Back to Journals » International Journal of Nanomedicine » Volume 13
A novel disulfide bond-mediated cleavable RGD-modified PAMAM nanocomplex containing nuclear localization signal HMGB1 for enhancing gene transfection efficiency
Authors Li J, Han Y, Lu Y, Song B, Zhao M, Hu H, Chen D
Received 3 August 2018
Accepted for publication 23 September 2018
Published 6 November 2018 Volume 2018:13 Pages 7135—7153
DOI https://doi.org/10.2147/IJN.S182445
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Linlin Sun
Ji Li, Yuting Han, Yue Lu, Baohui Song, Ming Zhao, Haiyang Hu, Dawei Chen
Department of Pharmaceutics, School of Pharmacy, Shenyang Pharmaceutical University, Shenyang 110016, China
Background: Polyamidoamine (PAMAM) dendrimers modified by polyethylene glycol (PEG) have frequently been investigated as a delivery carrier for gene therapy. However, modification of PAMAM with PEG using covalent linkage significantly reduces the cellular uptake rate and the transfection efficiency. How to conquer these barriers becomes a burning question in gene delivery.
Materials and methods: The present study constructed an effective disulfide bond-mediated cleavable RGD modified gene delivery system to overcome the aforementioned limitations. The disulfide bond was introduced between PAMAM dendrimers and PEG chains to realize the cleavage of PEG from the carrier system, whereas the arginine-glycine-aspartate (RGD) peptide was expected to promote the cellular uptake rate. A high mobility group Box 1 (HMGB1) protein containing nuclear localization signal (NLS) was simultaneously introduced to further promote gene expression efficiency. A pDNA/HMGB1/PAMAM-SS-PEG-RGD (DHP) nanocomplex was prepared via electrostatic interaction and characterized.
Results: The results showed that DHP generated small particles and was able to condense and protect pDNA against degradation. In addition, the RGD peptide could significantly promote the cellular uptake of a nanocomplex. Intracellular trafficking and in vitro expression study indicated that the DHP nanocomplex escaped from lysosomes and the disulfide bonds between PAMAM and PEG cleaved due to the high concentration of GSH in the cytoplasm, pDNA consequently became exclusively located in the nucleus under the guidance of HMGB1, thereby promoting the red fluorescence protein (RFP) expression. Importantly, an in vivo antitumor activity study demonstrated that the DHP nanocomplex had higher antitumor activity than any other reference preparation.
Conclusion: All these results confirm that DHP could be a new strategy for improving the transfection and expression efficiency in gene delivery.
Keywords: PAMAM dendrimers, disulfide bond, RGD, HMGB1, gene delivery
Introduction
A series of viral and nonviral vectors have been used for gene delivery in the past.1,2 In contrast to the viral vectors, nonviral carriers such as cationic polymers are promising alternatives, on account of their high safety and low immunogenicity. Polyamidoamine (PAMAM) dendrimers, as a representative cationic polymer, have frequently been investigated in the development of targeted gene delivery systems.3–5 PAMAM dendrimers can readily condense nucleic acids and have the ability to rapidly escape from endosomes by a “proton sponges” mechanism, resulting in efficient transfection and expression.6,7 Unfortunately, it has been demonstrated that cationic polymer/DNA complexes have a significantly short blood circulation time due to the reticuloendothelial system (RES), which brings about a lower targeting effect on tumors.8 To solve this problem, polyethylene glycol (PEG) has been used for the surface modification of PAMAM to protect the nanocarriers from being recognized by RES, thereby increasing their circulation time in the blood system and consequently enhancing the tumor accumulation via the enhanced permeability and retention effect (EPR).9–11 However, modification of PAMAM with PEG using covalent linkage strongly inhibits the interaction of the gene vector with the tumor cell surface. As a result, cellular uptake sharply decreases. Moreover, PEGylation also leads to a significantly lessened expression efficiency.12 Therefore, reduced cellular uptake and decreased expression efficiency are two main obstacles associated with the use of PEG, and how to conquer these barriers becomes a burning question in gene delivery.
To solve this matter, we are supposed to solve the predicament in low transfection efficiency first. A reduction-sensitivity strategy by coalescent disulfide bond between PAMAM dendrimers and PEG chains could be expected to realize the cleavage of PEG from the carrier system, thereby promoting the transfection. It is widely known that the concentration of reductive glutathione (GSH) in blood is only about 10 μM, which is far from enough to cleave the disulfide bond.13–15 However, the cleavage reaction happens instantly due to the high concentration (10 mM) of GSH in the cytoplasm. In another word, nanoparticles are stable in the blood circulation and, after being internalized by tumor cells, PEG chains will be cleaved from PAMAM in the cytoplasm.16–18 Therefore, this huge concentration difference makes it possible to solve the reduced expression efficiency, which was caused by modification of PAMAM with PEG. As far as we know, the transfection of exogenous DNA depends on three main steps: uptake by the cells, escape from the endosomes, and the nucleus import.19 There is no doubt that the nucleus import is a main restricted process, since the plasmid DNA is too large to be passively transported into the nucleus, and considerable efforts have been devoted to improving nuclear importation by using nucleoproteins. Using proteins with a nuclear localization signal (NLS), such as high mobility group box 1 (HMGB1), has been developed as a valid strategy to improve pDNA nuclear importation.20,21 HMGB1 consists of high mobility box A, box B, and C-terminal acidic regions. Positively charged residues of lysine and arginine in box A and box B regions can bind to pDNA via electrostatic interactions.22 HMGB1 is also known to have NLS, which can interact with the nucleus transport system and consequently promote the nucleus uptake and transfection of plasmid DNA.23
In order to further enhance the transfection efficiency, we use an arginine-glycine-aspartate (RGD)-modified active targeting strategy to promote the cellular uptake rate. As we all know, nanoparticles modified using active tumor targeting ligands specific to receptors overexpressed in cancer cells can further promote the accumulation of nanocarriers.24,25 The high-affinity interactions between RGD peptide and integrin ανβ3 have caught widespread attention to cancer targeting therapies.26,27 As reported previously, PAMAM linked with RGD peptide could enhance the active targeting of tumor cells.28,29 The ανβ3 receptor is known to be highly expressed on tumor cells, but rarely appears on normal blood vessels.30 Therefore, the highly selective expression of ανβ3 integrin on tumors can be regarded as an appropriate strategy to achieve active targeting and increase cellular uptake.
In this paper, we constructed an effective disulfide bond-mediated cleavable RGD modified gene delivery system (Figure 1). PAMAM-SS-PEG-RGD (PSSP-RGD) conjugate was synthesized and characterized by proton nuclear magnetic resonance spectroscopy (1H-NMR) and dynamic light scattering (DLS). The cleavage potential of disulfide bond was evaluated by monitoring change of the zeta potential and particle size in response to different concentration of GSH. Afterwards, PSSP-RGD conjugate was interacted with pDNA and HMGB1 to form a pDNA/HMGB1/PSSP-RGD (DHP) nanocomplex via electrostatic interaction (Figure 2). The important parameters of DHP, including size, morphology, DNA condensation, and protection efficiency, were assessed, respectively. The effect of the introduction of RGD ligands on cellular uptake as well as in vitro transfection and expression was also investigated. Furthermore, in vivo antitumor activity was carried out to evaluate the antitumor effect of the formulation.
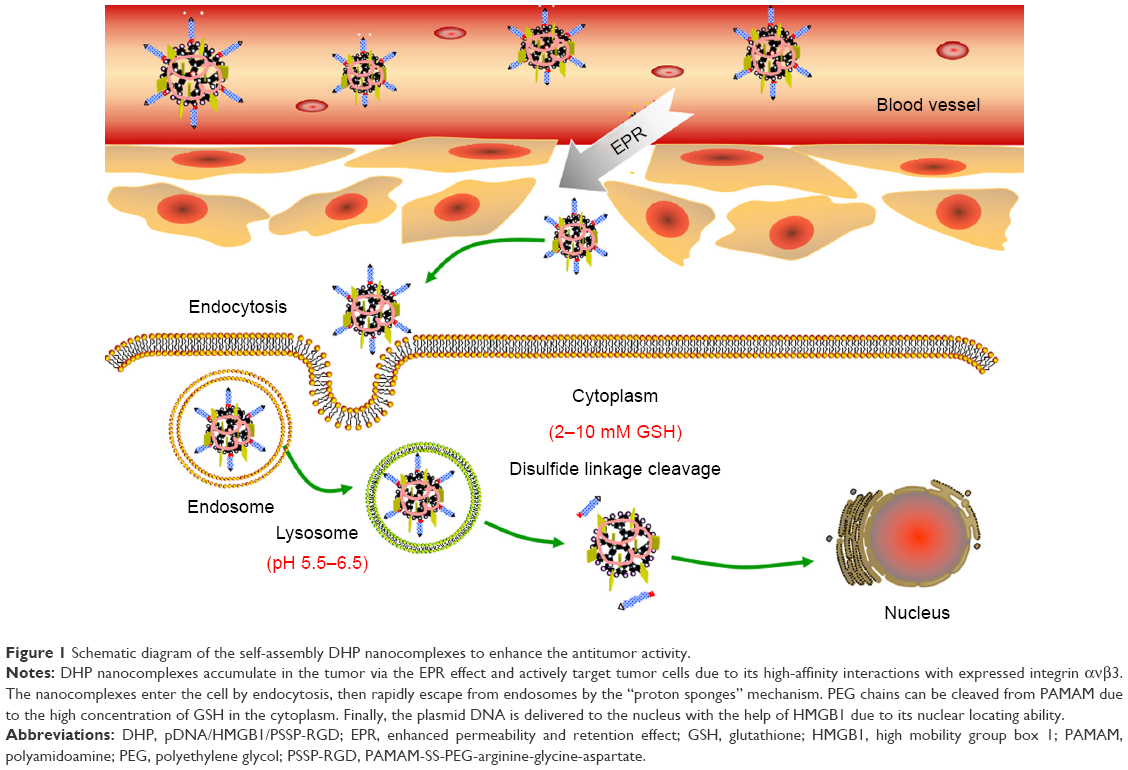 | Figure 1 Schematic diagram of the self-assembly DHP nanocomplexes to enhance the antitumor activity. |
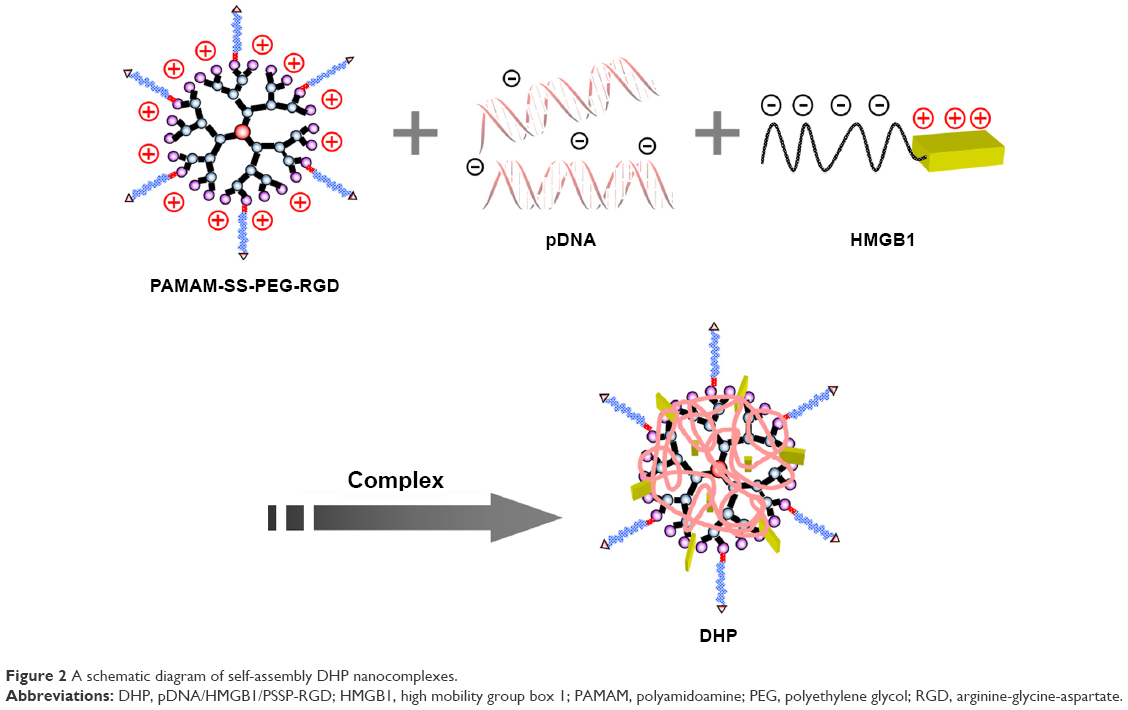 | Figure 2 A schematic diagram of self-assembly DHP nanocomplexes. |
Materials and methods
Materials and animals
PAMAM (G4.0), a high mobility group box 1 (HMGB1), fluorescein (FITC), and tetrazolium bromide (MTT) were obtained from Sigma-Aldrich (St Louis, MO). FBS and DMEM were purchased from Gibco (Waltham, MA, USA). cRGDyK was obtained from Shanghai Top-peptide Bio Co, Ltd (Shanghai, China). Agarose and ethidium bromide (EB) were obtained from Biowest (Nuaillé, France) and Gibco, respectively. Hoechst 33,258 and Lyso-Tracker Red were obtained from the Beyotime Institute of Biotechnology (Haimen, Jiangsu, China). N-succinimidyl 3-(2-pyridyldithio)-propionate (SPDP) and GSH were purchased from J&K Chemical Ltd. (Beijing, China). HS-PEG5000-COOH was obtained from Ponsure Biotech, Inc (Shanghai, China). Plasmid pDsRed-M-N1 and pGL3-control were a gift from Tsinghua University. pecmv-3× flag-trp53-m was purchased from miaoling.bio (Beijing, China). A549 and MCF-7 cells were obtained from the cell bank of the Chinese Academy of Sciences (Shanghai, China). All other buffer solution components and chemicals such as hydrochloric acid (HCL) and sodium hydroxide (NaOH) were commercially available reagents of analytical grade.
