Back to Journals » International Journal of Nanomedicine » Volume 9 » Supplement 1
A glimpse into the interactions of cells in a microenvironment: the modulation of T cells by mesenchymal stem cells
Authors Choi J ![]() , Hwang M, Lee J, Lee KH
, Hwang M, Lee J, Lee KH ![]()
Received 30 June 2013
Accepted for publication 20 July 2013
Published 7 May 2014 Volume 2014:9(Supplement 1) Pages 127—139
DOI https://doi.org/10.2147/IJN.S50767
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Jonghoon Choi,1,2 Mintai P Hwang,3 Jong-Wook Lee,3 Kwan Hyi Lee3,4
1Institute of Research Strategy and Development (IRSD), Seoul, Republic of Korea; 2Department of Bionano Engineering, Hanyang University, Ansan, Republic of Korea; 3Center for Biomaterials, Biomedical Research Institute, Korea Institute of Science and Technology, Seoul, Republic of Korea; 4Department of Biomedical Engineering, University of Science and Technology, Seoul, Republic of Korea
Abstract: Mesenchymal stem cells (MSCs) have been thought to hold potential as a mode of therapy for immuno-related pathologies, particularly for autoimmune diseases. Despite their potential, the interaction between MSCs and T cells, key players in the pathophysiology of autoimmune diseases, is not yet well understood, thereby preventing further clinical progress. A major obstacle is the highly heterogeneous nature of MSCs in vitro. Unfortunately, bulk assays do not provide information with regard to cell–cell contributions that may play a critical role in the overall cellular response. To address these issues, we investigated the interaction between smaller subsets of MSCs and CD4 T cells in a microwell array. We demonstrate that MSCs appear capable of modulating the T cell proliferation rate in response to persistent cell–cell interactions, and we anticipate the use of our microwell array in the classification of subpopulations within MSCs, ultimately leading to specific therapeutic interventions.
Keywords: mesenchymal stem cell, T cell, microwell array
Introduction
The immune system, comprised of multiple layers of complexity, can be simplified and thought of as a dynamic process seeking to maintain balance through any means possible. A key player in this system is the T cell, imparted with various modes of protective functionality. Often, however, T cells are negatively associated with pathological diseases; namely, those of the autoimmune type. To manage and reduce the sensitivity of the T cell mediated immune system, immunosuppressive medications, such as corticosteroids, cyclophosphamide, and tacrolimus, have unfortunately become a rather prosaic mode of treatment. Specifically, the attenuation of the entire immune system and the consequent overarching repercussions on the body have researchers and clinicians open to consider a different form of treatment.
Mesenchymal stem cells (MSCs) have recently been identified for their ability to target a site of inflammation, and thereby modulate the immune system in a biocompatible manner.1 Emerging as a potential mode of immunotherapy for many autoimmune diseases, MSCs have been verified in a limited set of preclinical models for their therapeutic capacity,2 opening up new areas of research.1,3–5 For instance, alloreactive immunity promoted by MSCs is thought to be a potential mode of therapy for the treatment and prevention of graft versus host diseases and allogeneic graft rejection.6,7 Specifically, results from recent literature indicate that the systemic infusion of allogeneic MSCs derived from the bone marrow of baboons can prolong the survival of allogeneic skin grafts.8 In addition, adipose tissue-derived MSCs expanded ex vivo exhibit a capacity to control graft versus host disease effects in mice transplanted with haploidentical stem cell grafts.9 Furthermore, MSCs demonstrate preferential engraftment at sites of tissue damage or tumor growth.10
Recent literature indicates that these results are likely a result of the interaction between MSCs and various immune cells through a range of mechanisms.1–5,11 Among several hypotheses that describe a possible mechanism for MSC-mediated immunomodulatory effects, the first suggests that the lack of costimulatory molecules on the MSC surface (ie, the hypoimmunogenicity of MSCs) enables them to avoid the direct allorecognition pathway.12 Furthermore, the capacity of dendritic cells, antigen-presenting cells, to induce peripheral tolerance is a potential mechanism for MSCs to escape T cell recognition. The second hypothesis is that MSCs interact with T cells directly to suppress their activation and proliferation, and consequently their alloreactivity.13 The third hypothesis is that the presence of MSCs interferes with the maturation and functionality of dendritic cells, consequently resulting in T cell clonal deletion and the expansion of regulatory T cells.4 The final hypothesis is that MSCs create an immunosuppressive environment through the modulation of many immune cells, resulting in a microenvironment composed of proteins and cytokines such as indoleamine 2,3-dioxygenase, prostaglandin E2 (PGE2), interleukin 10 (IL-10), and transforming growth factor 1 (TGF-1).1,3,4,11,13
These various hypotheses have become a crucial point of contention, as current studies continue to show highly controversial findings with no conclusive results in determining the key underlying mechanism of MSC-mediated T cell suppression. Furthermore, the implications of such uncertainty, as highlighted by the failure of two late-stage clinical trials by Osiris Therapeutics, the largest MSC therapeutic company,14 underscores the pressing need to better understand MSC-mediated immunomodulatory mechanisms and the development of technology that can facilitate this understanding. Unfortunately, most current technology and assays have the potential to be misleading, primarily because of their inability to generate cell–cell information from a larger bulk of cells. The problems resulting from the lack of cell–cell analytical capability are further magnified when considering that MSCs expanded in vitro are of a heterogeneous population with different physical properties and differentiation abilities.15 In essence, it is important to resolve cellular properties on a much smaller scale, which may then provide key insights into the regulation of stem cell maintenance and differentiation, and ultimately, the subtype identification of MSCs.16 This type of subprofiling would allow us to more effectively study MSC–T cell communication by providing an avenue to correlate various markers (soluble factors and cell surface markers) to different cellular functions (eg, T cell suppression by MSCs) on a level that cannot be found in a bulk assay. We propose herein a microwell array system with the capacity to take real-time cell–cell measurements, such as the proliferation rate and secretory profile of cytokines, in a high-throughput manner.17–20 We envision that such a system could lead to the cell–cell resolution of contributions by a specific MSC subset to its overall function.
