Back to Journals » International Journal of Nanomedicine » Volume 13
A niosome formulation modulates the Th1/Th2 bias immune response in mice and also provides protection against anthrax spore challenge
Authors Gogoi H, Mani R ![]() , Bhatnagar R
, Bhatnagar R
Received 3 October 2017
Accepted for publication 26 February 2018
Published 14 November 2018 Volume 2018:13 Pages 7427—7440
DOI https://doi.org/10.2147/IJN.S153150
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Thomas Webster
Himanshu Gogoi, Rajesh Mani, Rakesh Bhatnagar
Laboratory of Molecular Biology and Genetic Engineering, School of Biotechnology, Jawaharlal Nehru University, New Delhi, India
Introduction: In this study, we have investigated the immunogenicity and protective efficacy of a niosomal formulation encapsulating protective antigen (PA) and PA domain 4 (D4) of Bacillus anthracis.
Methods: Nonionic surfactant–based vesicles (NISV) + PA and NISV + D4 were prepared from span-60 and cholesterol by reverse-phase evaporation method and were evaluated for in vitro characteristics and immunological studies.
Results: Particle characterization using transmission electron microscopy and atomic force microscopy analysis showed that the niosomal formulation was spherical in shape. The entrapment efficiency values were calculated to be 58.5% and 44.75% for PA and D4, respectively. Confocal microscopy and flow cytometry studies showed an enhanced uptake of antigen in THP1 macrophages by niosome as compared to antigen only. An in vitro release assay showed a burst release of antigen from niosome within 24 hours followed by a gradual release for 144 hours. Immunological studies showed that both PA- and D4-encapsulated niosome elicited a robust IgG titer. Antibody isotyping and cytokine profile showed that NISV + PA and NISV + D4 enhanced both Th1 and Th2 responses in mice, suggesting a mixed Th1/Th2 response. Both NISV + PA and NISV + D4 elicited high levels of anti-inflammatory cytokine interleukin-10 with low levels of pro-inflammatory cytokine tumor necrosis factor-α, suggesting the anti-inflammatory property of niosome. Both the niosomal formulations were also able to confer protection against BA infection as compared to only PA and D4.
Conclusion: PA and D4 encapsulated NISV formulation could modulate both the Th1 and Th2 adaptive immune system and was found to be a better prophylactic against anthrax.
Keywords: Bacillus anthracis, niosome, protective antigen, protective antigen domain 4
Introduction
Bacillus anthracis (BA), the causative agent of anthrax, is a Gram-positive, rod-shaped, facultative anaerobic, and spore-forming bacterium. The ease of production and weaponization of BA spores make it apt to be used as a bioterror agent.1 The in vivo growth rate of BA being logarithmic makes its early detection nearly futile. A prolonged antibiotic treatment for at least 60 days is needed to prevent the disease.2
The pathogenesis of BA is attributed to a tripartite exotoxin–protein complex and an antiphagocytic poly-γ-d-glutamic acid capsule. The exotoxin component comprises protective antigen (PA), edema factor (EF), and lethal factor (LF). PA is the major immunogen in all the approved vaccines and elicits neutralizing antibodies against anthrax.3 PA comprises four domains. Domain 1 consists of the site for furin cleavage. Domains 2 and 3 are involved in heptamerization and pore formation for the translocation of LF and/or EF into the cytosol. Domain 4 (D4) is involved in binding of PA to macrophage cell receptors and has been reported to be the most immunodominant region.4 EF and LF bind to PA to form edema toxin and lethal toxin, respectively. On being secreted by BA during infection, PA (83 kDa) binds to the host cellular receptors TEM8 and CMG2,5,6 which results in the clipping-off of a 20-kDa region from the C-terminus of PA. The resulting PA63 moiety forms an LF-/EF-binding heptamer/octamer.7–11 The PA63 LF and/or EF complex is then internalized via a receptor-mediated endocytic mechanism.12 Acidic pH inside the endosome induces a change in the structure of PA63 prepore to a protein-conducting channel through which LF and EF are translocated inside the cells where they exert their deleterious effects. LF is a zinc metalloprotease.13 Lethal toxin evades the immune system by impairing dendritic cells and causes vascular leakage, hypoxia, and septic shock.14–16 EF is a calcium- and calmodulin-dependent adenylyl cyclase17 and converts adenosine triphosphate to cyclic adenosine monophosphate. Edema toxin increases the host cell susceptibility by suppressing polymorphonuclear neutrophil function.
Currently available prophylactic approach involves vaccination with alhydrogel-adsorbed cell-free culture filtrate of BA V770-NP1-R strain (anthrax vaccine adsorbed) and alum-precipitated culture supernatant of BA Sterne strain (anthrax vaccine precipitated). However, these currently licensed human vaccines suffer from certain drawbacks such as requirement of multiple booster doses over a prolonged period; severe side effects such as edema, fatigue, headache, myalgia, nasopharyngitis, and erythema at injection site; and elicitation of heterogeneous immune response among immunized population, which raised the requirement for a safer vaccine as well as provided a prolonged immunity against the disease.
Rehm and Schueler in 2005 had stated that nanotechnology is expected to revolutionize or at least bring a significant change in the pharma sector and life sciences.45 With the advent of nanotechnology, nanoscale vaccine formulations such as liposomes, virus-like particles, immune-stimulating complexes, and biodegradable polymers have received attention in the past decade. The conjugation of antigenic materials with these nanoparticles (NPs) has allowed the presentation of the immunogen in much the same way as would have been presented by the pathogen. By encapsulating antigenic material, NPs provide site-directed delivery of antigens along with increasing release time of the antigen. They also provide a method for delivering antigens which may otherwise degrade readily upon injection. With advances in nanotechnology, NPs with modified size, shape, targeted delivery, and surface properties have been designed for both therapeutic and prophylactic purposes. Due to the similarity of the size of the NPs with cellular components, they can enter the cells easily using cellular endocytosis mechanism.
