Back to Journals » Hepatic Medicine: Evidence and Research » Volume 6
A lipid-rich gestational diet predisposes offspring to nonalcoholic fatty liver disease: a potential sequence of events
Received 13 November 2013
Accepted for publication 27 December 2013
Published 6 March 2014 Volume 2014:6 Pages 15—23
DOI https://doi.org/10.2147/HMER.S57500
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Alexandria N Hughes, Julia Thom Oxford
Department of Biological Sciences, Biomolecular Research Center, Boise State University, Boise, ID, USA
Abstract: Nonalcoholic fatty liver disease (NAFLD) is the hepatic manifestation of metabolic syndrome. It affects 20%–30% of the US population, and it is increasing worldwide. Recently, the role of lipid-rich maternal gestational nutrition in spurring the development of NAFLD among offspring has been indicated. Fetal predisposition to NAFLD involves numerous physiological reroutings that are initiated by increased delivery of nonesterified fatty acids to the fetal liver. Hampered ß-oxidation, uncontrolled oxidative stress, increased triacylglycerol synthesis, and the endoplasmic reticulum unfolded protein response are all implicated in sculpting a hepatic phenotype with a propensity to develop NAFLD in the postnatal state. This review suggests a mechanism that integrates outcomes reported by a variety of studies conducted in an analysis of fetal hepatic metabolic capacity amid the maternal consumption of a high-fat diet. Potential preventive measures and therapies for use both as part of prenatal nutrition and for those at risk for the development of NAFLD are also discussed.
Keywords: nonalcoholic fatty liver disease, fetal–maternal diet, hepatocyte, oxidative stress
Introduction
Over the last few decades, metabolic syndrome has become a pervasive epidemic in the Western hemisphere, and perhaps surprisingly, this growth is not restricted to developed countries.1 Metabolic syndrome consists of a collection of risk factors for cardiovascular disease, encompassing such conditions as obesity, diabetes, and hypertension.1 Epidemiologists are now witnessing a worldwide spread of metabolic syndrome due to the industrialization of developing countries in conjunction with the globalization of Western dietary preferences.1 In the US, the National Health and Nutrition Examination Survey compiled during the years of 2003–2006 showed that 34% of adults over the age of 20 years met the criteria for metabolic syndrome, which is based on the clinical evaluation of abdominal obesity, plasma triglycerides, blood pressure, and fasting blood glucose levels.2 Recently, nonalcoholic fatty liver disease (NAFLD) has been adopted as the hepatic manifestation of metabolic syndrome,3 and it is clinically defined by triacylglycerol (TAG) storage exceeding 5% of the total liver mass.4
NAFLD affects an estimated 20%–30% of the US population, and 20% of those affected will eventually develop nonalcoholic steatohepatitis (NASH). Once an individual has NASH, fibrosis is visible in the liver tissue, and the continued presence of stressors may elicit the later emergence of cirrhosis or hepatocellular carcinoma.3,5 Although the etiology of NAFLD is poorly understood, a “two-hit hypothesis” was proposed in 1998 by Day and James6 to describe the development of NAFLD and its progression to NASH. According to this hypothesis, the first “hit” consists of hepatic insulin resistance or impaired β-oxidation of fatty acids, both of which contribute to hepatic lipid accumulation. The second “hit” consists of inflammation or oxidative stress, which may aggravate the existing steatosis and drive progression into NASH.6,7 Recently, it has been proposed that the first “hit” can occur in utero due to a maternal diet that is high in fat. Indeed, the offspring of such a condition have been shown to have a three-fold increase in hepatic TAG shortly after birth.4,8 This predisposes the offspring to the adult development of NAFLD, as only a second “hit” would be needed to spur the development of steatohepatitis.
The current literature regarding the contribution of prenatal nutrition to the development of NAFLD reveals several distinct phenotypic alterations. This review seeks to connect these seemingly disconnected outcomes into a plausible sequence of events. The underlying questions that this review addresses are: 1) in which ways does a lipid-rich nutritional environment furnished by the mother interact with the biochemical changes occurring within a developing fetus; and 2) how may these interactions result in changes in gene expression that contribute to an adult phenotype amenable to TAG accumulation in the liver? As NAFLD continues to extend its reach worldwide, seeking answers to these questions is essential.
Physiological background
In maternal circulation, plasma concentrations of nonesterified fatty acids (NEFAs) bound to albumin and TAG contained within chylomicrons gradually elevate throughout gestation.9 Placental uptake of NEFAs occurs either by diffusion or through a variety of fatty acid transport proteins. For chylomicrons, the placenta expresses lipoprotein lipase, which cleaves the contained TAG molecules into three NEFAs and a glycerol molecule.9 The NEFAs are then free to diffuse or be transported via a fatty acid transport protein into fetal circulation.
Upon reaching the developing liver, NEFAs diffuse into hepatocytes and they are rapidly converted into fatty acyl-coenzyme A (CoA).10 Primarily, fatty acyl-CoAs enter the mitochondria for β-oxidation. This is mediated by the rate-limiting enzyme, carnitine palmitoyltransferase 1 (CPT1), which exchanges the bound CoA for carnitine – yielding acyl-carnitine – which can then pass through the outer mitochondrial membrane.11 Remaining NEFAs that do not undergo β-oxidation will be stored by esterification at the endoplasmic reticulum (ER) as TAG.12 TAG may be exported within very low-density lipoproteins (VLDL), or TAG may be stored within the hepatocyte cytosol in lipid droplets.13,14 A lipid droplet is an organelle that functions to store TAG within a phospholipid membrane comprised chiefly of phosphatidylcholine (PC). Since NEFA accumulation is toxic to hepatocytes,15 VLDL secretion and lipid droplet formation can be regarded as protective measures against hepatotoxicity.16
If the liver as a whole attains more fatty acids than it eliminates via β-oxidation or VLDL secretion, TAG accumulates in lipid droplets,10 and this represents the defining characteristic of NAFLD. In a model of lipid-rich prenatal nutrition, the generalized pathway followed by NEFAs, as described above, is perturbed in a manner that affects the offspring’s physiology into adulthood, and induces a lifelong predisposition for NAFLD.
