Back to Journals » Drug Design, Development and Therapy » Volume 19
A Dual-Targeted Therapy with Fucoidan-Functionalized Thrombolytic Discoidal Microparticles for Pulmonary Thromboembolism
Authors Choi W, Cho H, Jang H, Park H, Youn I, Han S, Key J ![]()
Received 17 June 2025
Accepted for publication 16 September 2025
Published 21 November 2025 Volume 2025:19 Pages 10281—10297
DOI https://doi.org/10.2147/DDDT.S527596
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Anastasios Lymperopoulos
Wonseok Choi,1 Hyeyoun Cho,2 Hwijin Jang,2 Hyewon Park,1,3 Inchan Youn,1 Sungmin Han,1,4,5 Jaehong Key2
1Bionics Research Center, Biomedical Research Division, Korea Institute of Science and Technology (KIST), Seoul, Republic of Korea; 2Department of Biomedical Engineering, Yonsei University, Wonju, Republic of Korea; 3Department of Biomedical Engineering, Korea University, Seoul, Republic of Korea; 4Division of Bio-Medical Science & Technology, KIST School, Korea University of Science and Technology (UST), Seoul, Republic of Korea; 5KHU-KIST Department of Converging Science and Technology, Kyung Hee University, Seoul, Republic of Korea
Correspondence: Sungmin Han, Bionics Research Center, Biomedical Research Division, Korea Institute of Science and Technology (KIST), Seongbuk-gu, Seoul, 02792, Republic of Korea, Email [email protected] Jaehong Key, Department of Biomedical Engineering, Yonsei University, 1 Yonseidae-Gil, Wonju, Gangwon State, 26493, Republic of Korea, Email [email protected]
Background: Pulmonary thromboembolism, a pathological condition characterized by the occlusion of pulmonary vasculature by free-circulating thrombus, constitutes the third leading cause of cardiovascular-related mortality. Among conventional therapeutic approaches to manage the disease, systemic intravenous thrombolysis is hindered by inherent pharmacokinetic and pharmacodynamic limitations, including a short biological half-life, high requisite dosages, and an increased risk of hemorrhagic transformation. Given the critical need for prompt pulmonary reperfusion, this study introduces a dual-targeted therapeutic strategy employing fucoidan-functionalized, thrombolytic discoidal polymeric microparticles. This dual-targeted approach leverages the physicochemical properties of disc-shaped particles, which exhibit shape-dependent accumulation in the lungs, together with the biological binding affinity provided by the marine-derived component, fucoidan.
Methods: A top-down lithographic fabrication technique was employed to synthesize discoidal microparticle systems for physicochemical targeting to the pulmonary vasculature, providing precise control over the system’s geometry and uniform drug encapsulation efficiency. Furthermore, a PLGA polymeric matrix was positively modified to incorporate fucoidan onto its matrix surface, which is a sulfated polysaccharide with high-affinity interactions for P-selectin expressed on activated platelets in the nanomolar range. In vitro and in vivo thrombolysis assays were conducted to assess the therapeutic efficacy of microparticles.
Results: The proposed discoidal systems coupled with the fucoidan showed rapid accumulation due to their shape and selective interaction with activated platelets. Approximately 50% of the injected microparticles exhibited preferential accumulation within 15 minutes post-injection, and a significant portion remained over assay times. The fucoidan functionalization enhanced the targeting potential, yielding a 4.65- and 1.48-fold increase under static and dynamic flow assays, respectively (all p< 0.01). Although dramatic dissolution was not achieved using the proposed system in comparison with rtPA, alongside in vitro and in vivo investigations, the systems exhibited a more prolonged and dose-dependent lytic potential.
Conclusion: The proposed systems may offer an alternative to conventional systemic thrombolysis coupled with adjunctive pharmacological interventions.
Keywords: drug delivery system, pulmonary thromboembolism, discoidal polymeric particles, fucoidan
Introduction
Pulmonary thromboembolism, characterized by vessel occlusion in the lungs due to abnormal thrombus formation, is a third leading cause of mortality among cardiovascular diseases.1,2 Statistical data acquired in the United States demonstrate a gradual increase in morbidity and mortality rates associated with this condition.3 Additionally, the global spread of severe acute respiratory syndrome coronavirus 2 (COVID-19) has significantly heightened the risk of venous thromboembolism, further emphasizing the urgency of addressing this disease.4–6 Current standard therapies for pulmonary thromboembolism include thrombolytic agents such as recombinant tissue plasminogen activator (rtPA),7,8 anticoagulants (eg, unfractionated heparin, low-molecular-weight heparin, fondaparinux, warfarin, vitamin K antagonists), and reperfusion strategies.9 Among them, the administration of thrombolytic agents remains the most intuitive and powerful strategy for dissolving thromboembolic structures. However, these agents encounter significant limitations, including short half-lives, the requirement for high dosages, restricted applicability in patients with unstable hemodynamics, and an increased risk of hemorrhagic complications.10,11 These limitations restrict their therapeutic efficacy and clinical relevance.12 These challenges underscore the pressing need for innovative therapeutic strategies to improve the pharmacodynamics of these agents.
Drug delivery systems (DDS) offer a promising alternative to address these limitations. DDS refers to a specially designed platform that is intended to enhance the therapeutic efficacy of target drugs beyond the capabilities of conventional methods.13,14 By enabling controlled release,15–17 precise localization,16–21 and stabilization of therapeutic agents,20–22 thrombolytic DDS can ensure that lytic agents are delivered to their action site with optimal concentration and release rate. Criteria of effective thrombolytic DDS should (1) incorporate thrombolytic agents, (2) perform as targeted delivery within the lesion site, and (3) ensure rapid interaction with thromboembolic formations. Due to a rapid clearance of nanoparticles by the reticuloendothelial system (RES), larger microparticles, where they do not pose embolic risk themselves, have been proposed as more advantageous due to their slower clearance rates.23 Among nonspherical microparticles, discoidal particles with an approximate size of 3 µm have continuously demonstrated superior retention within the RES, particularly in the pulmonary system, due to their preferential accumulation property and vascular margination behavior.24–26 These particles tend to move alongside the vascular walls, thereby enhancing their interaction with elongated thromboembolic formations.27,28 Consequently, these physicochemical capabilities of discoidal particles could be beneficial for handling pulmonary thromboembolism.