Experiment
Cell preparation
We isolated peripheral blood mononuclear cells from fresh human blood using Ficoll-Paque (Sigma-Aldrich, St Louis, MO, USA), a highly branched hydrophilic polysaccharide. The isolated cells were run through a CD4 T cell enrichment kit (STEMCELL Technologies Inc., Vancouver, BC, Canada) to obtain a relatively pure population of CD4+ T cells. Purified CD4 T cells were cultured in Roswell Park Memorial Institute 1640, 10% fetal bovine serum, and other antibiotics at 37°C and 5% CO2. T cells were activated by adding magnetic beads coated with anti-CD3/anti-CD28 for 48 hours (Figure S1; Figure S2). Although they are a bulk population of human peripheral blood CD4+, T cells activated by CD3/CD28 are widely considered to be heterogeneous in their phenotype and function; such differences are well-documented compared with those of MSCs. Fresh peripheral blood mononuclear cells, as well as frozen-thawed ones, were compared for their activation efficiencies with microbeads for sample validation (Figure S3). Bone marrow-derived MSCs (Poietics™ hMSC human mesenchymal stem cells; Lonza, Walkersville, MD, USA) were cultured in Dulbecco’s Modified Eagle Medium supplemented with 10% knockout serum replacement, 2 mM L-glutamine, 1 mM nonessential amino acid, 4 ng/mL basic fibroblast growth factor, 50 U/mL penicillin-streptomycin, and 0.1 mM ß-mercaptoethanol (Life Technologies, Carlsbad, CA, USA).
Flow cytometry
CD25 and CD69 expression on T cells was analyzed via flow cytometry. Specifically, T cells were immunocytochemistry stained with each antibody pair, and live cell markers CellTracker™ Red, carboxyfluorescein succinimidyl ester-fluorescein isothiocyanate, and Calcein violet were used to track cells and determine cell viability. A BD LSR II flow cytometer was used and data analysis was performed with the FACS (fluorescent activated cell sorting) DiVa (BD Biosciences, San Jose, CA, USA) and FlowJo software packages (TreeStar, Ashland, OR, USA).
Microwell device fabrication
Arrays were prepared as previously described.19 Briefly, polydimethylsiloxane (Dow Corning, Washington, DC, USA) was injected into a custom-built mold and cured at 80°C for 2 hours. The arrays were then adhered directly onto a standard glass slide. Each array contains 4 × 4 microwells (300 μm × 300 μm × 300 μm). Fully prepared arrays were sterilized in an oxygen plasma chamber for 30 seconds immediately before use.
Detection of IL-10, PGE2, and TGF-1
Monoclonal antibodies certified for ELISpot were used to capture and detect IL-10, PGE2, and TGF-1 (R&D Systems, Minneapolis, MN, USA). To detect these cytokines, each antibody was conjugated with N-hydroxysuccinimide ester-activated fluorescent Alexa Fluor dyes (Life Technologies, Carlsbad, CA, USA) and purified by spin column (Piercenet, Rockford, IL, USA). The average degree of labeling was 3–4 fluorescent molecules per antibody, as determined by ultraviolet-visible spectroscopy.
Microwell array cell viability
The viability of MSCs and T cells was determined by adding 1 mM Calcein violet AM, carboxyfluorescein succinimidyl ester-fluorescein isothiocyanate, and CellTracker Red (Life Technologies, Carlsbad, CA, USA) to the cell suspension for 30 minutes in the dark. CD4 T cells were stained with Calcein AM (live cells) and Alexa 568-conjugated anti-CD4. After washing the MSCs and T cells with phosphate-buffered saline (PBS), MSCs were loaded onto the microwells first. After 5–10 minutes, the T cells were loaded onto the microwells. Cells settled randomly into the wells by gravity. The array was then imaged on a fully automated inverted epifluorescence microscope (Leica DMI6000B AFC; Leica Microsystems, Wetzlar, Germany). Transmitted light and epifluorescence micrographs were collected well by well.
Microengraving
Glass slides functionalized with protein G were coated with capture antibodies against IL-10, PGE2, or TGF-1 (10 μg/mL) in the presence of 10 μg/mL goat anti-human immunoglobulin G (Life Technologies, Carlsbad, CA, USA) to facilitate registration of arrays. After incubating the microwell array with cells at 37°C, 5% CO2, for 6 hours, the array was rinsed gently with media containing 1 ng/mL human immunoglobulin G and immediately applied onto a glass slide precoated with the first set of capture antibodies for 1 hour. The microwell array was then detached from the first glass slide, rinsed with media, and immediately applied onto a second glass slide coated with the second set of capture antibodies. After the second print, the microwell array was stored in media at 37°C, 5% CO2, for 15 hours after the initial distribution. This process was repeated for serial microengraving. The glass slides with captured cytokines were blocked with 3% fat-free milk and washed sequentially with PBS-T (PBS with 0.05% Tween) and PBS. The glass slides were then incubated with fluorescent dye conjugated detection antibodies (1 μg/mL) for 1 hour at room temperature. Next, the slides were briefly washed with PBS-T and PBS and spun dry. Reference slides were made at the end of each experiment with the same capture and detection antibodies used on the printed slides. Capture antibodies were coated on a poly-L-lysine glass slide, as described previously, and a dilution series of the target cytokines and chemokine were spotted onto the slides (1 μL/spot). After the slides were stored at 37°C, 5% CO2, for 30 minutes, they were washed as described earlier. Fluorescently labeled detection antibodies were then introduced to the slides as they were done in microengraving.
