Back to Journals » International Medical Case Reports Journal » Volume 10
A familial study of Hallermann–Streiff–François syndrome
Authors Epée E, Beleho D, Bitang AT, Njami VA, Bengondo C, Ebana Mvogo C ![]()
Received 2 June 2016
Accepted for publication 2 February 2017
Published 10 June 2017 Volume 2017:10 Pages 193—201
DOI https://doi.org/10.2147/IMCRJ.S114115
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Scott Fraser
E Epée,1 D Beleho,2 AT Bitang,3 VA Njami,4 C Bengondo,5 Côme Ebana Mvogo1
1Ophthalmology Department, Yaoundé University Teaching Hospital, Yaoundé, Cameroon; 2Ophthalmology Department, Okola District Hospital, Okola, Cameroon; 3Faculty of Medicine and Biomedical Sciences, University of Yaoundé I, Yaoundé, Cameroon; 4Higher Institute of Health Sciences, Université des Montagnes, Bangangté, Cameroon; 5Stomatology Department, Yaoundé University Teaching Hospital, Yaoundé, Cameroon
Abstract: Hallermann–Streiff–François syndrome is a rare sporadic genetic pathology characterized by a phenotype consisting of growth retardation, ocular abnormalities, and a “bird-like head”. We hereby report a case of this syndrome found in three generations of the same family – father, daughter, and grand-daughter – who presented with a short stature and facial dysmorphic features, nystagmus, cataract, and bilateral microphthalmia. The discussion is based on the clinical and genetic aspects, and the challenges in management of this oculo-mandibulo-facial syndrome. The association of congenital cataract, facial dysmorphic features, and microphthalmia, should guide the diagnosis of dysmorphic syndromes such as Hallermann–Streiff–François syndrome.
Keywords: Hallermann–Streiff–François syndrome, familial cataract, dysmorphic features, rare, Cameroon
Introduction
Hallermann–Streiff–François syndrome is a rare genetic disorder characterized by distinct cranio-facial abnormalities.1 Also called Francois’ Syndrome, Francois dyscephaly syndrome, Hallerman Syndrome, oculomandibulofacial syndrome, oculomandibulodyscephaly with hypotrichosis, Aubry 1 syndrome or Ulrich and Fremerey-Dohna syndrome, or finally François Type Mandibulo-facial dysmorphia,2–4 it is characterized by “bird-like” facies, dental abnormalities, hypotrichosis, skin atrophy, congenital cataract, bilateral microphthalmia and harmonious dwarfism.1,5
The first description of this syndrome was done in 1893 by Aubry6 and was completed by Hallermann7 in 1948 and Streiff8 in 1950. Hallermann and Streiff distinguished this syndrome from mandibulo-facial dysostosis described by Franceschetti and Zwahlen in 1944.9
In 1958, François, after analyzing 22 cases, concluded that the principal characteristics were: dyscephaly with a “bird-like head”, dental abnormalities, proportional dwarfism, skin atrophy (especially nasal),10,11 hypotrichosis, microphthalmia, and congenital cataract.11–13 Between 1965 and 1975, a number of authors completed describing this syndrome; Cohen studied 150 cases and analyzed the incidence of each sign while giving it a percentage.14 It is said that 100 cases have been described worldwide15,16 with a prevalence of less than 1/1,000,000 people.16 Despite the fact that many hypotheses on the genetic transmission have been raised,17–19 the etiology of this syndrome remains unknown1 and there is no predilection of gender.1,13
Clinical presentation
We report cases of Hallermann–Streiff–François syndrome found in three generations of the same family: father, daughter, and grand-daughter who came for a consultation during a cataract screening campaign for the management of a particular congenital cataract associated to other characteristic features of Hallermann–Streiff–François syndrome. As the patients are unable to read and write, the publication of the case details were explained in full to the patients and next-of-kin, and informed consent was received by two patients, and the next-of-kin on behalf of the third patient, who has since passed away. This included consent from the parents/guardians of the child whose image appears in this paper, and patient consent for the publication of all images and data.