Polymeric discoidal particles exhibit versatility to be modified with targeting moieties. One of the targeting moieties for circulating thromboembolic is a fucoidan. Fucoidan is a sulfated polysaccharide derived from marine sources and exhibits nanomolar affinity with P-selectin on activated platelets that are major components of thrombus.29,30 An underlying molecular basis of this interaction is the functional similarity of fucoidan to the natural ligand, P-selectin glycoprotein ligand-1 (PSGL-1).29 Typically, the structural features of Sialyl Lewis X (sLex) and sulfated tyrosine in PSGL-1 enable binding to P-selectin via its C-type lectin domain.31 Although fucoidan does not contain sLex or sulfated tyrosine residues, its sulfated fucose units can mediate strong interactions with P-selectin, thereby facilitating a high-affinity binding. Such sLex-mimicking molecule has been widely investigated for interacting with P-selectin, as well as other types of selectins.32,33 Current studies have demonstrated the efficacy of fucoidan-functionalized DDS combined with thrombolytic agents in lysing clots and reducing thromboembolic burden.34–36 Building upon this rationale, we developed a dual-targeted DDS that integrates both physicochemical and biological targeting mechanisms. The targeting capabilities of the proposed particles are derived from their unique discoidal shape and fucoidan functionalization, respectively. We hypothesize that this dual-targeted approach enhances the therapeutic efficacy of thrombolytic agents by enabling more precise and targeted drug delivery.
Material and Methods
Materials and Reagents
The poly(lactic-co-glycolic acid) (PLGA; 50:50; MW:25,000–35,000) was provided by Akina Inc (USA). Branched polyethyleneimine (branched-PEIs; MW: ~25,000), 1,3-dicyclohexylcarbodiimide (DCC), N-hydroxysuccinimide (NHS), dimethyl sulfoxide (DMSO), dichloromethane (DCM), poly(vinyl alcohol) (PVA; MW:9000–10,000), fucoidan from fucus vesiculosus, dialysis bags (molecular weight cut-off (MWCO):12,400), thromboplastin from rabbit (3–4 mg per vial), calcium chloride (CaCl2) dihydrate, Nile red, and collagen type I solution were purchased from Sigma-Aldrich (USA). Slide-A-Lyzer mini dialysis unit (MWCO:10,000) and bicinchoninic acid (BCA) assay kits were provided by Thermofisher Scientific (USA). Polydimethylsiloxane (PDMS) silicone elastomer base and its curing agent were purchased from Dow Corning (USA). FITC-labeled CD62p antibody solution was provided by BD Bioscience (USA). DyLight649-labelled IgG derivative against GPIbβ for labeling platelets in vivo (X649) was provided by Emfret analytic (Germany). The 4% paraformaldehyde (PFA) solution was purchased from Biosesang (South Korea).
Preparation of PLGA Bound to Branched PEIs (PLGA-PEIs)
We utilized the PLGA as the matrix of proposed particles. The PLGA was functionalized with branched PEIs (PLGA-PEIs) to electrostatically incorporate their matrix with fucoidan. More detailed, the PLGA-PEIs were synthesized using a typical DCC-NHS coupling reaction. The PLGA, DCC, and NHS (1:20:20, molar ratio) were dissolved in 5 mL of DMSO to activate the terminal carboxyl groups under 550 RPM for 1 hour at room temperature, followed by adding branched PEIs to couple with PLGA. The reaction was maintained for 24 hours at room temperature under a constant 550 RPM. The unreacted components were removed using a dialysis bag with a pore size of 12,400 of MWCO for 72 hours. The mixture was freeze-dried for 24 hours, and the final product was collected.
General Procedure of Lithography-Based Top-Down Fabrication
The proposed carriers, fucoidan-functionalized thrombolytic discoidal polymeric particles, were fabricated by a lithography-based top-down fabrication method found elsewhere.16,24 Briefly, this fabrication was performed as follows: (1) fabrication of master template, (2) fabrication of semi-permanent template using PDMS, (3) fabrication of disposable template using PVA, (4) loading components of particles into a disposable template, (5) and collecting particles. A more detailed description of each step is presented in the following subsections.
Synthesis of Control Discoidal Polymeric Particles
The silicon (Si) master template was fabricated via electron beam lithography, with each well designed to have a diameter of 3.0 µm and a height of 1.5 µm. A mixture of silicone elastomer base and curing agent (10:1, v/v) was prepared and cast into the master template to fabricate a semipermanent PDMS mold. The mold was then cured in an oven at 60 °C for 6 hours. Subsequently, 48 grams of PVA dissolved in 800 mL of distilled water were poured into the PDMS mold and incubated at 37 °C for 12 hours to synthesize disposable PVA film. For the control particles, referred to as discoidal polymeric particles (DPPs) in this article, 70 mg of PLGA-PEIs was dissolved in a co-solvent solution of 100 µL DCM and chloroform, then uniformly loaded onto the PVA film. The solvent mixture was evaporated in an oven at 37 °C. The dried PVA film was then immersed in 400 mL of distilled water with magnetic stirring (200 RPM) for 90 minutes to facilitate DPPs collection. A filtration was conducted sequentially with two membrane filters (100 µm nylon and 1.2 µm membrane) to remove PVA debris. The DPPs retained on the 1.2 µm membrane filter were collected by centrifugation at 4000 RPM for 90 seconds. The particle suspension was subsequently freeze-dried, and the final product was stored at 4 °C until further use.
Synthesis of Experimental Discoidal Polymeric Particles
The experimental discoidal polymeric particles included the following types: fucoidan-functionalized discoidal particles (FDPPs), rtPA-carrying thrombolytic discoidal particles (RDPPs), and fucoidan-functionalized rtPA-carrying thrombolytic discoidal particles (FRDPPs). Hereafter, all denotations for particles will be presented in their abbreviated forms unless otherwise specified. The procedures for synthesizing these particles were analogous to the above subsection, except for the loading components. More detailed, a 100 µL of rtPA solution (PBS; 15 mg mL−1) was prepared together with PLGA-PEIs solution for synthesizing overall rtPA-relevant thrombolytic particles. The fucoidan functionalization procedure was performed by adding the fucoidan solution (PBS; 1 mg mL−1) into each type of collected particle by rotating for 30 minutes to allow electrostatic binding. Each formulation was collected after centrifugation at 4000 RPM for 90 seconds. All particles were freeze-dried until further usage.
Characterization of Particles
Fourier Transform Infrared Spectroscopy (FT-IR)
A conjugation of branched PEIs with the PLGA matrix was confirmed using FT-IR spectra of dried formulation (FT-IR, Perkin Elmer, USA). Three different spectra of PLGA, branched PEIs, and PLGA-PEIs were compared with each other.