Since the first liposome colloidal system was prepared in 1964 by Alec D Bangham,18 tremendous progress has been made in the area for site-specific drug/protein delivery. Liposomes have been extensively explored due to their versatile ability to entrap both lipophilic and hydrophilic drugs. However, in dispersions, liposomes tend to undergo hydrolytic degradation, sedimentation, aggregation, or fusion during storage.19,20 These problems were addressed by Baillie et al21 and Uchegbu and Florence22 where they demonstrated the formation of liposome-like vesicles from a hydrated mixture of cholesterol and nonionic surfactant such as mono- or dialkylpolyoxyethylene ether. Niosomes or nonionic surfactant–based vesicles (NISVs) are self-assembling lamellar vesicles prepared from nonionic amphiphiles with/without cholesterol as an additive in aqueous media. These vesicular systems are similar to liposome; are biodegradable, biocompatible, and nonimmunogenic; and are capable of encapsulating and delivering bioactive compounds such as anticancer drugs, genes, and antigens with a wide range of solubility. Niosomes have been prepared from different classes of nonionic surfactants such as polyoxyethylene alkyl ethers (Brij®), sorbitan monoesters (span 20, 40, 60, and 80), and polyoxyethylene sorbitan monoesters (Tween 20, 60, and 80).23 Although the structure and properties of niosomes are similar to those of liposomes, their chemical stability and relatively low cost of raw materials make them more interesting for industrial manufacturers. In a study by Brewer and Alexander,24 bovine serum albumin (BSA) encapsulated within NISV was shown to be potent stimulators of Th1-mediated response than Freund’s complete adjuvant. Here, we have studied the immune response generated by noisome-encapsulated PA and D4, the stable immunogenic domain of PA in Swiss albino mice. Anthrax spore challenge was carried out to check the protective efficacy of the niosome-encapsulated antigen formulations against the disease.
Materials and methods
Materials
Sterile deionized water was used in making all buffers. Dimethyl sulfoxide (DMSO), ammonium chloride, span-60, cholesterol, lipopolysaccharide (LPS), sodium bicarbonate, sodium phosphate monobasic, and sodium phosphate dibasic were purchased from Sigma-Aldrich Co. (St Louis, MO, USA). L-glutamine, urea, and sodium chloride (NaCl) were purchased from Amresco (Solon, OH, USA). Isopropyl β-D-thiogalactopyranoside (IPTG) was purchased from HiMedia (Mumbai, India). HEPES, streptomycin, and ampicillin were purchased from USB (Cleveland, OH, USA), and chloroform was purchased from Thermo Fisher Scientific (Waltham, MA, USA). Fluorescein isothiocyanate (FITC) was purchased from MP Biomedicals (Solon, OH, USA). Nickel-nitrilotriacetic acid was purchased from Qiagen (Hilden, Germany). Bradford reagent was purchased from Bio-Rad Laboratories Inc. (Hercules, CA, USA). Tissue culture plate and micro-bicinchoninic acid (BCA) assay kit were purchased from Thermo Fisher Scientific. Fetal bovine serum (FBS) was purchased from Gibco® (Thermo Fisher Scientific). Horseradish peroxidase (HRP)-conjugated anti-mouse IgG, IgG1, and IgG2a were purchased from Santa Cruz Biotechnology Inc. (Dallas, TX, USA). 3,3′,5,5′-Tetramethylbenzidine (TMB) substrate was purchased from BD Biosciences Pharmingen (San Diego, CA, USA).
Purification of PA and D4
PA and D4 were purified as described by Manish et al.25 Briefly, PA/D4 expression plasmid-transformed Escherichia coli cells were grown to OD600nm of 0.8 at 37°C before inducing with 1 mM IPTG and allowed to grow in an incubator shaker for 6 hours. The cells were then harvested at 6,000 rpm for 10 minutes. The bacterial cell pellet was lysed and solubilized using denaturing lysis buffer (8 M urea, 0.1 M phosphate buffer [PB], pH 7.4, 250 mM NaCl) on a rotary shaker for 2 hours at room temperature (RT). The insoluble fraction of cell lysate was removed by centrifugation at 13,000 rpm for 30 minutes at RT. The supernatant was incubated with Nickel-nitrilotriacetic acid slurry for 2 hours. This mix was transferred to a propylene tube column- and slurry-bound PA/D4 was renatured by passing a gradient of urea solution of 8 M to 0 M (0.1 M PB, pH 7.4, 250 mM NaCl). The steps after 4 M urea gradient were carried out at 4°C. The column was washed with 10 bed volume of 10 mM, 20 mM, and 30 mM imidazole and 250 mM NaCl containing 0.1 M PB (pH 7.4). The column-bound protein was eluted using 300 mM imidazole containing 250 mM NaCl and 0.1 M PB. Protein content was quantified using Bradford reagent (Bio-Rad).
Preparation of niosome
PA- and D4-loaded niosomes were prepared by the reverse-phase evaporation method as described by Moghassemi et al.26 Briefly, 5.5 mg span-60 and 7.4 mg cholesterol were dissolved in 5 mL chloroform in a round-bottom flask. Subsequently, 1 mL PA/D4 (2 mg/mL) in 0.1 M PB (pH 7.4) was added to the flask and vortexed for 60 seconds. The mixture was then emulsified by sonicating in a water bath sonicator for 5 minutes with on/off pulse. The organic phase was evaporated in a rotary evaporator at 60 rpm at RT. The resulting thin film was resuspended in PBS. Unentrapped protein was separated by ultracentrifugation.
Characterization of niosome
The morphology of niosome was visualized by atomic force microscopy (AFM; Alpha300 RA; WITec, Ulm, Germany) and transmission electron microscopy (TEM; JEOL 2100F; Tokyo, Japan). The sizes of the NPs were measured by dynamic light scattering (DLS). Zeta potential was measured to evaluate the surface charge on the particles. Samples were diluted in double-distilled water for both DLS and zeta sizer and were analyzed in Malvern Zetasizer ZS 90 (Malvern Instruments, Malvern, UK). For TEM analysis, the samples were diluted in double-distilled water, loaded on copper grid, and kept under vacuum for drying. After drying, the samples were analyzed under JEOL 2100F. For the AFM analysis, the samples were diluted in double-distilled water, and 5 μL of the sample was loaded onto a mica sheet and dried. The samples were analyzed in WITEC ALPHA 300RA. The resonant frequency of the tip was 80 kHz with a spring constant of 40 N/m, and imaging was performed in air by tapping mode. The analysis of topography data was performed by Project 4 software.
Entrapment efficiency
In order to determine the entrapment efficiency, unencapsulated protein from the niosome formulation was removed by ultracentrifugation. The obtained pellet fraction was treated with 1% sodium dodecyl sulfate (SDS) and centrifuged at 13,000 rpm. The encapsulated protein was quantified by micro-BCA assay kit. Protein concentration was determined from the standard curve of BSA in 1% SDS. In order to get a qualitative view of the encapsulated protein, the 1% SDS-treated niosome sample was visualized by SDS–polyacrylamide gel electrophoresis (PAGE) analysis.
