Back to Journals » International Journal of Nanomedicine » Volume 15
A NAG-Guided Nano-Delivery System for Redox- and pH-Triggered Intracellularly Sequential Drug Release in Cancer Cells
Authors Liang Y ![]() , Zhang J, Tian B, Wu Z
, Zhang J, Tian B, Wu Z ![]() , Svirskis D
, Svirskis D ![]() , Han J
, Han J ![]()
Received 6 August 2019
Accepted for publication 10 December 2019
Published 5 February 2020 Volume 2020:15 Pages 841—855
DOI https://doi.org/10.2147/IJN.S226249
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Mian Wang
Yan Liang,1 Jing Zhang,1 Baocheng Tian,1 Zimei Wu,2 Darren Svirskis,2 Jingtian Han1
1School of Pharmacy, Binzhou Medical University, Yantai 264003, Shandong Province, People’s Republic of China; 2School of Pharmacy, Faculty of Medical and Health Sciences, The University of Auckland, Auckland 1023, New Zealand
Correspondence: Jingtian Han
School of Pharmacy, Binzhou Medical University, 346 Guanhai Road, Yantai 264003, People’s Republic of China
Tel +86-535-6913317
Fax +86-535-6913718
Email [email protected]
Darren Svirskis
School of Pharmacy, University of Auckland, 85 Park Road, Grafton, Auckland, New Zealand
Tel +64-9-923 1158
Email [email protected]
Aim: Sequential treatment with paclitaxel (PTXL) and gemcitabine (GEM) is considered clinically beneficial for non-small-cell lung cancer. This study aimed to investigate the effectiveness of a nano-system capable of sequential release of PTXL and GEM within cancer cells.
Methods: PTXL-ss-poly(6-O-methacryloyl-d-galactopyranose)-GEM (PTXL-ss-PMAGP-GEM) was designed by conjugating PMAGP with PTXL via disulfide bonds (-ss-), while GEM via succinic anhydride (PTXL:GEM=1:3). An amphiphilic block copolymer N-acetyl-d-glucosamine(NAG)-poly(styrene-alt-maleic anhydride)58-b-polystyrene130 acted as a targeting moiety and emulsifier in formation of nanostructures (NLCs).
Results: The PTXL-ss-PMAGP-GEM/NAG NLCs (119.6 nm) provided a sequential in vitro release of, first PTXL (redox-triggered), then GEM (pH-triggered). The redox- and pH-sensitive NLCs readily distributed homogenously in the cytoplasm. NAG augmented the uptake of NLCs by the cancer cells and tumor accumulation. PTXL-ss-PMAGP-GEM/NAG NLCs exhibited synergistic cytotoxicity in vitro and strongest antitumor effects in tumor-bearing mice compared to NLCs lacking pH/redox sensitivities or free drug combination.
Conclusion: This study demonstrated the abilities of PTXL-ss-PMAGP-GEM/NAG NLCs to achieve synergistic antitumor effect by targeted intracellularly sequential drug release.
Keywords: sequential release, redox-sensitive, pH-sensitive, synergistic efficiency, combination drug delivery, gemcitabine, paclitaxel
Background
The incidence of non-small-cell lung cancer (NSCLC) is increasing, with more effective and well-tolerated treatment options urgently needed.1–3 Two chemotherapeutic drugs commonly used in NSCLC therapy include gemcitabine (GEM) and paclitaxel (PTXL), with independent mechanisms of action and non-overlapping toxic effects.4,5 GEM is a hydrophilic molecule that interferes with DNA synthesis whereas PTXL is a hydrophobic molecule that blocks microtubule disassembly. The GEM-PTXL combinational therapy is recommended for advanced or metastatic NSCLC.6–9 Interestingly, the clearance of GEM from patients can be favorably reduced in the presence of PTXL.8 The sequence of administration of the two anticancer drugs can be critical to achieve the most beneficial effects.10 Studies demonstrate that PTXL can strengthen the metabolic and apoptotic index of GEM at the cellular level, and that administration of PTXL prior to GEM could maximize the anticancer benefits.5,7 However, a significant challenge for many chemotherapeutic drugs including PTXL and GEM is their lack of specificity, causing dose-dependent severe side effects to patients.11 Furthermore, it is a technical challenge to achieve timely sequential drug delivery in the same cancer cells as hydrophobic and hydrophilic drugs have different pharmacokinetic profiles and bio-distribution patterns to the cells, where GEM relies on drug transporters to get into the cells.12
In the drug delivery field, nanostructured lipid carriers (NLCs)13 have been employed as an anticancer drug delivery tool owing to their ability to encapsulate drug efficiently,14 reduce side effects15 and prolong the half-life of drugs,16 thus enhancing the therapeutic effects. Relatively rapid drug release can be achieved inside tumor cells by utilizing intracellular stimuli, such as redox and low pH as a single or dual-trigger.17,18 For example, biomaterials containing acid-labile bonds such as ester bond can be used to fabricate nanocarriers for targeted drug release to the acidic microenvironment of tumor, both intracellularly (pHin, endo-lysosomal lumen pH 5–6.5) and extracellularly (pHex; >6).18 Meanwhile, nano-structures containing a redox-sensitive disulfide bond (-ss-) have been utilized for rapid tumor-specific intracellular drug release by exploiting the reducing potential in tumor cells as a stimulus.19 The tumoral intracellular concentration of glutathione (GSH, 2–10 mM) is much higher concentrations than the extracellular levels (20–40 μM).20 Therefore, disulfide bonds are preferentially cleaved in the cancer cells, while remaining relatively stable in the extracellular space.21–23 In addition, tumor cells, including NSCLCs tend to have a high expression of glucose transporters (GLUT),24,25 therefore, N-acetyl-d-glucosamine (NAG) has been used as a targeting moiety to enhance glucose receptor-mediated endocytosis of nano-scaled drug delivery systems.26,27 We have previously found that 3:1 was the optimal molar ratio of GEM and PTXL in treating NSCLC mouse models.28
In this study, we aimed to investigate the feasibility and effectiveness of a dual-drug-polymer conjugate incorporated in an NLCs system for sequential release of PTXL and GEM within cancer cells using pHin and intracellular GSH as stimuli for treatment NSCLC. Specifically, drug–polymer conjugate PTXL-ss-poly(6-O-methacryloyl-d-galactopyranose)-GEM (PTXL-ss-PMAGP-GEM) was synthesized. The PTXL:GEM molar ratio of 1:3 was adopted during synthesis. An amphiphilic block copolymer NAG-poly(styrene-alt-maleic anhydride)58-b-polystyrene130 (NAG-P(St-alt-MA)58-b-PSt130) was used as the targeting moiety as well as an emulsifier for the preparation of NLCs. It was hypothesized that NAG would promote specific cellular uptake of the PTXL-ss-PMAGP-GEM/NAG NLCs by the cancer cells via NAG-mediated endocytosis, then sequential and intracellular drug release could be achieved where first, PTXL cleaved off in response to intracellular GSH, followed by GEM release via hydrolysis of ester bonds due to low pHin. The abilities of sequential drug release, cellular uptake and intracellular trafficking, and anti-tumor efficacy of the PTXL-ss-PMAGP-GEM/NAG NLCs were critically evaluated against various reference formulations.