Hydrodynamic Size and ζ-Potential
All particles were dissolved in distilled water for measuring hydrodynamic diameter and ζ-potential. The diameter was measured using the Multisizer (Multisizer 4e Coulter Counter; Beckman Coulter, USA) due to the nonspherical shape of discoidal particles. The ζ-potential of particles was evaluated using a standard Zetasizer (Nano ZS90; Malvern Instruments, UK).
Morphology
Morphology was acquired using a scanning electron microscope (SEM; JSM-7800F; JEOL, Japan) and standard fluorescence imaging due to its size in the microscale. More detailed for SEM imaging, the acceleration voltage was fixed at 1.0 kV with an emission current of ~89.8 μA, while the working distance was maintained at approximately 4.0 mm. The magnification ranged from 4000× to 15,000× depending on the sample, and the system was operated under high vacuum conditions (~1.9 × 10−4 Pa). Since the discoidal microparticles were synthesized with thrombolytic drugs covalently conjugated to Rhodamine B, fluorescence imaging was employed to assess their morphology characteristics at the microscale.
Encapsulation Efficiency and Drug Loading
The encapsulation efficiency (EE) and drug loading (DL) were quantified using a standard BCA assay. The investigated loading mass of rtPA was 0.5, 1.0, 1.5, and 2 mg. The drug was incorporated during the synthesis stage. In order to estimate the amount of drugs in the carriers, a 25 µL aliquot of each particle solution dissolved in organic solvent (1 mg mL−1) was reacted with BCA stock solutions. A standard curve was prepared using a serially diluted rtPA solution ranging from 500 to 0 µg. The amount of drugs was estimated by using the standard curve. All BCA reactions were conducted according to the manufacturer’s protocol. The optical density was measured at 562 nm using a multi-mode reader (Epoch, BioTek, USA) after a 30-minute incubation. EE was calculated as the percentage of the actual amount of drug encapsulated within the particles relative to the initial amount of drug used for loading. DL was determined as the percentage of the mass of the particles that consisted of the encapsulated drug.
Cumulative Release Profile of rtPA From Carriers
A cumulative release profile of rtPA from the particles was assessed using dialysis. A 300 µL aliquot of FRDPPs solution (PBS; 1 mg mL−1) was loaded into dialysis units with a cut-off size of 10,000 MWCO. The released drug was collected at the bottom of an Eppendorf tube filled with 1.4 mL of PBS. All measurements were conducted at 37 °C. Released profiles were evaluated throughout 48 hours. The released drugs were quantified using the BCA assay. The solution containing released drugs was reacted with the BCA stocks, and optical density was measured following the analogous procedure as described in the above Encapsulation efficiency and drug loading subsection.
Stability Test
The stability of the particles was assessed by monitoring changes in their hydrodynamic size. A 10 µL sample of particles (1 mg mL−1) was diluted with 20 mL of the specific buffer (ISOTON II Diluent). Two different media–PBS and 30% fetal bovine serum (FBS)–were prepared to investigate particle stability. The variations in hydrodynamic size were measured over time. Particle stability was quantified by comparing the initial particle size to subsequent measurements and presented as a percentage of the initial size.
Animal
All animal experiments were approved by the Institutional Animal Care and Use Committee of the Korea Institute of Science and Technology (KIST-IACUC-2024-054) and conducted in accordance with institutional guidelines for animal welfare. Experiments followed the Laboratory Animal Act and Animal Protection Act of Korea, and adhered to internationally recognized principles, including the 3Rs (Replacement, Reduction, and Refinement). All rats were housed in standardized cages measuring 280×220 × 130 mm3 (length × width × height). Each mouse cage accommodated five animals. Food and water were provided ad libitum via containers placed on the cage lids. In vitro blood clots were fabricated using blood acquired from Sprague-Dawley (SD) rats weighing 280 g through heart puncture. In vitro fucoidan-mediated static/dynamic targeting examinations were established using blood acquired from C57BL/6 mice. The pulmonary thromboembolism examinations were performed using 6-week-old female C57BL/6 mice.
In vitro Thrombolysis
In vitro blood clots were fabricated for the thrombolysis assay as previously described.15,16 More detailed, 70 µL of freshly collected blood was mixed with 20 U (1 kU mL−1) of thrombin and incubated in a shaking incubator at 70 RPM for 30 minutes to produce highly retracted blood clots. The retracted clots were gently washed with PBS to remove unreacted blood components. Blood clots exhibiting excessive hemolysis during the washing process were excluded from further thrombolysis analysis. For in vitro thrombolysis, a blood clot was placed in a well of a 24-well plate, with each well containing 1.5 mL of PBS. The doses of either naive rtPA or rtPA-loaded particles (ie, RDPPs and FRDPPs) were adjusted to deliver 10 µg of rtPA per well. The thrombolysis reaction was conducted at 70 RPM in a shaking incubator for 180 minutes. The optical density measured at 415 nm was utilized to estimate the extent of blood clot dissolution due to peak absorbance of hemoglobin leakage. The thrombolysis observations were recorded at intervals of 30 or 60 minutes.
In vitro Targeting Examinations
Static Targeting Capability
A static targeting potential of FRDPPs was evaluated via immunohistochemistry. Briefly, in vitro blood clots were prepared and fixed with 4% PFA solution overnight. The fixed clots were cryo-sectioned at a thickness of 20 µm using a cryostat (Leica CM1950, Germany). Each section was incubated with 190 µL of FITC-labeled, diluted antibody solution to identify P-selectin on the activated platelets and stored at 4 °C overnight. Excess and unbound antibodies were removed by washing the slides twice with PBS. To investigate fucoidan-mediated binding, two types of particles were prepared: DPPs as a control and FRDPPs as the final formulation. Both particles were loaded with Nile red (0.5 mg mL−1) to visualize under fluorescence imaging. A 60 µL sample from each type (PBS; 0.3 mg mL−1) was applied to the sectioned slide. After washing with PBS three times, the static capability of each particle type was assessed using a Nikon Eclipse TS100 fluorescence microscope (Nikon Instruments Inc., Japan). A mean fluorescence intensity (MFI) surrounding P-selectin on the activated platelets was quantified using ImageJ software.37
Dynamic Targeting Capability
The dynamic targeting capability of FRDPPs was evaluated using microfluidic channels. To induce shear stress-mediated platelet activation/aggregations, linear channels within the microfluidic chip were coated with 10 µL of collagen type I solution (5 mg mL−1) and stored at 4 °C overnight. Before dynamic experiments, all channels were rinsed with 1 mL of PBS to remove excess coating materials. Fresh blood acquired from mice was circulated through the channel for 5 minutes using a pulsatile pump to induce platelet activation. The channels were then washed with 50 mL of PBS for 1 minute to remove unreacted blood components. Subsequently, the FITC-labeled, diluted antibody solution (PBS; 20 µg mL−1) was circulated within the channel to directly label P-selectin. Two types of particles were prepared to assess the dynamic targeting capability of fucoidan-functionalized particles, as described above: DPPs and FRDPPs. A particle solution (PBS; 0.5 mg mL−1) was circulated through the channel for 5 minutes at a velocity of 57 cm⋅s−1, which was close to venous conditions in the pulmonary region. The evaluation of dynamic targeting was conducted following the procedure outlined in the above static targeting capability subsection. Quantification of dynamic targeting was performed by measuring the MFI within platelet aggregates using the same software.