Male Kunming mice (18–22 g) obtained from the Department of Experimental Animals, Shenyang Pharmaceutical University (Shenyang, China), were fed at 25°C and 55% humidity under natural light/dark conditions. All animal experiments were performed in accordance with guidelines evaluated and approved by the ethics committee of Shenyang Pharmaceutical University. The ethics committee of Shenyang Pharmaceutical University approved the experiments.
Synthesis of PSSP-RGD
Synthesis of PSSP
The synthetic process of PSSP-RGD conjugates is shown in Figure 3. In brief, PAMAM (40 mg, 2.8 μmol) and SPDP (7.0 mg, 22.4 μmol) were accurately weighed, respectively, and dissolved in 4 mL of methanol solution containing proper triethylamine and placed in a flask with N2 atmosphere protection. The mixture was stirred at room temperature for 6 hours. Subsequently, HS-PEG5000-COOH (112.5 mg, 22.4 μmol) was added into the solution to continue reaction for 24 hours. After the reaction, duct PAMAM-SS-PEG (PSSP16) was obtained.
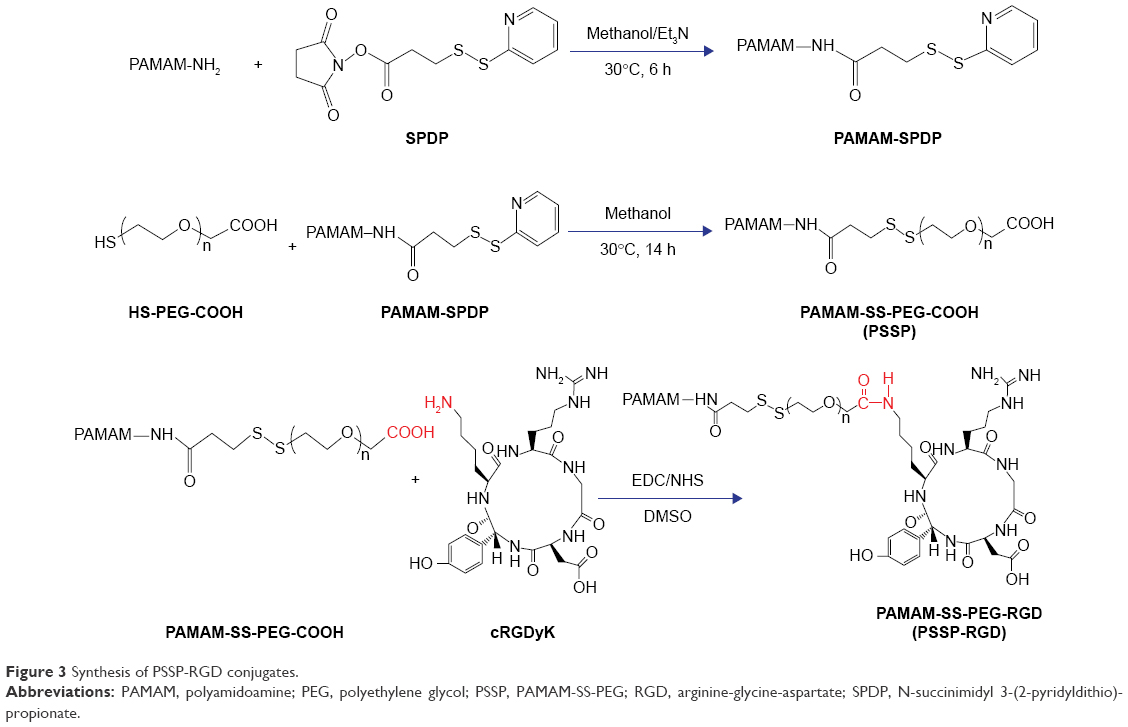 | Figure 3 Synthesis of PSSP-RGD conjugates. |
Synthesis of PSSP-RGD
PSSP16 (40.0 mg) was accurately weighed and dissolved in 4 mL of DMSO solution in a flask. EDC (6.8 mg, 35 μmol) and NHS (4.0 mg, 35 μmol) were added to activate the carboxyl for 6 hours. Afterwards, cRGDyK was added with the molar ratio (PSSP16:RGD) of 1:4, 1:8, and 1:16, respectively. It reacted in a thermostat water bath at 30°C for 24 hours with N2 atmosphere protection. After the reaction, the mixture was put into a dialysis membrane (MWCO: 3,500 Da) and dialyzed against deionized water for 2 days, followed by lyophilization. The products PSSP-RGD4, PSSP-RGD8, and PSSP-RGD16 were obtained, respectively.
Reduction-sensitive study
To investigate the reduction-sensitivity of vector, various concentrations of GSH solution were used to simulate the difference of GSH concentration between normal tissue and tumor cells. PSSP-RGD4, PSSP-RGD8, and PSSP-RGD16 conjugates were dissolved in 10 mM and 10 μM GSH solution separately. We assessed the stability of conjugates by monitoring change of the particle size and zeta potential. The zeta potential and particle size of each sample were measured after incubation at 37°C for 2 hours by DLS instrument (Zetasizer Nano ZS, Malvern Instruments Ltd, Malvern, UK).
Hemolytic test and in vitro stability assay
The hemolytic effect of the vector was evaluated using New Zealand rabbit erythrocytes. In brief, the plasma samples were centrifuged and diluted with normal saline to separate the red blood cells (RBCs 2%). Afterwards, 2 mL of RBCs was added to each sample. As a comparison, 2 mL of normal saline (negative control) and 2 mL of distilled water (positive control) were added. PAMAM, PSSP, and PSSP-RGD16 (0.5 mL, respectively) at various concentrations (100, 200, 400, 600, 800, and 1,000 μg/mL) were incubated with a 2 mL RBC suspension, and proper normal saline was added to maintain the same volume. All samples were kept at 37°C for 4 hours and then centrifuged at 5,000 rpm for 10 minutes. The absorbance of the supernatant was measured using a UV spectrophotometer at 570 nm. The hemolysis percentage of RBCs was determined using the following equation:
|
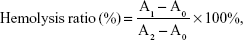
where A0, A1, and A2 represent the absorbance of negative control, samples, and positive control, respectively.
The stability of PSSP-RGD16 in 10% FBS was determined by DLS. Briefly, a proper amount of PSSP-RGD16 conjugate was weighed accurately and dissolved in PBS solution containing 10% FBS (pH 7.4). The zeta potential and particle size of each sample were measured by DLS after being incubated at 37°C for 24 hours, respectively.
Preparation of the DHP nanocomplexes
DHP nanocomplexes were prepared with pDNA, HMGB1, and PSSP-RGD conjugate by electrostatic interaction. The final concentration of pDNA was 1 mg/mL. In brief, HMGB1 was mixed with pDNA (weight ratio was 1:1), and then an appropriate amount of PSSP-RGD dissolved in the deionized water was added. The nanocomplex was consequently formed after incubation for 30 minutes at room temperature. pDNA/PSSP-RGD16 (DP16), pDNA/HMGB1/PSSP-RGD16 (DHP16), pDNA/HMGB1/PSSP-RGD8 (DHP8), and pDNA/HMGB1/PSSP-RGD4 (DHP4) nanocomplexes at a series of W/W ratios (weight ratio of vectors to pDNA) were prepared separately. pDNA/HMGB1/PAMAM-PEG (DHPP, PAMAM-PEG conjugate was synthesized before), pDNA/HMGB1/PSSP (DHPSSP), and pDNA/HMGB1/PSSP(DHPAMAM) nanocomplexes were also prepared as reference formulations.