Data analysis
The collection of fluorescent images was analyzed using a customized software program (created in MATLAB; Natick, MA, USA) that identifies the number of cells present in each well. Commercial software (GenePix Pro 6.1; Molecular Devices LLC, Sunnyvale, CA, USA) was used to determine the mean fluorescent intensity of each spot obtained from the microarray of captured secreted proteins. The microarray data were subsequently filtered to remove spots that contained saturated pixels (percentage saturation >1) or spots that exhibited a high degree of covariance (covariance >60). A standard curve constructed from the reference slide was used to convert the mean fluorescent intensities to a quantitative amount of captured target molecules during microengraving. The data for all measurements were combined on a well-by-well basis using unique well identifiers in each data set. Of the resulting array data, wells containing T cells were then selected for further computational analysis (transition matrices, probability gains). Raw data were plotted using Excel (Microsoft Corporation, Redmond, WA, USA), Graphpad (GraphPad Software, Inc., La Jolla, CA, USA), MATLAB, Igor (WaveMetrics, Inc., Portland, OR, USA), and GenePattern (Broad Institute; MIT and Harvard, Boston, MA, USA).20
Results and discussion
Preparation and activation of CD4 T cells
Human MSCs expanded in vitro are known to be highly heterogeneous in that they contain cells with different physical properties and differentiation potentials. We hypothesize that in a heterogeneous MSC population, different subpopulations of MSCs contribute differently toward the modulation of CD4 T cells through the suppression of their activation and proliferation potential. This process involves cross-talk between MSCs and T cells through an orchestrated secretion of cytokines and cell–cell contact. MSCs and T cells cocultured in 6-well plates were distinguished by labeling with cell-specific surface markers (MSC: CD90; T cell: CD4). T cells were activated using magnetic beads conjugated with anti-CD3 and anti-CD28, mimicking the activation of T cells by antigen-presenting cells in vivo (Figure S1; Figure S2). The T cell activation state was monitored by real-time immunostaining of CD25 and CD69 expression. Cell viability and proliferation were accessed by live/dead staining and manual cell counts. Figure 1A shows an experimental schematic of CD4 T cell isolation from the peripheral blood, their activation, and coincubation with bone marrow MSCs. When CD4 T cells were freshly isolated, more than 95% of the cells (of 5,000,000 cells) were positive for CD4 in the flow cytometry assay (data not shown). To confirm T cell activation, we followed two early markers of T cells: CD25 and CD69. CD25 constitutes the alpha chain of the IL-2 receptor, an important component in the proliferation of T cells. CD69 is a part of the T cell receptor complex, which appears after T cell-receptor activation. Before T cell activation, approximately 20% of the cells expressed CD25, whereas only a small number of cells expressed CD69. After the activation of T cells, two distinct populations with regard to the expression of CD25 and CD69 were observed. Approximately 50% of the activated cells were double-positive for CD25 and CD69 after 16 hours of incubation (Figure 1B). After 48 hours, a dramatic change was observed in the cell morphology compared with that from earlier times (ie, an increase in cell size), and about 80% of the cells were double-positive for both CD25 and CD69, indicative of their activation status and readiness for coculture with MSCs. Figure 1C shows coculturing of two types of cells in the assay to study their interactions.
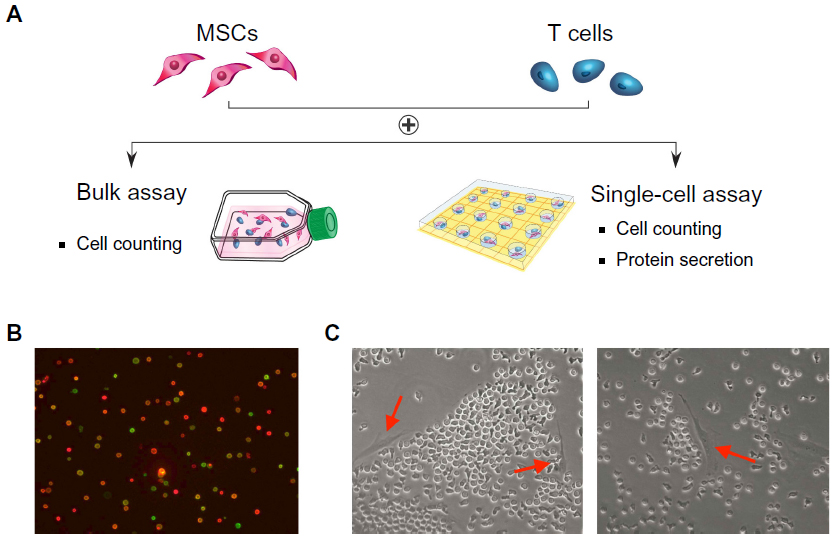 | Figure 1 Probing the immunosuppressive effects of mesenchymal stem cells (MSCs) on CD4 T cells. (A) Individual preparation of CD4 T cells and MSCs, followed by coculturing in a bulk or microwell assay. (B) Representative images of CD4 T cells after activation (imaged with anti-CD25 [green] and anti-CD69 [red] immunocytochemical staining). (C) Representative images of MSCs and CD4 T cells cocultured in 6-well plates (left arrow: CD4 T cell; right arrow: MSC). |
CD4 T cell and MSC coculture in a bulk assay
Figure 2A shows representative images of CD4 T cell and MSC cocultures. When T cells were not activated, they were seen randomly distributed around the MSCs. Activated T cells, however, had a tendency to be attracted toward MSCs and displayed adherence to MSCs (Figure 2A; Figure S4). In addition, activated T cells continued to proliferate, whereas less proliferation was observed in the presence of MSCs (Figure S5). Furthermore, bromodeoxyuridine data indicate that most of the cells either went through the cell cycle once or are in the process of DNA synthesis, suggesting that the T cell proliferation rate is negatively affected by the MSCs (Figure S6). To investigate the contribution of MSCs on the decreased rate of T cell proliferation, we altered the ratio of MSCs to T cells (1:1, 1:10, 1:100) and looked for changes in T cell proliferation rate at multiple times (0, 12, 24, 36, 48, and 60 hours) (Figure S7). The cell morphology for the 1:1 coculture group displayed a unique association between MSC colony formation and T cell distribution pattern. As the MSC:T cell ratio was increased (eg, from a 1:10 to 1:1 ratio of MSCs to T cells), the number of T cells significantly decreased. This phenomenon, however, was abolished when the MSC:T cell ratio was decreased (eg, from a 1:10 to 1:100 ratio of MSCs to T cells); the T cells, however, were more firmly attached to the MSCs (Figure 2A). Substantial cell proliferation was observed in the middle of the culture where most T cells had settled, indicating the T cell proliferation rate overcame any immunosuppressive stimuli released by the MSCs. At later incubation times (eg, 48 and 60 hours), the association between MSC colony formation and T cell distribution pattern in 1:1 groups was diminished completely. In the meantime, MSCs grew to almost 100% confluence at these times. In the 1:10 group at later times, T cells behaved differently around different MSC colonies. Although some MSCs appear to promote adhesion or seem to have no visible effect on T cell mobility, others seem to even repel T cells in wells. In the 1:100 group, the T cells completely outgrew the MSCs, suggesting the presence of a possible threshold for MSC immunosuppressive capacity. By adding MSCs to the culture at a 1:1 to 1:10 ratio, T cell proliferation was significantly suppressed (approximately 90% inhibition of proliferation). In contrast, when there were fewer MSCs in the culture (1:100 group), T cell proliferation persisted (Figure 2B). We also examined the effect of adding exogenous IL-2 to the activated T cells (Figure S8). On T cell activation, T cells naturally release IL-2 to induce proliferation and survival of the T cells. IL-2 can also be added exogenously to the culture medium to maintain T cell survival. The addition of IL-2 may reverse the immunosuppressive effects of MSCs, which suggests that the MSC immunosuppressive property may be transient. We found that although the addition of exogenous IL-2 accelerated the proliferation rate of T cells, the MSCs were still able to suppress T cell proliferation (Figure 2C).