Pulmonary Thromboembolic Rodent Model
A pulmonary thromboembolism model was established in 6-week-old female C57BL/6 mice, with slight modifications based on previous reports.38,39 Thromboplastin derived from rabbit was utilized to induce the model. Mice were anesthetized intraperitoneally with a mixture of Zoletil 50 and Rompun (3:1, v/v). A vial containing 3–4 mg of thromboplastin was dissolved in 4 mL of 10 mM CaCl2 solution to achieve a concentration of approximately 1 mg mL−1. Due to the low solubility of thromboplastin in the solvent, the thromboplastin solution was continuously sonicated using a standard bath sonicator to ensure complete dissolution before use. Several doses of thromboplastin (0–600 µg) were initially screened to determine critical and sub-lethal dose ranges for the model. Doses exceeding 100 µg were not further tested, as this range approached the critical threshold. A criterion for sub-lethal dose was based on the observation of macroscopic acute/severe symptoms of pulmonary thromboembolism, such as shock or respiratory distress. The thromboplastin solution was administered via tail vein injection.
In vivo Imaging System (IVIS) Analyses of Pulmonary Thromboembolism
The intensity of circulating platelets in vivo was measured using the IVIS. The mice were treated with thromboplastin solution (50 µg/100 µL) to induce thromboembolism, followed by being administered 100 µL of DyLight649-labeled, diluted X649 antibodies 15 minutes post-thromboplastin injection to label circulating platelets. We hypothesized that relatively high signals of platelets corresponded to thromboembolic formation. The dilution of the antibody solution depended on the weight of the mouse, following the manufacturer’s guidelines. Representative organs, including the heart, lungs, spleen, liver, and kidneys, were harvested 20 minutes post-injection of the labeling antibodies to confirm the distribution of embolism. The quantitative measurement of thromboembolism was defined as total radiant efficiency per organ, which was assessed using the IVIS Lumina Series III system (Perkin Elmer, USA).
To confirm the thromboplastin dose-dependent intensity of thromboembolism, mice were injected with thromboplastin at varying doses (0, 35, and 50 µg/100 µL). The subsequent procedures, including labeling, organ harvesting, and quantification of fluorescence intensity in the lungs, were conducted to the protocol described above paragraph.
The physicochemical accumulation of particles into the lungs was validated using Cy5.5-labeled FRDPPs. A 200 µL solution of particles (PBS; 2 mg mL−1) was administered intravenously. The time-dependent accumulation of labeled particles was analyzed in the ex vivo lungs at onset, 15, 30, and 60 minutes post-injection.
To assess the therapeutic potential of FRDPPs, a comparative study was conducted using rtPA and FRDPPs. The doses of therapeutic agents were calibrated to 6 and 10 µg of rtPA. Following the establishment of thromboembolic model mice, the animals were treated with either PBS, rtPA, or FRDPPs via tail vein injection. The lungs were harvested 30 minutes post-injection, and fluorescence intensities were measured using the IVIS.
Statistical Analyses
Almost all data were presented by the mean and standard error of the mean (mean±s.e.m) in the respective figure, unless otherwise stated. Statistical comparison among groups over 3 groups was verified with one-way ANOVA, followed by Tukey’s multiple comparison post hoc analyses using Python Scipy API. Statistical comparison between the two groups was performed by an unpaired t-test using the same API. Statistical significance was presented as *p<0.05, **p<0.01 in the respective figure.
Results
Particles Characterization
The coupling of PLGA with branched PEIs was achieved through a conventional DCC–NHS reaction. The FT-IR spectra of three substances confirmed the success of the binding process (Figure 1a). In the FT-IR spectra of PLGA-PEIs, characteristic peaks were observed at 1638 cm−1 and 1557 cm−1, corresponding to C=O stretching (Amide I) and N-H bending coupled with C-N stretching (Amide II), respectively. Additional peaks at 1371, 1334, and 1247 cm−1 were identified as characteristic of C-N stretching and N-H bending vibrations (Amide III). This result shows the formation of amide bonds between PLGA and branched PEIs and a successful reaction.
