrile in a ratio of buffer:acetonitrile (70:30). Buffer was prepared from KH2PO4 (0.1 M) and triethylamine (1%), and the pH was adjusted to 2.8 using phosphoric acid. Flow rate was adjusted to 0.5 mL/min, and the data were captured using Agilent Chemstation® software.
To calculate the EE and LE, the amount of nonencapsulated insulin in the supernatant of the centrifuged drug-loaded NPs suspension was determined. All the experiments were performed in triplicate, and the mean values were used to calculate EE% and LE% according to Equations 1 and 2, respectively:
|
|
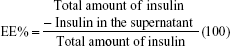
|
|

In vitro drug release studies
To investigate, more relevantly, the plausibility of the proposed delivery system for oral delivery of peptides and proteins, the rate and extent of the release of the model peptide (insulin) was investigated in both simulated intestinal fluid (SIF; pH 6.8, duodenum pH) and phosphate-buffered saline (pH 7.4, colon pH). To study the in vitro release behavior of insulin from these cross-linked NPs, a proper amount of lyophilized NPs equivalent to 30 mg of insulin was dispersed in 500 mL of SIF, shaking at 50 rpm. The temperature was set constant at 37°C±0.5°C. The release medium was chosen relatively large to ensure the sink condition. At predetermined time intervals, some specific aliquots (1 mL) were collected and replaced by preheated blank medium. The samples were centrifuged at 14,000 rpm for 20 min. The supernatant was investigated for insulin content, and the sediment was dispersed in 1 mL blank medium and returned to release medium instead of the addition of blank medium after withdrawal of aliquots. Therefore, the unreleased insulin in the withdrawn NPs was also released, because insulin release from the polymeric mesh of the NPs was not completed in the first few hours. The amount of insulin in the supernatant was determined using the HPLC method as mentioned earlier.
To elucidate the mechanism of peptide release from the developed nanoparticulate delivery system, the in vitro insulin release data were fitted to Ritger–Peppas model:
|
|
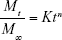
where Mt and M∞ are the cumulative release of insulin at time (t) and infinite time, respectively; K is a constant related to the structural and geometric characteristic of the device and n is an exponent reflecting the diffusion mechanism. Depending on the amount of the calculated values for n, the release mechanism was categorized. Accordingly, if n=0.45 the release mechanism is Fickian (case I) diffusion, if 0.45< n <0.89 the release mechanism is non-Fickian (anomalous) transport and if n=0.89 the release mechanism is diffusion and zero-order (case II) transport.
Caco-2 cell culture studies
Caco-2 cells were provided by Pasteur Institute at passage number 30–40 and grown in an incubator at 37°C in humidified atmosphere with 5% CO2 and 95% air. Cells were maintained in T-75 flasks using Modified Eagle’s Medium supplemented with 20% fetal bovine serum, 1% nonessential amino acids, 10,000 U/mL penicillin and 10,000 μg/mL streptomycin. Growth medium was changed every other day. After 8 days and confluency of 80%–90%, the cells were passaged using 0.25% trypsin/ethylenediaminetetra acetic acid solution.
In vitro cytotoxicity studies
The cells were seeded in 24-well plates at a density of 5×105 cells/well and cultured for 24 h in an incubator at 37°C under 5% CO2. The medium was replaced with drug-loaded NPs of either plain CS or cross-linked CPP-conjugated NPs from CS at a concentration of 5 mg/mL/well and incubated for 20 h. The particles were removed, and MTT assay was carried out.