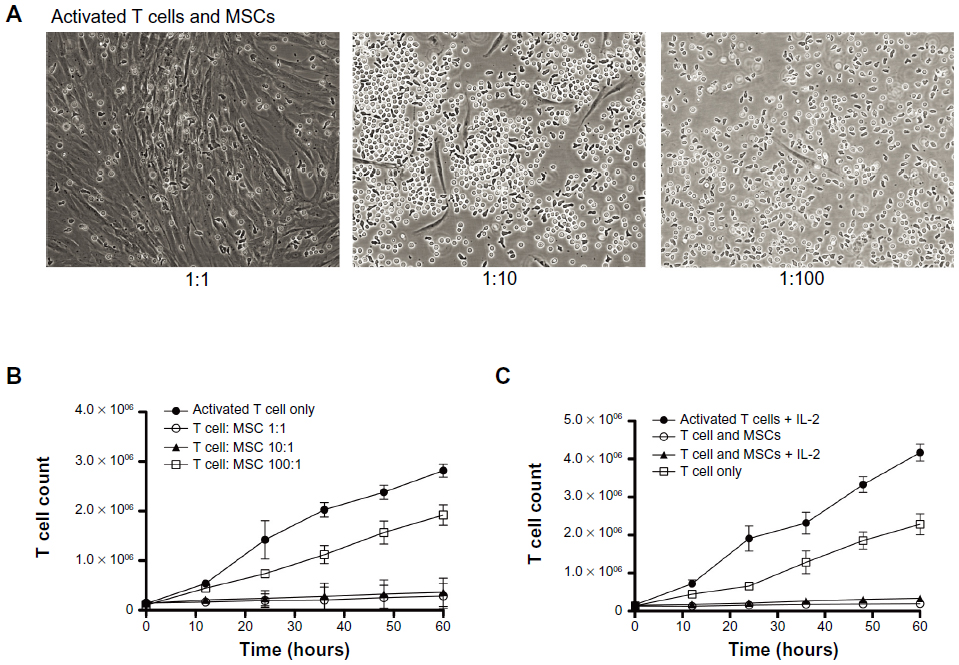 | Figure 2 Effect of mesenchymal stem cells (MSCs) on T cell proliferation. (A) Representative images of MSCs and CD4 T cells (MSC:T cell = 1:1, 1:10, 1:100). (B) T cell count after coculture with/without MSCs. Addition of MSCs to the culture significantly suppressed T cell proliferation (~90%) at 1:1 to 1:10 ratios. At a 1:100 ratio, there are fewer MSCs in the culture and T cell proliferation persisted. (C) T cell count after exogenous addition of interleukin 2 (IL-2). The addition of interleukin 2 significantly increased activated T cell proliferation. MSCs, however, still suppress T cell proliferation when interleukin 2 is added to the culture. |
CD4 T cell and MSC coculture in a cell–cell assay
We observed the inhibition of T cell proliferation by MSCs in the bulk assay format and confirmed the heterogeneity of MSCs with regard to their inhibition effects. We then devised a cell–cell assay to study the MSC and T cell interaction. Here, we wanted to demonstrate that smaller subsets of MSCs exhibit similar properties as those exemplified in the bulk assay, such as immunosuppression toward T cells. Figure 3A describes the experimental scheme of the cell assay performed. T cells and MSCs were cocultured in a microwell array, using a slightly modified protocol.19 The wells, which were 300 μm along all edges, were made using polydimethylsiloxane and coated with fibronectin on the bottom surface for optimal cell attachment. The MSCs were seeded first, followed by the addition of T cells 1 minute later. Thirty to 50 microwells were chosen, and the cells within were monitored (Figure 3B). Live-cell imaging for MSCs and T cells stained with different live-cell markers allowed us to track each cell type for up to 48 hours. Although we found that the number of T cells in microwells cocultured with MSCs for 48 hours did not change significantly (within ±2%), we also qualitatively determined that the number of T cells had a tendency to decrease in most of the selected microwells (Figure 3C).
 | Figure 3 Cell–cell analysis of mesenchymal stem cell (MSC)–T cell microenvironment interaction. (A) Schematic of microwell array assay for probing immunosuppressive effects of MSCs on cocultured CD4 T cells. (B) Representative image of CD4 T cells (round cells) and MSCs (yellow arrow) in microwells. (C) Immunosuppressive effect of MSCs on T cell proliferation. Percentage increase in each selected well is defined as the difference between the number of T cells at 48 hours and the initial number of T cells, divided by the initial number of T cells. (D) Representative plot of secretory responses measured by microengraving from microwells containing CD4 T cells cocultured with (+) or without (−) MSCs for 48 hours. Rates of secretion: *P < 0.05, **P < 0.01; one-tailed Mann–Whitney U test. Data are representative of three independent experiments. |
To investigate the key mechanism involved in the immunosuppressive process of MSCs on T cells, we employed the microwell cell–cell coculture system in conjunction with microengraving technology.17–19 Microengraving technology allows for multidimensional analysis of the rate and frequency of cytokine secretion. We tested three different soluble factors (IL-10, PGE2, and TGF-1) known to be associated with the immunosuppressive effects of MSCs. The average rates of secretion of the three soluble factors in the selected microwells were higher than those from microwells with only T cells (Figure 3D). Although not directly characterized here, similar measurements focusing on the secretory responses of MSCs could provide further information on the effect the evolution of microenvironments, created during cognate contact, has on both populations of cells. In addition, measuring cell–cell interactions between CD4 T cells and MSCs increases the dimensionality of data available and should further enable new criteria with which to discern key immunosuppressive signatures of MSCs and with which to construct models describing the behavior of cellular networks.