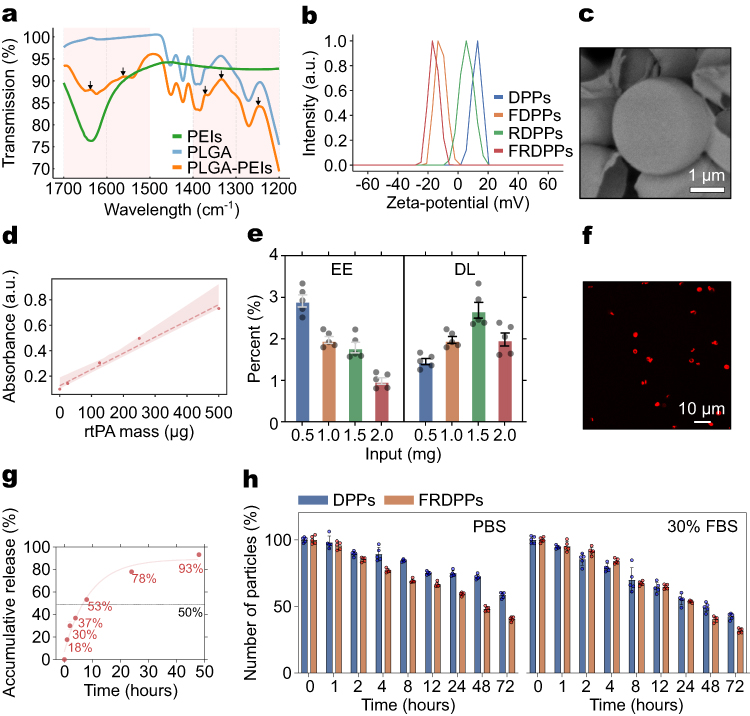 |
Figure 1 Characterization of particles. (a) FT-IR spectra of PLGA, PEIs, and PLGA-PEIs, confirming the successful integration of PEIs into the PLGA matrix. Black arrows indicate characteristic amide bond formations. (b) ζ-potential measurements of DPPs, FDPPs, RDPPs, and FRDPPs, illustrating the surface charge variations among different formulations, depending on loading materials. (c) Representative SEM image of FRDPPs, revealing a disc-like morphology. Scale bar: 1 µm. (d) Standard curve regression using serially diluted rtPA solution to estimate encapsulation efficiency (EE) and drug loading (DL). (e) EE and DL depending on the amount of added drugs (input), showing a highest DL when 1.5 mg of rtPA was loaded within the polymeric structure (all n=5). (f) Representative fluorescence image of FRDPPs containing fluorescently labeled rtPA, demonstrating even spatial distribution of rtPA. Scale bar: 10 µm. (g) Cumulative release profile of rtPA from the polymer over time due to natural polymer hydrolysis (n=5). A half-time for drug release from the carriers is approximately 7.13 hours. (h) Stability test based on size variations in PBS and 30% FBS, showing the most rapid degradation among all tested buffers in the FRDPPs group (all n=5). |
A top-down fabrication technique was employed to produce microparticles with highly uniform size and precise geometric characteristics. We designed the shape of particles as a disc. Fundamental properties of particles, such as hydrodynamic size, ζ-potential, and shape, were first measured. The hydrodynamic size of DPPs, FDPPs, RDPPs, and FRDPPs was 3005±464, 2824±423, 3028±520, and 2820±138 nm, respectively (mean±s.d; n=5). A similarity of hydrodynamic size across these formulations indicates that neither drug loading nor fucoidan functionalization had a statistically significant effect on their hydrodynamic size (all p>0.05). As shown in Figure 1b, the ζ-potential exhibited variations between groups depending on the loading materials. The ζ-potential of DPPs was 11.90±1.25 mV, revealing a positive charge. Functionalization with fucoidan resulted in a shift to a negative ζ-potential (−10.42±1.19 mV), which was attributed to the intrinsic negative charge of fucoidan. When rtPA was incorporated into the polymer matrix, the ζ-potential remained at a positive value (5.26±1.88 mV). The final formulation, FRDPPs, exhibited a ζ-potential of −15.10±0.90 mV.
The structural characteristics of FRDPPs were directly assessed via SEM imaging, confirming a disc-like morphology (Figure 1c). Based on the analysis of SEM images, the average diameter and pitch of these particles were 2.64±0.15 µm and 1.21±1.71 µm, respectively (mean±s.d; n=5). These values were closely aligned with specifications of the Si master template fabricated with a diameter of 3.0 µm and a pitch of 1.5 µm using the lithography technique. A slight discrepancy in such dimensions was likely due to the evaporation of organic solvents during the loading stage, as it promoted a compact loading of components into the wells in the PVA film. Importantly, the particle size and morphology remained controllable and showed minimal variations across different formulations using the top-down fabrication approach.
The quantification of EE and DL in the carriers was conducted using a BCA assay (Figure 1d and e). Interaction between BCA stocks and rtPA can induce a colorimetric change due to the nature of the protein, thereby facilitating the generation of the standard curve for estimation of drug loading. The regressed standard curve is depicted in Figure 1d. The particles were fully dissolved in organic solvent to allow drug release from the polymer matrix. The assay solution reacted with a solution containing leakage drugs and stocks resulted in a purple-like coloration, which was indicative of protein presence. The optical density exhibited a positive correlation with increasing loaded drug amounts. As a top-down fabrication approach offers the advantage of producing particles with uniform physicochemical properties rather than entrapping drugs within their structures, the estimated EE was relatively low, saturating at approximately 1–3% depending on the loading mass (Figure 1e, n=5). The highest DL was estimated at the loading mass of 1.5 mg. To further investigate the spatial distribution of rtPA within the polymeric matrix, a fluorescent dye was covalently conjugated to the drug molecules. The fluorescence image showed a homogeneous distribution of rtPA within the polymeric structures (Figure 1f). This observation suggests drug encapsulation and a uniform distribution of rtPA throughout the synthesized particles.
Given the critical urgency associated with pulmonary thromboembolism, the design of DDS prioritizes the efficient and rapid release of thrombolytic agents from carriers. Therefore, we investigated a release profile of the particles (Figure 1g). As a result, in vitro release kinetics of particles demonstrated that approximately 18% of the encapsulated drug was released within the first hour, primarily due to the hydrolytic degradation of the polymeric matrix. Overall, approximately 18%, 30%, 37%, 53%, 78%, and 93% of the drugs were released from the carriers at 1, 2, 4, 8, 24, and 48 hours under hydrolytic conditions, respectively (n=5). The stability assessments concerning the size variations revealed that the polymeric structures underwent a slightly accelerated degradation upon functionalization with fucoidan and rtPA (Figure 1h, n=5). This enhanced degradability suggests potential benefits in addressing the acute therapeutic demands of pulmonary thromboembolism. Irrespective of formulation type, all particles exhibited a pronounced tendency to degrade more rapidly in complex biological environments, such as FBS.