Case 1
The patient aged 77, a farmer, came for a consultation for the management of reported congenital bilateral cataract. It was his first ophthalmologic consultation and the patient could not provide information on his birthweight or the neonatal period and there was no consanguinity between his parents. We found the same clinical features in his daughter and grand-daughter. He could see hand movements with the right eye and could count fingers at 2m with the left eye.
In addition, the ophthalmologic exam showed strabismus and nystagmus with a palpebral fissure of 2.3mm in both eyes; microphthalmia, enophthalmia, and a micro-cornea 8mm in diameter (Figure 1). Slit-lamp examination showed, apart from the above mentioned abnormalities, a gerontoxon, a clear cornea, and the anterior chamber was calm and deep; the photomotor reflex was present and normal, with an obliterating and bilateral mature cortical and nuclear cataract (Figure 2). Intraocular pressure could not be measured but seemed to be increased on digital palpation. Fundoscopy was not possible.
 | Figure 1 Case 1: bird-like face with strabismus and nystagmus. |
 | Figure 2 Case 1: obliterating cortical and nuclear cataract. |
General examination found a height of 1.58m, skin atrophy, retrognathism (Figure 3), an arched palate with a slight palatine depression at buccal cavity exam, and a narrowness of the maxilla with respect to the mandible (Figures 4 and 5). We also noticed microdontia, macrodontia, and oligodontia on Xray (Figure 5).
 | Figure 3 Case 1: retrognathism. |
 | Figure 4 Case 1: arched palate and slight palatine depression. |
 | Figure 5 Case 1: narrowness of the maxilla with tooth agenesis, microdontia of tooth 12, loss of teeth 31 and 32, macrodontia of tooth 33, and apical gap of tooth 36, as shown on Xray. |
Case 2
The second patient, daughter of the previous patient, aged 39 years, a farmer as well, had similar complaints. It was her first ever ophthalmologic consultation and there was no consanguinity link between her parents; pregnancy and delivery context was not documented for it was done traditionally; the mother of the patient had leprosy but died without any visual abnormality; history of chronic sinusitis treated with traditional medication and leprosy treated medically. The siblings of the patient had no visual abnormality.
Ophthalmologic exam revealed a visual acuity of 4/10 and 5/10 for the right and left eye respectively, a bilateral horizontal search nystagmus, a palpebral fissure of 2.7mm for both eyes, enophthalmia, microphthalmia, and a bilateral micro-cornea of 8mm (Figure 6). There was a bilateral nuclear cataract (Figure 7) rendering fundoscopy impossible and ocular tension was normal with digital palpation.
 | Figure 6 Case 2: bird-like face with strabismus and nystagmus. |
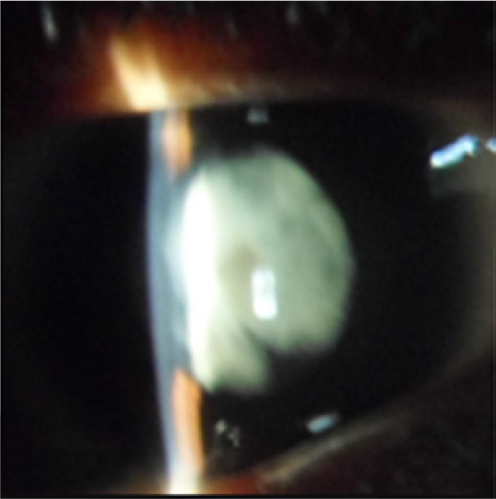 | Figure 7 Case 2: cortical and nuclear cataract. |
General examination found a height of 1.59m, retrognathism (Figure 8), skin atrophy (Figure 9); an arched palate with a slight palatine depression at buccal cavity exam, and narrowness of the maxilla with respect to the mandible (Figure 10).
 | Figure 8 Case 2: retrognathism. |
 | Figure 9 Case 2: nasal skin atrophy. |
 | Figure 10 Case 2: arched palate. |
Case 3
The third patient aged 19, was brought by her mother during the large scale screening campaign for management of a congenital bilateral cataract. There was no history of consanguinity between her parents either. The patient had a low birthweight according to the mother, but this information was not documented, since delivery was traditional. The patient had no chronic health conditions.