Hoechst 33,258 intercalation assay
The DNA condensation with PSSP-RGD and HMGB1 was evaluated using the Hoechst 33,258 intercalation assay. In brief, Hoechst 33,258 was dissolved in a Tris-NACL-EDT buffer (0.2 μg/mL). DHP16 nanocomplexes at different W/W ratios were mixed with 100 μL of the Hoechst 33,258 solution and incubated for 5 minutes at 37°C. The fluorescence intensity of each sample was determined at 353 nm (ex) and 457 nm (em). The fluorescence intensity of free DNA was used as a control. Encapsulation efficiency was obtained according to the following equation:
|
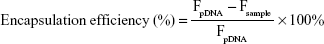
where Fsample and FpDNA represent the fluorescence intensity of DHP16 nanocomplexes and the fluorescence intensity of Hoechst 33,258 bound to free pDNA, respectively.
Gel electrophoresis
pDNA condensation
DHPP, DHPSSP, DP16, and DHP16 nanocomplexes at different W/W ratios ranging from 0 to 30 were loaded onto a 0.7% agarose gel and electrophoresed separately. The gel was dyed with EB (10 μg/mL) and photographed by a Tanon 2500R image system.
Serum stability
To evaluate the ability of nanocomplexes to resist anion replacement, the serum stability study was investigated. DHP16 nanocomplexes at different W/W ratios were prepared and cultured with FBS (v:v=1:1) at 37°C for 24 hours. Subsequently, nanocomplexes were incubated under acidic conditions (pH 5.5) for 30 minutes and evaluated via gel electrophoresis in a TBE buffer using 1% agarose gel.
Particle size, zeta potential, and transmission electron microscopy (TEM)
The particle size and zeta potential of nanocomplexes were examined by DLS, and the morphology of DHP16 nanocomplexes was visually assessed via TEM. In brief, the nanocomplexes were suspended in PBS solution and then placed on the copper grid. The excess liquid was blotted out and the grids were allowed to dry overnight at room temperature. Prepared samples were imaged using an FEI TecnaiG220 TEM (Gatanmodel 794 CCD, bottom mounted).
In vitro cytotoxicity
A549 cells were cultured in DMEM supplemented with 10% FBS and 1% penicillin/streptomycin at 37°C in a humidified atmosphere containing CO2 (5%, v/v). The cells were seeded in 96-well plates (1×104 cells/well) and subsequently cultured overnight. Cells were treated with: 1) DHP16 nanocomplexes at different W/W ratios; 2) PAMAM, PSSP, and PSSP-RGD conjugates at various concentrations; and (3) DP16, DHPP, DHPSSP, DHP16, DHP8, and DHP4 nanocomplexes at a W/W ratio of 8. When cells were incubated for 24, 48, and 72 hours, 10 μL of fresh MTT (5 mg/mL) was added to each well and further incubated for another 4 hours, and then 150 μL of DMSO was added to dissolve the formazan crystals formed in the live cell. The absorbance was measured at 490 nm. Cells without treatment served as the control group, and the results were expressed as a percentage viability of control cells.
Cell uptake study
To investigate the effect of the introduction of RGD ligands on cellular uptake, nanocomplexes were labeled with FITC. Briefly, FITC in anhydrous acetone was added into DHP solution and stirred for 24 hours without light. Excess FITC was removed by dialysis using a dialysis membrane (MWCO 3.5 kDa).
A549 and MCF-7 cells were seeded on glass cover slips in 6-well plates (4×105 cells/well). After growing to about 80% confluence, the cells were washed twice with PBS and then different formulations of FITC labeled nanocomplexes were added to each well. After incubation for 2 hours, the cells were carefully washed with cold PBS, trypsinized, and resuspended in the fresh medium. Subsequently, the cells were measured by flow cytometry using a BD FACS Calibur flow cytometer (BD Bioscience, Bedford, MA).
Intracellular trafficking study
The intracellular distribution of FITC labeled DHP16 nanocomplexes was evaluated using a laser scan confocal microscope (LSCM, Olympus, Japan). A549 cells were seeded on glass cover slips in 6-well plates (1×105 cells/well) and cultured in DMEM medium overnight. The medium was replaced with 2 mL fresh serum-free medium and cells were treated with FITC labeled DHP16 nanocomplexes (10 μg/mL). At different time points, cells were treated with 50 nM Lyso-tracker red for 30 minutes and rinsed three times with PBS again before soaking in 4% paraformaldehyde. After that, the cells were rinsed three times again with PBS in advance of staining with Hoechst 33,258 (10 μg/mL). Confocal images of cells were obtained by LSCM.
In vitro expression study
Fluorescence intensity of RFP by LSCM
The transfection efficiency of the DHP nanocomplexes at different W/W ratios and different nanocomplexes at a W/W ratio of 8 were evaluated in A549 cells separately. The cells were seeded in 6-well plates (3×105 cells per well) and cultured overnight. The medium was replaced with 2 mL fresh serum-free medium. Subsequently, the cells were incubated with 100 μL DHP nanocomplexes containing 500 ng pDsRed-M-N1 plasmid for 4 hours. After incubation, cells were washed twice with PBS and treated with Hoechst 33,258 (10 μg/mL) at 37°C for 30 minutes, followed by rinsing with PBS (pH 7.4) and soaking in 4% paraformaldehyde for another 30 minutes. The transfection and expression of red fluorescence protein was evaluated by LSCM.
Luciferase activity assay
The cells were seeded in 6-well plates (3×105 cells/well) and cultured overnight. The medium was subsequently replaced with 2 mL fresh serum-free medium and cells were treated with 100 μL nanocomplex of different formulations containing 500 ng pGL3-control pDNA. After a 4 hours transfection, the serum-free medium was replaced with fresh complete medium and the cells were incubated at 37°C for an additional 24 hours. Subsequently, cells were rinsed twice with PBS. The luciferase activity was measured using a Luciferase Assay System (E1500, Promega, WI) and an Infinite 200 Pro luminometer (Tecan, Switzerland).
In vivo antitumor activity studies
p53 gene is one of the most important tumor suppressor genes, which acts as a sensor of DNA damage and can induce cell cycle arrest and subsequent DNA repair, senescence, or apoptosis, depending on the damage and cellular environment.31 Herein, we use the p53 gene to evaluate the in vivo antitumor activity. In brief, S180 cells were obtained and diluted to 5×106 cells/mL with normal saline. Subsequently, S180 cells (0.2 mL) were carefully injected subcutaneously into the right limb armpit of the mice. When the tumor size reached approximately 100 mm3, the mice were randomly separated into six groups (n=6), as follows: saline, free DNA, DHPP, DHPSSP, DHP4, and DHP16. Tumor volume was calculated by the following formula:
|
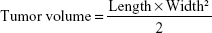
The experimental groups were injected through the tail vein every 2 days for 14 days, the dosage of p53 gene was 0.3 mg/kg. The body weight and tumor size of the mice were assessed every 2 days. Animal euthanization was carried out on the 14th day, and the tumors were excised and weighed. The inhibition rate (IR%) was calculated using the formula:
|
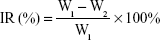
where W1 represents the tumor weight of the saline group, and W2 represents the treatment group. All the animal experiments were performed in accordance with the Experimental Animal Administrative Committee of Shenyang Pharmaceutical University.
Statistics
All experiments were carried out at least three times. Quantitative data are presented as the mean±SD. The statistical significance of data was analyzed using the Student’s test, with P<0.05 as the minimal level of significance.
Results and discussion
Synthesis and characterization of PSSP-RGD conjugates
PSSP and PSSP-RGD conjugates were characterized by 1H-NMR. In the 1H-NMR spectrum of PSSP (Figure 4A), signals in accordance with both PAMAM (δ=2.47, 2.61, 2.88, and 3.25 ppm) and PEG (δ=3.34 and 3.63 ppm) were detectable, indicating that PEG chains were successfully conjugated to PAMAM dendrimers. Meanwhile, in the 1H-NMR spectrum of PSSP-RGD (Figure 4B), on the basis of both PAMAM and PEG characteristic peaks, the proton peaks at 6.76 ppm and 7.05 ppm were attributable to the aromatic ring of cRGDyK. These results above suggested that the PSSP-RGD conjugates were successfully synthesized. Additionally, the conjugated number of cRGDyK peptide was calculated by the proton integration method and was assessed to be 2.8, 5.3, and 10.1, when the molar ratio of cRGDyK and PSSP was 4:1, 8:1, and 16:1, respectively. In addition, the particle size and zeta potential of PSSP-RGD conjugates were investigated, respectively. Table 1 showed that the particle size and zeta potential all increased slightly as the conjugated number of RGD increased. The conjugate has good size distribution (Figure 4C).