In vitro Thrombolytic Potential of Particles
The thrombolytic efficacy of particles was evaluated using an in vitro blood clot dissolution model (Figure 2). The blood clots exhibiting spontaneous hemolysis under washing steps were excluded to ensure experimental reliability. The size, morphology, and uniformity of the fabricated blood clots were meticulously standardized to minimize variability using a tube model.15,16
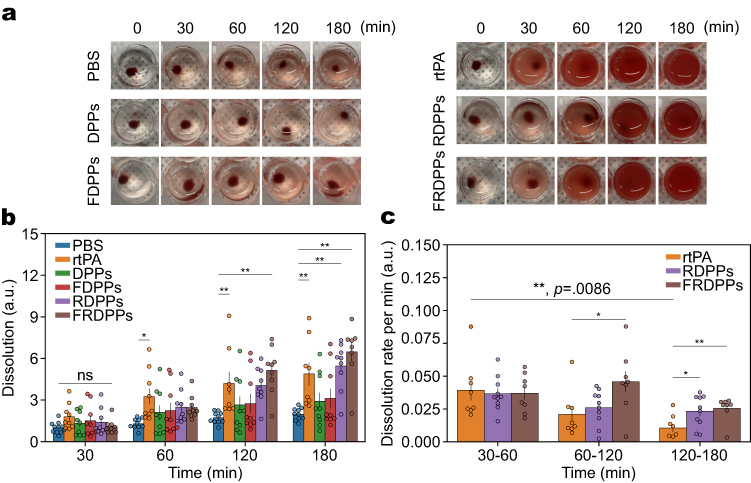 |
Figure 2 In vitro thrombolytic potential of the synthesized particles. Various particle formulations were evaluated for in vitro thrombolysis. (a) Representative thrombolysis images over time (0, 30, 60, 120, and 180 minutes) following treatment with PBS, rtPA, DPPs, RDPPs, FDPPs, and FRDPPs. Dose of rtPA was maintained at 10 µg across all groups. (b) Quantification of blood clot dissolution over the incubation period. Sample sizes were n=10–11 for PBS, n=8–9 for rtPA and FRDPPs, and n=9 for DPPs, FDPPs, and RDPPs, with outliers excluded. (c) Quantification of the clot dissolution rate per minute. While a rapid decline in lytic potential was observed in the rtPA group over the time, that of FRDPPs tended to be maintained over the investigated period. Sample sizes included n=8 for rtPA and FRDPPs, and n=10 for RDPPs, with outliers removed. Statistical significance was determined using one-way ANOVA followed by Tukey’s post hoc analysis. Significance levels are indicated as *p<0.05, **p<0.01, and ns (no statistical difference). All data are presented as mean±s.e.m. |
As anticipated, treatment with PBS resulted in negligible thrombolytic activity, with dissolution values remaining relatively constant across all time points. The slight reddish coloration observed in the assay medium across investigated time points could be attributed to mechanical agitation rather than the thrombolytic action of drugs (Figure 2a; along 30–180 minutes). The typical thrombolysis snapshots of PBS, DPPs, and FDPPs (left panel of Figure 2a) showed a minimal lytic potential. In contrast, the formulations containing rtPA—namely, rtPA solution, RDPPs, and FRDPPs—showed significantly enhanced thrombolytic activity, as depicted in the right panel of the same figure This result suggests that the incorporation of rtPA was the primary factor for thrombolysis in our design.
A degree of initial dissolution across investigated groups was not significantly different, supporting initial control over fabricated blood clots was well established (Figure 2b; 30 minutes; p=ns). A comprehensive quantitative analysis revealed that direct rtPA treatment induced a rapid thrombolytic reaction within the initial 60 minutes, while RDPPs and FRDPPs exhibited relatively lower thrombolytic activity during this early phase (Figure 2b; 60 minutes). However, both particle-based formulations achieved thrombolytic potential comparable to naive rtPA after 120 minutes. The primary advantage of these formulations lies in their ability to sustain drug activity over time, as depicted in Figure 2c. Although the initial dissolution rate per minute of all groups was similar, the thrombolytic activity of rtPA declined sharply over the assay time, showing approximately 73% of its initial thrombolytic activity lost by 120 minutes. In contrast, both rtPA-carrying particles, RDPPs and FRDPPs, tended to maintain consistent thrombolytic activity and led to significant differences in the dissolution rate compared to the rtPA group beyond 60 minutes post-treatment. The value for FRDPPs remained consistently high during the 60–120 minute interval and started to decline over time. As a prolonged thrombolytic effect was not observed in the RDPPs group, it could be interpreted that the fucoidan functionalization contributed to the protracted thrombolytic effect through expected biological interactions. Unfortunately, all rtPA-containing formulations did not present considerable blood clot dissolution; however, modification with fucoidan offered superior thrombolytic efficacy by extending the therapeutic duration of rtPA.
In vitro Biological Targeting Potential of Particles
Although our previous in vitro results demonstrated that fucoidan functionalization tended to aid in sustaining thrombolytic potential over time more effectively than the naive rtPA, a significant enhancement in clot dissolution was not observed in terms of quantifiable dissolution. This complicates the interpretation of the biological binding effects of fucoidan. Therefore, it was necessary to validate whether the proposed particles exhibited direct biological binding interactions with clots (Figure 3).
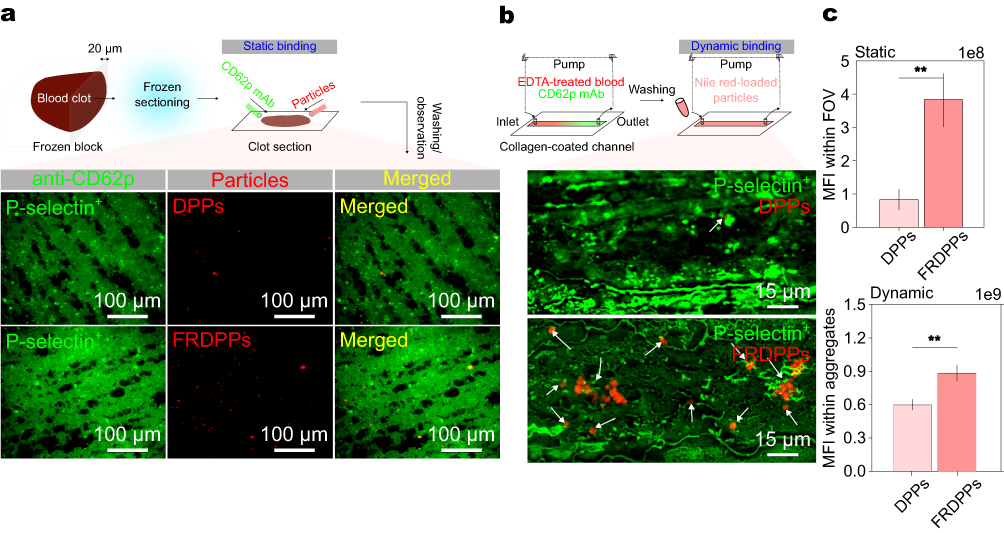 |
Figure 3 In vitro biological targeting potential of proposed particles. (a) Representative images depicting static targeting of particles toward P-selectin on the platelets, showing an increased affinity of FRDPPs in clot sections. Scale bar: 100 µm. (b) Representative images of dynamic targeting toward P-selectin, even highlighting the superior targeting efficiency of FRDPPs under fluidic conditions. White arrows for DPPs and FRDPPs binding on the activated platelets. Scale bar: 15 µm. (c) Quantitative analysis of MFI for static and dynamic binding. The upper and lower panels correspond to MFI values within field of view (FOV) for clot sections and platelet aggregates, respectively (n=10 and n=12 for static binding; n=6 and n=5 for dynamic binding for DPPs and FRDPPs). Statistical significance was assessed using an unpaired t-test; **p <0.01. All data are presented as mean±s.e.m. |
In order to investigate the fucoidan-mediated binding capability of FRDPPs, two distinct in vitro binding assays were conducted: static and dynamic binding evaluations. The static binding assay was designed using transverse sections of blood clots (Figure 3a). The fixed and frozen-sectioned clots with a thickness of 20 µm were incubated overnight with anti-CD62p antibodies before exposure to the formulated particles for visualizing activated platelets within the section. Multiple rigorous washing steps were performed to eliminate potential biases arising from nonspecific and gravitational binding. We hypothesized that biological binding capability could be confirmed even after the samples were labeled with these antibodies, with high nanomolar affinity. As a qualitative analysis, typical static examination revealed a substantial enrichment of FRDPPs in labeled clot sections, even after extensive washing, indicating their superior targeting affinity compared to non-functionalized counterparts (Figure 3a).