Ophthalmic exam showed a visual acuity of 1/20 of the right eye and counting fingers at 4m for the left eye; the palpebral fissure was 2.8mm in both eyes, and we noticed a bilateral search nystagmus, less amplified than the mother’s and grand-father’s, an alternate converging strabismus, microphthalmia, and a bilateral micro-cornea of 8mm in diameter (Figure 11). There was a bilateral nuclear and cortical dense cataract (Figure 12) rendering fundoscopy inaccessible and intraocular ocular pressure was normal at digital palpation.
 | Figure 11 Case 3: esotropia. |
 | Figure 12 Case 3: obliterating cortical and nuclear cataract. |
General examination found a height of 1.6m, skin atrophy, retrognathism (Figure 13); an arched palate with a slight palatine depression at buccal cavity exam (Figure 14), and narrowness of the maxilla with respect to the mandible (Figure 15). Globally, the patient presented fewer signs than her grand-father or mother. Cardiopulmonary exam was normal. Ocular ultrasound showed an apparently normal retina non-detached with axial length of 22.7mm and 23.2mm in the right and left eye, respectively.
 | Figure 13 Case 3: retrognathism. |
 | Figure 14 Case 3: arched palate with a slight palatine depression. |
 | Figure 15 Case 3: narrowness of the maxilla with respect to the mandible as shown on Xray. |
She was the mother of a 4-year old girl who is normal (Figure 16).
 | Figure 16 Case 3: obvious microcornea of the grand-daughter’s eyes, while her first child looks normal. |
Discussion
Hallermann–Streiff–François syndrome is a rare genetic condition where there is a high frequency of parental consanguinity,2,19 which is not the case with our family. Koliopoulos and Palimeris described a family while observing five cases in three generations with a father-to-son transmission.20 Guyard in 1962, reported a case of father-to-daughter transmission;21 later, Fraser and Friedmann defended the theory of dominant transmission, however a long history of consanguinity introduced the possibility of pseudo-dominance.22 Ponte, in 1962, reported the case of a woman and a man both of whom had the syndrome but did not transmit the trait to their offspring.23 Bueno-Sanchez found this syndrome in two brothers amongst three siblings from a marriage with consanguinity, defending the theory of a recessive autosomal transmission, the karyotypes were normal.24 Finally, in 1991, Cohen studied the cases of Ponte in the presence of corroborating and conflicting abnormalities in homozygote twins to affirm that the cases were sporadic.14
Father-to-daughter transmission of the phenotype, the presence of normal individuals amongst the siblings of the mother and the daughter and the disappearance of the phenotype at the fourth generation (great grand-daughter) in our case, can suggest an autosomal recessive transmission or dominant variable expressivity, but the sudden appearance of the syndrome in the first generation (father) does not clear the hypothesis of a new mutation which could have occurred during the embryo formation of the father. We could not proceed with further research by conducting genetic investigations since this was not accessible.
According to the literature, the potential causes of this syndrome are: an asymmetric anomaly of the second brachial arch which occurs during the fifth and sixth weeks of gestation, maternal viral infections, exposure to toxins, and paternal age.17 In our case, a history of leprosy was a common denominator between the daughter and her mother but not in the grand-father’s case. Leprosy is caused by a bacterial infection; it can therefore not be implicated as the cause of Hallermann–Streiff–François syndrome in this family.