Materials and Methods
Materials
Paclitaxel (PTXL), gemcitabine (GEM) and 3,3ʹ-dithiodipropionic acid (DPA) were purchased from Aladdin (Shanghai, China). 4-Dimethylaminopyridine (DMAP) and 1-ethyl-3-(3-dimethylaminopropyl)-carbodiimide hydrochloride (EDC) were from Dojindo Laboratories (Tokyo, Japan). The targeting moiety NAG-P(St-alt-MA)58-b-PSt130 was synthesized according to our previous work.29 All other chemicals were analytical reagent grade.
For cell culture studies, human pulmonary adenocarcinoma cell lines A549, LTEP-a-2, and L929 mouse fibroblasts were purchased from Bena Culture Collection (Beijing, China) and reagents, 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) and Hoechst 33342 were from Sigma-Aldrich (St Louis, MO, USA). Fetal bovine serum (FBS), 1% penicillin-streptomycin, RPMI-1640 medium and 0.25% trypsin/EDTA were purchased from Hyclone (Utah, USA). Fluorescein isothiocyanate (FITC) was purchased from Fanbo Biochemicals (Beijing, China).
For animal studies, male BALB/c nude mice (18–20 g) were purchased from Beijing Vital River Laboratory Animal Technology Co., Ltd. (Beijing, China). 1,1ʹ-Dioctadecyl-3,3,3ʹ,3ʹ-tetramethylindotricarbocyanine iodide (DiR) was purchased from Fanbo Biochemicals (Beijing, China). Paclitaxel injections Medaxol (5 mL, 30 mg) were purchased from Beijing Union Pharmaceutical Factory (Beijing, China).
The animal experiments, care and handling of animals were performed followed the National Institute of Health Guide for the Care and Use of Laboratory Animals in China, and were approved by the Experimental Animals Administrative Committee of Binzhou Medical University.
Synthesis and Characterization of PTXL-ss-PMAGP-GEM
Synthesis of PMAGP
The copolymer poly(6-O-methacryloyl-1,2;3,4-di-O-isopropylidene-D-galactopyranose) (PMaIPG) was synthesized via reversible addition-fragmentation chain-transfer (RAFT) polymerization according to our previous report.28 Briefly, PMaIPG (200 mg) was dissolved in 80% methanoic acid (20 mL) and stirred for 48 h. Distilled water (3 mL) was then added and stirred. The pure PMAGP was obtained by dialyzing followed by freeze-drying (FDU-2110; Rikakikai, Tokyo, Japan).
Synthesis and Characterization of PTXL-ss-DPA
EDC (1.5 mmol) and DMAP (1.5 mmol) were added to CH2Cl2 (20 mL) with DPA (1.5 mmol) at 0°C. In order to activate the carboxyl group of DPA, the reaction was stirred for 0.5 h under nitrogen. Then, CH2Cl2 (5 mL) containing PTXL (0.5 mmol) was added dropwise to the solution. After stirring for 24 h at room temperature, thin-layer chromatography (TLC) was used to monitor the reaction with chloroform/acetone (1:1, v/v) as eluent. The product was washed three times with 0.01 mM HCl and distilled water, respectively. PTXL-ss-DPA was collected after vacuum-drying. Proton nuclear magnetic resonance (1H-NMR) spectra were recorded on an AM-400 spectrometer (Bruker Optik GmbH, Ettlingen, Germany) using deuterated DMSO-d6 as the solvent.
Synthesis and Characterization of PTXL-ss-PMAGP-GEM
Succinic anhydride (0.89 mM) and GEM•HCl (0.44 mM) reacted in anhydrous DMF containing TEA (62 μL) for 24 h. After 24 h, the progress was monitored by TLC (chloroform/methanol, 2:1, v/v). The solvent was evaporated under vacuum and recrystallized from a diethyl ether-chloroform system (1:3 v/v). Finally, 2ʹ-succinyl-GEM was obtained by filtrating followed by vacuum-drying for 48 h.
Defined amounts of PTXL-ss-DPA, DCC (0.08 mmol) and DMAP (4 mmol) were mixed in 10 mL DMF with PMAGP (100 mg) under nitrogen. TLC (chloroform/methanol, 5:3, v/v) was applied after stirring at 70°C for 24 h. 2ʹ-Succinyl-GEM was added to the mixture and stirred at 70°C for another 24 h, followed by TLC (chloroform/acetone, 1:1, v/v). Finally, the product was dialyzed with deionized water (MWCO = 4,000 Da), and PTXL-ss-PMAGP-GEM conjugates were obtained after freeze-dried. An optimal molar ratio of GEM and PTXL 3:1 was chosen28 for the PTXL-ss-PMAGP-GEM conjugates, and was confirmed by double-wavelength ultraviolet spectrophotometry, following the principle reported in the literature.30,31 The 1H-NMR spectra were recorded using deuterated DMSO-d6 as the solvent.
The thermal behaviors of PTXL, GEM, a physical mixture of PTXL and GEM and PTXL-ss-PMAGP-GEM conjugates were studied by a differential scanning calorimeter (DSC, Mettler Toledo).
As controls, non-disulfide functionalized PTXL-PMAGP-GEM conjugates were also synthesized according to our previous report.28
Fabrication and Characterization of NLCs
Preparation of NLCs
The emulsification and solvent evaporation was used to prepare the PTXL-ss-PMAGP-GEM/NAG NLCs. Briefly, PTXL-ss-PMAGP-GEM (20 mg), glycerol monostearate (250 mg), NAG-P(St-alt-MA)58-b-PSt130 (250 mg), liquid lipid (100 μL), soy lecithin (100 mg), Tween 80 (40 mg) and methanol (3 mL) were mixed in a glass bottle at 70°C. Sodium deoxycholate (30 mg) and poloxamer 188 (220 mg) were dissolved in distilled water and heated to the same temperature. The aqueous phase was then spread evenly on the organic phase after the methanol can completely evaporate. The dispersions were emulsified for 40 min at 70°C and cooled in an ice-box for 30 min. Finally, the PTXL-ss-PMAGP-GEM/NAG NLCs were filtrated through a membrane filter (0.45 μm) and obtained by washing with an Amicon Ultra centrifugal filter (MWCO 100 kDa).