The dynamic binding assay was conducted using a microfluidic line-channel system to simulate physiological shear conditions. Freshly drawn blood from the mice was circulated within a collagen-precoated channel to induce platelet activation under flow (Figure 3b). Given the somewhat high velocity (v = 57 cm⋅s−1), passive sedimentation of particles was deemed negligible, thereby allowing for an accurate assessment of specific binding interactions. As presented in Figure 3b, the trend of dynamic binding was similar to that of the static assay, showing enhanced affinity for shear-activated platelets compared to non-functionalized counterparts (see white arrows for FRDPPs binding on activated platelets).
Consistently, the quantitative analysis of both static and dynamic binding assays demonstrated that FRDPPs enhanced the biological targeting efficiency in comparison with DPPs. FRDPPs exhibited a 4.65-fold increase in binding affinity under static conditions and a 1.48-fold increase under dynamic flow conditions, as shown in Figure 3c (all p<0.01). These results further emphasize the role of fucoidan functionalization in promoting efficient platelet targeting, which could enhance thrombolytic therapy by facilitating selective clot dissolution while minimizing off-target effects. These findings underscore the role of fucoidan functionalization in improving the targeting efficiency of polymeric DDS, thereby enhancing their potential for selective thrombolytic therapy under physiologically relevant conditions.
In vivo Pulmonary Thrombolysis of Particles
The pulmonary thrombolysis was performed using the thromboplastin-inducible pulmonary thromboembolic model (Figure 4). A bolus injection of thromboplastin is a well-documented method for inducing pulmonary thromboembolism.38,39 A preliminary dose-escalation study was conducted to determine the sub-lethal threshold of thromboplastin dose for 6-week-old female mice (Figure 4a). The dose criteria were to induce a survival rate of up to 50%. The thromboplastin solution was prepared at a concentration of 1 mg mL−1 and administered via tail vein injection. As a result, the survival profile revealed that the dose of 50 µg (100 µL; 50 µL of stock plus the remaining 50 µL of PBS buffer) induced a moderate state of pulmonary thromboembolism. All mice administered the dose above 70 µg exhibited acute symptoms and ultimately succumbed to the conditions (ie, all died). Therefore, the thromboplastin dose for subsequent studies was determined at 50 µg with the same volume as described above. This dosage was considered to be the optimal dose for our model, as evidenced by an approximate 60% survival rate among the experimental cohort.
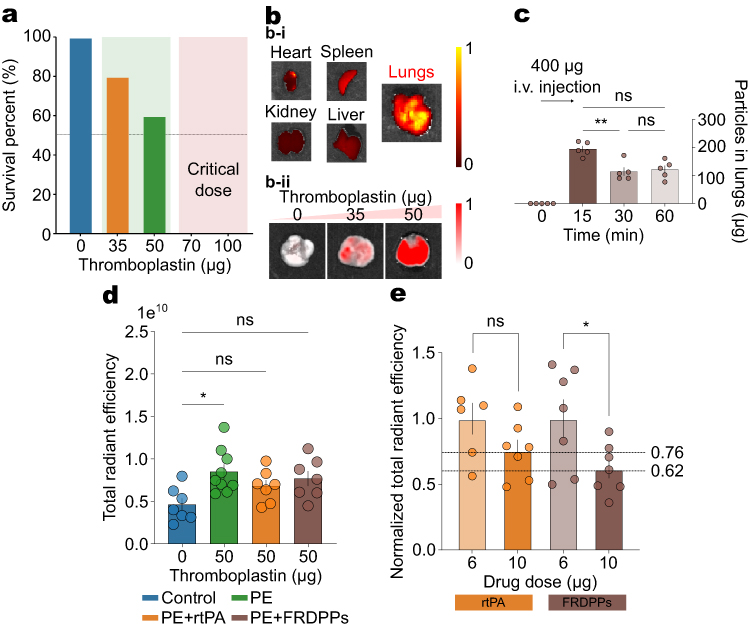 |
Figure 4 In vivo pulmonary thrombolysis potential of FRDPPs. (a) Survival rate depending on thromboplastin dose, indicating that doses exceeding 50 µg approach the sub-lethal threshold. All mice administered doses above 70 µg exhibited acute symptoms of pulmonary thromboembolism. The determined dose for this study was 50 µg. (b) Representative IVIS images. (b-i) Visualized IVIS intensity of harvested major organs, showing the lungs displayed the highest signal intensity among all examined organs. (b-ii) Visualized IVIS intensity of lungs from mice treated with varying thromboplastin doses (0, 35, and 50 µg), revealing dose-dependent thromboembolic accumulation in the lungs. All organ images in (b-i and b-ii) are displayed with the same radiant efficiency scale (min–max fixed) to enable direct comparison. (c) A preferential accumulation of disc-shaped FRDPPs in the lungs, peaking at 15 minutes post-injection and remaining at elevated levels up to 60 minutes, as shown by the quantification of particle retention in the lung tissues (all n=5). (d) Quantification of total radiant efficiency in the lungs. Administration of 50 µg thromboplastin significantly increased thromboembolic signals (p=0.011). Treatment with either rtPA or FRDPPs reduced the efficiency levels to values comparable to the control group. PE: pulmonary thromboembolism-induced group. (e) Dose-dependent reduction in radiant efficiency in rtPA and FRDPP-treated groups. Data for 6 µg cases were normalized to directly compare with 10 µg cases. Statistical significance was determined using one-way ANOVA followed by Tukey’s post hoc analysis or unpaired t-test; *p<0.05, **p <0.01, and ns (no statistical difference). All data are presented as mean±s.e.m. |
In order to confirm the extent of thromboembolic burden in the lungs following thromboplastin injection, fluorescently labeled antibodies that targeted circulating platelets were administered to model mice (Figure 4b). These antibodies, conjugated with DyLight649 immunoglobulin derivatives, have been extensively utilized for real-time in vivo thrombus visualization.40,41 We hypothesized that the fluorescence intensity of these antibodies in the major organs correlated with thromboembolic accumulation, as thromboembolic formations coincide with enriched and activated platelets. Representative IVIS imaging of ex vivo major organs showed that the most prominent signal was localized to the lungs (Figure 4b–i), supporting that the thromboplastin could induce pulmonary thromboembolism. Furthermore, a dose-dependent escalation in thromboembolic deposition was evident across the tested dose range (Figure 4b–ii). The strongest pulmonary thromboembolic signal was observed in the 50 µg group under standardized imaging parameters, reinforcing the dose-dependent progression of thromboplastin-induced thromboembolism.