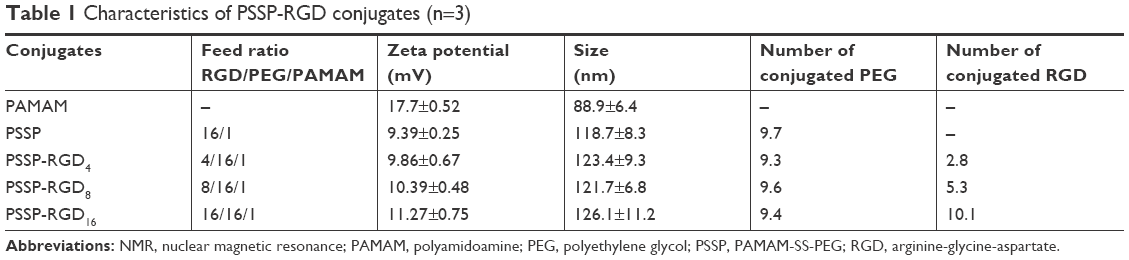 | Table 1 Characteristics of PSSP-RGD conjugates (n=3) |
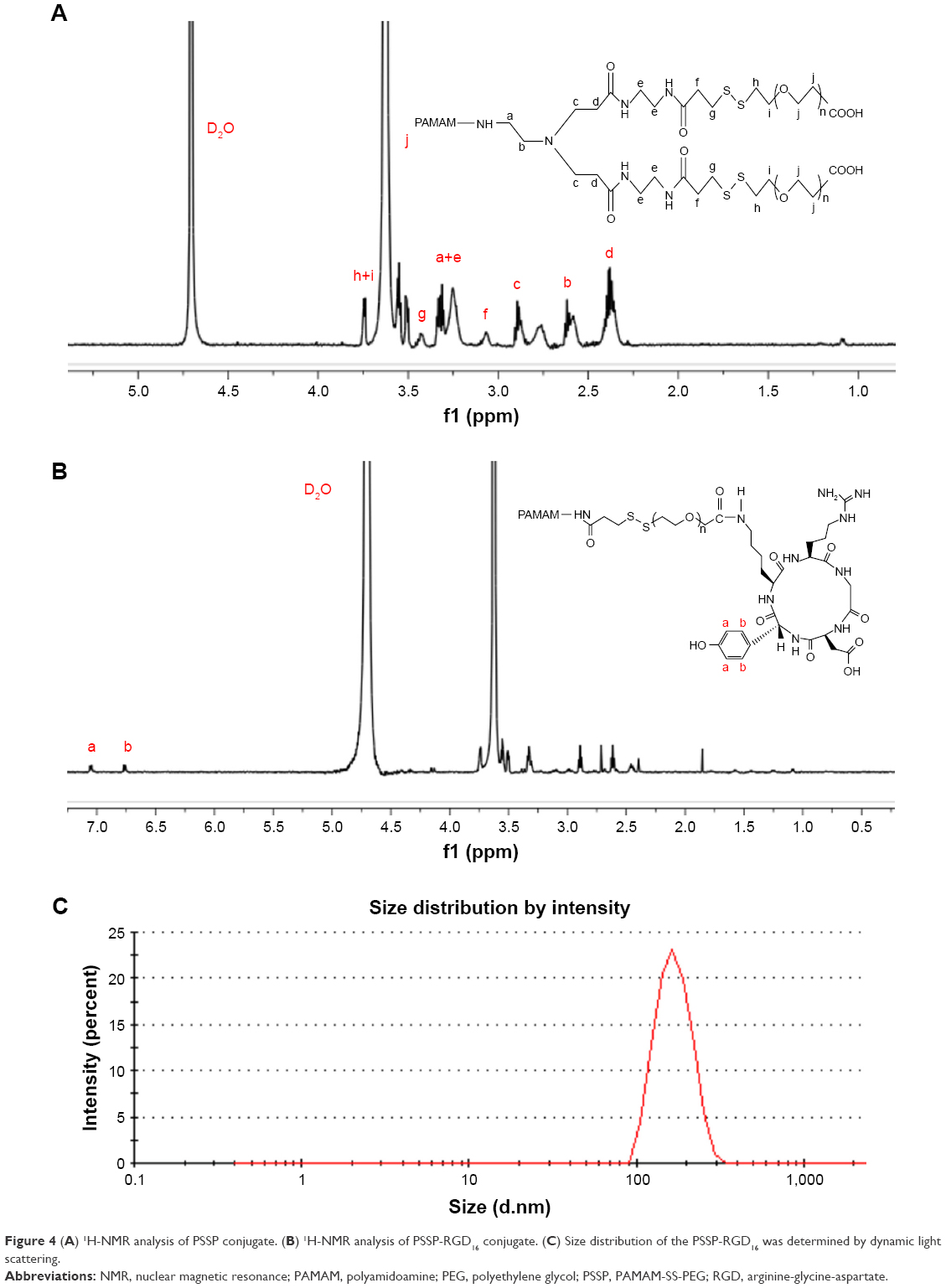 | Figure 4 (A) 1H-NMR analysis of PSSP conjugate. (B) 1H-NMR analysis of PSSP-RGD16 conjugate. (C) Size distribution of the PSSP-RGD16 was determined by dynamic light scattering. |
Moreover, the serum stability assay and hemolytic test were carried out separately. Since it has been reported that nanoparticles with high polymer content may bring about gelation or coagulation in vivo,32 it was necessary to measure the stability of conjugates. After incubation for 24 hours, there was no significant change in particle size (Figure 5A) or zeta potential (Figure 5B), indicating that PSSP-RGD16 conjugates were relatively stable in serum. On the other hand, the hemolytic test result is shown in Figure 5C, the hemolysis rate of PAMAM solution gradually enhanced when the concentration increased from 100 to 1,000 μg/mL, suggesting that it had strong hemolytic toxicity. Nevertheless, after PEGylation, the hemolytic toxicity of PSSP and PSSP-RGD16 were significantly reduced. In addition, as far as we know, the high buffering capacity of nonviral vectors plays an important role in the transfection because they could facilitate the endosomal escape of complexes into the cytoplasm. So, we also investigated the buffering capacity of the conjugates using an acid-base titration test, and the results showed that PSSP-RGD conjugates exhibited good buffering capacity (Figure S1).
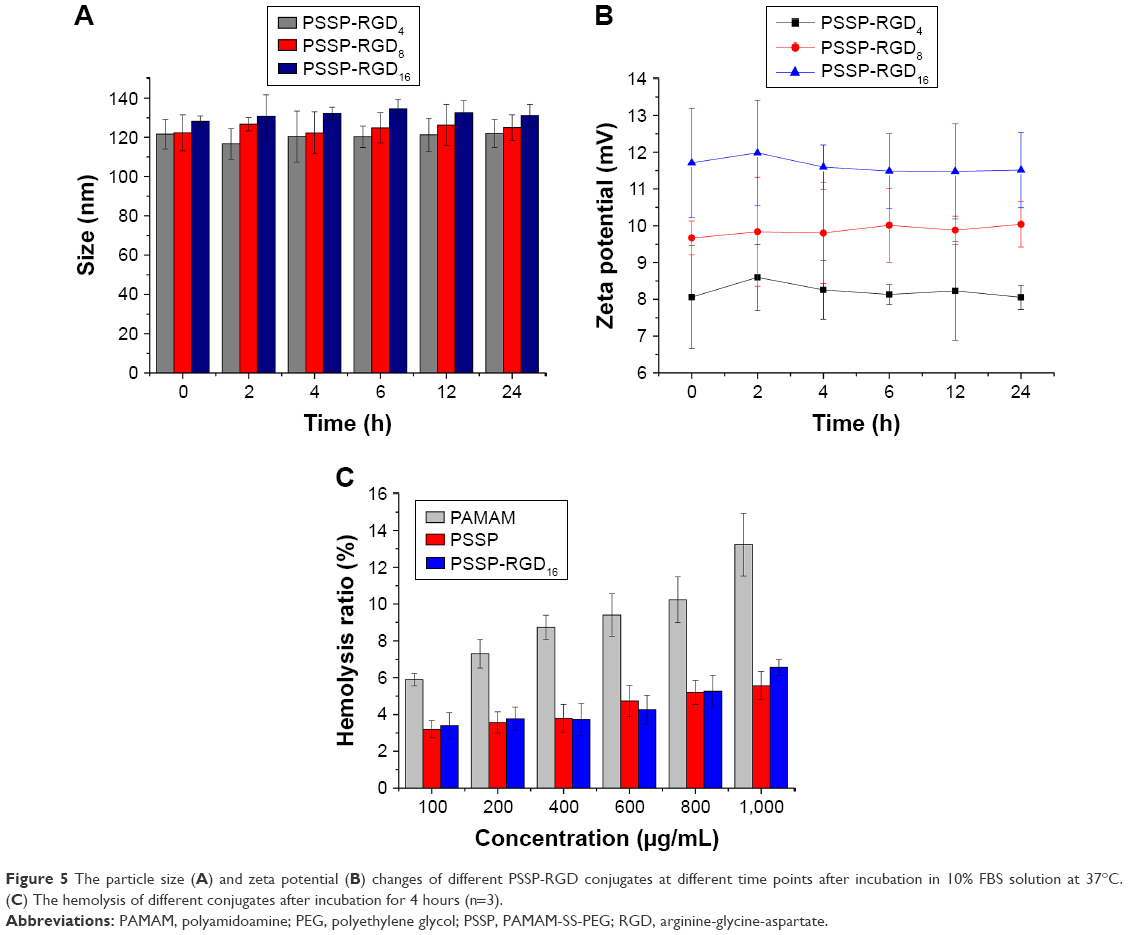 | Figure 5 The particle size (A) and zeta potential (B) changes of different PSSP-RGD conjugates at different time points after incubation in 10% FBS solution at 37°C. (C) The hemolysis of different conjugates after incubation for 4 hours (n=3). |
Furthermore, to investigate the reductive-sensitivity of PSSP-RGD conjugates, various concentrations of GSH solution were used to simulate the different concentration of GSH between normal tissue and tumor cells. We assessed the stability of conjugates by monitoring change of the particle size and zeta potential. As exhibited in Figure 6A, the zeta potential and particle size of PSSP-RGD16 conjugates did not change significantly in 120 minutes under the condition of 10 mM GSH, indicating that the conjugates could be stable in the blood circulation. However, when GSH was 10 μM, the particle size of PSSP-RGD16 decreased from 130.9±6.54 nm to 80.2±7.35 nm, and the zeta potential increased from 10.63±0.45 mV to 13.58±0.27 mV (Figure 6B). This was probably because the disulfide bond between PAMAM dendrimers and PEG chains broke down at high concentrations of GSH in tumor cells, and the PEG chains detached from the PAMAM surface, resulting in decreased particle size. At the same time, part of the positive charges on PAMAM surface masked by the PEG chains were re-exposed, leading to increased zeta potential.33,34 These results suggest that the PSSP-RGD conjugate has an obvious reductive sensitivity, it can be stable at normal physiological environment, and, after being internalized by tumor cells, PEG chains will be cleaved from PAMAM in the cytoplasm due to the high concentration of GSH.
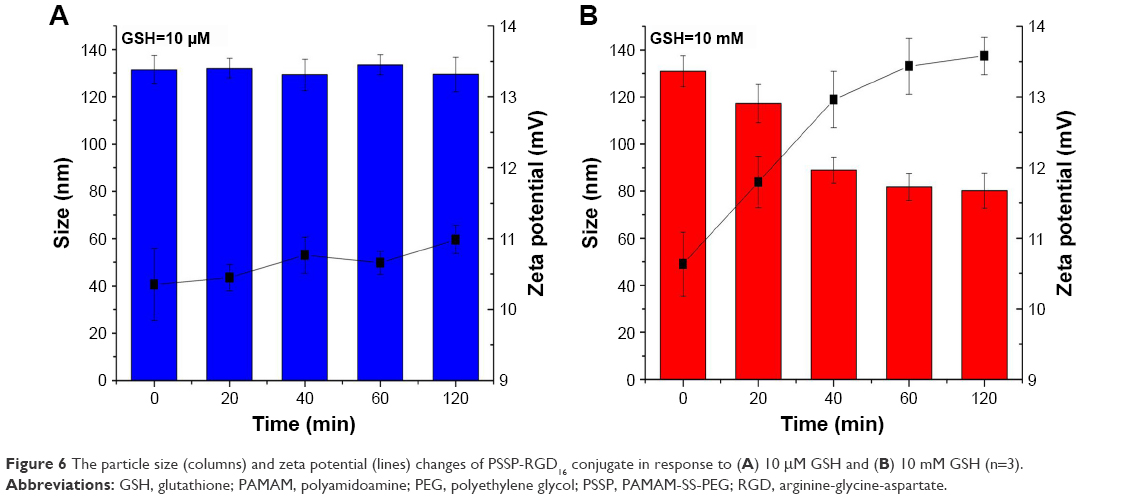 | Figure 6 The particle size (columns) and zeta potential (lines) changes of PSSP-RGD16 conjugate in response to (A) 10 μM GSH and (B) 10 mM GSH (n=3). |
From the results above we can conclude that PSSP-RGD conjugate was successfully synthesized. It has good serum stability and low hemolytic toxicity, as well as reduction sensitivity, laying a solid foundation for building a complete gene delivery system.