In vitro release study
Unencapsulated PA/D4 was separated from niosomal formulation by ultracentrifugation. The obtained pellet was resuspended in 0.1 M PB (pH 7.4). The samples were incubated at 37°C with gentle shaking; 1 mL sample was drawn at an interval of 1, 2, 6, 12, 24, 48, 72, 96, 120, and 144 hours and centrifuged, and the supernatant collected to be estimated for protein content. The amount of collected supernatant was made up by adding 1× PBS. The amount of protein released was measured by micro-BCA method.
FITC labeling of protein
Briefly, 5 mg/mL D4 in carbonate buffer was mixed with 1 mg/mL FITC in DMSO and incubated for 4 hours at RT. FITC-bound D4 was eluted by 1× PBS using G-25 Sepharose® beads and used for encapsulation into niosome.
Cell culture
Human monocytic cell line THP1 was purchased from National Center for Cell Science, Pune, India, and cultured in Roswell Park Memorial Institute (RPMI)-1640 media supplemented with 10% FBS. L-glutamine (2 mM), HEPES (20 mM), penicillin (100 units/mL), streptomycin (100 μg/mL), and sodium bicarbonate (2 g/L) at 5% CO2 and 95% humidified environment. Prior to the experiment, the monocytes were differentiated using phorbol myristate acetate (PMA) at a concentration of 20 ng/mL.
In vitro uptake study
Confocal microscopy
The in vitro cellular uptake of niosome was observed in THP1 cell line. Briefly, 1×106 cells/mL were seeded on coverslips in a six-well plate and differentiated using PMA. The cells were maintained at 5% CO2 and 37°C and in a humidified atmosphere. After 48 hours, media were replaced with fresh incomplete media, and niosome-encapsulated FITC–D4 was added to the cells and incubated for 30 minutes in a CO2 incubator. Media were aspirated, and the cells were washed thoroughly with 1× PBS three times, fixed with 4% formaldehyde, and washed three times with 1× PBS. The uptake of niosome-encapsulated FITC-D4 was analyzed under confocal microscopy, and 4′,6-diamidino-2-phenylindole was used for nuclear staining.
Flow cytometry
Flow cytometry was performed in order to get a quantitative measurement of the in vitro uptake of the NPs by THP1 cells. Briefly, 1×106 THP1 cells/mL were seeded in a Petri plate, and PMA was added for differentiation. The cells were maintained at 5% CO2 and 37°C and in a humidified atmosphere. After 48 hours, media were replaced with fresh incomplete media and incubated with niosome-encapsulated FITC-D4 or FITC-D4 alone for the predetermined period. The culture media were removed, and the cells were washed three times with precooled PBS and then resuspended in 0.5 mL PBS. Experiments were carried out in Beckman Coulter MoFlo cytomation (Brea, CA, USA). For each analysis, 15,000 events were collected. The cells with no treatment were taken as control.
In vivo studies
Female Swiss albino mice (4–6 weeks old) were used for in vivo studies. Each group consisted of five mice each. Mice were injected with PBS, NISV only, PA, D4, NISV + PA, and NISV + D4. Subsequent boosters were given on Days 15 and 29 with PBS, NISV only, PA, D4, NISV + PA, and NISV + D4. Blood samples were collected on Days 0, 14, 28, and 42 via retro-orbital route. Plasma was separated by centrifugation and stored at −20°C for further use. The animal experiments were approved by Central Laboratory Animal Resources, Jawaharlal Nehru University.
Antibody titer determination
Evaluation of IgG response
Antibody end point titer was measured using indirect enzyme-linked immunosorbent assay (ELISA). The absorbance of pre-immune sera at 1:1,000 dilution plus 2.5 times its SD was taken as the cutoff. Briefly, a 96-well, flat-bottomed polystyrene plate (Thermo Fisher Scientific) was coated with 500 ng of PA/D4 and allowed to incubate overnight in a humidified atmosphere at 4°C. Protein was aspirated, and the plate washed with 1× PBS 0.1% Tween 20 (PBST) three times. Wells were then blocked with 2% BSA in 1× PBS and incubated at 37°C for 1 hour. The plate was then washed three times with 1× PBST. Individual serum samples were serially diluted twofold in PBS, added to the wells in triplicates, and incubated at 37°C for 2 hours. After aspiration, the plate was washed five times with PBST. HRP-conjugated anti-mouse IgG (Santa Cruz Biotechnology Inc.) was added at a dilution of 1:10,000 and incubated at 37°C for 1 hour. After washing five times with 1× PBST, TMB substrate reagent was added and incubated for 20 minutes in the dark. Reaction was stopped using 50 uL 1 N HCl, and absorbance reading was noted at 450 nm.
Evaluation of IgG1 and IgG2a response
The differential IgG subtype response was measured by determining the levels of IgG1 and IgG2a. IgG1 and IgG2a levels were determined by the same method as described for total IgG, except that the secondary antibody used was HRP-conjugated anti-mouse IgG1 and IgG2a, respectively. The secondary antibody was added at a dilution of 1:10,000. The response was evaluated in terms of antibody titer employing serial dilution of serum rather than using a single dilution of the serum.
Evaluation of secreted cytokines after in vitro stimulation of splenocytes
On the 49th day postimmunization, spleens from mice of different groups were harvested. Spleens were aseptically removed, and splenocytes were isolated using frosted slides. Red blood cells were lysed using 0.9% ammonium chloride. Splenocytes were washed in incomplete RPMI-1640 and suspended in RPMI-1640 media supplemented with 10% FBS. Splenocytes were plated at 2×105 cells/well in a 24-well tissue culture plate. The cells were stimulated with 5 μg/mL LPS (positive control), 10 μg/mL PA/D4 (test sample), or only media (negative control). Culture supernatants were collected after 48 hours of incubation, and cytokine levels were evaluated for Th1/Th2 immune response using opt-EIA kit (BD Biosciences Pharmingen) as per the manufacturer’s instructions. Interleukin (IL)-4 and interferon (IFN)-γ levels were measured for Th1/Th2 immune response. To evaluate the inflammatory property of NISV + PA and NISV + D4, levels of tumor necrosis factor (TNF)-α (pro-inflammatory cytokine) and IL-10 (anti-inflammatory cytokine) were measured in a splenocyte supernatant.