Transepithelial electrical resistance (TEER) and insulin transport studies
The Caco-2 cell lines were grown in transmembrane inserts with 0.4 μm pore size (Millipore) and cell density of 5×105 cells/well. TEER studies were carried out according to a method reported elsewhere.24 The concentration of the plain CS NPs or CPP-conjugated CS NPs used was 10 mg/mL/well, and the TEER of the cell monolayer was investigated at predetermined time intervals at 37°C. In addition to recombinant human insulin, aspart insulin (a new and short-acting analog of human insulin) was also loaded in either the plain CS NPs or CCP-conjugated CS NPs with the same concentration, because there are some reports25,26 that aspart insulin has more potential to pass through the lumen epithelium due to its monomeric nature and linear conformation in aqueous medium. To evaluate this potential and to investigate whether this nanoparticulate system has a better outcome with aspart insulin, we studied both human insulin and aspart insulin in the same condition and compared the results. Simple solution of both human insulin and aspart insulin was applied to the donor chamber of the cell layer as control. For insulin transport studies, the medium of donor chamber was replaced with fresh medium containing the insulin-loaded plain CS NPs or CPP-conjugated CS NPs (10 mg/well). Aliquots were taken from the receiver chamber at predetermined time intervals and continued for 4 h, and the samples were investigated for insulin content with a special ELISA kit specific and sensitive to both regular human insulin and aspart insulin (Alpha Diagnostic International). All cases were studied in triplicate, and the average values were reported. The following equation:
|
|
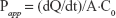
was used to calculate the apparent permeability coefficient (Papp) of insulin. In this equation, dQ/dt is the rate of permeability, A is the surface area of the filter membrane and C0 is the initial concentration of insulin in the apical chamber.
Results and discussion
Physical characteristics of cross-linked NPs before conjugation with CPP
Dispersion of the NPs in DI water readily results in a transparent solution, indicating the water-dispersible nature of NPs and their independent nature at a particular pH. As far as NPs are freely water dispersible, the size reported by photon correlation spectroscopy is the size of NPs in swollen state. The average particle size reported by SEM (dry state) was 12±4.2 nm (Figure 3), while the particle size in swollen state (reported by DLS) was around 100 nm, indicating a 6- to 8-fold increase in size in swollen state. As depicted in Figure 3, the NPs are spherical in shape and highly monodispersed with smooth surface that is unique to the nano- and microemulsion technique. In addition to simplicity and robustness of this technique, monodispersity and ultrasmall size of the NPs are the other advantages of this optimized technique. As shown in Figure 3, among the monodispersed NPs, there are some large particles (100–500 nm) that are assemblies of ultrasmall and individually monodispersed NPs that are closely associated with each other to form large particles. The presence of these relatively large aggregates is the reason why the polydispersity index reported by DLS is always very high for CS NPs, and CS NPs are reported to have very wide size distribution in the literature.23,30 The development of ultrasmall and monodispersed NPs is because of the compartmentalization of CS polymer chains inside the very tiny water droplet of nanoemulsion that acts as a “nano-container”. The compartmentalization process can be attributed to some main factors such as the confined environment of the nano-container and the relative neutralization of the charged primary amine groups (“charge shielding”) of CS because of the interaction with the nanoemulsion components such as surfactant and cosurfactant molecules at the interface of W/O.23 As far as the formation of the NPs is due to the covalent bond between the amine groups of CS and the carboxylic groups of PEG dicarboxylic acid, integrity of the formed NP is guaranteed at all pH ranges. The hydrophilic PEG has a dual action here, as it not only acts as the cross-linker to form the NPs but also makes the CS NPs water dispersible in all pH ranges by incorporating several polyoxyethylene groups in the structure of NPs. Zeta potential analysis of NP in DI showed a zeta potential value of +5 mV, considerably lower than the typical zeta potential values reported for plain CS NPs (+20 to +35 mV).30 Presence of PEG in the structure of CS NPs reduces the surface charge of CS NPs by introducing a hydrophilic component in the structure of NPs and also a hydrophilic shell around them making them water soluble and improving their biocompatibility. But still zeta potential is a positive value, indicating a positive surface charge, confirming the presence of free amine groups.
 | Figure 3 Particle size and shape of CS-PEG NPs in dry (not swollen) state taken by SEM. |
Physical characteristics of cross-linked NPs after conjugation with CPP
The same procedures were applied to investigate the size, shape and swelling properties of NPs after conjugation with CPP sequences. After conjugation with CPP, the size of the NPs increased in both shrank (Figure 4) and swelled states. But the shape of the NPs was not changed considerably. The swelling ratio of the CPP-conjugated NPs was also increased (>10-fold) compared to the swelling ratio of the initial NPs (6- to 8-fold). After conjugation of CPP sequences with the CS NPs, the zeta potential sharply increased reaching an average value of +32 mV, while the zeta potential of PEG-cross-linked CS NPs was +5 before conjugation with the CPP because this CPP is rich in cationic amino acids arginine and lysine.