PTXL-PMAGP-GEM/NAG NLCs, without the disulfide bond, and blank PMAGP/NAG NLCs, without drug, were also obtained by a similar process except replacing PTXL-ss-PMAGP-GEM with PTXL-PMAGP-GEM or PMAGP, respectively.
Characterization of NLCs
The size and ζ-potential of NLCs were detected by dynamic light scattering (DLS, Malvern Instruments, UK) at room temperature. The morphologies of NLCs were observed by transmission electron microscopy (TEM, JEM-1230; JEOL, Tokyo, Japan).
To measure encapsulation efficiency (EE) and drug loading (DL), drug-loaded NLCs without washing were first diluted and placed in an Amicon Ultra centrifugal filter (molecular weight cutoff 100 kDa). After centrifugation at 8,000 rpm for 20 min, the bottom liquid was collected and free drug content determined. The amounts of PTXL-ss-PMAGP-GEM or equivalent to free drugs were quantitated by double-wavelength ultraviolet spectrophotometer, and EE and DL were calculated using the Equations:
(1) (2)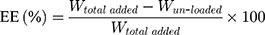

where Wtotal added and Wun-loaded are the total weight of drug conjugate added during NLCs preparation and the amount of un-loaded PTXL-ss-PMAGP-GEM, respectively. Dtotal and Dfree are the amount of total added and un-loaded drug in PTXL-ss-PMAGP-GEM, while Wlipid is the weight of lipids.
Characterization of Drug Release Properties
The in vitro drug release properties of PTXL-ss-PMAGP-GEM/NAG NLCs were examined under different conditions. Briefly, 2 mL of PTXL-ss-PMAGP-GEM/NAG NLCs (containing 40 μg/mL total dual-drugs) were sealed in membrane dialysis bags (MWCO = 6,000–8,000 Da) and dialyzed against 40 mL PBS (10 mM, containing 0.2% Tween 80) at pH 6.0 (to simulate the tumor intracellular environment) or 7.4 (to simulate blood and normal tissue environment) with 0 mM, 10 mM (matching the intracellular tumor concentration), or 20 μM of GSH. The drug release process took place in a shaker incubator at 37°C (BS-2F; Zhengji Instruments, Changzhou, China). Periodically, 4 mL of release media was collected from outside the membrane dialysis bags and replaced with 4 mL of fresh releasing media. The amounts of GEM and PTXL in the release media were quantified using high-performance liquid chromatography (Agilent 1260) equipped with a C18 column (4.6 × 250 mm internal diameter, 5 μm). The chromatographic conditions were provided for GEM (acetonitrile: water, 5:95, 269 nm) and PTXL (acetonitrile; water, 60:40, 227 nm), respectively.
In vitro Cytotoxicity Assay
Human non-small-cell lung cancer cell lines A549 and LTEP-a-2, and mouse fibroblast cell lines L929 were cultured in RPMI-1640 with 10% fetal bovine serum. All cell lines were grown in a humidified incubator under 37°C, 5% CO2 atmosphere.
MTT assay was used to test the biocompatibility of blank PMAGP/NAG NLCs, three types of cell lines were seeded at a density of 1 × 104 cells/well in 96-well plates for 24 h. Cells were treated with blank PMAGP/NAG NLCs with varying polymer dosages from 0.128 to 400 μg/mL. After 72 h treatment, cells were washed, and 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) was added. Another 4 h later, 150 μL of DMSO was substituted to dissolve the formazan crystals generated. The relative cell viability (%) was defined as absorbance values at 490 nm with microplate spectrophotometry (SpectraMax M2, Molecular Devices, CA, USA). IC50 (the concentration at which cell viability was reduced by half) values was calculated.
The cytotoxicity profiles of different formulations against A549 and LTEP-a-2 cells were studied following the methodology above. The total concentration of PTXL and GEM was from 0.0128 to 40 μg/mL, and the incubation time was 72 h. The combination index (CI50) used to quantify the extent of synergism was estimated using the Chou’s method.32 Values of CI50 < 1, CI50 = 1 and CI50 > 1 indicate a synergism, additive and antagonistic effect, respectively, in the drug combination.
In vitro Cellular Uptake and Intracellular Drug Release
To investigate the ability for the NAG-targeted formulations to enhance cellular uptake and intracellular trafficking (distribution in cytoplasm), FITC-labeled PTXL-ss-PMAGP-GEM/NAG NLCs and FITC-labeled PTXL-ss-PMAGP-GEM NLCs were prepared. Briefly, 50 mg of each NLCs and FITC (5 mg) were dissolved in DMF and reacted at ambient temperature (about 20°C) for 24 h, followed by dialyzing and freeze-drying.
To quantify cellular uptake, A549 and LTEP-a-2 cells (which overexpress glucose receptors) were plated in six-well plates at a density of 2 × 105 cells/well, and treated with FITC-labeled PTXL-ss-PMAGP-GEM/NAG NLCs or FITC-labeled PTXL-ss-PMAGP-GEM NLCs with total drug concentrations of 5, 10, and 20 μmol/L, respectively. Untreated cells served as a control. Following incubation of 0.5, 2 and 4 h, respectively, cells were detached and washed with PBS three times. The cells were spread into 0.5 mL PBS and tested by flow cytometry (Epics XL; Beckman Coulter, Brea, CA, USA). Similarly, control cells L929 fibroblasts (with little or no glucose receptors) representing normal cells were also treated with 10 μmol/L FITC-PTXL-ss-PMAGP-GEM/NAG NLCs.
To evaluate the cellular uptake visually, confocal laser scanning microscopy (CLSM) images of A549 cells were acquired. A549 cells were seeded in six-well plates and incubated with FITC-labeled PTXL-ss-PMAGP-GEM/NAG NLCs and FITC-labeled PTXL-ss-PMAGP-GEM NLCs (10 and 20 μmol/L), respectively. After exposure for 1, 2 and 4 h, cells were washed and fixed by paraformaldehyde. After washing again, Hoechst 33342 (10 mg/mL) was added to stain cell nuclei, and the images of cells were observed via confocal microscopy (TCS SPE; Leica Microsystems, Wetzlar, Germany).
Animal Studies
An NSCLC tumor xenograft model was obtained by the subcutaneous injection of A549 cells (1 × 107 cells/mL, 0.2 mL in PBS) into the left leg limb of each mouse. The tumor volume (V) was monitored every second day and determined by the equation: V=length × width2/2.