Anthrax spore challenge
For efficacy testing, PBS, blank NISV, PA only/D4 only, and NISV + PA/NISV + D4, immunized female Swiss albino mice (each group, n=10) were injected with 0.5×103 spores of BA virulent strain on 42nd day by intraperitoneal (IP) route and observed for 14 days for morbidity and death. All the infected mice were kept in an animal isolator in a Biosafety Level-3 facility. Mice immunized with alhydrogel + PA and alhydrogel + D4 were used as positive control. Mice experiments were conducted according to the protocol approved by Institutional Animal Ethical Committee, Jawaharlal Nehru University. Protective efficacy was determined in terms of percentage survival by plotting Kaplan–Meier curve.
Statistical analysis
Experimental data were analyzed by Sigma Plot/Microsoft Excel and expressed as ±SD. Comparisons between individual data points were made using either one-way or two-way analysis of variance (ANOVA) followed by Tukey’s multiple comparison test. Antigen alone–immunized mice group were compared with niosome-encapsulated antigen to calculate the P-value. P-value <0.05 was considered as significant (*<0.05, **<0.01, ***<0.001, ****<0.0001, ns = nonsignificant).
Results
Physical characterization of niosome
The particles were spherical in shape as observed using TEM and AFM (Figure 1). The average particle size of blank niosome determined using DLS was found to be 107 nm. The entrapment of PA and D4 resulted in an increase in the size of the niosome to 182 and 160 nm, respectively, as obtained from DLS (Figure 2). The zeta potential of blank niosome was observed to be −54.4 mV, while those for the PA- and D4-encapsulated niosome were observed to be −59 and −58.5 mV (Figure 3).
 | Figure 1 Surface morphology of niosome formulation was visualized using AFM and TEM: (A) AFM image of NISV–D4; (B) AFM image of NISV–PA; (C) TEM image of NISV–D4; and (D) TEM image of NISV–PA. |
 | Figure 2 DLS of the niosome nanoparticles: (A) blank niosome; (B) NISV–PA; and (C) NISV–D4. |
 | Figure 3 Zeta potential of niosome nanoparticles: (A) blank niosome; (B) NISV–PA; and (C) NISV–D4. |
Entrapment efficiency
The encapsulation of PA and D4 in the niosome formulation was calculated by ultracentrifugation method and was calculated as the fraction of total protein entrapped in the niosome. The percentages of PA and D4 encapsulated in the niosome as calculated by micro-BCA assay kit were found to be 58.5 and 44.75, respectively. The qualitative analysis of PA/D4 encapsulated in niosome was analyzed using SDS-PAGE (Figure 4), which confirmed the encapsulation of the proteins in niosome.
 | Figure 4 Encapsulation of PA and D4 in niosome. |
In vitro release assay
The evaluation of in vitro release is a necessary parameter to understand the release kinetics of the protein. It is an important analytical tool, which gives an idea of the rate of in vivo protein release profile. The release of PA and D4 from niosome was carried out at 37°C in 1× PBS; 1 mL of sample was aliquoted at different time periods as mentioned in the “Materials and Methods” section. The content was centrifuged, and the supernatant was analyzed for protein content by micro-BCA assay kit. The release profile of PA and D4 shows an initial burst release during 24 hours followed by a gradual release of the proteins until 1 week as observed in Figure 5.
 | Figure 5 In vitro release profile of PA and D4 from NISV. |
In vitro uptake study and localization
In order to elicit an immune response, it is critical for the niosome formulation to be taken up by antigen-presenting cells.
In order to obtain a qualitative view of niosome-enhanced D4 uptake by THP1 cells, we performed laser confocal microscopy of THP1 cells incubated with niosome-encapsulated FITC-labeled D4. Confocal images showed an enhanced uptake of niosome-encapsulated D4 in comparison with only D4 (Figure 6) with the particles localized on the cytoplasm of the cell.
 | Figure 6 Confocal microscopy image of in vitro uptake of (A) niosome-encapsulated FITC-labeled D4 by THP1 cells in 30 minutes and (B) FITC-labeled D4 only by THP1 cells. |
To evaluate the extent to which niosome is taken up by macrophage, D4-labeled FITC-encapsulated inside niosome was incubated with THP1 cells for 30 minutes and analyzed quantitatively using flow cytometry. As shown in Figure 7, 46.75% niosome-encapsulated D4 was uptaken by THP1 in 30 minutes as compared to only 19.65% D4, which suggests an increase in the uptake of niosome-encapsulated D4 as compared to only D4.
 | Figure 7 Comparative in vitro uptake study of FITC-labeled D4 (FITC-D4) and niosome-encapsulated FITC–D4 by flow cytometry: (A) THP1 cells only; (B) THP1 cells incubated with FITC–D4 only; and (C) THP1 cells incubated with niosome-encapsulated FITC–D4. |
The results obtained from confocal microscopy and flow cytometry suggest that niosome-mediated antigen delivery enhances the uptake of the antigen by macrophages and may assist in augmenting the immune response of an antigen as compared to that generated by an antigen alone.
Assessment of immune response
Niosome encapsulating 25 μg of PA (NISV + PA) and 25 μg PAD4 (NISV + D4) were injected in Swiss albino mice via IP route. Individual sera from each mouse were collected on the 14th, 28th, and 42nd day, and antibody titer was determined by indirect ELISA. The immune correlates of the NISV + PA and NISV + D4 immunized mice were compared with that of antigen only. Mice immunized with PA and D4 only did not show much enhancement in antibody titer even after two booster doses (Figures 8 and 9). The administration of NISV + PA and NISV + D4 elicited a robust antibody titer. Antibody titer elicited by NISV + PA after prime was determined to be 5,900, which increased to 94,000 and 502,000 after the first and second booster, respectively, whereas mice immunized with PA alone did not elicit significant antibody titers, with titers at only 1,090 and 3,500 after the 28th and 42nd day. Antibody response specific to D4 as elicited by NISV + D4 was also measured and observed to be 2,800, 106,000, and 246,000 when measured on the 14th, 28th, and 42nd day after the prime, first booster, and second booster, with titer of D4 alone-immunized mice eliciting antibody titer of 1,500 and 2,000 after the first and second booster dose. PBS and blank NISV did not elicit significant antibody titer.
 | Figure 8 Anti-PA titers elicited by Swiss albino mice immunized intraperitoneally with PBS, blank NISV, PA, and NISV + PA. |
 | Figure 9 Anti-D4 titers elicited by Swiss albino mice immunized intraperitoneally with PBS, blank NISV, D4, and NISV + D4. |
Differential IgG subtype response has been shown to be a major immune correlate in BA infection. Humoral response elicited by NISV + antigen was also measured in the sera of the immunized mice by evaluating IgG1 and IgG2a subtype level. Mice immunized with PA and D4 alone elicited low level of IgG1 only even after two booster doses (Figures 8 and 9). However, immunization with NISV + PA elicited both IgG1 and IgG2a, with higher level of IgG1 titer as compared to IgG2a; the titer levels for IgG1 and IgG2a being 272,000 and 77,000 on Day 42, suggesting a Th2-biased response (Figures 8 and 9). Similarly, for NISV + D4, it was observed that IgG1 antibody titer was predominant as compared to IgG2a antibody titer, with antibody titer levels of 246,000 and 29,000, suggesting a strong Th2 immune response. However, elicitation of significant level of IgG2a antibody titer by both NISV + PA and NISV + D4 as compared to only PA and D4 suggests a mixed Th1/Th2 response with Th2-type immune response skewing toward Th1 type.