The principal characteristics of Hallermann–Streiff–François syndrome (not to be mistaken for OMIM 221800 François syndrome) are dyscephaly with facio-mandibular malformations, bilateral congenital cataract, and dental abnormalities.2 The most frequently found characteristics are hypotrichosis, skin atrophy, microphthalmia, harmonious dwarfism, or small stature. Inconstant ophthalmologic signs are iris atrophy, blue sclera, angle malformation, peripheral anterior synechia, posterior synechia, papilla pallor, and glaucoma. Psychomotor development is usually normal, but exceptions do exist11,25 like in our case of the father, who presented a slight mental retardation. Complications, like respiratory difficulties and cardiac failure, were not found in this family. Seven diagnostic criteria exist for the oculo-mandibulo-facial syndrome,10,11,13 illustrated in Table 1.
 | Table 1 Diagnostic criteria of Hallermann–Streiff–François syndrome |
In our family, small stature being relative, we found six positive criteria concerning the father (1, 2, 4, 5, 6, 7) and the daughter (1, 3, 4, 5, 6, 7) and four criteria for the grand-daughter (1, 5, 6, 7). Table 2 represents all the characteristics of the syndrome found in our three patients with their frequency according to the literature. Meanwhile, the signs of the syndrome which are not part of this criteria are also grouped on the same table. Warburg in 1971 affirmed that the diagnosis of Hallermann–Streiff–François syndrome will be doubtful in the absence of cataract or microphthalmia;16 our three patients presented with cataract and microphthalmia, which are arguments in favor of the diagnosis. The dental examination is important since there are facial and craniofacial malformations.4,18,26,27 There may be mandible hypoplasia, lower jaw movement restriction, high-arched palate (palatine vault is curved), microdontia (narrow teeth root), presence of neonatal teeth (teeth present from birth), supernumerary teeth,18 dental growth restriction,26 and at times hypodontia (loss of a few teeth), or partial anodontia (absence of a series of teeth). Enamel hypoplasia may also be frequent, causing dental cavities; there may also be an abnormal dental alignment in the majority of cases27 or some ghost teeth.13,28 Stomatologic abnormalities in our patients were principally: an arched palate with a palatine depression and mandible hypoplasia, microdontia, macrodontia, and oligodontia in the father’s case with abnormal teeth alignment. Our three cases had a great variability of signs like several cases described in the literature. Lee et al in Korea, reported a case of Hallermann–Streiff–François syndrome with aphakia in 2008.5 Thomas et al, in India, reported a case of Hallermann–Streiff–François syndrome without cataract in 2013, stating that the lens, often described as white and liquefied, spontaneously resorbs in certain cases of Hallermann–Streiff–François syndrome.27
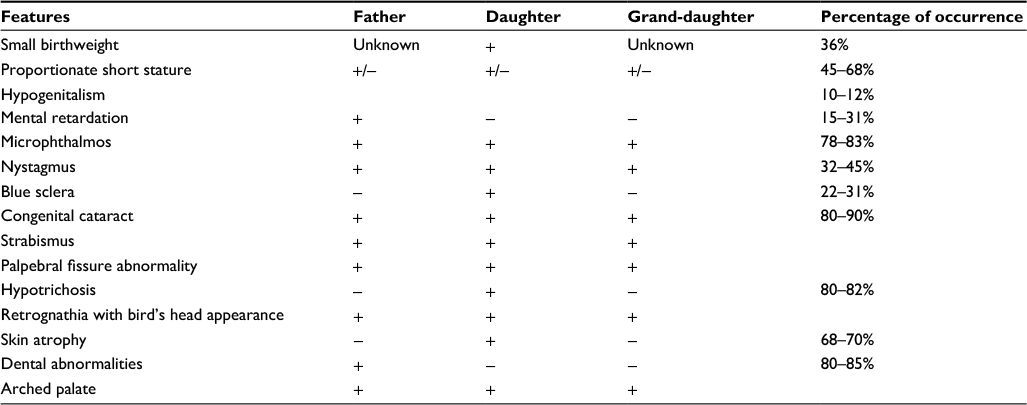 | Table 2 Frequency of Hallermann–Streiff–François syndrome signs found in father, daughter, and grand-daughter |
Daka et al, in 2013 in Kosovo, reported a case of Hallermann–Streiff–François syndrome without cataract, but there was glaucoma, corneal dystrophy, and a colobomatous iris.2 More recently, Le Merrer, in 2011, in France, found an abnormally folded retina with a retinal detachment peroperatively, complicating the surgery of an obliterating congenital cataract in a case of Hallermann–Streiff–François syndrome.15 A folded macula has been reported in the literature.18 A skull Xray would have revealed a mandible13 or a large skull with prominent frontal bumps;5 a chest Xray would have revealed distorted S clavicles12 or filiform ribs,18 and a limb Xray would have shown gracile long bones;26 biological exams are often normal.26
Differential diagnoses are oculo-dento-digital dysplasia (also called oculo-dento-bone dysplasia or Meyer-Schwickerath syndrome or Hallermann–Streiff pseudo-progeroid syndrome (PHS),13 mandibulo-facial dysostosis (Franceschetti syndrome or its less severe form, Treacher Collins syndrome), cleidocranial dysostosis or dysplasia, progeria and other progeroid syndromes.17 In Hallermann–Streiff PHS, or oculo-dento-digital dysplasia, we observe a severe spastic quadriplegia with apparently normal appearance at birth except for the absence of eyelashes and eyebrows.13 Progeria or Hutchinson-Gilford syndrome differs from Hallermann–Streiff–François syndrome in terms of the appearance of early arteriosclerosis, ungueal dystrophy, acromicria, and chronic arthritis, with normal eyes; Wiedemann-Rautenstrauch syndrome is a progeroid syndrome which differs from Hallermann–Streiff–François syndrome in terms of larger hands and feet.