In vivo Imaging Analysis
When the tumors were grown to ~700 mm3, tumor-bearing mice were applied for imaging analysis. For this study, the DiR-labeled PTXL-ss-PMAGP-GEM/NAG NLCs and PTXL-ss-PMAGP-GEM NLCs were prepared by incorporation of DiR at final concentration of 500 μg/mL.
The mice bearing A549 tumor were injected with 0.2-mL DiR-labeled PTXL-ss-PMAGP-GEM/NAG NLCs, DiR-labeled PTXL-ss-PMAGP-GEM NLCs and free DiR (500 μg/mL) via tail vein, respectively. The fluorescent images were taken on Carestream Molecular Imaging FX PRO (Carestream Health, Inc., USA) at 8 h post-injection. The fluorescence intensities were analyzed by Molecular Imaging Software version 5.X. At 8 h, the mice treated with the same method were sacrificed, the major organs (heart, liver, spleen, lung and kidney) as well as tumor were collected for ex vivo imaging.
In vivo Antitumor Efficiency
The mice bearing A549 tumor were allowed to grow to a volume of ~50 mm3 before treatment. The mice were divided to five groups (n = 6): (i) saline as control; (ii) free drug combination, GEM and PTXL; (iii) PTXL-ss-PMAGP-GEM NLCs; (iv) PTXL-PMAGP-GEM/NAG NLCs; or (v) PTXL-ss-PMAGP-GEM/NAG NLCs were administered via tail vein, all at a dose of 4.74 mg/kg GEM and 5.12 mg/kg PTXL. The animals were treated once a day for a total of 5 injections. Tumor volume and body weight were measured every other day to evaluate the treatment. On day 21, the mice were sacrificed, and the tumors were excised, weighed, and photographed.
Statistical Analysis
The differences between experimental groups were analyzed with Student’s t-test or one-way analysis of variance (ANOVA), and P < 0.05 indicated significant differences.
Results
Synthesis and Characterization of PTXL-ss-PMAGP-GEM Conjugates
The synthesis of PTXL-ss-PMAGP-GEM conjugates was conducted as illustrated in Figure 1.
 |
Figure 1 Synthesis route of the PTXL-ss-PMAGP-GEM conjugates (via steps A, B, C, and D). |
After 24 h of reaction, no free PTXL was detected by TLC, indicating the completion of the reaction. The 1H-NMR spectra (Figure 2A) showed the characteristic peaks at 2.47–2.95 ppm for DPA (-O-CO-CH2-CH2-), indicating that DPA was successfully conjugated with PTXL. Comparing the spectra of PMAGP (Figure 2B), the formation of the PTXL-ss-PMAGP-GEM was confirmed by the combined characteristic peaks at 7.2–8.4 ppm (PTXL phenyl ring) and 9.25–9.45 ppm (GEM 4C-NH2) (Figure 2C).
The DSC thermographs (Figure 2D) indicate the physical mixture maintained the typical melting peaks of PTXL and GEM, while the peaks of PTXL and GEM were absent in the thermal profiles of PTXL-ss-PMAGP-GEM. It could be concluded that the two drugs were grafted onto the polymer by chemical bonding rather than a physical mixture. Collectively, the results of 1H-NMR and DSC demonstrate the synthesis of PTXL-ss-PMAGP-GEM conjugates.
 |
Figure 2 The 1H-NMR spectra of PTXL-ss-DPA (A), PMAGP (B), and PTXL-ss-PMAGP-GEM in DMSO-d6 (C). (D) Differential scanning calorimetric (DSC) thermographs of (a) PTXL, (b) GEM, (c) physical mixture of PTXL and GEM, (d) and PTXL-ss-PMAGP-GEM (exo up). |
Characterization of NLCs
PTXL-ss-PMAGP-GEM/NAG NLCs were obtained with the emulsification and solvent evaporation method. DLS measurements revealed that the resulting PTXL-ss-PMAGP-GEM/NAG NLCs had a narrow-size distribution with a mean diameter of 119.6 nm ± 0.31 nm (Figure 3A). TEM imaging revealed monodispersed particles with a similar size (Figure 3B).
The physicochemical properties of PTXL-ss-PMAGP-GEM/NAG NLCs and PTXL-PMAGP-GEM/NAG NLCs are shown in Table 1.
 |
Figure 3 Characteristics of the PTXL-ss-PMAGP-GEM/NAG NLC nanoparticles. (A) Size distribution measured by DLS; (B) morphology by TEM; (C) the tumor micro-environmental triggered release profiles of PTXL, showing faster release rate in the presence of GSH (10 mM) particularly when pH is low (endosomal pH 6) than that in the extracellular environment (pH 7.4, and low GSH); and (D) the delayed release of GEM in the medium of pH 6 containing GSH 10 mM compared to PTXL. |
 |
Table 1 Characteristics of Different NLC Formulations. Data are Means ± SD (n = 3) |
In vitro Drug Release Studies
The drug release kinetics and sequential drug release property of PTXL-ss-PMAGP-GEM/NAG NLCs were determined using a membrane dialysis method under different pH and redox conditions. As shown in Figure 3C and D, both drugs showed sustained drug release patterns in normal extracellular environments (pH 7.4 and low GSH). However, the release rate of PTXL from PTXL-ss-PMAGP-GEM/NAG NLCs was significantly faster in the medium with 10-mM GSH compared to 0- or 20-μM GSH, particularly when the pH was lower which mimicked the tumoral cell environments (Figure 3C). In comparison, the release of GEM was also promoted by the low pH and 10-mM GSH, but at a much slower rate than PTXL (Figure 3D). The cumulative release (%) of PTXL at 12 h was approximately 35% and about 55% over the test duration of 24 h under pH 6.0 and 10-mM GSH conditions; however, it required an extra 24 h for GEM to achieve the similar proportion of release.
In vitro Cytotoxicity Analysis
The viability of LTEP-a-2, L929 and A549 cells all remained above 90% after incubating with blank PMAGP/NAG NLCs at different particle concentrations for 72 h, with no significant differences from the negative control group (P > 0.05) (Figure 4A). The results demonstrate that blank PMAGP/NAG NLCs showed negligible toxicity and good biocompatibility.
The cytotoxicity analysis of different formulations (free GEM + free PTXL, PTXL-ss-PMAGP-GEM NLCs, PTXL-ss-PMAGP-GEM/NAG NLCs, PTXL-PMAGP-GEM/NAG NLCs) was performed in A549 cells and LTEP-a-2 cells by MTT assay. As shown in Figure 4B and C, all formulations displayed cytotoxicity in a dose-dependent fashion. Compared to free drug combination in solution, NLCs enhanced cytotoxicity (Table 2). The IC50 of PTXL-ss-PMAGP-GEM/NAG NLCs was significantly lower than that of the PTXL-ss-PMAGP-GEM NLCs (without NAG). It was deduced that the targeting ligand NAG on the surface of NLCs could selectively bind with tumor cells overexpressing glucose receptor enhancing the drug effects. In addition, the cell-killing ability of PTXL-ss-PMAGP-GEM/NAG NLCs was greater than the non-pH- or redox-sensitive PTXL-PMAGP-GEM/NAG NLCs. NAG and the dual responsiveness appeared to increase cytotoxicity to the same extent.