We envision that these data could be used to evaluate the delay of proliferation of T cells when they are cocultured with MSCs or the presence of immunosuppressive soluble factors secreted from MSCs at a level beyond conventional methods. The cell–cell coculture assay described here presents new opportunities to examine how cell–cell communication regulates intercellular cytokine-mediated signaling pathways and also has the potential to determine the plasticity and breadth of immunosuppressive responses that may not be fully understood in bulk assays. Further technical developments for the in situ tracking of each cell type with in-well intracellular cytometry may allow for future studies, such as determining the role of physical contact or elucidating an optimal cell–cell distance between MSCs and T cells for an observed immunosuppressive effect. Ultimately, we hope that by understanding the role of endogenous MSCs in the modulation of alloreactive immunity, we can better understand the principles behind MSC-based therapies for the prevention of allogeneic graft rejections or for the treatment of various autoimmune diseases.
Conclusion
We observed different types of interactions between the MSCs and T cells from our results, which strongly support the idea that MSCs are highly heterogeneous and that MSCs contribute differently to the modulation of CD4 T cell proliferation. In the bulk assay, we observed varying MSC and T cell association patterns across different wells, thereby supporting our hypothesis that the interaction between MSCs and T cells may be not identical but, rather, composed of different mechanisms. The “state” and “number” of MSCs (eg, colony formation, proliferation, self-renewal population versus differentiated population) may play a role in defining these mechanisms. We subsequently propose a microwell-based method to identify candidate target factors that play a role in the immunosuppressive capacity of MSCs. Using this methodology, we have presented here evidence that MSCs may modulate the proliferation rate of activated T cells with/without the addition of IL-2, mediated through the dynamic secretion of various types of soluble factors as well as through physical contact with T cells in the microenvironment. For both bulk and microwell assays, our results place an emphasis on the relative number of MSCs available to individual T cells. Notably, the importance of both soluble factors and physical contact between two cells in the modulation of the T cell proliferation rate has been documented1,3 and underscores the significance of the cellular microenvironment. Using our system, we are