 | Figure 4 Particle size and shape of CS-PEG-CPP NPs in dry (not swollen) state taken by SEM. |
1H NMR studies revealed the successful cross-linking of CS with PEG as well as the attachment of the peptide sequence to the polysaccharide structure of CS polymer (Figure 5). The chemical shift at δ 7.2 is related to the aromatic protons of the phenylalanine moiety in the structure of CPP confirming the presence of CPP in the structure of CPP-conjugated CS NPs. The multiple peaks of oxymethyl groups in PEG at δ 3.3–3.7 cover the signals from pyranose ring in the structure of CS in CS-N-CONH-PEG-CPP confirming the presence of PEG in the structure of CS, implying the successful covalent cross-linking of CS with PEG dicarboxylic acid. The weak and broad peaks at δ 4.2–4.4 are from the protons of –NH–CH(CH2)–CO– in the CPP. The peak at δ 2.8–3.1 belongs to the protons of –CH2–NH–NH–NH2 of arginine groups in the structure of CPP, and the weak and multiple peaks at δ 1.3–1.9 are from the –CH2–CH2–CH2–NH–NH–NH2 in arginine in the structure of CPP conjugated to CS. The amount of ligand attached to the NPs was quantified by the determination of the residual unreacted CPP in conjugation medium. With these calculations, the amount of CPP conjugated to the NPs was found to be 0.12% w/w of NPs.
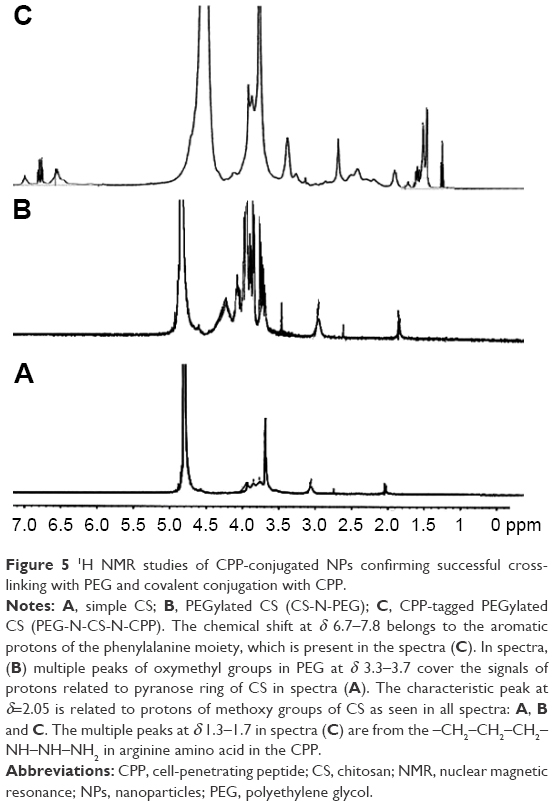 | Figure 5 1H NMR studies of CPP-conjugated NPs confirming successful cross-linking with PEG and covalent conjugation with CPP. |
EE and LE of CPP-conjugated CS NPs
Any polymeric nanoparticulate delivery system intended to be used for oral delivery of peptides (insulin) needs to demonstrate sufficient insulin encapsulation and good drug LE as a basic parameter. Because poor insulin EE would prevent the nanoparticulate delivery system to achieve the desired therapeutic functions following oral administration with a rationale and practical amount of polymeric NPs. Therefore, the insulin-loading capacity of the NPs produced with different weight ratios of CS/PEG (4:1, 3:1, 2:1 and 1:1) was investigated upon the production of NPs. Preliminary studies showed that in the weight ratio of CS:PEG, 3:1, swelling ratio of the NPs as well as drug loading capacity is optimum, while keeping the other factors of nanoemulsion system constant. As far as the initial concentration of insulin in the loading medium is important, the LE is influenced not only by the capacity of the NPs but also by the initial concentration of insulin, after the determination of the optimized weight ratio of polymer and cross-linking agent to produce NPs with optimized swelling ratio; different weight ratios of insulin to NPs were investigated for maximum LE. Four different insulin weight ratios of NPs (1:10, 2:10, 3:10 and 4:10) were investigated for maximum LE. NPs with a weight ratio of 3:1, polymer:PEG and in loading condition with insulin, NPs weight ratio of 2:10 with 12 h incubation time represent the highest LE and EE of 16 and 92%, respectively. Transmission electron microscopy studies revealed that after loading with insulin, the size of the NPs drastically increased but still the spherical shapes of the NPs were preserved (Figures 6 and 7).