Methods
Many of the studies reviewed here used mice as their model organism and the researchers apportioned a maternal prenatal diet to one group of dams with a higher composition of dietary lipids than that received by the control group. Although not uniform amongst all studies, control groups were fed a commercial “standard chow” diet containing macronutrients distributed aptly for the nutritional needs of mice. Increased consumption of dietary lipids is a frequent nutritional misallocation in the common Western diet; hence, what can be learned from these studies has the potential to be readily implemented and appreciated.
However, a significant and inherent limitation of this research is that increasing the quantity of one macronutrient requires decreasing that of another. If the total percentage of lipids increases, then the percentage of other nutrients falls relative to the total - it is difficult to know whether the experimental results can be attributed to an increase of one or a decrease in another, or the combination. Additionally, it is unlikely that an average “unbalanced” Western diet errs only in its total composition of lipids. Although the implications of a maternal high-fat (HF) diet explored in this review may apply to those mothers who, for example, additionally have a vitamin deficiency, whether there exists synergism or antagonism between certain dietary imbalances has yet to be explored thoroughly, especially in the context of prenatal nutrition.
Disruptions resulting from elevated β-oxidation
In a study by Bruce et al,17 the hepatic metabolic capacities of four groups of mice that received either a HF or control (C) prenatal and postnatal diet were analyzed. The groups were designated as C/C, C/HF, HF/C, and HF/HF, in reference to their prenatal/postnatal nutrition.17 This study revealed that levels of CPT1, which is the enzyme that mediates the transfer of fatty acyl-CoA through the outer mitochondrial membrane in the initiation of β-oxidation,11 did not differ among HF/C, C/HF, and HF/HF mice, although all three of these groups had marginally elevated CPT1 compared to the C/C group.17 This suggests that in the presence of increased cytosolic fatty acids, there is an upper limit to the amount of CPT1 that can be produced to accommodate the influx; CPT1 is the rate-limiting enzyme in this process.11
The increase in NEFAs in fetal circulation, which is a result of the quantity of maternal dietary lipids, was met with a corresponding slight escalation in fetal β-oxidation, as described by Byrne et al.18 Strikingly, this study determined that at 15 weeks of age, the HF/C and HF/HF groups exhibited a 3.7- and 3.2-fold respective decrease in electron transport chain (ETC) complex I activity compared to the C/C group, and the C/HF group did not vary significantly from the C/C group in this regard.18
Complex I of the ETC is notorious for the leakage of reactive oxygen species (ROS) (Figure 1).19 Byrne et al18 hypothesize that increased ROS leakage from complex I, which directly results from the increased rates of fetal β-oxidation, causes mitochondrial deoxyribonucleic acid (mtDNA) damage. This could potentially reduce the postnatal mitochondrial capacity for β-oxidation, as mtDNA lacks the shielding histone structure that nuclear deoxyribonucleic acid (DNA) is afforded.18 Albeit reasonable, this explanation neglects to consider the C/HF group’s unaffected complex I activity – if higher β-oxidation is the initiating event that results in downstream damage, as the authors assert, it remains unexplained why the C/HF group did not display decreased ETC complex I activity comparable to that of the HF/C and HF/HF groups.