DNA condensation analysis
As shown in Figure 1, DHP nanocomplexes were obtained with plasmid DNA, HMGB1, and PSSP-RGD by electrostatic interaction. The pDNA encapsulation efficiency at different W/W ratios was investigated using the gel retardation assay and Hoechst 33,258 intercalation. As manifested in Figure 7A, the encapsulation efficiency of DHP16 was W/W ratio dependent and showed an increased tendency as the W/W ratio increased. When the W/W ratio was 8, the encapsulation efficiency of DHPP, DHPSSP, DP16, and DHP16 were all above 90%, and there was no obvious difference among them (Figure 7B), suggesting that the introduction of HMGB1 and RGD peptides had no significant influence on the compression result. To further identify the encapsulation efficiency, agarose gel electrophoresis was also carried out. As shown in Figure 7C, when the W/W ratio was above 8, there was no significant fluorescence among DHPP, DHPSSP, DP16, and DHP16, indicating that pDNA was completely compressed, which was in keeping with the Hoechst 33,258 intercalation analysis.
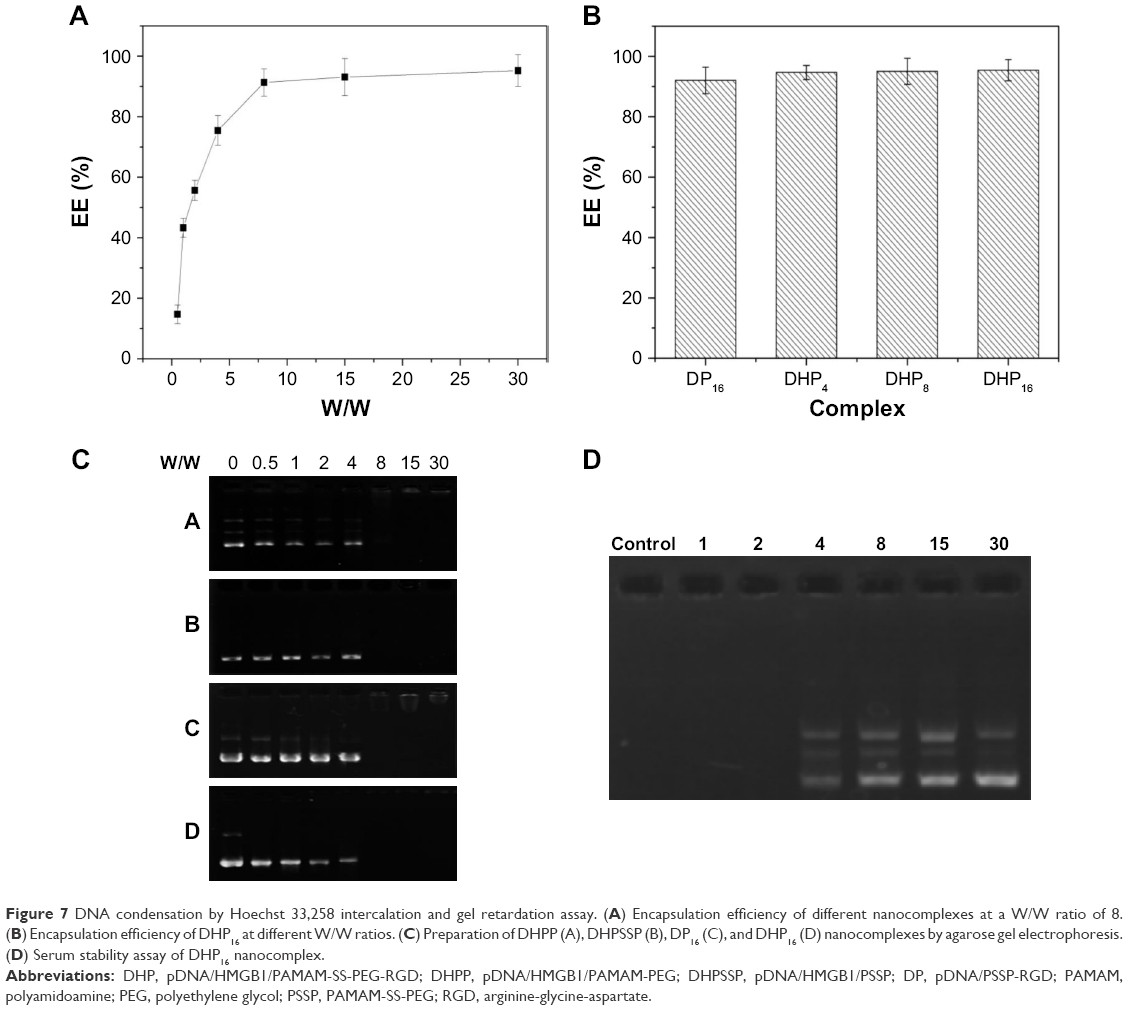 | Figure 7 DNA condensation by Hoechst 33,258 intercalation and gel retardation assay. (A) Encapsulation efficiency of different nanocomplexes at a W/W ratio of 8. (B) Encapsulation efficiency of DHP16 at different W/W ratios. (C) Preparation of DHPP (A), DHPSSP (B), DP16 (C), and DHP16 (D) nanocomplexes by agarose gel electrophoresis. (D) Serum stability assay of DHP16 nanocomplex. |
Stability analysis
Serum stability of pDNA could forecast the stability of the nanoparticles under physiological environment,35 and it was an important element for gene vector. DHP16 nanocomplexes at different W/W ratios were analyzed using gel electrophoresis after being incubated with 50% FBS at 37°C for 24 hours.36 As shown in Figure 7D, when the W/W ratio was below 2, there were no distinct DNA bands, indicating that pDNA had been degraded after serum treatment. However, pDNA encapsulated in the nanocomplexes at the W/W ratios ranging from 4 to 30 remained stable after incubation with serum, which suggested that PSSP-RGD conjugates could completely condense pDNA and were able to protect pDNA against degradation to some extent.
Size and zeta potential of the nanocomplex
Particle size and size distribution are crucial parameters for the development of a suitable nanocomplex, and they could affect the in vivo distribution and targeting ability of the nanocomplex system.37–39 On the other hand, the zeta potential of a nanocomplex is one of the most vital factors when the nanomedicine is for intravenous administration.40 Therefore, the change of particle size and zeta potential at different W/W ratios were investigated. As shown in Figure 8A, when the W/W ratio of nanocomplexes ranged from 0.5 to 30, the zeta potential increased from −2.4±0.75 mV to 19.0±1.5 mV, whereas the particle size decreased from 179.4±7.89 nm to 67.4±2.54 nm. This may be the reason that the ratio of cationic polymer once increased, and the compression effect of nanocomplex on pDNA enhanced accordingly. This change could make the structure of nanocomplex more tight, so that the particle size consequently decreased.
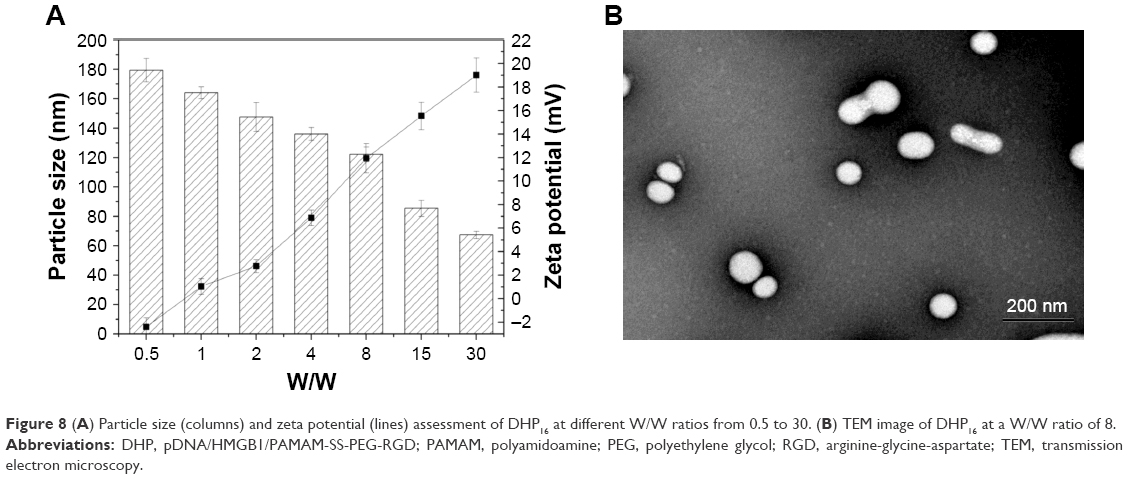 | Figure 8 (A) Particle size (columns) and zeta potential (lines) assessment of DHP16 at different W/W ratios from 0.5 to 30. (B) TEM image of DHP16 at a W/W ratio of 8. |
On the other hand, the morphology of the DHP16 was visually observed via TEM. As manifested in Figure 8B, when the W/W ratio was 8, the spherical nanocomplexes were in well-defined shapes. The size of the nanocomplexes was around 100 nm, which was in accordance with the results obtained by DLS. Such an appropriate size can have a positive effect on the in vivo distribution and passive targeting ability of the gene delivery system.
Cytotoxicity assay
In this study, the cytotoxicity of nanocomplexes was evaluated on A549 cells using an MTT assay. As shown in Figure 9B, the cytotoxicity of DHP16 was W/W ratio dependent, it gradually enhanced with the W/W ratio ranging from 0.5 to 30, and, the longer the action time, the stronger the cytotoxicity. When the W/W ratio was 30, the cell growth inhibition rate exceeded 20% after incubation for 72 hours. On the other hand, as shown in Figure 9A, there was no significant difference among DP16, DHP16, and DHPSSP, indicating that the introduction of HMGB1 and RGD peptide had no significant influence on cell viability. In addition, the cell viability of PAMAM, PSSP, and PSSP-RGD16 conjugates were investigated. As shown in Figure 9C, PAMAM exhibited obvious cytotoxicity as the concentration gradually increased, and the cell survival rate reached less than 30% at 800 μg/mL, whereas PSSP and PSSP-RGD were still over 80%. All the results above suggested that the DHP nannocomplex was a safe gene delivery system with low cytotoxicity.