able to generate a microenvironment with a high probability of cell–cell interactions because of the optimal volume and surface area. Putting our results into context, individual MSCs appear capable of dynamically modulating the T cell proliferation rate in response to persistent cell–cell interactions. Finally, based on the identification of soluble factors secreted during cell–cell interactions, we anticipate an ability to screen for biomarkers that play a key role in the induction or enhancement of the immunosuppressive ability of MSCs and envision widespread repercussions on the development of drugs, particularly on MSC-based cellular therapies for the treatment of alloreactive immune-related diseases.
Acknowledgments
We thank Professor Sun W Song from Inha University Hospital, Incheon, South Korea, for kindly providing bone marrow-derived MSCs. We also thank the editorial board of the IEEE International Conference on Nano/Molecular Medicine and Engineering (Bangkok, Thailand) for providing us an opportunity to present our preliminary findings as a conference proceeding paper.21 This work was supported by the Korea Institute of Science and Technology (KIST Institutional Project 2E23720 and KIST-Global Research Lab 2Z03720) and the National Research Foundation (NRF) grant funded by the Korean government (MSIP) (No. 2008-0061891).
Disclosure
The authors declare no conflicts of interest in this work.
References
Soleymaninejadian E, Pramanik K, Samadian E. Immunomodulatory properties of mesenchymal stem cells: cytokines and factors. Am J Reprod Immunol. 2012;67(1):1–8. | |
Uccelli A, Moretta L, Pistoia V. Mesenchymal stem cells in health and disease. Nat Rev Immunol. 2008;8(9):726–736. | |
Shi M, Liu ZW, Wang FS. Immunomodulatory properties and therapeutic application of mesenchymal stem cells. Clin Exp Immunol. 2011;164(1):1–8. | |
Yagi H, Soto-Gutierrez A, Parekkadan B, et al. Mesenchymal stem cells: Mechanisms of immunomodulation and homing. Cell Transplant. 2010;19(6):667–679. | |
Sotiropoulou PA, Papamichail M. Immune properties of mesenchymal stem cells. Methods Mol Biol. 2007;407:225–243. | |
Sato K, Ozaki K, Mori M, Muroi K, Ozawa K. Mesenchymal stromal cells for graft-versus-host disease : basic aspects and clinical outcomes. J Clin Exp Hematop. 20150(2):79–89. | |
Tolar J, Villeneuve P, Keating A. Mesenchymal stromal cells for graft-versus-host disease. Hum Gene Ther. 2011;22(3):257–262. | |
Bartholomew A, Sturgeon C, Siatskas M, et al. Mesenchymal stem cells suppress lymphocyte proliferation in vitro and prolong skin graft survival in vivo. Exp Hematol. 2002;30(1):42–48. | |