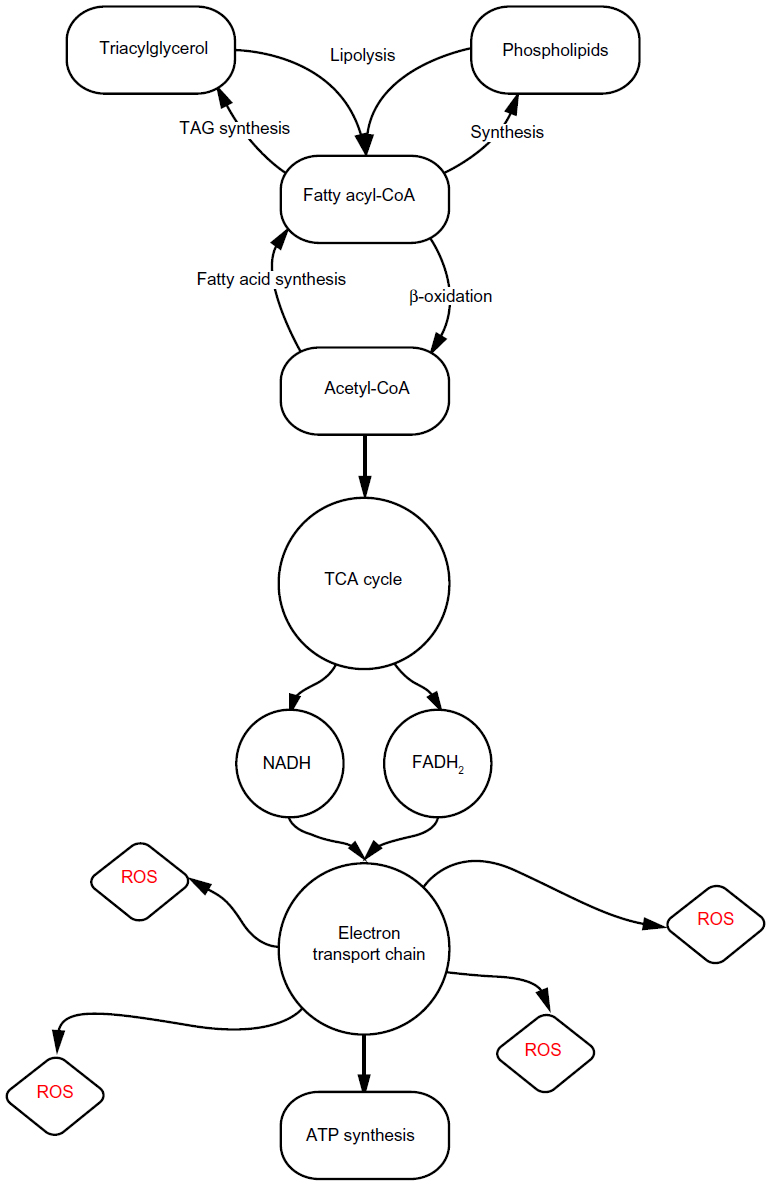 | Figure 1 Generalized pathway of lipid metabolism. |
Cytotoxic damage produced by sufficient ROS is often referred to as oxidative stress. In several models of oxidative stress, it has been demonstrated that a cell may selectively shunt metabolites from the methionine cycle (Figure 2), down the transsulfuration pathway at the expense of s-adenosylmethionine in order to increase the synthesis of glutathione (GSH), a scavenger of ROS, in an effort to combat oxidative stress.20,21 Independently, Bravo et al22 showed that in rats fed a HF diet, expression of the enzymes cystathionine β-synthase (CBS) and cystathionine γ-lyase, which participate in the transsulfuration pathway, were significantly downregulated.22 CBS is a necessary enzyme that is involved in the redirection of homocysteine from the methionine cycle toward GSH synthesis, which occurs in the first step of the transsulfuration pathway.21 In conjunction with pressure to generate GSH to neutralize ROS, homocysteine levels are elevated (Figure 2).
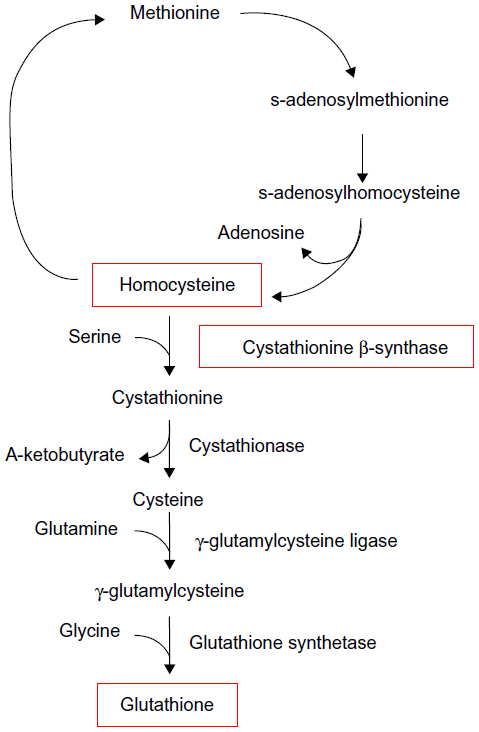 | Figure 2 The methionine cycle and transsulfuration pathway. |
Increased homocysteine levels lead to the downregulation of many antioxidant enzymes,23 including GSH peroxidase 1, by directly interfering with translation.24 In 2011, Zhang et al25 determined that offspring of mice fed a HF diet during gestation had decreased expression of the antioxidant enzymes GSH peroxidase 1, superoxide dismutase 1, and paraoxonases 1, 2, and 3. Although this group did not suggest a reason for this downregulation, they did expound upon the consequences of the resultant lowered threshold for oxidative stress in the absence of adequate endogenous defenses. It is possible that mild hyperhomocysteinemia, due to the downregulation of CBS and the diversion of methionine cycle metabolites down the transsulfuration pathway, is responsible for the downregulation of antioxidant enzymes, as noted by Zhang et al.25 In the study by Byrne18 discussed earlier, the mice fed a C diet in utero and a HF diet postnatally (C/HF) did not display altered ETC complex I activity, and it is possible that this is because their levels of ROS-neutralizing antioxidant enzymes were not downregulated during development and, consequently, mtDNA damage did not occur.
Disruptions resulting from triacylglycerol storage
In hepatocytes, NEFAs that are not accommodated by CPT1 will be esterified to form TAG, which may be packaged into a VLDL particle and exported, or they may be stored within the cytosol of the hepatocyte in a lipid droplet.13,14 The storage of TAG within a hepatocyte may be precipitated by four independent factors: 1) increased NEFAs in hepatocytes due to increased delivery of NEFAs or de novo lipogenesis; 2) increased rates of TAG synthesis; 3) reduced levels of VLDL export; or 4) decreased capacity for β-oxidation.11 Given that TAG accumulation exceeding 5% of the total liver weight is the defining criterion for the presence of NAFLD set forth by the American Association for the Study of Liver Diseases,4 presented here is a potential mechanism that connects excess hepatocyte cytosolic NEFAs with the derailing of lipid synthesis regulation by the ER, resulting in the accumulation of TAG within lipid droplets.