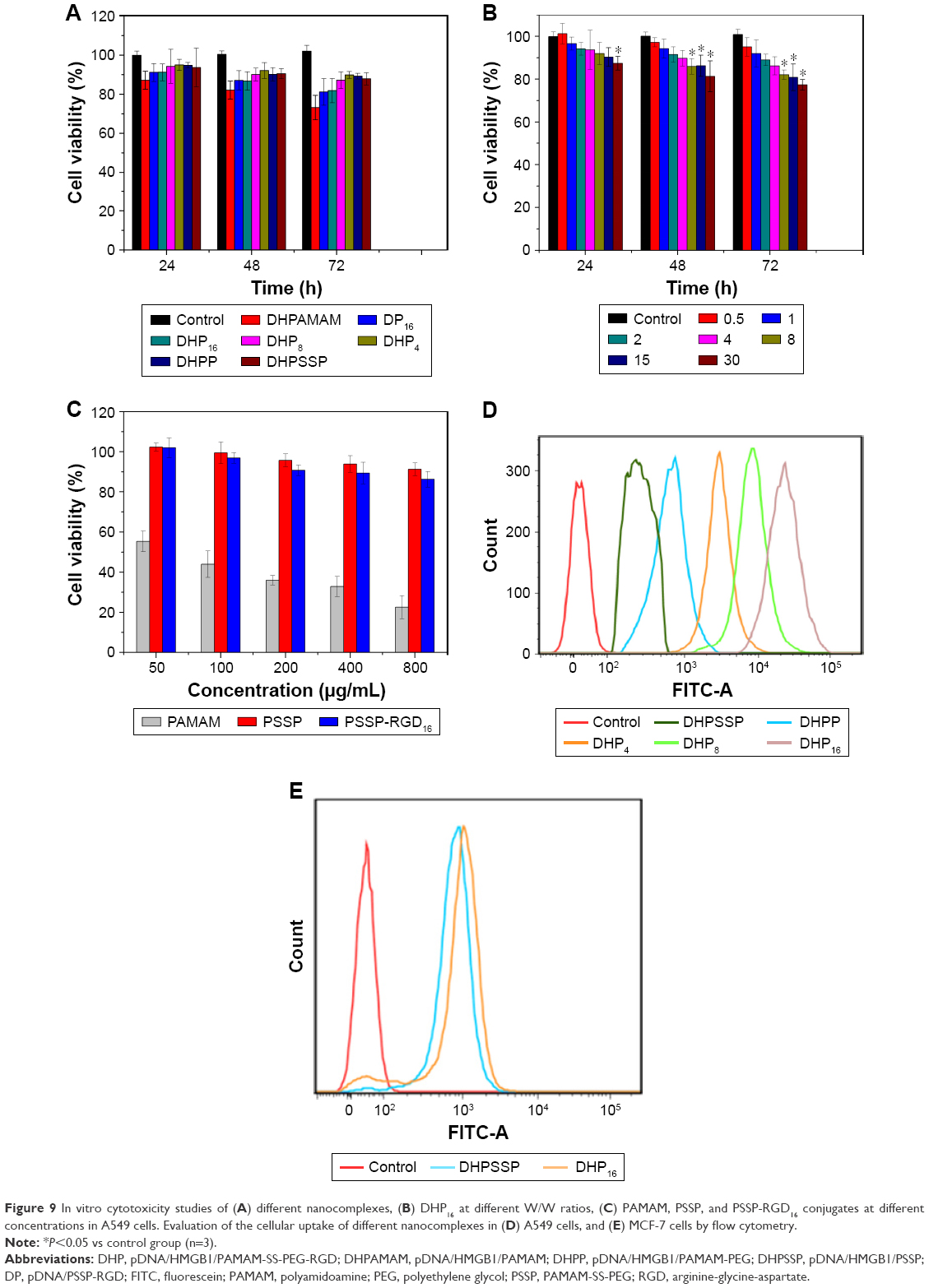 | Figure 9 In vitro cytotoxicity studies of (A) different nanocomplexes, (B) DHP16 at different W/W ratios, (C) PAMAM, PSSP, and PSSP-RGD16 conjugates at different concentrations in A549 cells. Evaluation of the cellular uptake of different nanocomplexes in (D) A549 cells, and (E) MCF-7 cells by flow cytometry. |
In vitro cellular uptake
To study the effect of the introduction of RGD ligands on cellular uptake, A549 cells were incubated with FITC-labeled nanocomplexes. The mean fluorescence intensity of different nanocomplexes was determined by flow cytometry. As shown in Figure 9D, DHP16 showed the strongest fluorescent signal among all DHP nanocomplexes, indicating that cellular uptake enhanced as the number of conjugated RGD ligands increased. The cellular uptake rate of DHPSSP was similar to DHPP, which suggested that the introduction of disulfide bond had no significant effect on cellular uptake. Compared with all DHP nanocomplexes, DHPSSP had the lowest fluorescence intensity. This was probably because that DHP nanocomplex could actively target tumor cells, due to its high-affinity interactions between RGD peptide and highly expressed integrin ανβ3, thereby having better cellular uptake efficiency. As we know, there was almost no expression of integrin receptors on the surface of MCF-7 cells.41 DHPSSP had similar cellular uptake with DHP16 in MCF-7 cells (Figure 9E). All of these results demonstrated that the introduction of RGD peptide could clearly promote the cellular uptake of the nanocomplex.
Intracellular trafficking
To further explore the intracellular distribution of the nanocomplex, DHP16 was labeled with FITC. As shown in Figure 10, the lysosomes and nucleus were labeled with red and blue fluorescence, respectively. As reported previously, nanocomplexes are generally entrapped in endosomes after being internalized, and tend to either recycle their contents back to the cell surface or fuse with the acidic lysosomes later, leading to no access to the cytoplasm or nucleus.42,43 After 30 minutes, part of the nanocomplexes labeled with green fluorescence (FITC) had already entered the cell, the vivid green color was gradually enhanced with the extension of time. At the same time, the orange color that was merged by green color and red color was highly luminescent. The intensity and quantity of orange fluorescence had decreased at 2 hours, and the individual green fluorescence was found. At 4 hours, only a small amount of orange fluorescence was observed and the green fluorescence ratio increased significantly, as well as having a tendency to cover up red fluorescence. Moreover, the green fluorescence was partially overlapped with the blue fluorescence of the nucleus. These results suggested that nanocomplexes were entrapped in lysosomes first, escaped from lysosomes later and consequently transferred to the nucleus. The rapid escape from lysosomes might be largely induced by the “proton sponge” hypothesis of PAMAM; the buffering capacity of PAMAM leads to osmotic swelling and rupture of endosomes, resulting in the release of the vector into the cytoplasm.
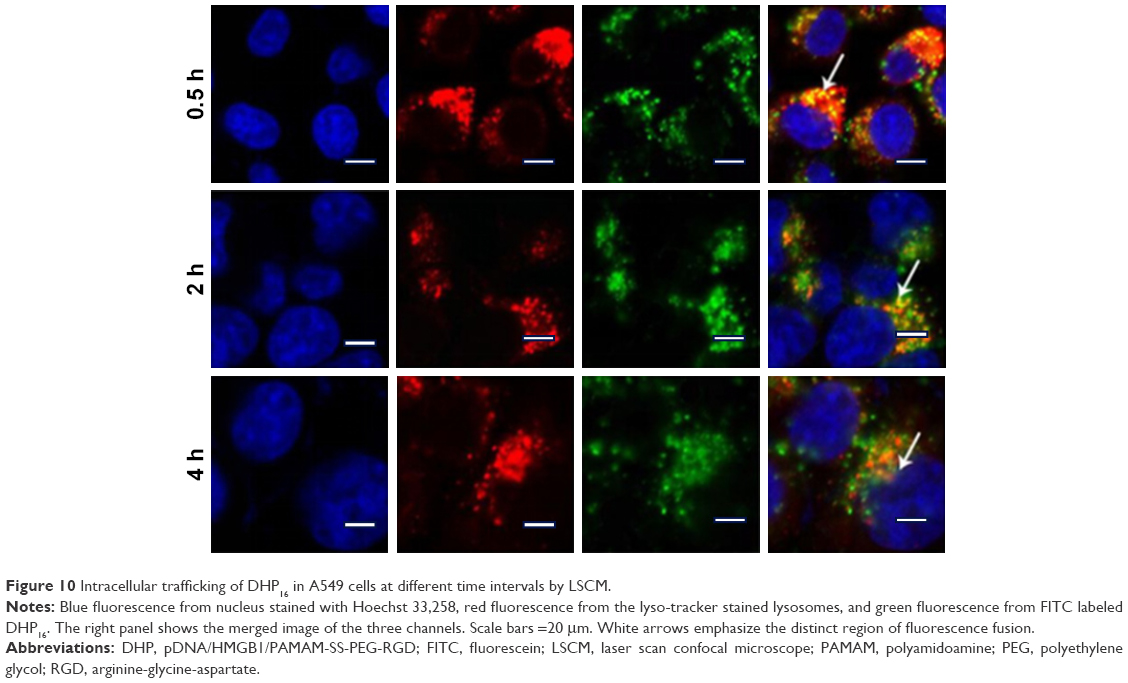 | Figure 10 Intracellular trafficking of DHP16 in A549 cells at different time intervals by LSCM. |
In vitro transfection and expression study
In vitro transfection and expression of RFP was qualitatively analyzed by LSCM. Figure 11A shows the results of the RFP expression after the cells were treated with the DHP16 nanocomplexes at different W/W ratios for 24 hours. The fluorescence intensity of DHP16 nanocomplexes significantly increased as the W/W ratios ranged from 0 to 30, and it was clear that the fluorescence intensity of the free pDNA was invisible. As shown in Figure 11B, DHP16 produced a higher expression level than that of DP16. This was probably attributed to the nuclear locating ability of HMGB1. HMGB1, as an NLS, could effectively target the nucleus, resulting in promoted gene expression efficiency.22 Compared with DHPSSP, DHP16 showed a higher fluorescent signal, probably due to its better cellular uptake, thereby gaining enhanced expression. Meanwhile, the expression level of DHPSSP was higher than that of DHPP, which indicated that the introduction of disulfide bond between PAMAM and PEG could overcome the aforementioned limitation of lessened expression efficiency caused by PEG chains to some extent. On the other hand, to further confirm the ability of gene delivery, luciferase activity was measured. As exhibited in Figure 11C and D, the luciferase activity showed similar results with the fluorescence intensity assessed by LSCM. The increase in the fluorescence intensity at a high W/W ratio could be due to the high pDNA encapsulation efficiency. DHP16 expressed more than any other formulation. In addition, there was no significant difference between DHPSSP and DHP16 in MCF-7 cells (Figure 11E), which stressed again the importance of the RGD peptide. These results suggest that DHP16 could significantly enhance the transfection and expression efficiency, thereby giving us great confidence in using DHP16 as an effective gene delivery.