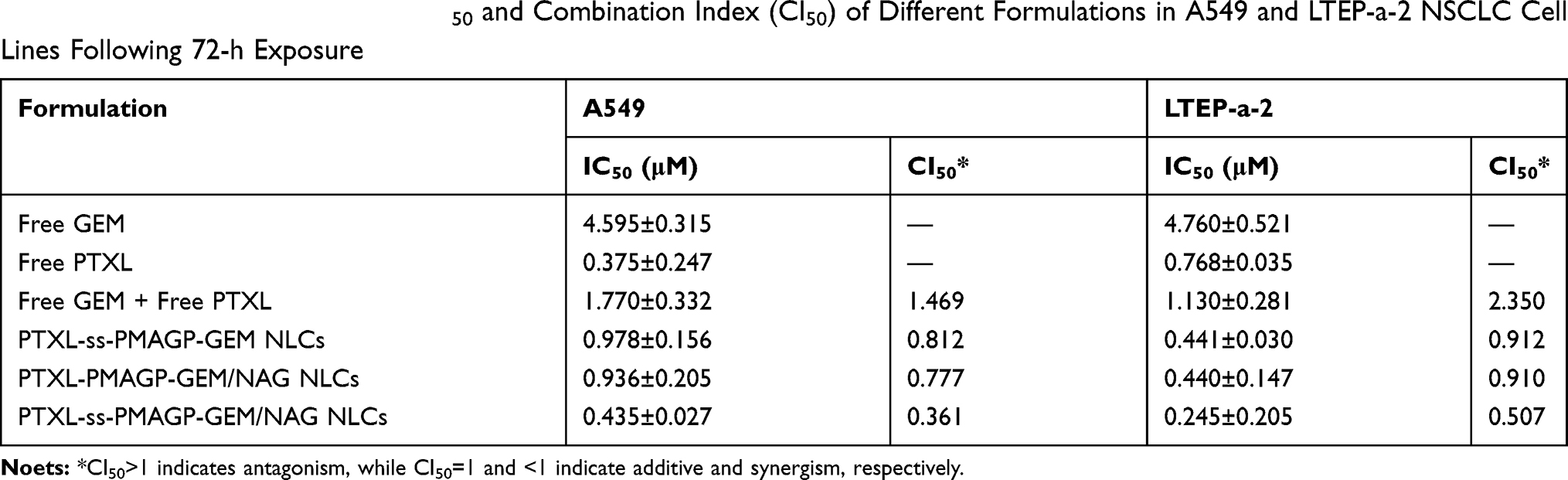 |
Table 2 Cytotoxicity Expressed as IC50 and Combination Index (CI50) of Different Formulations in A549 and LTEP-a-2 NSCLC Cell Lines Following 72-h Exposure |
In both cancer A549 cells, and LTEP-a-2 cells, the CI50 values were all lower than 1 for NLCs (Table 2), indicating synergistic cytotoxicity, but higher than 1 for free GEM + free PTXL, indicating antagonism. The CI50 values of PTXL-ss-PMAGP-GEM/NAG NLCs were significantly less than PTXL-PMAGP-GEM/NAG NLCs in both cancer cell lines.
 |
Figure 4 The biocompatibility study of blank PMAGP/NAG NLCs against LTEP-a-2 cells, A549 cells and L929 cells for 72 h (A). Cytotoxicity against (B) A549 cells and (C) LTEP-a-2 cells for 72 h: (a) free GEM + free PTXL, (b) PTXL-ss-PMAGP-GEM NLCs, (c) PTXL-PMAGP-GEM/NAG NLCs, and (d) PTXL-ss-PMAGP-GEM/NAG NLCs. Data are mean ± SD (n = 6). ***P < 0.001 compared PTXL-ss-PMAGP-GEM/NAG NLCs with free drugs. |
In vitro Cellular Uptake
The flow-cytometry analysis was conducted to examine the cellular uptake efficiency of FITC-labeled NLCs. As shown in Figure 5A, the fluorescence intensity of FITC increased with particle concentration of NLCs. As presented in Figure 5B and C, the mean fluorescence intensity improved with the increase of time and dosage. A stronger mean fluorescent intensity was observed in FITC-PTXL-ss-PMAGP-GEM/NAG NLCs compared to FITC-PTXL-ss-PMAGP-GEM NLCs at each time point or concentration level in both A549 and LTEP-a-2 cells (Figure 5D), possibly due to the glucose receptor–NAG interaction that enhanced the uptake of NAG-modified NLCs into the tumor cells. In contrast, the cellular uptake of normal L929 cells was negligible.
 |
Figure 5 (A) Flow-cytometry analysis of FITC-PTXL-ss-PMAGP-GEM/NAG NLCs and FITC-PTXL-ss-PMAGP-GEM NLCs in A549 cells and LTEP-a-2 cells. Mean fluorescence intensity of A549 cells (B) and LTEP-a-2 cells (C) incubated with different formulations for 0.5, 2 and 4 h. (D) The mean fluorescence intensity in NSCLC cells A549 and LTEP-a-2 compared with normal cell fibroblasts L929 following treatment with FITC-PTXL-ss-PMAGP-GEM/NAG NLCs (concentration 10 µmoL/L). Data are mean ± SD (n = 3). |
Consistent with the data of flow-cytometry analysis, CLSM showed the cellular uptake of FITC-PTXL-ss-PMAGP-GEM/NAG NLCs and FITC-PTXL-ss-PMAGP-GEM NLCs into A549 cells were both concentration- and time dependent (Figure 6). However, FITC-PTXL-ss-PMAGP-GEM/NAG NLCs treatment resulted in much stronger intensity than FITC-PTXL-ss-PMAGP-GEM NLCs throughout the time course.