 | Figure 6 TEM image of cross-linked NPs. |
 | Figure 7 TEM image of the insulin-loaded NPs. |
In vitro release study
Figure 8 shows the in vitro release profile of a model peptide (insulin) from the optimized nanoparticulate delivery system loaded with the embedded peptide in a post-loading procedure.31 The release profile was investigated in both SIF (pH 6.8) and phosphate-buffered saline (pH 7.4). As shown in Figure 8, there was a burst release of insulin in the first 30 min for both mediums and then the release rate slowed down but continued steadily during 24 h. During this burst release period, ~32%±5% of the total insulin was released at pH =7.4 and 19%±2.3% was released at pH 6.8. During optimization studies, we found that NPs produced under specific concentration of polymer and cross-linker (CS: 7.5 mg/mL and PEG: 2.5 mg/mL) had the best LE, release behavior and total release extent. As shown in Figure 8, insulin release from the optimized NPs was nearly completed in 24 h and during this time ~85% of the embedded insulin was released in phosphate-buffered saline (pH 7.4). But in SIF (pH =6.8), both the burst and total release of insulin were lower compared to phosphate-buffered medium (19%±2.3% burst release and 77%±6.4% total release). At pH 6.8, a few portions of primary amine group of CS may be still protonated and keep their positive charge,31 hence attracting the negatively charged insulin and slowing their release. At pH 7.4, a very high burst and total release were seen for this formulation, a property not usually seen in CS NPs.31,32 But in this formulation, the presence of hydrophilic PEG made the polymer water soluble in all pH ranges. In addition to complete solubility, CS lost its positive charge completely in this pH, therefore not attracting and keeping the negatively charged insulin. This fast release of peptide from the polymeric mesh of the NPs is beneficiary for peptides with short half-life like insulin to enable the delivery system to maintain therapeutic effects.33
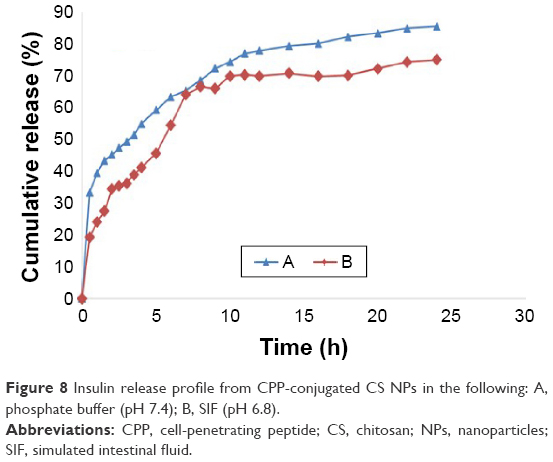 | Figure 8 Insulin release profile from CPP-conjugated CS NPs in the following: A, phosphate buffer (pH 7.4); B, SIF (pH 6.8). |
According to the results from other studies,29 the mechanism of drug release from erodible, hydrophilic polymer matrices is a complex process, because numerous factors are involved, including penetration of water into the polymeric matrix, solubilization/erosion of the polymeric formulation, swelling of the polymer and dissolution of the drug from the swollen matrix. Drug release from polymeric formulations with swelling properties usually follows a non-Fickian (anomalous) behavior, but for large molecules such as peptides and proteins especially when it comes to the interaction of ionic charges between the carrier polymer and the embedded moiety the story may be different. To investigate the release mechanism of insulin (as a peptide with ionic charge) from the developed NPs, the parameter “n” for Ritger–Peppas equation was calculated. The correlation coefficient values for the optimized formulation were found to be 0.8213 (R ≥0.99); this clearly shows that the release data are well fitted to the empirical equation. The “n” release exponent was between 0.86 and 0.89, indicating a non-Fickian (anomalous) transport (0.45< n <0.89).