The ER produces acyl-CoA synthetase, which activates NEFAs in the cytosol by converting NEFAs into fatty acyl-CoAs to allow for entrance into the ER, where the fatty acyl-CoAs will serve as substrates for lipid synthesis.12 Furthermore, due to the increased delivery of cytoplasmic NEFAs that results from a maternal lipid-rich diet, some of the TAG will be stored in lipid droplets adjacent to the ER.14 For reasons incompletely understood, sustained increases in cytosolic NEFA concentration – and especially increases in saturated NEFAs – beget ER stress and, subsequently, the induction of the unfolded protein response (UPR) in both mice and in the yeast, Saccharomyces cerevisiae.26 The existing discussion of NEFA-induced ER stress in the literature often elects to cover the downstream effects of ER stress, rather than investigate the mechanism responsible for its appearance; here, both will be addressed.
When TAG is synthesized for storage in a lipid droplet, this presents to the hepatocyte the additional challenge of producing PC for the lipid droplet membrane.27 While the ER membrane contains a specific ratio of PC and phosphatidylethanolamine (PE), the preferential phospholipid coat for lipid droplets is PC; due to its properties as a surfactant, PC maintains the integrity and prevents the coalescence of adjacent lipid droplets. When PC is replaced with PE, neighboring lipid droplets rapidly merge.28 Fu et al27 assert that in order to produce PC, the genes Pemt and Pcyt1a are activated. Pemt is a s-adenosylmethionine-dependent methyltransferase that converts existing PE into PC by way of sequential methylation, and it is only significantly expressed in the liver.27 Pcyt1a encodes the more ubiquitous synthesizer of PC, CTP:phosphocholine cytidylyltransferase.29
Membrane composition is a determinant of many biophysical properties of a cell or an organelle,30 and upon producing PC for lipid droplets, the specific PC/PE ratio in the ER membrane is threatened and may rise.15,28 Although the mechanism is incompletely understood, an elevated PC/PE ratio in the ER membrane causes spontaneous dysfunction of sarco/ER calcium adenosine triphosphatase (SERCA), and returning the ratio to its initial state improves SERCA function.31 SERCA maintains a significant calcium gradient; the concentration of calcium within the ER is nearly 10,000 times greater than in the cytoplasm.15 Proper SERCA function is crucial not only for maintaining low cytosolic calcium for the purpose of signaling events that may include a controlled release of calcium, but it is also important because many of the chaperone proteins in the ER lumen – such as binding immunoglobulin protein (also known as GRP78), calreticulin, and calnexin – are calcium-dependent.32,33 Impaired SERCA function thus impedes the ability of calcium-dependent chaperones to properly fold proteins.34,35 Additionally, the increase in cytosolic calcium concentration further aggravates mitochondria and contributes to additional oxidative stress.32
The accumulation of unfolded or misfolded proteins in the ER lumen defines ER stress and activates the UPR, a series of intracellular signaling events that function to restore ER homeostasis.36 These events culminate in reducing the ability of proteins to enter the ER for posttranslational modifications, upregulation of chaperone proteins and, if homeostasis cannot be restored, apoptosis.15 ER stress and the UPR have two implications that directly contribute to the predisposition of offspring to NAFLD:
- Upon induction of the UPR, activating transcription factor-6 (ATF6) – one of the proteins that participates in UPR intracellular signal transduction – is activated by intramembrane proteolysis. ATF6 shares its proteolytic machinery with sterol response element binding protein 1c (SREBP-1c),36 so upon cleavage of ATF6, SREBP-1c is also cleaved and is able to translocate to the nucleus.37 SREBP-1c is a transcriptional activator for the lipogenic proteins, acetyl-CoA carboxylase (ACC) and glucokinase.10
- ER stress also activates the c-Jun N-terminus kinase (JNK) pathway, which leads to the phosphorylation of certain serine residues in insulin receptor substrate-1 (IRS-1). Given that the docking of IRS-1, the subsequent tyrosine phosphorylation, and the release of IRS-1 is a necessary occurrence in successful insulin signaling, serine phosphorylation of IRS-1 blocks signal transduction and contributes to hepatic insulin resistance.38,39 Hepatic insulin resistance has been independently demonstrated to overactivate SREBP-1c.40
SREBP-1c activates the transcription of ACC and glucokinase, which both contribute to postnatal fatty acid synthesis.10,37,40 However, the presence of both ACC and glucokinase are complicated in a HF intrauterine environment. ACC converts acetyl-CoA into malonyl-CoA which, in addition to playing a key role in de novo lipogenesis, is a potent inhibitor of CPT1 and thus decreases the rate of β-oxidation (Figure 3).10 Additionally, it has been shown that the fetal liver is more sensitive than the liver of a newborn to the inhibitory effects of malonyl-CoA.9 In fact, in a maternal HF diet experiment performed by Ashino et al,4 82-day-old offspring of dams who consumed a diet deriving 45% of its caloric value from nondescript fats had higher levels of activated ACC than did offspring of mice fed a control diet, which indicates that the elevation of ACC persists in the postnatal state. Since Nagle et al11 included decreased β-oxidation as one of the four factors that directly contributes to TAG accumulation in the liver, it is probable that malonyl-CoA contributes to the predisposition of offspring to NAFLD.