Importantly, by 4 h, the fluorescence of both FITC-labeled NLCs was seen distributed homogenously in the cytoplasm, rather than remained entrapped as “dots” near the cell membranes.
 |
Figure 6 CLSM images of A549 cells incubated with FITC-PTXL-ss-PMAGP-GEM NLCs without NAG (A) in comparison with FITC-PTXL-ss-PMAGP-GEM/NAG NLCs (B) (equivalent to total drug concentrations 10 and 20 μmol/L) for 1, 2, 4 h, respectively. The scale bars correspond to 25 μm in all the images. |
Animal Studies
In vivo Imaging Analysis
The antitumor ability of PTXL-ss-PMAGP-GEM/NAG NLCs was evaluated through an in vivo imaging study. Fluorescence images of A549 tumor-bearing nude mice at 8 h post-injection were observed. As shown in Figure 7A, the fluorescence of free DiR was concentrated in the liver due to quick interception of free DiR by the reticuloendothelial system (RES), free DiR did not cluster at the tumor site. Importantly, the fluorescence intensity at the tumor site of DiR-labeled PTXL-ss-PMAGP-GEM/NAG NLCs group was stronger than that of DiR-labeled PTXL-ss-PMAGP-GEM NLCs group which lacked NAG. These results indicated that NAG has increased the accumulation of NLCs at the tumor site.
 |
Figure 7 (A) Representative in vivo images of A549 tumor-bearing nude mice and (B) excised tumors and other organs at 8 h following i.v. injection of free DiR, DiR-labeled NCLs with or without NAG (ie, PTXL-ss-PMAGP-GEM/NAG NLCs and PTXL-ss-PMAGP-GEM NLCs). |
In vivo Antitumor Efficacy
The in vivo antitumor efficiencies of various drug formulations on A549 human lung tumor-bearing nude mice were investigated. Despite the tumor volumes in the saline group increasing rapidly, the tumor growth of all other groups was inhibited to different degrees (Figure 8A). Negligible body weight loss (Figure 8B) suggested that all the formulations had insignificant toxicity in vivo. Of all the treatment groups, the greatest suppression of tumor growth was observed in the PTXL-ss-PMAGP-GEM/NAG NLCs group. The tumor volume in this group was only 20.1% of the control group at the end of the experiment and was 2.6-fold, 1.6-fold and 1.8-fold smaller than those treated with free drug combination, PTXL-ss-PMAGP-GEM NLCs and PTXL-PMAGP-GEM/NAG NLCs, respectively. A similar trend was observed in average tumor weight (Figure 8C and D).
 |
Figure 8 Anti-tumor efficacy in A549 xenograft models: (A) Tumor growth, (B) body weight profiles; (C) photographs of tumor tissues; and (D) tumor weight. The mice were treated with various formulations by i.v. injection at a dose of 4.74 mg/kg GEM and 5.12 mg/kg PTXL. **P < 0.01; ***P < 0.001. |
Discussion
In the current study, we successfully synthesized PTXL-ss-PMAGP-GEM for the sequential delivery of hydrophobic PTXL followed by hydrophilic GEM for the potential beneficial combination therapy of NSCLC. In order to accelerate the release of PTXL by exploiting the redox potential in the cancer cells, DPA was introduced as a cleavable linker to form PTXL-ss-DPA. The esterification occurred at 2ʹ-OH of PTXL, not at the 7ʹ-OH.33,34 The success of synthesis was confirmed with 1H-NMR which showed the key peaks for the PTXL-ss-PMAGP-GEM conjugate while DSC differentiated the conjugate from the physical mixture (Figure 2).
PTXL-ss-PMAGP-GEM/NAG NLCs and reference formulation (non-pH/redox-sensitive) PTXL-PMAGP-GEM/NAG NLCs (both about 120 nm) were obtained with the emulsification and solvent evaporation method. The amphiphilic block copolymer (NAG-P(St-alt-MA)58-b-PSt130) acted as a targeting moiety as well as the emulsifier. Both types of NLCs had modest negative surface charges of approximately −20 mV and similar EE levels (~83%).
In vitro study confirmed faster release of both drugs occurred from PTXL-ss-PMAGP-GEM/NAG NLCs at the lower pH (pHin) than at pH 7.4 (Figure 3). This is favorable and predicts a great potential for intracellular drug release. In addition, there was an accelerates drug release of PTXL in the presence of 10-mM GSH. PTXL release was most likely mediated through disruption of both the ester and disulfide bonds, indicating its ability for faster release in the redox conditions inside tumor cells. The release rate of GEM at pH 6.0 and 10-mM GSH conditions was much slower than PTXL with an approximate 24-h delay. The sulfhydryl generated could form hydrogen bonds with groups on GEM (eg, amidogen, hydroxyl, fluorine, etc.) which containing lone pairs of electrons. The hydrogen-bonding interactions reduced the energy of the system and stabilized the structure of PMAGP-GEM, therefore slowed down the release of GEM.32,35 As the extension of time, ester bonds were hydrolyzed and the “stable” structure was destroyed, GEM was progressively released, however, with a lag time compared with PTXL.
As introduced earlier, treatment with PTXL prior to GEM could significantly increase the intratumoral concentration of GEM, and thus further improves the synergistic effect.5,7,12 Figure 3D shows an approximate 24 h faster release of PTXL prior to GEM from PTXL-ss-PMAGP-GEM/NAG NLCs. Therefore, the PTXL-ss-PMAGP-GEM/NAG NLCs would provide synergistic drug effects in the tumor suppression36,37 with strengthened effects owing to the PTXL-GEM sequential delivery,10 particularly when the two drugs are simultaneously delivered into the same cancer cells (shown in Table 2). The results showed both NAG and -ss-bond improved the combination index.
The flow-cytometry results demonstrated that FITC-PTXL-ss-PMAGP-GEM/NAG NLCs could internalize much more quickly into both NSCLC cell models, A549 and LTEP-a-2, than non-cancer cell L929 fibroblasts (Figure 5D). The specific tumor cell uptake of NAG functionalized nanocarriers was postulated to be enhanced through glucose receptor (GLUT)-mediated endocytosis as GLUT is widely reported to be over-expressed on cancer cells. This was further confirmed by the comparative cellular uptake of FITC-PTXL-ss-PMAGP-GEM/NAG NLCs and FITC-PTXL-ss-PMAGP-GEM (without NAG) NLCs into A549 cells by CLSM (Figure 6). Notably, CLSM showed that by 4 h, the fluorescence of FITC-labeled PTXL-ss-PMAGP-GEM/NAG NLCs and PTXL-ss-PMAGP-GEM NLCs in A549 cells were both seen distributed homogenously in the cytoplasm rather than presented as “dots” as initially, indicating the redox/pH-sensitive nanocarriers were no more remained entrapped in endosomes.18 This would enhance the drug availability to their DNA target. High expression of glucose transporters (GLUT) was recently found to be significantly associated with adverse clinical outcomes in NSCLC patients,24 making NAG an ideal ligand in this case.