TEER and insulin transport studies
MTT cytotoxicity studies showed that neither plain CS NPs nor CPP-conjugated CS NPs had significant cytotoxic effects on the cultured Caco-2 cell lines. In comparison to control (medium), the particles showed nearly 100% cell viability. As shown in Figure 9, after 2 h from the exposure of either the plain drug-loaded CS NPs or CPP-conjugated drug-loaded CS NPs to Caco-2 cells with a dose of 10 mg/mL/well, there was a significant reduction in TEER, ~85 and 40% reduction compared to the initial values of CPP-conjugated CS NPs or plain CS NPs, respectively. As it can obviously be understood from Figure 9, TEER values were reduced significantly in the presence of both native CS NPs and CPP-conjugated CS NPs regardless of their embedded peptide (regular human insulin or aspart insulin), but there was no significant change in TEER values in the presence of either simple solution of regular human insulin or simple solution of aspart insulin. TEER reduction due to the presence of CS particles is a well-established scientific finding and is reported in different studies.34–36 But a very interesting phenomenon found in this study is the significant and extreme difference between the TEER reduction in the presence of plain CS NPs and CPP-conjugated CS NPs. As shown in Figure 9, TEER reduction in the presence of CPP-conjugated CS NPs was more than 2-fold compared to TEER reduction in the presence of plain NPs from CS. For more investigation of this interesting finding and to clarify if this phenomenon was due to the special properties of the CPP sequence or special characteristics of the newly developed peptide derivative of CS, we carried out another round of TEER measurement with the same condition and duration, but in this time we exposed the apical chamber of transwells to simple solution of CPP (10 μM). As shown in Figure 9, there was only a negligible reduction in TEER values in the presence of simple solution of CPP. This finding about the effect of L-penetratin (our CPP is a new analog of L-penetratin) on TEER values is in accordance with the results from other researchers.37 Although for some kinds of CPPs such as poly-arginine, the reduction in TEER is proposed as a possible mechanism for enhancing the pulmonary absorption of insulin,15 where other studies do not support the idea of TEER reduction for CPP. In a recent study carried out for elucidating the cell permeation mechanism of CPPs, such a mechanism was not proposed for CPPs and the effects of L-penetratin as well as poly-arginine on TEER values were not significant.37
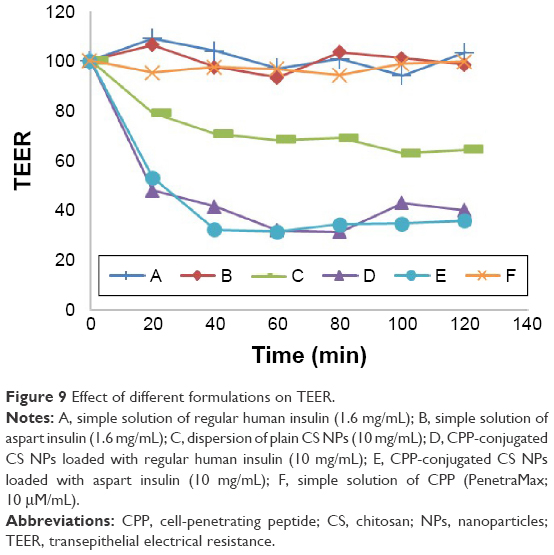 | Figure 9 Effect of different formulations on TEER. |
From these results, we can hypothesize that chemical conjugation between CS NPs and the CPP sequence leads to a new derivative of CS with extremely enhanced reducing effect on TEER and possibly opening the tight junctions. Figure 10 shows the permeation of insulin from apical chamber to basolateral chamber of transwells from different formulations in the same condition (37°C). The concentration of insulin in samples withdrawn from the receptor chamber of transwells was quantified via an ELISA-based kit specific for regular human insulin and aspart insulin. After the determination of cumulative transported insulin in the receiver chamber and comparing it to the initial concentration of insulin in the donor chamber, it was found that the translocation of insulin across the cell monolayer was ~0% for simple solutions of either regular or aspart insulin, around 5% for simple CS NPs loaded with regular human insulin, 5.4% for simple CS NPs loaded with aspart insulin, ~18% for CPP-conjugated CS NPs loaded with regular human insulin and ~16.7% for CPP-conjugated CS NPs loaded with aspart insulin (Figure 10). This is the highest level of insulin translocated from apical to basolateral sides (donor to receiver) of Caco-2 cells in cell culture studies till now.