Yañez R, Lamana ML, García-Castro J, Colmenero I, Ramírez M, Bueren JA. Adipose tissue-derived mesenchymal stem cells have in vivo immunosuppressive properties applicable for the control of the graft-versus-host disease. Stem Cells. 2006;24(11):2582–2591. | |
Studeny M, Marini FC, Champlin RE, Zompetta C, Fidler IJ, Andreeff M. Bone marrow-derived mesenchymal stem cells as vehicles for interferon- beta delivery into tumors. Cancer Res. 2002;62(13):3603–3608. | |
Akiyama K, Chen C, Wang D, et al. Mesenchymal-stem-cell-induced immunoregulation involves FAS-ligand-/FAS-mediated T cell apoptosis. Cell Stem Cell. 2012;10(5):544–555. | |
Ryan JM, Barry FP, Murphy JM, Mahon BP. Mesenchymal stem cells avoid allogeneic rejection. J Inflamm (Lond). 2005;2:8. | |
English K, Ryan JM, Tobin L, Murphy MJ, Barry FP, Mahon BP. Cell contact, prostaglandin E(2) and transforming growth factor beta 1 play non-redundant roles in human mesenchymal stem cell induction of CD4+CD25(High) forkhead box P3+ regulatory T cells. Clin Exp Immunol. 2009;156(1):149–160. | |
Macmillan ML, Blazar BR, DeFor TE, Wagner JE. Transplantation of ex-vivo culture-expanded parental haploidentical mesenchymal stem cells to promote engraftment in pediatric recipients of unrelated donor umbilical cord blood: results of a phase I-II clinical trial. Bone Marrow Transplant. 2009;43(6):447–454. | |
Hope K, Bhatia M. Clonal interrogation of stem cells. Nat Methods. 2011;8(Suppl 4):S36–S40. | |
Underhill GH, Bhatia SN. High-throughput analysis of signals regulating stem cell fate and function. Curr Opin Chem Biol. 2007; 11(4):357–366. | |
Zhao W, Schafer S, Choi J, et al. Cell-surface sensors for real-time probing of cellular environments. Nat Nanotechnol. 2011;6(8):524–531. | |
Choi J, Love KR, Gong Y, Gierahn TM, Love JC. Immuno-hybridization chain reaction for enhancing detection of individual cytokine-secreting human peripheral mononuclear cells. Anal Chem. 2011;83(17):6890–6895. | |
Gong Y, Ogunniyi AO, Love JC. Massively parallel detection of gene expression in single cells using subnanolitre wells. Lab Chip. 2010;10(18):2334–2337. | |
Reich M, Liefeld T, Gould J, Lerner J, Tamayo P, Mesirov JP. GenePattern 2.0. Nat Genet. 2006;38(5):500–501. | |
Choi J, Hwang MP, Lee JW, Lee KH. Interactions Between Mesenchymal Stem Cells and T Cells on a Single Cell Level. Proceedings of the 6th IEEE International Conference on Nano/Molecular Medicine and Engineering. 2012;111–116. |
Supplementary materials
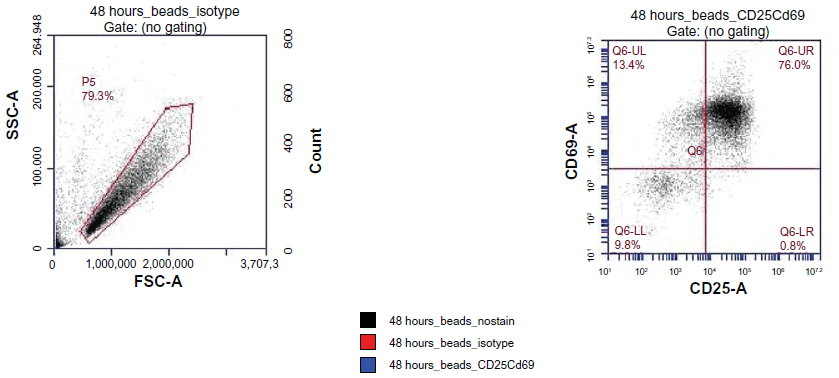 | Figure S1 T cells are activated via coincubation with anti-CD3 and anti-CD28 microbeads for 48 hours. Of the cells, 76% are double-positive for both markers (CD25 and CD69) on activation. |