Antigen-specific cytokine in splenocyte
The mixed IgG1 and IgG2a response elicited by NISV + PA and NISV + D4 led us to explore the Th1 and Th2 heterogeneity offered by the niosome formulations. T lymphocytes are the major source of cytokines and display antigen-specific receptors on their cell surface to recognize foreign pathogens. The two main subsets of T lymphocytes differentiated on the basis of the surface markers displayed on the cells are CD4 and CD8. T lymphocytes displaying CD4 molecules on their cell surface are called Th cells and are the regarded as the most prolific cytokine producers. Th cells are further differentiated into Th1 and Th2 cells. Naïve T cells after the recognition of antigen peptide on the surface of major histocompatibility molecule II (MHC II) through T-cell receptor are differentiated into two major phenotypically distinct memory Th cell populations, Th1 and Th2. Th1 subset is a subset that secretes cytokines that are associated with inflammation, such as IFN-γ and TNF-α and induces cell-mediated response. On the other hand, Th2 subset helps in the proliferation of B cells by secreting cytokines such as IL-4 and IL-5 and is associated with humoral response.
Upon infection, BA relies on phagocytes; mostly dendritic cells to evade the immune system to reach the regional lymph nodes. The spores germinate inside the macrophages and become vegetative cells, which are released from the macrophages and multiply in the lymphatic system and enter the bloodstream where they multiply and cause massive septicemia. On being released into the bloodstream, the bacteria secrete its toxin disrupting cellular activities.27
Although PA-specific antibodies are capable of neutralizing anthrax toxin activity,28 it has been reported that antibodies of IgG2a type or Th1 response is able to initiate powerful effector activities such as complement fixation and antibody-dependent cellular cytotoxicity to prevent the germination of spores and lead to their destruction through sporicidal or germination inhibitory mechanisms.29 Hence, for an anthrax vaccine, it is desirable that it is able to elicit both humoral and cellular immunity.
A mixed response is assumed to confer protection against BA infection.30 Spleens from three mice of each group were isolated, and splenocyte was harvested and stimulated in vitro with D4 or PA and with LPS for positive control and only media for negative control. Supernatants were collected after 48 hours and analyzed for IL4 (Th2 stimulatory cytokine, humoral-biased immune response), IFN-γ (Th1 stimulatory cytokine, evident for strong cell-mediated immune response), IL-10 (anti-inflammatory cytokine), and TNF-α (pro-inflammatory). The level of Th2 stimulatory cytokine, IL-4, secreted by NISV + PA (Figure 10) was 200±18 pg/mL, while the level of Th1 stimulatory cytokine, IFN-γ, was 102±18 pg/mL. The level of IL-4 elicited by splenocytes of mice immunized with PA only was 42±3 pg/mL, while the level of IFN-γ was 30±1 pg/mL. For mice immunized with NISV + D4 (Figure 11), the level of Th2 stimulatory cytokine, IL-4, secreted by splenocytes on stimulating with D4 was 141±3 pg/mL, while the level of Th1 stimulatory cytokine, IFN-γ, was 85±13 pg/mL. While, for the mice injected with D4 only, the level of IL4 was 37±1 pg/mL, and that of IFN-γ was 17±2 pg/mL. These results indicated a mixed Th1/Th2 response. In order to get a conclusive idea about the inflammatory property of niosome, TNF-α and IL-10 levels were measured in splenocyte culture supernatant after stimulation with PA and D4, respectively. The levels of IL-10 were 170±19 pg/mL for NISV + PA-immunized mice and 121±12 pg/mL for NISV + D4-immunized mice. The level of TNF-α was calculated to be 20±3 pg/mL for NISV + PA-immunized mice, while for NISV + D4-immunized mice, it was found to be 22±3 pg/mL. The results of IL-10 and TNF-α of NISV-encapsulated PA and D4 suggest anti-inflammatory property of niosome.
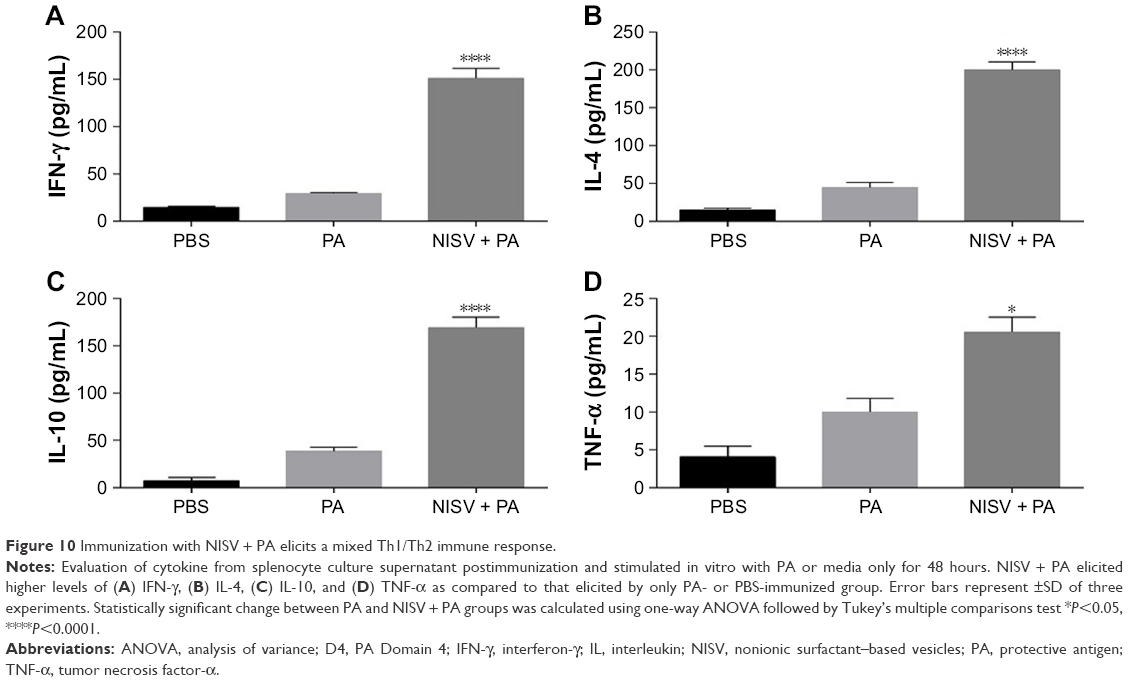 | Figure 10 Immunization with NISV + PA elicits a mixed Th1/Th2 immune response. |
 | Figure 11 Immunization with NISV + D4 elicits a mixed Th1/Th2 immune response. |
Protective efficacy of NISV + PA and NISV + D4 against anthrax spore challenge
The protective efficacy of NISV + PA and NISV + D4 against BA spore challenge was evaluated in Swiss albino mice, as they suit better as a model to correlate to human vaccine response.31 The route of spore injection was IP as spores were observed to evade phagocytosis by this route, by germinating into vegetative cells in the peritoneal cavities.32 All mice groups were injected with spores of a virulent BA strain as mentioned in the “Materials and Methods” section. As a control, mice immunized with alhydrogel + PA and alhydrogel + D4 were injected with the same number of spores. Infected mice were kept under observation for 14 days, and survival percentage was calculated using Kaplan–Meir curve.