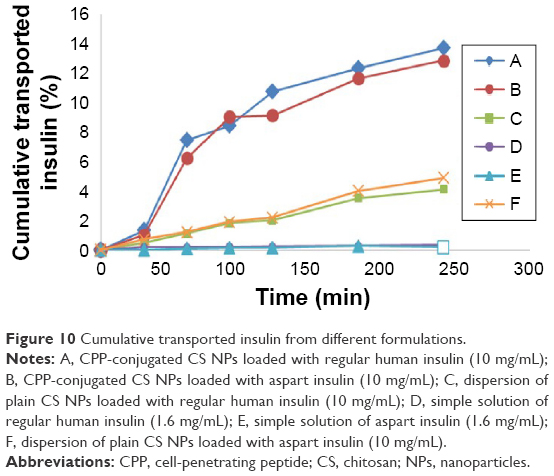 | Figure 10 Cumulative transported insulin from different formulations. |
These findings as well as release studies show that it is basically the nanoparticulate system and its special characteristics that determine how much insulin is transported across the cell monolayer not the kind of insulin. This study does not confirm the results of another study hypothesizing that aspart insulin has better chance for oral delivery due to its monomeric configuration.38 Another interesting finding in this study is that during 24 h after the translocation of insulin is stopped (withdrawing the insulin containing samples from the receiver chamber and incubation in fresh micro tubs), the concentration of assayable insulin is increased by the time. This finding is in good accordance with the results from in vitro release studies in which the release of insulin is almost completed after 24 h. Therefore, it is hypothesized that a greater portion of insulin is translocated via the translocation of NPs, and with the translocation of every single NPs numerous embedded insulin molecule is translocated from the apical chamber to basolateral chamber and then the release of insulin will continue from the meshes of the cross-linked NPs. As mentioned earlier, once this preformed nanoparticulate system is developed, it could easily be loaded with a great number of peptides, proteins, nucleotides and any other large and hydrophilic molecules in a very simple and mild post-loading process. After the peptide-loaded NPs are exposed to biological barriers, the embedded peptide will be translocated due to nanoparticulate properties, muco-adhesion and tight-junction opening properties of CS and direct and energy-independent37 penetration potential of CPP. The results show that this delivery system is very wisely developed, harnessing the outstanding tight-junction opening of CS and unique cell permeation potentials of CPPs. This nanoparticulate system seems to be very promising in the field of noninvasive delivery of peptides, proteins, vaccines and nucleotides as well as drug delivery to blood–brain barrier (BBB) and the delivery of diagnostics and contrast media for imaging from compartments inside the BBB.
Conclusion
The CS NPs fabricated by this optimized W/O nanoemulsion technique are ultrasmall (<15 nm), monodispersed, symmetric and spherical in shape and water dispersible in all pH ranges with extensive swelling properties (6- to 10-fold). Conjugation of the powerful CPP, PenetraMax, with the surface of the NPs gives them the capability to penetrate the monolayer Caco-2 cells through both the para-cellular and intracellular pathways. TEER is decreased drastically for NPs conjugated to CPP compared to plain CS NPs, indicating profound tight-junction opening properties for this newly developed peptide-conjugated derivative of CS. Translocation of the model peptide (insulin) is increased up to 18% for CPP-conjugated NPs compared to simple insulin solution. These functional NPs can easily be loaded with any kinds of peptides, nucleotides and hydrophilic large molecules for oral administration. The preliminary cell culture results are very promising in this study, and ex vivo and animal studies are under progress and will be published separately.
Disclosure
The authors report no conflicts of interest in this work.