In Seckel syndrome, we can see prominent eyes without cataract, malformed ears with a normal temporo-mandibular joint and skin atrophy with absence of hypotrichosis. In mandibulo-facial dysostosis, lower eyelid coloboma is common with ear abnormalities and in mandibulo-acral dysplasia, we note shortened distal phalanx acro-osteolysis. Oculo-facio-cardio-dental syndrome is a syndrome of multiple congenital abnormalities characterized by dental radiculomegaly, congenital cataract, facial dysmorphism and a congenital cardiac abnormality.29 Patients with Hallermann–Streiff–François syndrome might also have complications due to anatomical abnormalities of the upper respiratory tract such as swallowing difficulties, congestion of the respiratory tract, sleep apnea and cor pulmonale as a result of tracheomalacia.5,27 In our family case, there was no cardio-respiratory lesion.
According to the literature, there is no treatment for Hallermann–Streiff–François syndrome,13 management is symptomatic with particular attention to ophthalmologic, dental, and respiratory problems, right up to cranio-facial surgery.17 Recommendations exist in as much as respiratory issues are concerned, on the use of general anesthesia, ocular abnormalities, and on rhinoplasty, but data on the management of mandible malformations are not satisfactory.11 Concerning the management of cataract, piggyback type intra-ocular implantations have been described by Gayton and Sanders in 1993 in a patient with microphthalmia and cataract.30
Since then, a good number of surgeons adopted that technique which gave an adequate pseudophakic optic correction. This technique gives rise to a reduced number of spherical aberrations and gives a better image when the optical centers of the implants are well aligned only when we use an implant of great power. Nevertheless, complications like inter-lenticular opacification by Elschnig pearls are possible, or a membrane formation leading to a reduction of the best corrected visual acuity; this can occur three years after implantation. A potential complication can be endotracheal intubation which may cause difficulty during anesthesia.5 Shen et al, in China, in 2010, opted for cataract surgery in a patient with microphthalmia who did not present any vital risk using piggyback intra-ocular implants which gave good results.17 Lee et al, in Korea, conducted ocular surgery on both eyes, one after another, of a little girl with Hallermann–Streiff–François syndrome with aphakia but intra-ocular lens implantation corresponding to her age could not be done because of microphthalmia with a greater risk of post-operative glaucoma.5 The little girl finally received a prescription for corrective glasses. The risk of abnormally folded retina discovery and a peroperative retinal detachment should be considered during surgery according to Marc et al.15 A multidisciplinary follow-up, preventive health care programs, meticulous oral hygiene, a nutritional follow-up, and parent information are essential measures for patients with Hallermann–Streiff–François syndrome.17 Genetic counseling should be provided to the patients.13 The outcome is often bad; it is therefore crucial for the ophthalmologist to recognize the characteristics of Hallermann–Streiff–François syndrome and provide immediate care for these patients in order to improve their quality of life, and at best, save their lives.17
In our case, only a large scale screening campaign permitted us to discover and diagnose this rare disease, it is therefore difficult for the ophthalmologist in his/her routine practice to encounter these dysmorphic syndromes or to rapidly recognize them; since these patients live far away, in absence of primary eye care, and they often adapt to their condition. Furthermore, our family of patients with Hallermann–Streiff–François syndrome could not afford cataract surgery as a result of poverty which would also affect the follow-up after surgery.