In the A549 tumor-bearing nude mice, strong fluorescence was detected at the tumor site 8 h post-injection of DiR-labeled PTXL-ss-PMAGP-GEM NLCs and DiR-labeled PTXL-ss-PMAGP-GEM/NAG NLCs (Figure 7). These results also indicated that NAG increased the accumulation of NLCs at the tumor site. Antitumor studies (Figure 8) suggested that all the NLCs formulations had insignificant toxicity in vivo. Enhanced antitumor effect was observed for the PTXL-ss-PMAGP-GEM/NLCs group could be attributed to both the prolonged half-lives of the drugs and the simultaneous delivery of the two drugs in an ideal sequence in the cancer cells to achieve a synergistic effect. The drug release sequence enhanced the synergism of PTXL and GEM compared to the simple combination as free drugs or in the non-reductive/pH-sensitive delivery system. Moreover, NAG significantly further increased the tumor accumulation and thus the efficacy. In addition, the drug molar ratio (PTXL:GEM=1:3) was also favorable for treatment, as shown in many other studies.38
It is worth noting that this study highlighted the feasibility of designing polymer-drug conjugates to achieve intracellular and sequential drug release by exploring the tumor micro-environments as internal stimuli. However, the efficacy data presented in this research were based on subcutaneously implanted NSCLC animal models which may not predict clinical translation.39,40 A better understanding of the nature and complexity of tumors in establishing strategies is needed to enhance tumor targeting for clinical use.41
Conclusion
Combination drug therapy has shown great potential to enhance the therapeutic efficacy. The rationale behind it is to use different mechanisms for targeting cancer cells. By exploiting the internal stimuli in cancer cells, PTXL-ss-PMAGP-GEM/NAG NLCs demonstrated promising drug delivery systems for intracellularly sequential drug release to enhance therapeutic efficacy, and further strengthen the tumor-targeting therapy by synchronizing the delivery of two drugs into the same cells.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Yuan ZQ, Chen WL, You BG, et al. Multifunctional nanoparticles co-delivering EZH2 siRNA and etoposide for synergistic therapy of orthotopic non-small-cell lung tumor. J Control Release. 2017;268:198–211. doi:10.1016/j.jconrel.2017.10.025
2. Li Y, Xuan J, Song Y, et al. Nanoporous glass integrated in volumetric bar-chart chip for point-of-care diagnostics of non-small cell lung cancer. ACS Nano. 2015;10(1):1640–1647. doi:10.1021/acsnano.5b07357
3. Bradley CJ, Yabroff KR, Mariotto AB, Zeruto C, Tran Q, Warren JL. Antineoplastic treatment of advanced-stage non-small-cell lung cancer: treatment, survival, and spending (2000 to 2011). J Clin Oncol. 2017;35(5):529–535. doi:10.1200/JCO.2016.69.4166
4. Miao L, Guo S, Zhang J, Kim WY, Huang L. Nanoparticles with precise ratiometric co-loading and co-delivery of gemcitabine monophosphate and cisplatin for treatment of bladder cancer. Adv Funct Mater. 2014;24(42):6601–6611. doi:10.1002/adfm.201401076
5. Kroep JR, Giaccone G, Tolis C, et al. Sequence dependent effect of paclitaxel on gemcitabine metabolism in relation to cell cycle and cytotoxicity in non-small-cell lung cancer cell lines. Br J Cancer. 2000;83(8):1069–1076. doi:10.1054/bjoc.2000.1399
6. Baykara M, Coskun U, Berk V, et al. Gemcitabine plus paclitaxel as second-line chemotherapy in patients with advanced non-small cell lung cancer. Asian Pac J Cancer Prev. 2012;13(10):5119–5124. doi:10.7314/apjcp.2012.13.10.5119
7. Kroep JR, Smit EF, Giaccone G, et al. Pharmacology of the paclitaxel-cisplatin, gemcitabine-cisplatin, and paclitaxel-gemcitabine combinations in patients with advanced non-small cell lung cancer. Cancer Chemother Pharmacol. 2006;58(4):509–516. doi:10.1007/s00280-006-0191-z
8. Shord SS, Faucette SR, Gillenwater HH, et al. Gemcitabine pharmacokinetics and interaction with paclitaxel in patients with advanced non-small-cell lung cancer. Cancer Chemother Pharmacol. 2003;51(4):328–336. doi:10.1007/s00280-002-0560-1
9. Fogli S, Danesi R, De Braud F, et al. Drug distribution and pharmacokinetic/pharmacodynamic relationship of paclitaxel and gemcitabine in patients with non-small-cell lung cancer. Ann Oncol. 2001;12(11):1553–1559. doi:10.1023/a:1013133415945
10. Shim G, Kim MG, Kim D, Park JY, Oh YK. Nanoformulation-based sequential combination cancer therapy. Adv Drug Deliv Rev. 2017;115:57–81. doi:10.1016/j.addr.2017.04.003
11. Tapeinos C, Battaglini M, Ciofani G. Advances in the design of solid lipid nanoparticles and nanostructured lipid carriers for targeting brain diseases. J Control Release. 2017;264:306–332. doi:10.1016/j.jconrel.2017.08.033
12. Gesto DS, Cerqueira NM, Fernandes PA, Ramos MJ. Gemcitabine: a critical nucleoside for cancer therapy. Curr Med Chem. 2012;19(7):1076–1087. doi:10.2174/092986712799320682
13. Vitorino C, Almeida J, Gonçalves LM, Almeida AJ, Sousa JJ, Pais AA. Co-encapsulating nanostructured lipid carriers for transdermal application: from experimental design to the molecular detail. J Control Release. 2013;167(3):301–314. doi:10.1016/j.jconrel.2013.02.011
14. Scioli Montoto S, Sbaraglini ML, Talevi A, et al. Carbamazepine-loaded solid lipid nanoparticles and nanostructured lipid carriers: physicochemical characterization and in vitro/in vivo evaluation. Colloids Surf B. 2018;167:73–81. doi:10.1016/j.colsurfb.2018.03.052
15. Li X, Jia X, Niu H. Nanostructured lipid carriers co-delivering lapachone and doxorubicin for overcoming multidrug resistance in breast cancer therapy. Int J Nanomedicine. 2018;13:4107–4119. doi:10.2147/IJN.S163929
16. Beloqui A, Solinís MÁ, Rodríguez-Gascón A, Almeida AJ, Préat V. Nanostructured lipid carriers: promising drug delivery systems for future clinics. Nanomedicine. 2016;12(1):143–161. doi:10.1016/j.nano.2015.09.004