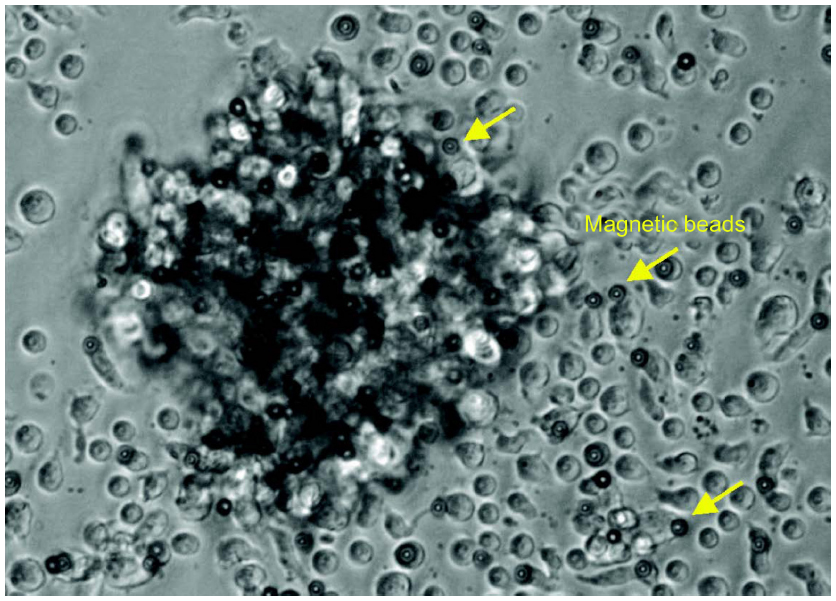 | Figure S2 The representative morphology of activated T cells coincubated with anti-CD3 and anti-CD28 microbeads is observed. Clusters of T cells and magnetic beads are formed, and cells are irregularly shaped and larger in size compared with nonactivated T cells. |
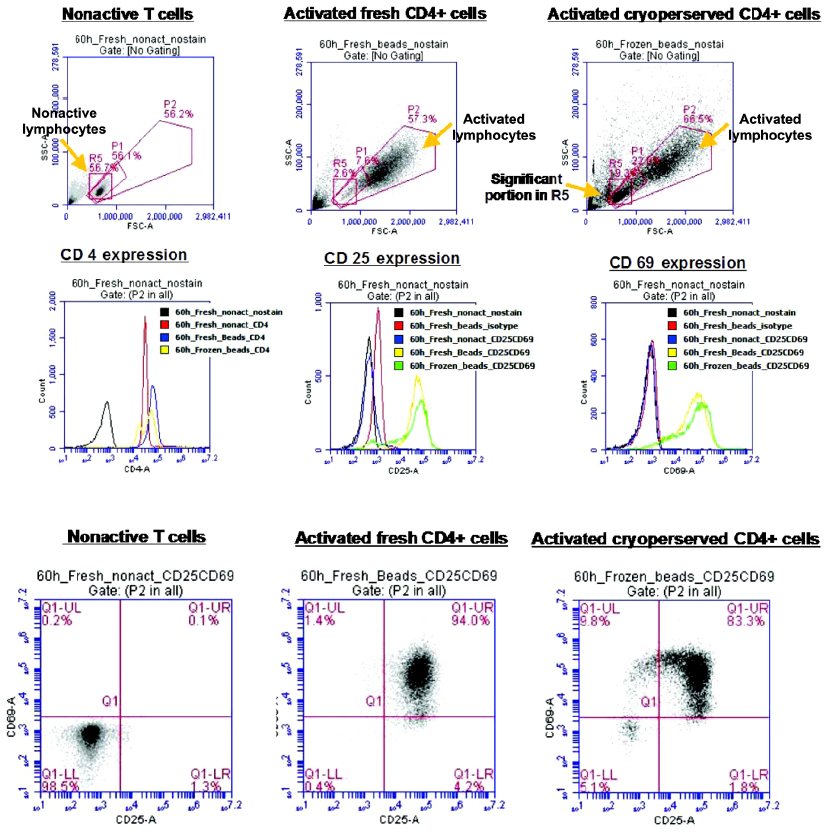 | Figure S3 T cell activation using isolated CD4+T cells from fresh peripheral blood mononuclear cells (PBMCs) and those from cryopreserved PBMCs is compared. Although cryopreservation potentially affects the activation of T cells, fresh PBMCs isolated from different donors may result in different relativities against anti-CD3 and anti-CD28 antibodies. |
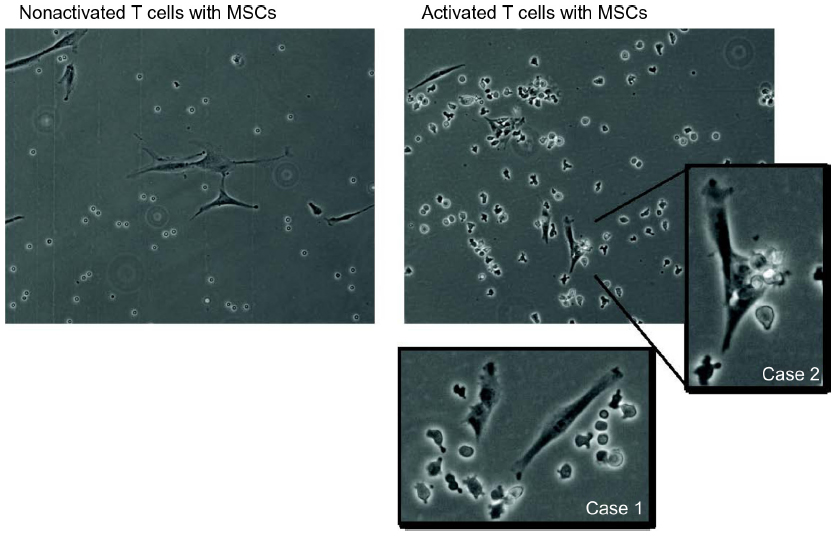 | Figure S4 The effect of mesenchymal stem cell (MSC) coculture on activated/nonactivated T cell proliferation is examined. Nonactivated T cells display a random distribution around MSCs, whereas activated T cells exhibit attraction (Case 1) or adherence (Case 2) to MSCs. |
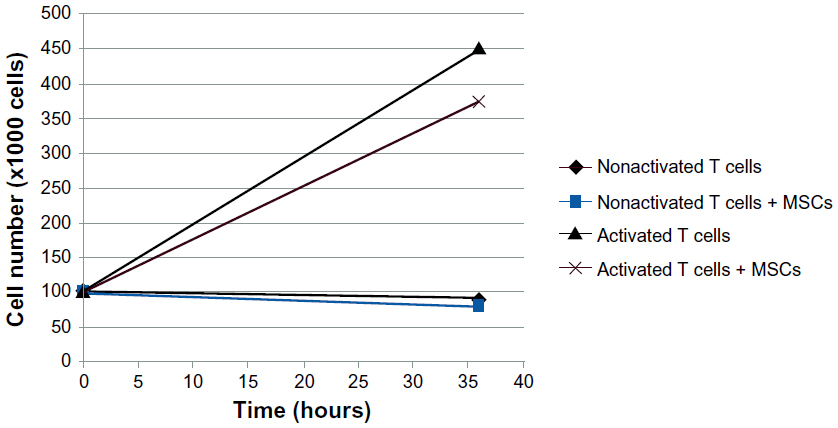 | Figure S5 A proliferation assay of T cells cocultured with or without mesenchymal stem cells (MSCs) for 36 hours indicates a lower number of T cells in the presence of MSCs. |
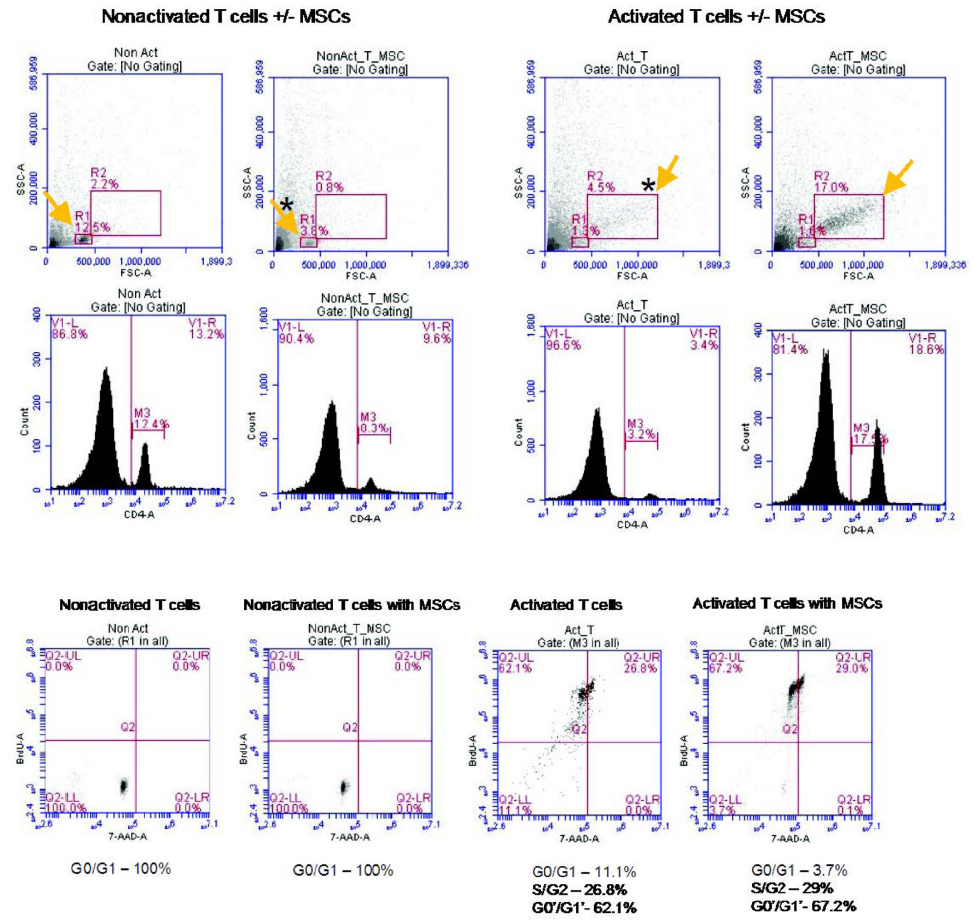 | Figure S6 Cell proliferation and cell cycle analysis are assessed using a bromodeoxyuridine proliferation assay. While activated T cells are actively proliferating, there is no significant difference in cell cycle position between groups with and without mesenchymal stem cells (MSCs). This may be a result of the prolonged incubation of bromodeoxyuridine, which could result in a full cell cycle for the majority of the cells at the time of analysis. |
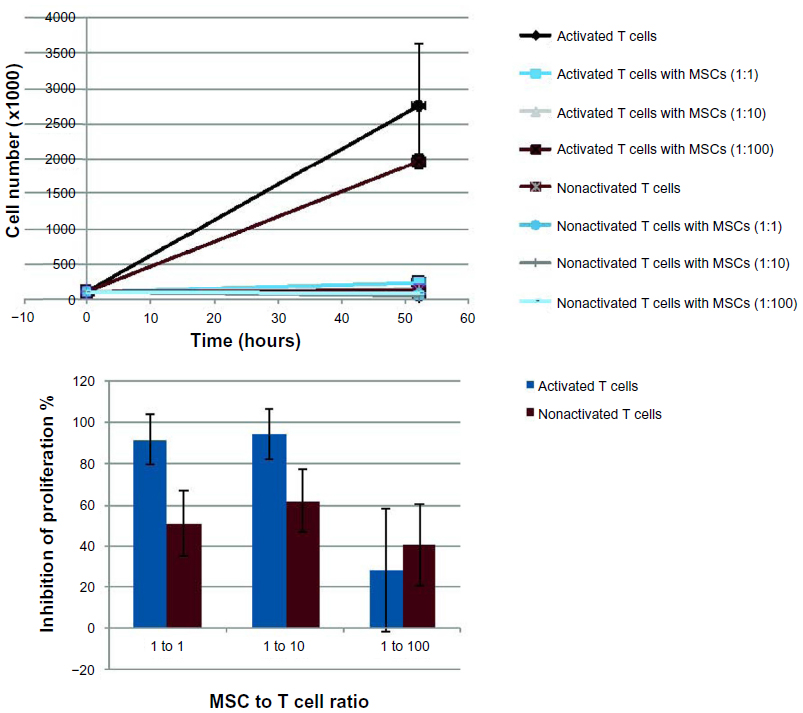 | Figure S7 The dose-dependent effect of mesenchymal stem cells (MSCs) in suppressing T cell proliferation is examined. The addition of MSCs to cultures of T cells at 1:1 to 1:10 ratios (MSC:T cell) significantly suppresses the T cell proliferation rate: approximately 90% proliferation inhibition is observed. At lower ratios of MSCs to T cells (1:100), T cell proliferation persists. |
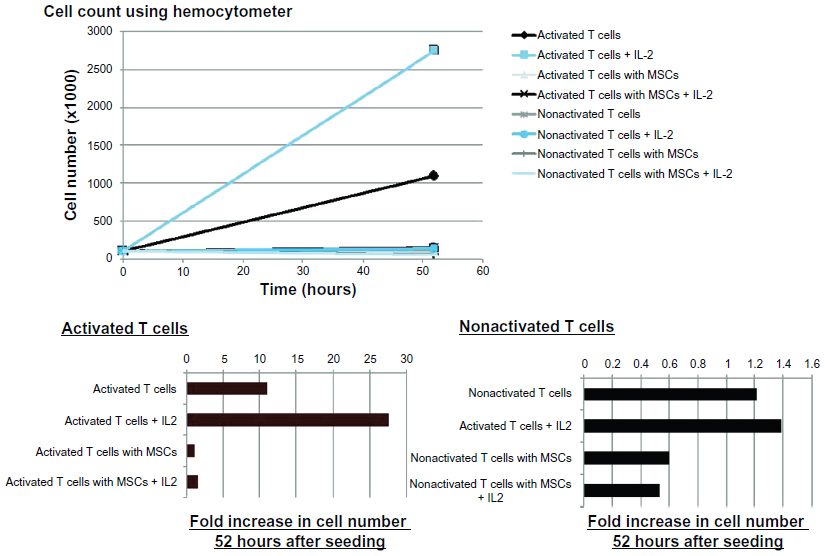 | Figure S8 The effect of exogenously adding interleukin 2 (IL-2) on the mesenchymal stem cell (MSC) suppression of T cell proliferation is examined. Although interleukin 2 addition significantly increases activated T cell proliferation in the absence of MSCs, it has no effect in the presence of MSCs as T cell proliferation suppression is observed. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.