Conclusion
Hallermann–Streiff–François syndrome is a rare disease. Its diagnosis is not easy and the circumstances of diagnosis showcase the value of outreach programs and resource-limited community strategies in an environment where all rare diseases are considered evil, without any hope outlet or solutions. Hallermann–Streiff–François syndrome is characterized by “bird-like” facies, dental abnormalities, hypotrichosis, skin atrophy, congenital cataract, bilateral microphthalmia, and harmonious dwarfism stature. Its management is multidisciplinary, and often difficult, with a poor prognosis most of the time as a result of poor vision or blindness. Nevertheless, symptomatic treatment of isolated manifestations, associated with a corneal sensitization could improve quality of life. Genetic tests are often unavailable in our context, despite the fact that genetic counseling is invaluable.
Disclosure
The authors report no conflicts of interest in this work.
References
Robotta P, Schäfer E. Hallermann–Streiff syndrome: case report and literature review. Quintessence Int. 2011;42(4):331–338. | ||
Daka Q, Miftari A, Vuciterrna A, Shiroka L, Bucinca S, Capuni-Brestovci M. Hallermann–Streiff syndrome without cataract: CASE REPORT FROM Kosova. Med Arch. 2013;67(5):378–380. | ||
vulgaris-medical.com [homepage on the Internet]. Syndrome-d-hallerman-streiff-francois Glossaire médical. Encyclopedie-medicale. Available from: http://www.vulgaris-medical.com/encyclopedie-medicale/syndrome-de-francois. Updated on August 23, 2016. Accessed February 22, 2017. | ||
Parikh S, Gupta S. Orodental findings in Hallermann–Streiff syndrome. Indian J Dent Res. 2012;23(1):124. | ||
Lee MC, Choi IJ, Jung JW. A case of Hallermann–Streiff syndrome with aphakia. Korean J Pediatr. 2008;51(6):646–649. | ||
Aubry M. Variété singulière d’alopécie congénitale:alopécie suturale [A singular variety of congenital alopecia: sutural alopecia]. Ann Dermatol Synphiligr. 1893;4:899–900. French. | ||
Hallermann W. Vogelgesicht und Cataracta Congenita [Bird face and congenital cataract]. Klin Montatsbl Augenh. 1948;113:315–318. German. | ||
Streiff EB. Dysmorphie Mandibulo-faciale (tete d’oiseau) et alterations oculaire [Mandibulofacial dysmorphia with ocular abnormalities]. Ophthalmologica. 1950;120:79–83. French. | ||
Franceschetti A, Zwahlen P. Un syndrome nouveau : De la dysostose mandibulo-faciale [A new syndrome: the mandibulo-facial dysostosis]. Bull Acad Suisse Sci Méd. 1944;1:60. French. | ||
Francois J. A new syndrome; dyscephalia with bird face and dental anomalies, nanism, hypotrichosis, cutaneous atrophy, microphthalmia, and congenital cataract. AMA Arch Ophthalmol. 1958;60(5):842–862. | ||
Kirzioğlu Z, Ceyhan D. Hallermann–Streiff Syndrome: a case report from Turkey. Med Oral Patol Oral Cir Bucal. 2009;14(5):E236–E238. | ||
el Massri A. Dyscephaly with congenital cataract. Br J Ophthalmol. 1967;51(5):352–355. | ||
Jain V, Sethi U, Dua S, Ahuja A, Wali BG. Hallermann–Streiff syndrome: a rare case report. J Indian Acad Oral Med Radiol. 2011;23(3):237–240. | ||
Cohen MM Jr. Hallermann–Streiff syndrome: a review. Am J Med Genet. 1991;41(4):488–499. | ||