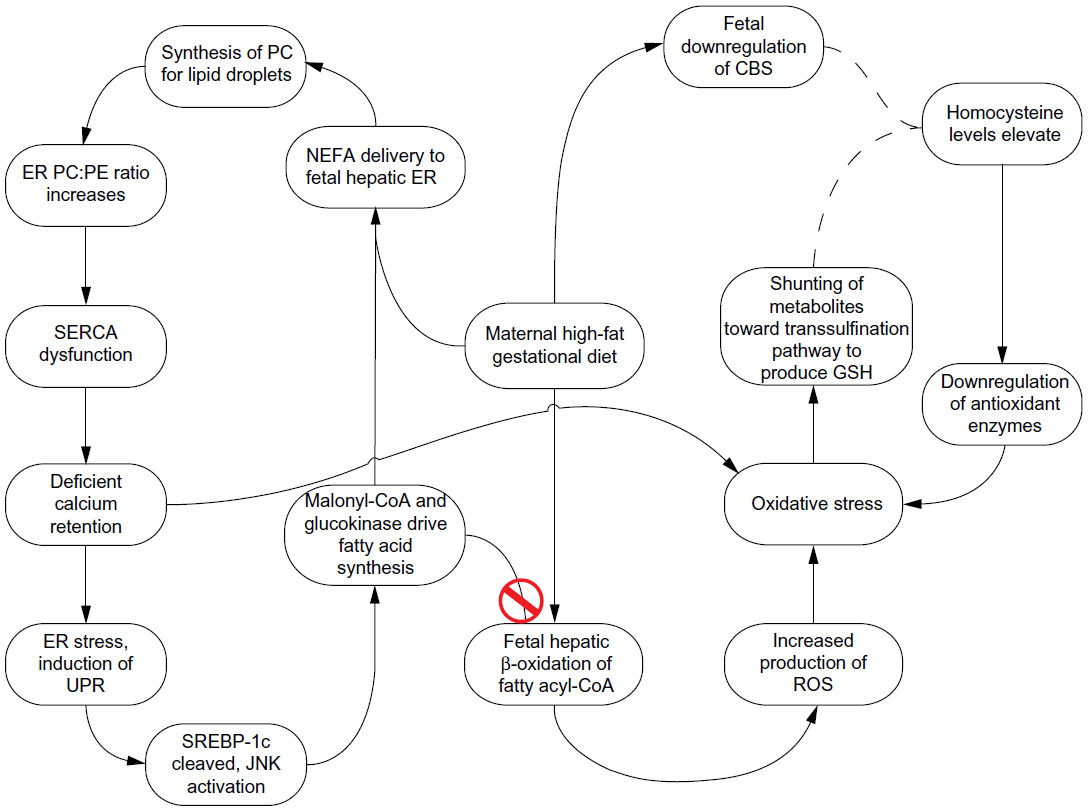 | Figure 3 Sequence of physiological changes resulting from a maternal lipid-rich diet. |
Glucokinase participates in the first step of fatty acid synthesis by phosphorylating glucose, resulting in glucose-6-phosphate.41 Trophoblasts ordinarily express numerous glucose transporters from maternal to fetal circulation, such that glucose is readily available for the fetus.9 Additionally, it has been demonstrated in a rodent model that the placentas of mothers who consume a HF diet express a five-fold increase in glucose transporter-1, which facilitates the diffusion of glucose into the fetal circulation.42 The increased expression of glucokinase in conjunction with an elevated influx of glucose may additionally contribute to de novo lipogenesis.
As NEFAs are produced by the de novo lipogenesis, in which both malonyl-CoA and glucokinase participate, and since ER stress was initiated with excessive NEFAs entering the ER for lipid synthesis, this process can be perceived as cyclic. Certainly, the NEFAs produced by de novo lipogenesis could instead undergo β-oxidation, but as their synthesis requires nicotinamide adenine dinucleotide phosphate,9 this would be more energetically expensive than to reserve β-oxidation for exogenous, maternally-derived NEFAs. Additionally, the inhibition of β-oxidation by malonyl-CoA, and the potential mtDNA damage caused by rampant ROS, both encourage the continuation of NEFAs in the cycle.
Management of NAFLD: prenatal and postnatal, present and future
Given the multifaceted nature of TAG accumulation borne in utero, it may seem likely that many opportunities exist for therapeutic interjection. Presently, omega-3 polyunsaturated fatty acids (n-3 PUFAs) and genistein have evidence to support their intake as part of preventative maternal prenatal nutrition. Novak et al43 performed a study whereby two groups of dams were fed the same total amount of dietary fat, but the composition of dietary fats differed between the groups. Each received different fats but both had the same total amount of dietary fat. On postnatal day 3, the offspring of the group that received adequate n-3 PUFAs displayed higher levels of proteins that were integral to mitochondrial β-oxidation, including CPT1, and they downregulated genes associated with fatty acid synthesis, compared to the group that received inadequate n-3 PUFAs.43 It appears that supplementation of n-3 PUFAs as part of proper prenatal nutrition, which is reflective of this finding, may be a possible preventative route for NAFLD. However, in conflict with this proposition is evidence proposed by Dennery in 2010,44 who claimed that maternal PUFA supplementation causes oocyte mitochondria to increase their ROS production, which leads to impaired embryonic development. In adults with NAFLD, supplementation with docosahexaenoic acid, an n-3 PUFA, has been shown to reduce the rates of de novo lipogenesis via the negative regulation of SREBP-1c transcription.45 Perhaps a conciliatory solution lies in the timing of n-3 PUFA supplementation; questions with such therapeutic potential warrant additional investigation.