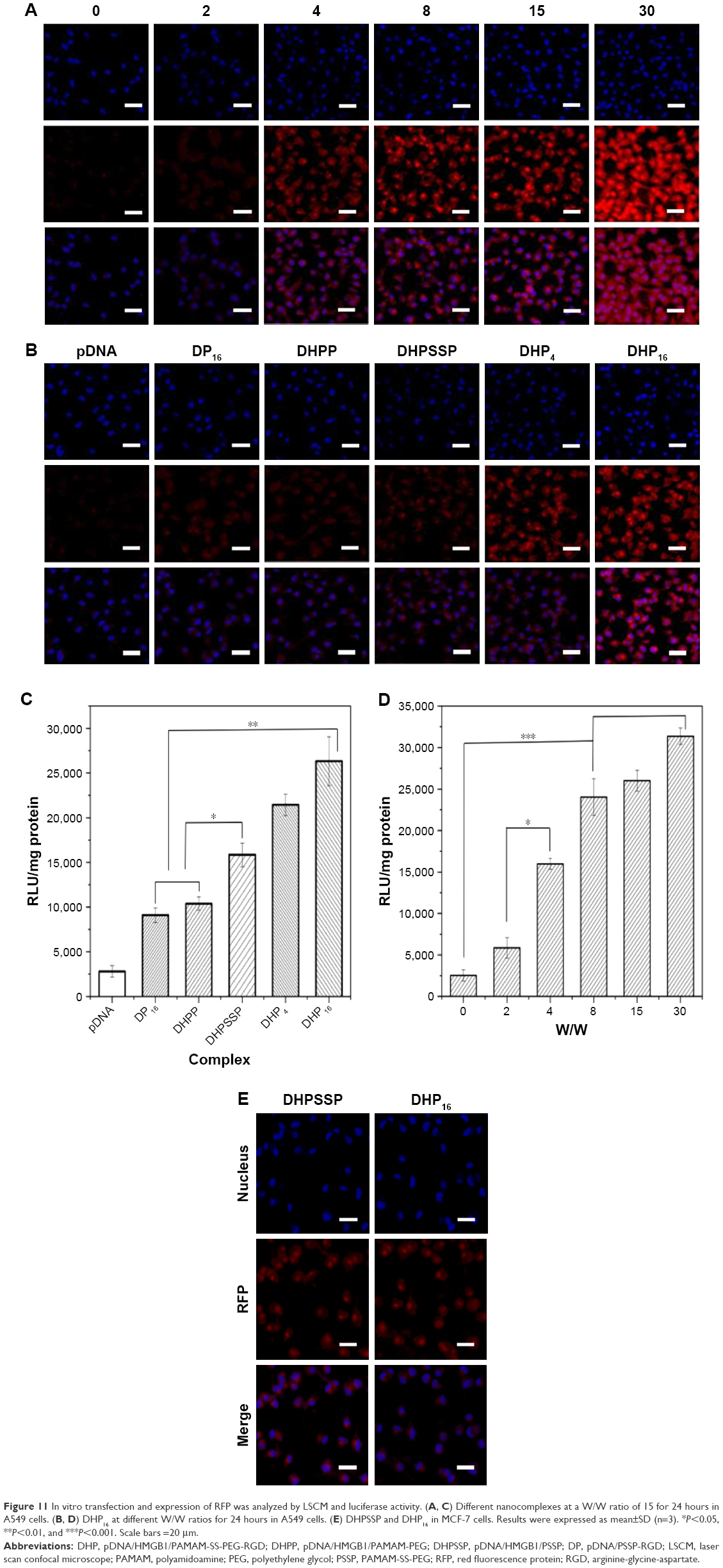 | Figure 11 In vitro transfection and expression of RFP was analyzed by LSCM and luciferase activity. (A, C) Different nanocomplexes at a W/W ratio of 15 for 24 hours in A549 cells. (B, D) DHP16 at different W/W ratios for 24 hours in A549 cells. (E) DHPSSP and DHP16 in MCF-7 cells. Results were expressed as mean±SD (n=3). *P<0.05, **P<0.01, and ***P<0.001. Scale bars =20 μm. |
In vivo antitumor activities
The in vivo antitumor effect of nanocomplexes was investigated using a S180 tumor xenografts model on Kunming mice. Nanocomplexes were intravenously injected into the mice every 2 days at a 3 mg/kg p53 gene dose (n=6). As shown in Figure 12A, all treatment groups except for free DNA exhibited no significant difference in tumor volume in the first 6 days, while treatment groups inhibited the tumor growth to different degrees after 6 days. As shown in Figure 12C and D, the DHPP group displayed moderate antitumor efficiency (34.1%), whereas the DHPSSP and DHP4 groups showed similar tumor inhibition effects (47.3% and 52.9%, respectively) and the DHP16 group showed the highest inhibition rate (70.3%), emphasizing again the importance of high-affinity interactions between RGD peptide and highly expressed integrin ανβ3 on the tumor surface. This is consistent with the result of cellular uptake and the expression study. However, compared with other treatment groups, the tumor inhibition rate of the free DNA group (11.1%) was relatively low, this probably because the naked DNA will be almost degraded in the blood and consequently have an inconspicuous effect on tumors. This distinct contrast also demonstrated that the DHP nanocomplex could escape the capture of the RES system via the PEGylation of its surface and accumulate successfully in tumors by the EPR effect, thereby having an outstanding tumor inhibition effect. In addition, the HMGB1/p53 complex could affect the cytoplasmic localization of the reciprocal binding partner, thereby regulating subsequent levels of autophagy and apoptosis.44 The link between HMGB1 and p53 is still to be studied in the future. The changes in body weight of mice were also known as a direction for safety. As Figure 12B shows, there was no significant difference in the body weight of all groups during the treatment process, indicating the DHP nanocomplex was a safe formulation. All these results suggest that a self-assembly DHP nanocomplex can accumulate in the tumor via the EPR effect and actively target tumor cells due to its high-affinity interactions with expressed integrin ανβ3, thereby enhancing the antitumor activity. DHP might be regarded as a desired delivery system to transport DNA.
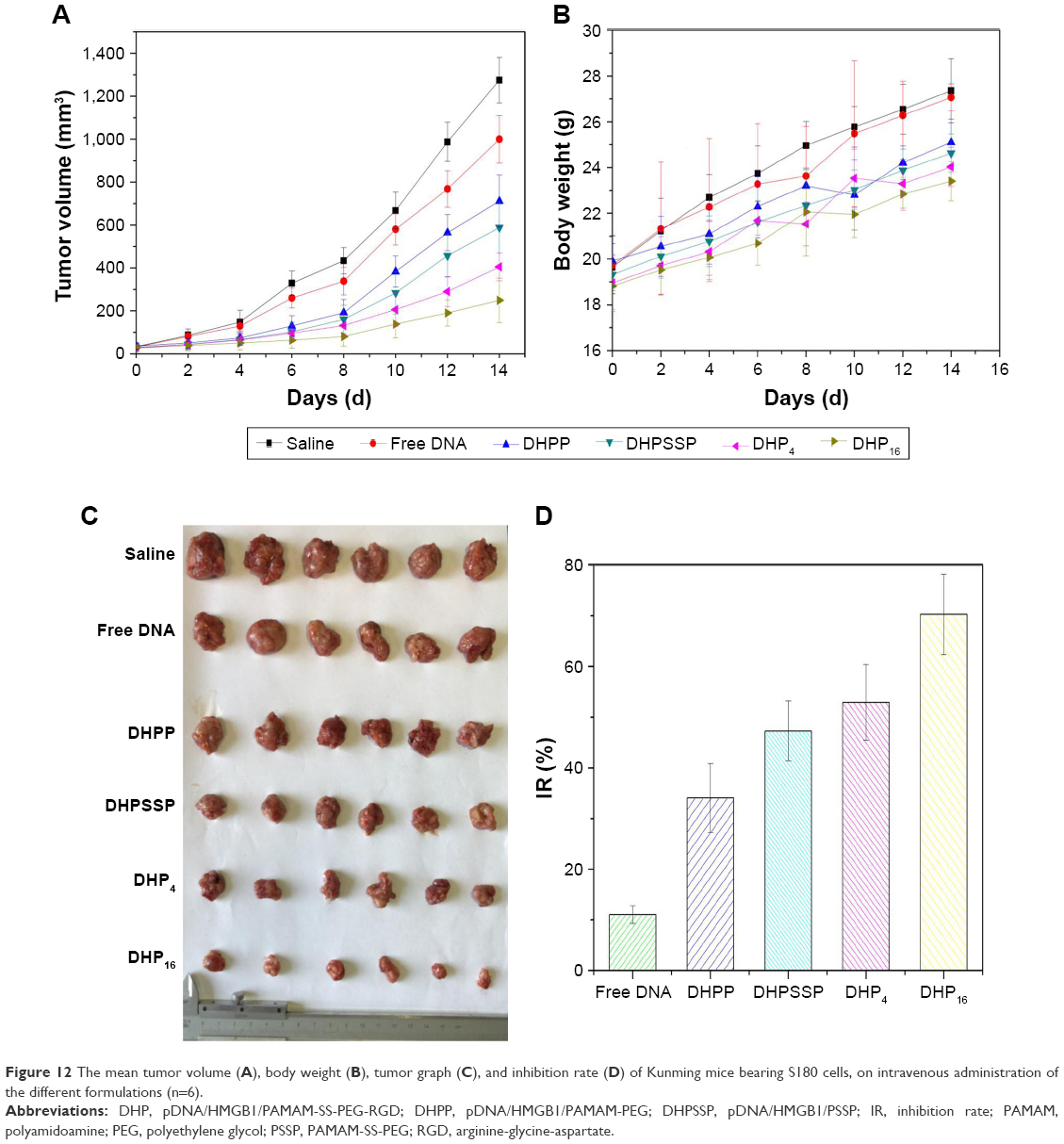 | Figure 12 The mean tumor volume (A), body weight (B), tumor graph (C), and inhibition rate (D) of Kunming mice bearing S180 cells, on intravenous administration of the different formulations (n=6). |
Conclusions
In this study, we successfully constructed an effective disulfide bond-mediated cleavable RGD modified gene delivery system. DHP nanocomplexes could effectively facilitate prolonged gene circulation and accumulate in tumor tissues via the EPR effect. Afterwards, DHP nanocomplexes actively targeted tumor cells due to the high-affinity interactions between RGD peptide and highly expressed integrin ανβ3, thereby clearly promoting the cellular uptake rate. DHP nanocomplexes could escape from lysosomes and PEG chains cleaved from PAMAM due to the high concentration of GSH in the cytoplasm. The cleavage of PEG from the carrier system could enhance expression efficiency to some extent, and the transfection and expression efficiency were further promoted under the guidance of NLS HMGB1. More importantly, in vivo antitumor activity study demonstrated that DHP16 had highest antitumor activity among all preparations. All these results enhance our confidence that DHP could be a suitable gene delivery system for enhancing the transfection and expression efficiency.
Disclosure
The authors report no conflicts of interest in this work.