Marc C, Guigou S, Boulicot C, Denis D. Anomalie rétinienne et chirurgie de la cataracte dans le syndrome d’Hallermann Streiff François: à propos d’un cas [Retinal anomaly and cataract surgery in Hallermann Streiff François syndrome]. J Fr Ophtalmol. 2011;34(2):118–121. French. | ||
orpha.net [homepage on the Internet]. Syndrome de Hallermann–Streiff. ORPHA 2108, Orphanet. Available from: http://www.orpha.net/consor/cgi-bin/OC_Exp.php?Lng= FR&Expert=2108. Updated on March 26, 2016. Accessed February 22, 2017. | ||
Shen MQ, Yuan F, Li L, Wang LY. Hallermann–Streiff syndrome: a case report. Chin Med J (Engl). 2010;123(22):3356–3357. | ||
Le Merrer M. Syndromes malformatifs ectodermiques [Ectodermal malformation syndromes]. Chapitre 31, [e-book] Février 2009.P491–495. Avalaible from: http://www.sfo.asso.fr/sites/sfo.prod/files/files/Rapports/2005 Oeil%20et%20genetique.pdf. Accessed March 24, 2017. | ||
omim.org [homepage on the Internet]. Hallermann–Streiff-Syndrome; HSS. MIM 234100. On line Mendelian Inheritance in Man. Available from: http://www.omim.org/entry/234100. Updated on November 22, 2010. Accessed February 22, 2017. | ||
Koliopoulos J, Palimeris G. A typical Hallermann–Streiff-Francois syndrome in three successive generations. J Pediatr Ophthal. 1975;12:235–239. | ||
Guyard M, Perdriel G, Ceruti F. Sur deux cas de syndrome dyscephalique à tête d’oiseau [On 2 cases of cranial dysostosis with ‘bird head’]. Bull Soc Ophtal Franc. 1962;62:443–447. French. | ||
Fraser GR, Friedmann AI. The Causes of Blindness in Childhood. A Study of 776 Children with Severe Visual Handicaps. Baltimore, MD: John Hopkins Press; 1967:89. | ||
Ponte F. Further contributions to the study of the syndrome of Hallermann and Streiff. (Congenital cataract with «bird’s face»). Ophthalmologica. 1962;143:399–408. | ||
Bueno-Sanchez M. Sindrome de Hallermann–Streiff-Francois [Hallermann–Streiff-Francois’ syndrom. About a familial case]. A proposito de una presentacion familiar. Boll Soc Vasco-Navarra. 1966;1:21–35. Italian. | ||
Francois J, Pierard J. The François dyscephalic syndrome and skin manifestations. Am J Ophthalmol. 1971;71(6):1241–1250. | ||
Yüksel B, Ozer G, Yilmaz M. Hallermann–Streiff syndrome: a case report. Gazi Medical Journal. 1995;6:87–90. | ||
Thomas J, Ragavi BS, Raneesha PK, Ahmed NA, Cynthia S, Manoharan D, Manoharan R. Hallermann–Streiff syndrome. Indian J Dermatol. 2013;58(5):383–384. | ||
Zhang, XX, Feng, HL. Hallermann-Streiff Syndrome: two typical cases with dental treatment and primary aetiological investigation. Chin J Dent Res. 2008;11(2)137–141. Available from: http://cjdr.quintessenz.de/index.php?doc=abstract&abstractID=14254. Accessed March 24, 2017. | ||
chu-rouen.fr [homepage on the Internet]. Syndrome oculo-facio-cardio-dentaire. CISMEF. Available from: http://www.chu-rouen.fr/page/doc/ DOC_56734. Updated December 10, 2007. Accessed March 24, 2017. | ||
Gayton JL, Sanders VN. Implanting two posterior chamber intraocular lenses in a case of microphthalmos. J Cataract Refract Surg. 1993; 19(6):776–777. |
 © 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.