17. Yang Q, Li L, Sun W, Zhou Z, Huang Y. Dual stimuli-responsive hybrid polymeric nanoparticles self-assembled from POSS-based starlike copolymer-drug conjugates for efficient intracellular delivery of hydrophobic drugs. ACS Appl Mater Interfaces. 2016;8(21):13251–13261. doi:10.1021/acsami.6b02403
18. Kanamala M, Wilson WR, Yang M, Palmer BD, Wu Z. Mechanisms and biomaterials in pH-responsive tumour targeted drug delivery: a review. Biomaterials. 2016;85:152–167. doi:10.1016/j.biomaterials.2016.01.061
19. Hu YW, Du YZ, Liu N, et al. Selective redox-responsive drug release in tumor cells mediated by chitosan based glycolipid-like nanocarrier. J Control Release. 2015;206:91–100. doi:10.1016/j.jconrel.2015.03.018
20. Fleige E, Quadir MA, Haag R. Stimuli-responsive polymeric nanocarriers for the controlled transport of active compounds: concepts and applications. Adv Drug Deliv Rev. 2012;64(9):866–884. doi:10.1016/j.addr.2012.01.020
21. Deng B, Ma P, Xie Y. Reduction-sensitive polymeric nanocarriers in cancer therapy: a comprehensive review. Nanoscale. 2015;7(30):12773–12795. doi:10.1039/c5nr02878g
22. Liu X, Wu M, Hu Q, et al. Redox-activated light-up nanomicelle for precise imaging-guided cancer therapy and real-time pharmacokinetic monitoring. ACS Nano. 2016;10(12):11385–11396. doi:10.1021/acsnano.6b06688
23. Chi Y, Yin X, Sun K, et al. Redox-sensitive and hyaluronic acid functionalized liposomes for cytoplasmic drug delivery to osteosarcoma in animal models. J Control Release. 2017;261:113–125. doi:10.1016/j.jconrel.2017.06.027
24. Koh YW, Lee SJ, Park SY. Differential expression and prognostic significance of GLUT1 according to histologic type of non-small-cell lung cancer and its association with volume-dependent parameters. Lung Cancer. 2017;104:31–37. doi:10.1016/j.lungcan.2016.12.003
25. Pawar S, Vavia P. Glucosamine anchored cancer targeted nano-vesicular drug delivery system of doxorubicin. J Drug Target. 2015;24(1):68–79. doi:10.3109/1061186X.2015.1055572
26. Kumar P, Tambe P, Paknikar KM, Gajbhiye V. Folate/N-acetyl glucosamine conjugated mesoporous silica nanoparticles for targeting breast cancer cells: a comparative study. Colloids Surf B Biointerfaces. 2017;156:203–212. doi:10.1016/j.colsurfb.2017.05.032
27. Pawar SK, Badhwar AJ, Kharas F, Khandare JJ, Vavia PR. Design, synthesis and evaluation of N-acetyl glucosamine (NAG)-PEG-doxorubicin targeted conjugates for anticancer delivery. Int J Pharm. 2012;436(1–2):183–193. doi:10.1016/j.ijpharm.2012.05.078
28. Liang Y, Tian B, Zhang J, et al. Tumor-targeted polymeric nanostructured lipid carriers with precise ratiometric control over dual-drug loading for combination therapy in non-small-cell lung cancer. Int J Nanomedicine. 2017;12:1699–1715. doi:10.2147/IJN.S121262
29. Tian B, Ding Y, Han J, Zhang J, Han Y, Han J. N-Acetyl-D-glucosamine decorated polymeric nanoparticles for targeted delivery of doxorubicin: synthesis, characterization and in vitro evaluation. Colloids Surf B Biointerfaces. 2015;130:246–254. doi:10.1016/j.colsurfb.2015.04.019
30. Lotfy HM, Hegazy MA, Rezk MR, Omran YR. Comparative study of novel versus conventional two-wavelength spectrophotometric methods for analysis of spectrally overlapping binary mixture. Spectrochim Acta a Mol Biomol Spectrosc. 2015;148:328–337. doi:10.1016/j.saa.2015.04.004
31. Saad AS. Novel spectrophotometric method for selective determination of compounds in ternary mixtures (dual wavelength in ratio spectra). Spectrochim Acta a Mol Biomol Spectrosc. 2015;147:257–261. doi:10.1016/j.saa.2015.03.095
32. Musialik M, Kuzmicz R, Pawłowski TS, Litwinienko G. Acidity of hydroxyl groups: an overlooked influence on antiradical properties of flavonoids. J Org Chem. 2009;74(7):2699–2709. doi:10.1021/jo802716v
33. Yin S, Huai J, Chen X, et al. Intracellular delivery and antitumor effects of a redox-responsive polymeric paclitaxel conjugate based on hyaluronic acid. Acta Biomater. 2015;26:274–285. doi:10.1016/j.actbio.2015.08.029
34. Yin T, Wu Q, Wang L, Yin L, Zhou J, Huo M. Well-defined redox-sensitive polyethene glycol-paclitaxel prodrug conjugate for tumor-specific delivery of paclitaxel using octreotide for tumor targeting. Mol Pharm. 2015;12(8):3020–3031. doi:10.1021/acs.molpharmaceut.5b00280
35. Lakshmipriya A, Chaudhary M, Mogurampelly S, Klein ML, Suryaprakash N. The intramolecular hydrogen bonding appetency for conformational penchants in oxalohydrazide fluoro derivatives: NMR, MD, QTAIM and NCI studies. J Phys Chem A. 2018;122(10):2703–2713. doi:10.1021/acs.jpca.8b00913
36. Kanda S, Goto K, Shiraishi H, et al. Safety and efficacy of nivolumab and standard chemotherapy drug combination in patients with advanced non-small-cell lung cancer: a four arms phase Ib study. Ann Oncol. 2016;27:2242–2250. doi:10.1093/annonc/mdw416
37. Quoix E, Lena H, Losonczy G, et al. TG4010 immunotherapy and first-line chemotherapy for advanced non-small-cell lung cancer (TIME): results from the phase 2b part of a randomised, double-blind, placebo-controlled, phase 2b/3 trial. Lancet Oncol. 2016;17(2):212–223. doi:10.1016/S1470-2045(15)00483-0
38. Houdaihed L, Evans JC, Allen C. Codelivery of paclitaxel and everolimus at the optimal synergistic ratio: a promising solution for the treatment of breast cancer. Mol Pharmaceut. 2018;15(9):3672–3681. doi:10.1021/acs.molpharmaceut.8b00217
39. Lu ZR, Qiao P. Drug delivery in cancer therapy, quo vadis? Mol Pharmaceut. 2018;15(9):3603–3616. doi:10.1021/acs.molpharmaceut.8b00037
40. Venditto VJ, Szoka FC
41. Golombek SK, May JN, Theek B, et al. Tumor targeting via EPR: strategies to enhance patient responses. Adv Drug Deliv Rev. 2018;130:17–38. doi:10.1016/j.addr.2018.07.007
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.