References
Lv H, Zhang S, Wang B, Cui S, Yan J. Toxicity of cationic lipids and cationic polymers in gene delivery. J Control Release. 2006;114(1):100–109. | ||
Luo D, Saltzman WM. Synthetic DNA delivery systems. Nat Biotechnol. 2000;18(1):33–37. | ||
Kesharwani P, Jain K, Jain NK. Dendrimer as nanocarrier for drug delivery. Prog Polym Sci. 2014;39(2):268–307. | ||
Zhu J, Shi X. Dendrimer-based nanodevices for targeted drug delivery applications. Journal of Materials Chemistry B. 2013;1(34):4199–4211. | ||
Han H, Chen W, Yang J, et al. Inhibition of cell proliferation and migration through nucleobase-modified polyamidoamine-mediated p53 delivery. Int J Nanomedicine. 2018;13:1297–1311. | ||
Wu HM, Pan SR, Chen MW, et al. A serum-resistant polyamidoamine-based polypeptide dendrimer for gene transfection. Biomaterials. 2011;32(6):1619–1634. | ||
Varkouhi AK, Scholte M, Storm G, Haisma HJ. Endosomal escape pathways for delivery of biologicals. J Control Release. 2011;151(3):220–228. | ||
Tseng WC, Jong CM. Improved stability of polycationic vector by dextran-grafted branched polyethylenimine. Biomacromolecules. 2003;4(5):1277–1284. | ||
Kibria G, Hatakeyama H, Sato Y, Harashima H. Anti-tumor effect via passive anti-angiogenesis of PEGylated liposomes encapsulating doxorubicin in drug resistant tumors. Int J Pharm. 2016;509(1–2):178–187. | ||
Harris JM, Chess RB. Effect of pegylation on pharmaceuticals. Nat Rev Drug Discov. 2003;2(3):214–221. | ||
Guillaudeu SJ, Fox ME, Haidar YM, Dy EE, Szoka FC, Fréchet JM. PEGylated dendrimers with core functionality for biological applications. Bioconjug Chem. 2008;19(2):461–469. | ||
Hatakeyama H, Akita H, Harashima H. A multifunctional envelope type nano device (MEND) for gene delivery to tumours based on the EPR effect: a strategy for overcoming the PEG dilemma. Adv Drug Deliv Rev. 2011;63(3):152–160. | ||
Kakizawa Y, Harada A, Kataoka K. Glutathione-sensitive stabilization of block copolymer micelles composed of antisense DNA and thiolated poly(ethylene glycol)-block-poly(L-lysine): a potential carrier for systemic delivery of antisense DNA. Biomacromolecules. 2001;2(2):491–497. | ||
Nguyen DH, Lee JS, Bae JW, et al. Targeted doxorubicin nanotherapy strongly suppressing growth of multidrug resistant tumor in mice. Int J Pharm. 2015;495(1):329–335. | ||
Nguyen Thi TT, Tran TV, Tran NQ, Nguyen CK, Nguyen DH. Hierarchical self-assembly of heparin-PEG end-capped porous silica as a redox sensitive nanocarrier for doxorubicin delivery. Mater Sci Eng C Mater Biol Appl. 2017;70(Pt 2):947–954. | ||
Li J, Huo M, Wang J, et al. Redox-sensitive micelles self-assembled from amphiphilic hyaluronic acid-deoxycholic acid conjugates for targeted intracellular delivery of paclitaxel. Biomaterials. 2012;33(7):2310–2320. | ||
Sun Y, Huang Y, Bian S, Liang J, Fan Y, Zhang X. Reduction-degradable PEG-b-PAA-b-PEG triblock copolymer micelles incorporated with MTX for cancer chemotherapy. Colloids Surf B Biointerfaces. 2013;112:197–203. | ||
Wen HY, Dong HQ, Xie WJ, et al. Rapidly disassembling nanomicelles with disulfide-linked PEG shells for glutathione-mediated intracellular drug delivery. Chem Commun. 2011;47(12):3550–3552. | ||
Wang W, Li W, Ma N, Steinhoff G. Non-viral gene delivery methods. Curr Pharm Biotechnol. 2013;14(1):46–60. | ||
Kaouass M, Beaulieu R, Balicki D. Histonefection: Novel and potent non-viral gene delivery. J Control Release. 2006;113(3):245–254. | ||
Jin YC, Kim SW, Cheng F, et al. The effect of biodegradable gelatin microspheres on the neuroprotective effects of high mobility group box 1 A box in the postischemic brain. Biomaterials. 2011;32(3):899–908. | ||
Shen Y, Peng H, Deng J, et al. High mobility group box 1 protein enhances polyethylenimine mediated gene delivery in vitro. Int J Pharm. 2009;375(1–2):140–147. | ||
Foldvari M, Chen DW, Nafissi N, Calderon D, Narsineni L, Rafiee A. Non-viral gene therapy: Gains and challenges of non-invasive administration methods. J Control Release. 2016;240:165–190. | ||
Henne WA, Kularatne SA, Hakenjos J, Carron JD, Henne KL. Synthesis and activity of a folate targeted monodisperse PEG camptothecin conjugate. Bioorg Med Chem Lett. 2013;23(21):5810–5813. | ||
Kim TH, Jo YG, Jiang HH, et al. PEG-transferrin conjugated TRAIL (TNF-related apoptosis-inducing ligand) for therapeutic tumor targeting. J Control Release. 2012;162(2):422–428. | ||
Morlieras J, Dufort S, Sancey L, et al. Functionalization of small rigid platforms with cyclic RGD peptides for targeting tumors overexpressing αvβ3-integrins. Bioconjug Chem. 2013;24(9):1584–1597. | ||
Wu PH, Onodera Y, Ichikawa Y. Targeting integrins with RGD-conjugated gold nanoparticles in radiotherapy decreases the invasive activity of breast cancer cells. Int J Nanomedicine. 2017;12:5069–5085. | ||
Boswell CA, Eck PK, Regino CA, et al. Synthesis, characterization, and biological evaluation of integrin alphavbeta3-targeted PAMAM dendrimers. Mol Pharm. 2008;5(4):527–539. | ||
Waite CL, Roth CM. PAMAM-RGD conjugates enhance siRNA delivery through a multicellular spheroid model of malignant glioma. Bioconjug Chem. 2009;20(10):1908–1916. | ||
Dunehoo AL, Anderson M, Majumdar S, Kobayashi N, Berkland C, Siahaan TJ. Cell adhesion molecules for targeted drug delivery. J Pharm Sci. 2006;95(9):1856–1872. | ||
Rufini A, Tucci P, Celardo I, Melino G. Senescence and aging: the critical roles of p53. Oncogene. 2013;32(43):5129–5143. | ||
Cai G, Zhang H, Liu P, Wang L, Jiang H. Triggered disassembly of hierarchically assembled onion-like micelles into the pristine core-shell micelles via a small change in pH. Acta Biomater. 2011;7(10):3729–3737. | ||
Liu Y, Bryantsev VS, Diallo MS, Goddard WA. PAMAM dendrimers undergo pH responsive conformational changes without swelling. J Am Chem Soc. 2009;131(8):2798–2799. | ||
Han L, Huang R, Liu S, Huang S, Jiang C. Peptide-conjugated PAMAM for targeted doxorubicin delivery to transferrin receptor overexpressed tumors. Mol Pharm. 2010;7(6):2156–2165. | ||
Xie Y, Qiao H, Su Z, Chen M, Ping Q, Sun M. PEGylated carboxymethyl chitosan/calcium phosphate hybrid anionic nanoparticles mediated hTERT siRNA delivery for anticancer therapy. Biomaterials. 2014;35(27):7978–7991. | ||
Zhou C, Yu B, Yang X, et al. Lipid-coated nano-calcium-phosphate (LNCP) for gene delivery. Int J Pharm. 2010;392(1–2):201–208. | ||
Han L, Zhou X. Synthesis and characterization of liposomes nano-composite-particles with hydrophobic magnetite as a MRI probe. Appl Surf Sci. 2016;376:252–260. | ||
Shen Y, Guo J, Chen G, et al. Delivery of Liposomes with Different Sizes to Mice Brain after Sonication by Focused Ultrasound in the Presence of Microbubbles. Ultrasound Med Biol. 2016;42(7):1499–1511. | ||
Ullrich M, Haša J, Hanuš J, Šoóš M, Štěpánek F. Formation of multi-compartmental particles by controlled aggregation of liposomes. Powder Technol. 2016;295:115–121. | ||
Qiu L, Hu Q, Cheng L, et al. cRGDyK modified pH responsive nanoparticles for specific intracellular delivery of doxorubicin. Acta Biomater. 2016;30:285–298. | ||
Li B, Zhang XX, Huang HY, et al. Effective deactivation of A549 tumor cells in vitro and in vivo by RGD-decorated chitosan-functionalized single-walled carbon nanotube loading docetaxel. Int J Pharm. 2018;543(1–2):8–20. | ||
Khalil IA, Kogure K, Akita H, Harashima H. Uptake pathways and subsequent intracellular trafficking in nonviral gene delivery. Pharmacol Rev. 2006;58(1):32–45. | ||
Nam HY, Kwon SM, Chung H, et al. Cellular uptake mechanism and intracellular fate of hydrophobically modified glycol chitosan nanoparticles. J Control Release. 2009;135(3):259–267. | ||
Livesey KM, Kang R, Vernon P, et al. p53/HMGB1 complexes regulate autophagy and apoptosis. Cancer Res. 2012;72(8):1996–2005. |
Supplementary materials
We have supplemented the acid-base titration test. The result is shown in Figure S1. The buffering capacity of PSSP-RGD from pH 7.4 to 5.1 was determined by acid base titration. Briefly, PSSP-RGD conjugates were dissolved in 0.01 M NaCl (4 mg/mL), and the solution was adjusted to pH 12 with 1 M NaOH. The diluted solution was titrated by the stepwise addition of 0.01 M HCl to obtain the titration profile. The pKa of PSSP-RGD was determined by the primary derivative method, and the buffering capacity (BC%) was defined as the percentage of amino groups from pH 7.4 to 5.1, as previously described. The buffering capacity was 27.36%.
|
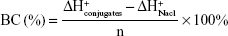
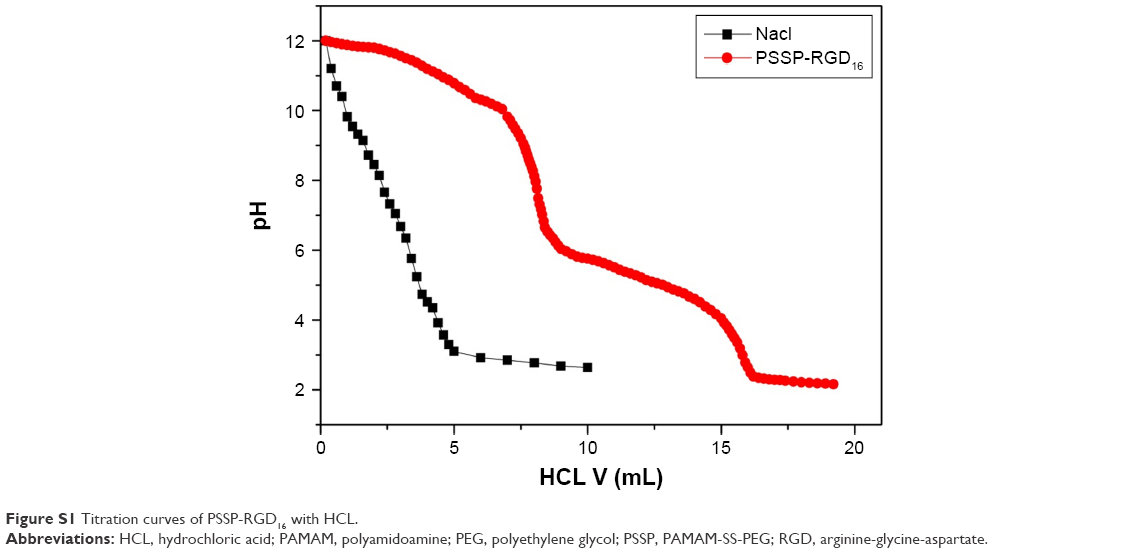 | Figure S1 Titration curves of PSSP-RGD16 with HCL. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.