As shown in Figure 12, the anthrax spore challenge experiment revealed that mice immunized with PA alone were able to provide only 20% protection, while mice immunized with D4 alone were not able to provide any protection. Mice immunized with alhydrogel + PA and alhydrogel + D4 showed protection of 80% and 30%, respectively. Both NISV + PA and NISV + D4 were able to provide protection against BA infection in Swiss albino mice and showed an increase in mean survival after infection. NISV + PA offered 80% survivability, while for NISV + D4, the percentage survival was 50%. This shows that niosome can be used as a potential vaccine adjuvant against anthrax, as it could provide similar/better protection against anthrax spore challenge in mice when compared to alhydrogel.
 | Figure 12 NISV + PA and NISV + D4 protect Swiss albino mice against anthrax spore challenge. |
Discussion
Aluminum hydroxide and alum are still the choice of adjuvants in the currently available vaccines against anthrax, with aluminum salts being able to elicit robust Th2-based immune response.33 However, these vaccines suffer from drawbacks such as requirement of multiple booster doses over a long regime to maintain protective antibody titers and adjuvant side effects such as fatigue, rashes, joint pains, and sleep disturbances.34 Therefore, much effort toward the generation of better vaccine against anthrax has been invested, like PLGA, which is a US Food and Drug Administration–approved polymer and has been studied as a potential vaccine adjuvant against anthrax, eg, PLGA-encapsulated D4,25 and PLGA–dendron NP-based PA–DNA vaccine,35 dual function viral NP,36 where the authors have developed a chimeric particle by multivalent display of the anthrax toxin cell receptor binding region on the surface of an icosahedral insect nodavirus, Flock House virus, to act as an antitoxin and vaccine candidate and CpG-Ficoll NP adjuvant encapsulating PA,37 which is based on linking sucrose polymer Ficoll to synthetic oligonucleotides containing CpG-rich motifs (CpG-oligodeoxynucleotide) and has been shown to provide protection against inhaled anthrax in monkeys.
A versatile adjuvant capable of enhancing the immune response over a wide range of antigens is desirable. In this study, we have investigated the immune response generated by niosome encapsulating PA and D4 and its protective efficacy against anthrax spore challenge for the first time. Niosome were prepared using span-60 and cholesterol via reverse-phase evaporation technique as this method has been reported to encapsulate hydrophilic molecules with relatively higher efficiency.38 It has also been reported that niosome prepared from span-60 has been reported to show higher entrapment efficiency as compared to its counterparts such as span-80 and span-40.39 In-process stability of the encapsulated PA and D4 was assessed by SDS-PAGE analysis, which revealed that the antigens maintained their integrity during the preparation of niosome. In vitro uptake study in macrophage showed an enhanced uptake of D4-encapsulated niosome, suggesting antigen cleavage within the macrophage and presented through MHC class I pathway leading to Th1 immunity, while the Th2 immune response could be due to the initial protein released by the enzymatic degradation of niosome and presented by B cells through MHC class II pathway to Th2 cells.
A study by Bielinska et al40 has shown that IgG2a is better neutralization of toxins than IgG1. Similar studies by Merkel et al29 and Cote et al41 showed that anti-PA IgG2a or Th1 response can initiate the compliment fixation and antibody-dependent cellular toxicity, which can be sporicidal or alternatively inhibit spore germination. Although humoral response is the prime requirement for the clearance of toxin against anthrax infection, cell-mediated immune response is also essential to combat intracellular stage of infection, which is mediated by cytokines activated by activated T cells. In our study, the niosome formulations along with a heightened IgG response were also able to modulate the quality of immune response, as observed from IgG1 and IgG2a titers of both PA- and D4-encapsulated niosome. However, the production of IgG1 antibody titer was significantly higher than that of IgG2a, which indicated the Th2-dominant immune response. IgG1 and IgG2a titers give an idea of the contribution of Th1- and Th2-mediated immune response elicited by the formulation. The results from our study showed a significant increase in IgG1 and IgG2a antibody titers elicited by both PA- and D4-encapsulated niosome, as compared to only PA and D4, which were able to induce low levels of IgG1 and relatively no IgG2a antibody titer. This demonstrates that niosomes were able to modulate the quality of immune response of the antigen from a predominantly Th2 response along with a mixed Th1/Th2 type immune response.
In order to probe the involvement of Th cell-derived cytokines in the adjuvant activity of niosome, we analyzed the cytokine profile secreted by splenic T cells from immunized mice. Splenocytes of both PA- and D4-encapsulated niosome mice secreted a high quantity of IL-4 on stimulation with PA and D4, respectively, which is consistent with the high levels of IgG1 antibody titer. The level of IFN-γ was also high in the splenocyte culture supernatant of both NISV + PA and NISV + D4 as compared to only PA- and D4-immunized mice. Hence, both cytokine profiling and antibody isotyping confirm a mixed Th1/Th2 immune response by both PA- and D4-encapsulated niosome.