Genistein, a soy isoflavone found in soybeans, has received more consistent favorable literature reviews than n-3 PUFAs. When provided concurrently with a maternal HF diet and comprising 0.2% (by weight) of total maternal nutrition, the offspring displayed significantly less hepatic TAG accumulation and elevated levels of GSH when compared to offspring that solely received a maternal HF diet.46 As GSH production is regulated in response to ROS levels, it is possible that genistein supplementation ameliorated these defenses and prevented the downstream hepatotoxic effects of uncontrolled ROS. Additionally, in an adult HF feeding study conducted by Ji et al,47 genistein supplementation embodied the qualities of an anti-inflammatory compound and appreciably decreased the serum levels of the cytokines, tumor necrosis factor-alpha and interleukin 6. As inflammation provides the “second hit” that drives the transition from NAFLD into NASH, the anti-inflammatory effects of genistein may prevent or attenuate the development of NASH. Ji et al47 also noted that genistein is a tyrosine kinase inhibitor, and they implicated this characteristic as the cause of the marked inhibition of the JNK pathway observed in response to genistein supplementation. Activation of the JNK pathway is implicated in the development of hepatic insulin resistance and subsequent overactivation of SREBP-1c.39,40 Preventing these outcomes in utero by supplementing the prenatal diet with genistein may improve the offspring’s hepatic phenotype. It is also encouraging that these improvements in inflammation occurred in the postnatal environment, as they represent a potential degree of reversibility for those born at risk for the development of NAFLD.
Certainly with the number of factors that contribute to the development of NAFLD, additional therapeutic avenues for preventing TAG accumulation have yet to be discovered. An experimental approach of conjugating the lipophilic cation, triphenylphosphonium, to an antioxidant such as vitamin E or ubiquinone has been shown to permit the delivery of the antioxidant moiety through the mitochondrial membrane.48,49 When this approach was described by Murphy and Smith in 2007,48 it was their intention to employ the lipophilicity of triphenylphosphonium to ensure diffusion of the antioxidant into the mitochondria, where ROS neutralization is most useful in relegating oxidative stress. Recently, Finichiu et al49 confirmed that this form of antioxidant delivery does indeed reach mitochondria in the tissues with high levels of oxidative stress. Although this mechanism has only been investigated in general models of oxidative stress, given the role of ROS in the development of NAFLD, future advances may render this an applicable method of antioxidant delivery for those affected by NAFLD.
There is a need for a clearly defined, ideal prenatal diet that considers the negative implications of the HF diet discussed here, and there is also the need for a diet for those affected by NAFLD. The American Dietetic Association does not currently suggest a recommended daily allowance of fats for pregnant women.50 Current research is exploring many different diet and exercise regimes for those living with NAFLD, with mixed results.51,52 Future work needs to ascertain the maximum daily allowance for dietary fats when undesirable outcomes for the fetus arise. This will allow for the development of dietary guidelines that could easily be followed by expecting mothers.
The diagnostic approach to NAFLD in childhood has recently been developed by the European Society of Paediatric Gastroenterology, Hepatology, and Nutrition committee.53 The gold standard for the detection of NAFLD is through liver biopsy, but this should only be performed after less invasive procedures to rule out other conditions. Testing for appropriate biomarkers and imaging by magnetic resonance imaging or ultrasound are noninvasive and fairly reliable diagnostic tools for adults and older children, and these methods may also be developed for infants.
Conclusion
Education aimed at promoting a balanced prenatal diet may be an effective intervention in convincing expecting mothers to consider nutrition as equally important to the health of a fetus as abstaining from tobacco use and alcohol consumption. Previous efforts to educate women on the harmful consequences of these activities during pregnancy have been met with success.9 A voluntary decrease in smoking and drinking alcohol by pregnant women was demonstrated following these efforts, which was indicative of a willingness by women to modify their lifestyle for the benefit of their babies.9 As the metabolic syndrome has garnered significant attention from public health organizations in Western countries, the recent acceptance of NAFLD as the hepatic manifestation of metabolic syndrome will likely spur its inclusion in future public health educational efforts.
Of adults who present with NAFLD, most display significant mtDNA abnormalities and mutations, and it has been estimated that about 26% of hepatic TAG in NAFLD patients is derived from de novo lipogenesis.10,21 In the context of elevated maternal dietary lipids, it is clear that both de novo lipogenesis and esterification of delivered NEFAs are encouraged in the fetus via multiple mechanisms discussed herein: damaged mtDNA and associated reduced capacity for β-oxidation; the inhibition of CPT1 by malonyl-CoA; and the inadvertent activation of SREBP-1c during the unfolded protein response. The resultant phenotype of the offspring whose mothers consumed a lipid-rich diet during pregnancy shares many commonalities with that of adult NAFLD, and as illustrated in this review, it predisposes offspring for the later development of NAFLD.
Acknowledgments
The authors acknowledge the editorial assistance of Kristen Mitchell, Ken Cornell, Lindsey Comstock, and Stephanie Hamilton. The research reported in this publication was supported by an Institutional Development Award (IDeA) from the National Center for Research Resources and the National Institute of General Medical Sciences of the National Institutes of Health under grant numbers P20 RR016454 and P20 GM103408, and the Department of Biological Sciences (BIOL451).
Disclosure
The authors report no conflicts of interest in this work.