References
Moroz E, Matoori S, Leroux J-C. Oral delivery of macromolecular drugs: where we are after almost 100 years of attempts. Adv Drug Deliv Rev. 2016;101:108–121. | ||
Khafagy E-S, Morishita M, Onuki Y, Takayama K. Current challenges in non-invasive insulin delivery systems: a comparative review. Adv Drug Deliv Rev. 2007;59(15):1521–1546. | ||
Sousa F, Castro P, Fonte P, Sarmento B. How to overcome the limitations of current insulin administration with new non-invasive delivery systems. Ther Deliv. 2015;6(1):83–94. | ||
He H, Ye J, Sheng J, et al. Overcoming oral insulin delivery barriers: application of cell penetrating peptide and silica-based nanoporous composites. Front Chem Sci Eng. 2013;7(1):9–19. | ||
Ross P, Milburn J, Reith D, Wiltshire E, Wheeler B. Clinical review: insulin pump-associated adverse events in adults and children. Acta Diabetol. 2015;52(6):1017–1024. | ||
Kamei N, Nielsen EJB, Nakakubo T, et al. Applicability and limitations of cell-penetrating peptides in noncovalent mucosal drug or carrier delivery systems. J Pharm Sci. 2016;105(2):747–753. | ||
Lopes M, Simões S, Veiga F, Seiça R, Ribeiro A. Why most oral insulin formulations do not reach clinical trials. Ther Deliv. 2015;6(8):973–987. | ||
Aguirre T, Teijeiro-Osorio D, Rosa M, Coulter I, Alonso M, Brayden D. Current status of selected oral peptide technologies in advanced preclinical development and in clinical trials. Adv Drug Deliv Rev. 2016;106(pt B):223–241. | ||
Rekha M, Sharma CP. Simultaneous effect of thiolation and carboxylation of chitosan particles towards mucoadhesive oral insulin delivery applications: an in vitro and in vivo evaluation. J Biomed Nanotechnol. 2015;11(1):165–176. | ||
Lopes MA, Abrahim-Vieira B, Oliveira C, et al. Probing insulin bioactivity in oral nanoparticles produced by ultrasonication-assisted emulsification/internal gelation. Int J Nanomedicine. 2015;10:5865. | ||
Baker JC, Hanquier JM. Acylated insulin analogs. Google Patents. 1999. | ||
Kamei N, Takeda-Morishita M. Brain delivery of insulin boosted by intranasal coadministration with cell-penetrating peptides. J Control Release. 2015;197:105–110. | ||
Jones SW, Christison R, Bundell K, et al. Characterisation of cell-penetrating peptide-mediated peptide delivery. Br J Pharmacol. 2005;145(8):1093–1102. | ||
Deshayes S, Morris M, Divita G, Heitz F. Cell-penetrating peptides: tools for intracellular delivery of therapeutics. Cell Mol Life Sci. 2005;62(16):1839–1849. | ||
Patel LN, Wang J, Kim K-J, Borok Z, Crandall ED, Shen W-C. Conjugation with cationic cell-penetrating peptide increases pulmonary absorption of insulin. Mol Pharm. 2009;6(2):492–503. | ||
Zorko M, Langel Ü. Cell-penetrating peptides: mechanism and kinetics of cargo delivery. Adv Drug Deliv Rev. 2005;57(4):529–545. | ||
Kristensen M, Nielsen HM. Cell-penetrating peptides as carriers for oral delivery of biopharmaceuticals. Basic Clin Pharmacol Toxicol. 2016;118(2):99–106. | ||
Khafagy E-S, Kamei N, Nielsen EJB, Nishio R, Takeda-Morishita M. One-month subchronic toxicity study of cell-penetrating peptides for insulin nasal delivery in rats. Eur J Pharm Biopharm. 2013;85(3):736–743. | ||
Dung TH, Lee S-R, Han S-D, et al. Chitosan-TPP nanoparticle as a release system of antisense oligonucleotide in the oral environment. J Nanosci Nanotechnol. 2007;7(11):3695–3699. | ||
Bayat A, Dorkoosh FA, Dehpour AR, et al. Nanoparticles of quaternized chitosan derivatives as a carrier for colon delivery of insulin: ex vivo and in vivo studies. Int J Pharm. 2008;356(1):259–266. | ||