The introduction of nanomaterials to biomedicine since the 1970s has paved the way to early detection, diagnosis, and targeted treatment of the disease. However, the nanomaterials, being in nanoscale range and having high surface-to-volume ratio, can enter the circulatory and lymphatic system causing irreversible injuries through oxidative or nitric oxide stress and are critical mediators of inflammation. Hence, in order to investigate the inflammatory property of PA- and D4-loaded niosome, we checked for the presence of pro- and anti-inflammatory cytokines (TNF-α and IL-10) in the splenocyte culture supernatant. The stimulation of the splenocyte with the respective antigen induced a significant level of IL-10 with very low level of TNF-α production, suggesting the anti-inflammatory behavior of niosome. IL-10 has been shown to play a role in pathogen clearance by protecting the innate immune cells from apoptosis by the downregulation of TNF-α and elevated production of IFN-γ by natural killer cells at times of high pathogen load,42 which can also be a factor for the protective efficacy of the niosome formulations. Although it has been reported that positively charged particles are more likely to induce inflammatory reactions and activate the cells of the immune system as compared to anionic and neutral particles,43 the enhanced immune response may be due to the enhanced phagocytosis of the negatively charged niosome particles by macrophage cells in vivo as reported by Lunov et al,44 that anionic polystyrene NPs are phagocytosed by macrophages as compared to positively charged polystyrene NPs.
Irrespective of the immune response elicited, the protective efficacy offered by a vaccine formulation is of critical importance for it to proceed for clinical trials. In order to determine the efficacy of the two vaccine formulations, we performed a challenge study in the immunized mice and compared the protective efficacy as compared to unimmunized mice. While PA and D4 were able to provide only 20% and 0% protection against anthrax spore challenge, niosome-encapsulated PA and D4 enhanced the protective efficacy up to 80% and 50% respectively. This can be attributed to toxin neutralization by the synergistic effect of IgG2a antibodies along with IgG1 antibodies, as it has been shown earlier that IgG2a was able to neutralize anthrax toxins better as compared to IgG1 antibodies.29 The protective correlation between PA- and D4-encapsulated niosome can be attributed to the fact that PA being a full-length molecule is a better immunogen as compared to D4.
In conclusion, this study shows that niosome is a powerful adjuvant which is able to induce a mixed Th1/Th2 response against both PA and D4 along with a robust IgG response and also provides protection against anthrax spore challenge in mice model. This improved efficacy of the niosome formulation can be attributed to the synergistic effect of enhanced cellular uptake of the niosomes as compared to only PA/D4 along with a slow and prolonged release of the antigen. This proposed system is economical and potentially safe as it does not induce inflammatory cytokines and can be regarded as a delivery vehicle of antigen against anthrax. However, the adjuvant potential and protective efficacy of niosome against other pathogens have also to be probed into.
Acknowledgments
We are grateful to the University Grants Commission for providing financial assistance during the tenure of the work. We would like to extend our sincere thanks to the staff of the Advanced Instrumental Research Facility: Gajender Saini (TEM), Saroj Kumar Jha (AFM), Ashok Kumar Sahu (confocal microscopy), and Smita Sundaram (flow cytometry). We would like to thank Department of Science and Technology (DST)/SR/PURSE PHASE II/11 for financial assistance to publish this work.
Disclosure
The authors report no conflicts of interest in this work.
References
Inglesby TV, O’Toole T, Henderson DA, et al. Anthrax as a biological weapon, 2002: updated recommendations for management. JAMA. 2002;287(17):2236–2252. | ||
Friedlander AM, Welkos SL, Pitt ML, et al. Postexposure prophylaxis against experimental inhalation anthrax. J Infect Dis. 1993;167(5):1239–1242. | ||
Kaur M, Bhatnagar R. Recent progress in the development of anthrax vaccines. Recent Pat Biotechnol. 2011;5(3):148–159. | ||
Flick-Smith HC, Walker NJ, Gibson P, et al. A recombinant carboxy-terminal domain of the protective antigen of Bacillus anthracis protects mice against anthrax infection. Infect Immun. 2002;70(3):1653–1656. | ||
Scobie HM, Rainey GJ, Bradley KA, Young JA. Human capillary morphogenesis protein 2 functions as an anthrax toxin receptor. Proc Natl Acad Sci U S A. 2003;100(9):5170–5174. | ||
Bradley KA, Mogridge J, Mourez M, Collier RJ, Young JA. Identification of the cellular receptor for anthrax toxin. Nature. 2001;414(6860):225–229. | ||
Mogridge J, Cunningham K, Lacy DB, Mourez M, Collier RJ. The lethal and edema factors of anthrax toxin bind only to oligomeric forms of the protective antigen. Proc Natl Acad Sci U S A. 2002;99(10):7045–7048. | ||
Kintzer AF, Thoren KL, Sterling HJ, et al. The protective antigen component of anthrax toxin forms functional octameric complexes. J Mol Biol. 2009;392(3):614–629. | ||
Milne JC, Furlong D, Hanna PC, Wall JS, Collier RJ. Anthrax protective antigen forms oligomers during intoxication of mammalian cells. J Biol Chem. 1994;269(32):20607–20612. | ||
Mogridge J, Cunningham K, Collier RJ. Stoichiometry of anthrax toxin complexes. Biochemistry. 2002;41(3):1079–1082. | ||
Cunningham K, Lacy DB, Mogridge J, Collier RJ. Mapping the lethal factor and edema factor binding sites on oligomeric anthrax protective antigen. Proc Natl Acad Sci U S A. 2002;99(10):7049–7053. | ||
Abrami L, Lindsay M, Parton RG, Leppla SH, van der Goot FG. Membrane insertion of anthrax protective antigen and cytoplasmic delivery of lethal factor occur at different stages of the endocytic pathway. J Cell Biol. 2004;166(5):645–651. | ||