References
Strakovsky RS, Pan YX. In utero oxidative stress epigenetically programs antioxidant defense capacity and adulthood diseases. Antioxid Redox Signal. 2012;17(2):237–253. | |
Ervin RB. Prevalence of metabolic syndrome among adults 20 years of age and over, by sex, age, race and ethnicity, and body mass index: United States, 2003–2006. Natl Health Stat Report. 2009;5(13):1–7. | |
Henao-Mejia J, Elinav E, Jin C, et al. Inflammasome-mediated dysbiosis regulates progression of NAFLD and obesity. Nature. 2012;482(7384):179–185. | |
Ashino NG, Saito KN, Souza FD, et al. Maternal high-fat feeding through pregnancy and lactation predisposes mouse offspring to molecular insulin resistance and fatty liver. J Nutr Biochem. 2012;23(4):341–348. | |
Farrell GC, Larter CZ. Nonalcoholic fatty liver disease: from steatosis to cirrhosis. Hepatology. 2006;43(2 Suppl 1):S99–S112. | |
Day CP, James OF. Steatohepatitis: a tale of two “hits”? Gastroenterology. 1998;114(4):842–845. | |
Rector RS, Thyfault JP, Uptergrove GM, et al. Mitochondrial dysfunction precedes insulin resistance and hepatic steatosis and contributes to the natural history of non-alcoholic fatty liver disease in an obese rodent model. J Hepatol. 2010;52(5):727–736. | |
McCurdy CE, Bishop JM, Williams SM, et al. Maternal high-fat diet triggers lipotoxicity in the fetal livers of nonhuman primates. J Clin Invest. 2009;119(2):323–335. | |
Rebholz SL, Burke KT, Yang Q, Tso P, Woollett LA. Dietary fat impacts fetal growth and metabolism: uptake of chylomicron remnant core lipids by the placenta. Am J Physiol Endocrinol Metab. 2011;301(2):E416–E425. | |
Kawano Y, Cohen DE. Mechanisms of hepatic triglyceride accumulation in non-alcoholic fatty liver disease. J Gastroenterol. 2013;48(4):434–441. | |
Nagle CA, Klett EL, Coleman RA. Hepatic triacylglycerol accumulation and insulin resistance. J Lipid Res. 2009;50 Suppl:S74–S79. | |
Li J, Huang J, Li JS, Chen H, Huang K, Zheng L. Accumulation of endoplasmic reticulum stress and lipogenesis in the liver through generational effects of high fat diets. J Hepatol. 2012;56(4):900–907. | |
Rolo AP, Teodoro JS, Palmeira CM. Role of oxidative stress in the pathogenesis of nonalcoholic steatohepatitis. Free Radic Biol Med. 2012;52(1):59–69. | |
Brasaemle DL, Wolins NE. Packaging of fat: an evolving model of lipid droplet assembly and expansion. J Biol Chem. 2012;287(4):2273–2279. | |
Fu S, Watkins SM, Hotamisligil GS. The role of endoplasmic reticulum in hepatic lipid homeostasis and stress signaling. Cell Metab. 2012;15(5):623–634. | |
Choi SS, Diehl AM. Hepatic triglyceride synthesis and nonalcoholic fatty liver disease. Curr Opin Lipidol. 2008;19(3):295–300. | |
Bruce KD, Cagampang FR, Argenton M, et al. Maternal high-fat feeding primes steatohepatitis in adult mice offspring, involving mitochondrial dysfunction and altered lipogenesis gene expression. Hepatology. 2009;50(6):1796–1808. | |
Byrne CD, Olufadi R, Bruce KD, Cagampang FR, Ahmed MH. Metabolic disturbances in non-alcoholic fatty liver disease. Clin Sci (Lond). 2009;116(7):539–564. | |
Chen Q, Vazquez EJ, Moghaddas S, Hoppel CL, Lesnefsky EJ. Production of reactive oxygen species by mitochondria: central role of complex III. J Biol Chem. 2003;278(38):36027–36031. | |
Hitchler MJ, Domann FE. An epigenetic perspective on the free radical theory of development. Free Radic Biol Med. 2007;43(7):1023–1036. | |
Mantena SK, King AL, Andringa KK, Eccleston HB, Bailey SM. Mitochondrial dysfunction and oxidative stress in the pathogenesis of alcohol- and obesity-induced fatty liver diseases. Free Radic Biol Med. 2008;44(7):1259–1272. | |
Bravo E, Palleschi S, Aspichueta P, et al. High fat diet-induced non alcoholic fatty liver disease in rats is associated with hyperhomocysteinemia caused by down regulation of the transsulphuration pathway. Lipids Health Dis. 2011;10:60. | |
Werstuck GH, Lentz SR, Dayal S, et al. Homocysteine-induced endoplasmic reticulum stress causes dysregulation of the cholesterol and triglyceride biosynthetic pathways. J Clin Invest. 2001;107(10):1263–1273. | |
Handy DE, Zhang Y, Loscalzo J. Homocysteine down-regulates cellular glutathione peroxidase (GPx1) by decreasing translation. J Biol Chem. 2005;280(16):15518–15525. | |
Zhang X, Strakovsky R, Zhou D, Zhang Y, Pan YX. A maternal high-fat diet represses the expression of antioxidant defense genes and induces the cellular senescence pathway in the liver of male offspring rats. J Nutr. 2011;141(7):1254–1259. | |
Pineau L, Colas J, Dupont S, et al. Lipid-induced ER stress: synergistic effects of sterols and saturated fatty acids. Traffic. 2009;10(6):673–690. | |
Fu S, Yang L, Li P, et al. Aberrant lipid metabolism disrupts calcium homeostasis causing liver endoplasmic reticulum stress in obesity. Nature. 2011;473(7348):528–531. | |