Bodnar M, Hartmann JF, Borbely J. Preparation and characterization of chitosan-based nanoparticles. Biomacromolecules. 2005;6(5):2521–2527. | ||
Banerjee T, Mitra S, Singh AK, Sharma RK, Maitra A. Preparation, characterization and biodistribution of ultrafine chitosan nanoparticles. Int J Pharm. 2002;243(1):93–105. | ||
Tallury P, Kar S, Bamrungsap S, Huang Y-F, Tan W, Santra S. Ultra-small water-dispersible fluorescent chitosan nanoparticles: synthesis, characterization and specific targeting. Chem Commun. 2009;17:2347–2349. | ||
Zhi J, Wang Y, Luo G. Adsorption of diuretic furosemide onto chitosan nanoparticles prepared with a water-in-oil nanoemulsion system. React Funct Polym. 2005;65(3):249–257. | ||
Kitchens KM, Kolhatkar RB, Swaan PW, Eddington ND, Ghandehari H. Transport of poly (amidoamine) dendrimers across Caco-2 cell monolayers: influence of size, charge and fluorescent labeling. Pharm Res. 2006;23(12):2818–2826. | ||
Home P, Barriocanal L, Lindholm A. Comparative pharmacokinetics and pharmacodynamics of the novel rapid-acting insulin analogue, insulin aspart, in healthy volunteers. Eur J Clin Pharmacol. 1999;55(3):199–203. | ||
Sonaje K, Lin K-J, Wey S-P, et al. Biodistribution, pharmacodynamics and pharmacokinetics of insulin analogues in a rat model: oral delivery using pH-responsive nanoparticles vs subcutaneous injection. Biomaterials. 2010;31(26):6849–6858. | ||
Chen C-C, Rajagopal B, Liu XY, et al. A mild removal of Fmoc group using sodium azide. Amino Acids. 2014;46(2):367–374. | ||
Mukhopadhyay P, Kundu P. Chitosan-graft-PAMAM–alginate core–shell nanoparticles: a safe and promising oral insulin carrier in an animal model. RSC Adv. 2015;5(114):93995–94007. | ||
Illum L, Fisher A, Jabbal-Gill I, Davis S. Bioadhesive starch microspheres and absorption enhancing agents act synergistically to enhance the nasal absorption of polypeptides. Int J Pharm. 2001;222(1):109–119. | ||
Qin C, Li H, Xiao Q, Liu Y, Zhu J, Du Y. Water-solubility of chitosan and its antimicrobial activity. Carbohydr Polym. 2006;63(3):367–374. | ||
Pillai C, Paul W, Sharma CP. Chitin and chitosan polymers: chemistry, solubility and fiber formation. Prog Polym Sci. 2009;34(7):641–678. | ||
Chalasani KB, Russell-Jones G, Yandrapu SK, Diwan PV, Jain SK. A novel vitamin B 12-nanosphere conjugate carrier system for peroral delivery of insulin. J Control Release. 2007;117(3):421–429. | ||
Thanou M, Kotze A, Scharringhausen T, et al. Effect of degree of quaternization of N-trimethyl chitosan chloride for enhanced transport of hydrophilic compounds across intestinal Caco-2 cell monolayers. J Control Release. 2000;64(1):15–25. | ||
Kotzé AR, Lueβen HL, de Leeuw BJ, Verhoef JC, Junginger HE. N-trimethyl chitosan chloride as a potential absorption enhancer across mucosal surfaces: in vitro evaluation in intestinal epithelial cells (Caco-2). Pharm Res. 1997;14(9):1197–1202. | ||
Kotzé AF, Lueßen HL, de Leeuw BJ, et al. Comparison of the effect of different chitosan salts and N-trimethyl chitosan chloride on the permeability of intestinal epithelial cells (Caco-2). J Control Release. 1998;51(1):35–46. | ||
Kamei N, Onuki Y, Takayama K, Takeda-Morishita M. Mechanistic study of the uptake/permeation of cell-penetrating peptides across a Caco-2 monolayer and their stimulatory effect on epithelial insulin transport. J Pharm Sci. 2013;102(11):3998–4008. | ||
Damgé C, Socha M, Ubrich N, Maincent P. Poly (ε-caprolactone)/eudragit nanoparticles for oral delivery of aspart-insulin in the treatment of diabetes. J Pharm Sci. 2010;99(2):879–889. |
 © 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.