Klimpel KR, Arora N, Leppla SH. Anthrax toxin lethal factor contains a zinc metalloprotease consensus sequence which is required for lethal toxin activity. Mol Microbiol. 1994;13(6):1093–1100. | ||
Duesbery NS, Webb CP, Leppla SH, et al. Proteolytic inactivation of MAP-kinase-kinase by anthrax lethal factor. Science. 1998;280(5364):734–737. | ||
Vitale G, Bernardi L, Napolitani G, Michèle M, Montecucco C. Susceptibility of mitogen-activated protein kinase kinase family members to proteolysis by anthrax lethal factor. Biochemical Journal. 2000;352(3):739–745. | ||
Vitale G, Pellizzari R, Recchi C, Napolitani G, Mock M, Montecucco C. Anthrax lethal factor cleaves the N-terminus of MAPKKs and induces tyrosine/threonine phosphorylation of MAPKs in cultured macrophages. Biochem Biophys Res Commun. 1998;248(3):706–711. | ||
Leppla SH. Anthrax toxin edema factor: a bacterial adenylate cyclase that increases cyclic AMP concentrations of eukaryotic cells. Proc Natl Acad Sci U S A. 1982;79(10):3162–3166. | ||
Bozzuto G, Molinari A. Liposomes as nanomedical devices. Int J Nanomedicine. 2015;10:975. | ||
Hunt CA, Tsang S. α-Tocopherol retards autoxidation and prolongs the shelf-life of liposomes. Int J Pharm. 1981;8(2):101–110. | ||
Wong M, Thompson TE. Aggregation of dipalmitoylphosphatidylcholine vesicles. Biochemistry. 1982;21(17):4133–4139. | ||
Baillie AJ, Florence AT, Hume LR, Muirhead GT, Rogerson A. The preparation and properties of niosomes – non-ionic surfactant vesicles. J Pharm Pharmacol. 1985;37(12):863–868. | ||
Uchegbu IF, Florence AT. Nonionic surfactant vesicles (niosomes): physical and pharmaceutical chemistry. Adv Colloid Interface Sci. 1995;58(1):1–55. | ||
Kumar GP, Rajeshwarrao P. Nonionic surfactant vesicular systems for effective drug delivery – an overview. Acta Pharm Sin B. 2011;1(4):208–219. | ||
Brewer JM, Alexander J. The adjuvant activity of non-ionic surfactant vesicles (niosomes) on the BALB/c humoral response to bovine serum albumin. Immunology. 1992;75(4):570. | ||
Manish M, Rahi A, Kaur M, Bhatnagar R, Singh S. A single-dose PLGA encapsulated protective antigen domain 4 nanoformulation protects mice against Bacillus anthracis spore challenge. PLoS One. 2013;8(4):e61885. | ||
Moghassemi S, Hadjizadeh A, Omidfar K. Formulation and characterization of bovine serum albumin-loaded niosome. AAPS PharmSciTech. 2017;18(1):27–33. | ||
Tournier JN, Rossi Paccani S, Quesnel-Hellmann A, Baldari CT. Anthrax toxins: a weapon to systematically dismantle the host immune defenses. Mol Aspects Med. 2009;30(6):456–466. | ||
Brenneman KE, Doganay M, Akmal A, et al. The early humoral immune response to Bacillus anthracis toxins in patients infected with cutaneous anthrax. FEMS Immunol Med Microbiol. 2011;62(2):164–172. | ||
Merkel TJ, Perera PY, Kelly VK, et al. Development of a highly efficacious vaccinia-based dual vaccine against smallpox and anthrax, two important bioterror entities. Proc Natl Acad Sci U S A. 2010;107(42):18091–18096. | ||
Mcbride BW, Mogg A, Telfer JL, et al. Protective efficacy of a recombinant protective antigen against Bacillus anthracis challenge and assessment of immunological markers. Vaccine. 1998;16(8):810–817. | ||
Hughes HR, Crill WD, Chang GJ. Manipulation of immunodominant dengue virus E protein epitopes reduces potential antibody-dependent enhancement. Virol J. 2012;9(1):115. | ||
Welkos SL, Keener TJ, Gibbs PH. Differences in susceptibility of inbred mice to Bacillus anthracis. Infect Immun. 1986;51(3):795–800. | ||
Lindblad EB. Aluminium compounds for use in vaccines. Immunol Cell Biol. 2004;82(5):497–505. | ||
Unwin C, Blatchley N, Coker W, et al. Health of UK servicemen who served in Persian Gulf War. Lancet. 1999;353(9148):169–178. | ||
Ribeiro S, Rijpkema SG, Durrani Z, Florence AT. PLGA-dendron nanoparticles enhance immunogenicity but not lethal antibody production of a DNA vaccine against anthrax in mice. Int J Pharm. 2007;331(2):228–232. | ||
Manayani DJ, Thomas D, Dryden KA, et al. A viral nanoparticle with dual function as an anthrax antitoxin and vaccine. PLoS Pathog. 2007;3(10):e142. | ||
Kachura MA, Hickle C, Kell SA, et al. A CpG-Ficoll nanoparticle adjuvant for anthrax protective antigen enhances immunogenicity and provides single-immunization protection against inhaled anthrax in monkeys. J Immunol. 2016;196(1):284–297. | ||
Szoka F, Papahadjopoulos D. Procedure for preparation of liposomes with large internal aqueous space and high capture by reverse-phase evaporation. Proc Natl Acad Sci U S A. 1978;75(9):4194–4198. | ||
Kamboj S, Saini V, Bala S, Sharma G. Formulation and characterization of drug loaded niosomal gel for anti-inflammatory activity. Int Sch Sci Res Innov. 2013;2013(7):541–545. | ||
Bielinska AU, Janczak KW, Landers JJ, et al. Mucosal immunization with a novel nanoemulsion-based recombinant anthrax protective antigen vaccine protects against Bacillus anthracis spore challenge. Infect Immun. 2007;75(8):4020–4029. | ||
Cote CK, Rossi CA, Kang AS, Morrow PR, Lee JS, Welkos SL. The detection of protective antigen (PA) associated with spores of Bacillus anthracis and the effects of anti-PA antibodies on spore germination and macrophage interactions. Microb Pathog. 2005;38(5–6):209–225. | ||
Londoño D, Marques A, Hornung RL, Cadavid D. IL-10 helps control pathogen load during high-level bacteremia. J Immunol. 2008;181(3):2076–2083. | ||
Dobrovolskaia MA, Mcneil SE. Immunological properties of engineered nanomaterials. Nat Nanotechnol. 2007;2(8):469–478. | ||
Lunov O, Syrovets T, Loos C, et al. Differential uptake of functionalized polystyrene nanoparticles by human macrophages and a monocytic cell line. ACS Nano. 2011;5(3):1657–1669. | ||
Rehm B, Schueler D. Small, smaller, smallest. In: Kreysa G, Marquardt R, editors. Biotechnology 2020: From the Transparent Cell to the Custom-Designed Process. Brussels: European Commission; Directorate-General for Research; 2005:58–63. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.