Krahmer N, Guo Y, Wilfling F, et al. Phosphatidylcholine synthesis for lipid droplet expansion is mediated by localized activation of CTP:phosphocholine cytidylyltransferase. Cell Metab. 2011;14(4):504–515. | |
RL, Devlin C, Tabas I, Vance DE. Targeted deletion of hepatic CTP: phosphocholine cytidylyltransferase alpha in mice decreases plasma high density and very low density lipoproteins. J Biol Chem. 2004;279(45):47402–47410. | |
Hoile SP, Irvine NA, Kelsall CJ, et al. Maternal fat intake in rats alters 20:4n-6 and 22:6n-3 status and the epigenetic regulation of Fads2 in offspring liver. J Nutr Biochem. 2013;24(7):1213–1220. | |
Li Y, Ge M, Ciani L, et al. Enrichment of endoplasmic reticulum with cholesterol inhibits sarcoplasmic-endoplasmic reticulum calcium ATPase-2b activity in parallel with increased order of membrane lipids: implications for depletion of endoplasmic reticulum calcium stores and apoptosis in cholesterol-loaded macrophages. J Biol Chem. 2004;279(35):37030–37039. | |
Ji C, Kaplowitz N. ER stress: can the liver cope? J Hepatol. 2006;45(2):321–333. | |
Berridge MJ, Bootman MD, Roderick HL. Calcium signalling: dynamics, homeostasis and remodelling. Nat Rev Mol Cell Biol. 2003;4(7):517–529. | |
Burdakov D, Petersen OH, Verkhratsky A. Intraluminal calcium as a primary regulator of endoplasmic reticulum function. Cell Calcium. 2005;38(3–4):303–310. | |
Coe H, Michalak M. Calcium binding chaperones of the endoplasmic reticulum. Gen Physiol Biophys. 2009;28 Spec No Focus:F96–F103. | |
Ron D, Walter P. Signal integration in the endoplasmic reticulum unfolded protein response. Nat Rev Mol Cell Biol. 2007;8(7):519–529. | |
Ferré P, Foufelle F. Hepatic steatosis: a role for de novo lipogenesis and the transcription factor SREBP-1c. Diabetes Obes Metab. 2010; 12 Suppl 2:83–92. | |
Heerwagen MJ, Miller MR, Barbour LA, Friedman JE. Maternal obesity and fetal metabolic programming: a fertile epigenetic soil. Am J Physiol Regul Integr Comp Physiol. 2010;299(3):R711–R722. | |
Dara L, Ji C, Kaplowitz N. The contribution of endoplasmic reticulum stress to liver diseases. Hepatology. 2011;53(5):1752–1763. | |
Begriche K, Massart J, Robin MA, Bonnet F, Fromenty B. Mitochondrial adaptations and dysfunctions in nonalcoholic fatty liver disease. Hepatology. 2013;58(4):1497–1507. | |
Peter A, Stefan N, Cegan A, et al. Hepatic glucokinase expression is associated with lipogenesis and fatty liver in humans. J Clin Endocrinol Metab. 2011;96(7):E1126–E1130. | |
Bruce KD, Hanson MA. The developmental origins, mechanisms, and implications of metabolic syndrome. J Nutr. 2010;140(3):648–652. | |
Novak EM, Keller BO, Innis SM. Metabolic development in the liver and the implications of the n-3 fatty acid supply. Am J Physiol Gastrointest Liver Physiol. 2012;302(2):G250–G259. | |
Dennery PA. Oxidative stress in development: nature or nurture? Free Radic Biol Med. 2010;49(7):1147–1151. | |
Byrne CD. Fatty liver: role of inflammation and fatty acid nutrition. Prostaglandins Leukot Essent Fatty Acids. 2010;82(4–6):265–271. | |
Lee YM, Choi JS, Kim MH, Jung MH, Lee YS, Song J. Effects of dietary genistein on hepatic lipid metabolism and mitochondrial function in mice fed high-fat diets. Nutrition. 2006;22(9):956–964. | |
Ji G, Yang Q, Hao J, et al. Anti-inflammatory effect of genistein on non-alcoholic steatohepatitis rats induced by high fat diet and its potential mechanisms. Int Immunopharmacol. 2011;11(6):762–768. | |
Murphy MP, Smith RA. Targeting antioxidants to mitochondria by conjugation to lipophilic cations. Annu Rev Pharmacol Toxicol. 2007;47:629–656. | |
Finichiu PG, James AM, Larsen L, Smith RA, Murphy MP. Mitochondrial accumulation of a lipophilic cation conjugated to an ionisable group depends on membrane potential, pH gradient and pK(a): implications for the design of mitochondrial probes and therapies. J Bioenerg Biomembr. 2013;45(1–2):165–173. | |
Kaiser L, Allen LH; American Dietetic Association. Position of the American Dietetic Association: nutrition and lifestyle for a healthy pregnancy outcome. J Am Diet Assoc. 2008;108(3):553–561. | |
Oakes JL, Ideraabdullah FY. Maternal nutrition and epigenetic perturbation: modeling trends to translation. Curr Pediatr Rep. 2013;1(4):257–265. | |
Bradford V, Dillon JF, Miller MH. Lifestyle interventions for the treatment of non-alcoholic fatty liver disease. Hepat Med. 2013;2014(6):1–10. | |
Giorgio V, Prono F, Graziano F, Nobili V. Pediatric non alcoholic fatty liver disease: old and new concepts on development, progression, metabolic insight and potential treatment targets. BMC Pediatr. 2013;13:40. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.