The shape-dependent physicochemical targeting efficiency of FRDPPs was analyzed using the IVIS (Figure 4c). Model mice were administered 200 µL of Cy5.5-conjugated FRDPPs (PBS; 2 mg mL−1), and pulmonary accumulation was evaluated at 0, 15, 30, and 60 minutes post-injection. Quantitative analyses revealed that 48.7±6.27, 28.7±8.44, and 30.3±8.11% of the injected particles accumulated in the lungs at these respective time points (mean±s.d; all n=5). A rapid accumulation of discoidal particles was observed within the 15 minutes post-injection, and also a large portion of the injected carriers remained in the lung vasculature over the investigated time (p=ns between 30 and 60 minutes). These results further support that a substantial proportion of disc-shaped particles rapidly localize to the pulmonary vasculature, likely contributing to the sustained degradation of pulmonary thromboembolic through prolonged retention within the lung region.
A precise control of dose between rtPA and FRDPPs was performed to compare in vivo pulmonary thrombolysis efficacy. Since circulating platelets are naturally present in the pulmonary vasculature, the average baseline total radiant efficiency in the lungs was quantified at approximately 4.6×109 (Figure 4d; Control). The induction of moderate pulmonary thromboembolism was achieved using 50 µg of thromboplastin (Figure 4d; PE), resulting in an approximately twofold increase in radiant efficiency compared to the control group (p=0.011). Unfortunately, the treatment of rtPA or FRDPPs only exhibited a decrease in the mean signal intensity relative to the 50 µg thromboplastin-treated group and did not show statistical significance (all p=ns). Consistent with in vitro observations, a pronounced therapeutic effect was only observed when comparing across different dose groups (Figure 4e). At a reduced dose of 6 µg, neither rtPA nor FRDPPs demonstrated a significant thrombolytic effect, with statistical analysis indicating no meaningful differences between the 50 µg thromboplastin group and the 6 µg rtPA- or FRDPPs-treated groups. To allow for intra-group comparison, values measured at 6 µg were normalized within their respective groups before direct comparison with the 10 µg dose. While the dose of rtPA to 10 µg did not show any dose-escalation reduction in the thromboembolism (p=ns), administration of 10 µg FRDPPs resulted in a decrease to 62% of the corresponding 6 µg values (p=0.036). These findings suggest that dose escalation of FRDPPs may be essential for eliciting an effective thrombolytic response, in contrast to rtPA, which failed to demonstrate enhanced efficacy under equivalent dosing conditions.
Discussion
In this study, a dual-targeted thrombolysis system was developed utilizing fucoidan-functionalized thrombolytic discoidal particles, and their thrombolytic potential was investigated. The formulated FRDPPs were designed to exhibit dual-targeting capabilities for abnormal thromboembolic formations by leveraging both physicochemical and biological targeting mechanisms. Each targeting potential resulted from their discoidal shape, favorable to target the lungs physically24–26 and additional fucoidan functionalization,29,30,34–36 respectively.
The effectiveness of this system can be largely attributed to the utilization of PLGA as the particle matrix, which enables precise modulation of the release profile of therapeutic agents through adjustments in the lactic-to-glycolic acid ratio.24 In general, the release behavior of PLGA-based microparticles is strongly influenced by this ratio, thereby affecting both encapsulation efficiency and release kinetics. Previous studies have demonstrated that PLGA with a 50:50 ratio undergoes faster degradation than other common formulations (eg, 75:25, 65:35), likely due to its balanced hydrophilic–hydrophobic composition.42,43 The higher glycolic acid content in the 50:50 formulation contributes to increased hydrophilicity, leading to more rapid and preferential degradation. Considering the time-sensitive nature of pulmonary thromboembolism, PLGA with a higher glycolic acid content represents a more suitable matrix candidate. Furthermore, the biocompatible and biodegradable properties of PLGA make it highly suitable for a broad range of biomedical applications, including thrombolysis,16,44,45 despite the inherent hemorrhagic risks associated with the therapeutic agent, rtPA.10,11 Notably, the chemical versatility of PLGA polymers allows for the incorporation of diverse functional moieties, thereby expanding their potential for targeted drug delivery. In this study, cationic PLGA-PEI polymers were synthesized to integrate a biological targeting moiety, specifically the polysaccharide fucoidan. This modification was accomplished via a conventional DCC–NHS coupling reaction, which facilitated the introduction of positive charges into the PLGA matrix through the conjugation of branched PEIs. The formation of amide bonds within the spectral range of 1200–1700 cm−1 confirmed its successful reaction (Figure 1a). The binding capability of fucoidan-functionalized particles was further validated by ζ-potential measurements comparing DPPs with fucoidan-functionalized particles (FRDPPs or FDPPs), where the functionalized groups exhibited slightly negative surface charges (Figure 1b). The resulting particles displayed well-defined physicochemical characteristics, demonstrating uniformity in size (Figure 1c and f). The physicochemical uniformity of particles suggests their potential efficacy in treating lung-related diseases.
In our top-down fabrication process, the direct loading of protein-based drugs into polymeric structures is not inherently relevant to a “closed packing of payloads”.16 Due to the porous nature of PLGA polymers, the hydrophilic regions of rtPA inevitably remain exposed on the outer surface of the system during synthesis, resulting in a reduced drug loading capacity per particle (Figure 1d and e). This observation is consistent with our previous finding, which reported significantly low encapsulation efficiency when utilizing this fabrication protocol.16 The partial exposure of rtPA has also been documented in the study employing 1 µm-sized particles, where tPA was chemically conjugated to the PLGA matrix.46 In this case, drug entrapment occurred both within the polymeric matrix and on its surface. Although our fabrication method showed a lower EE compared to the approach involving chemical conjugation, it offers a key advantage by eliminating the need for a purification step. By directly loading materials onto a disposable PVA film, our approach significantly simplifies the fabrication process. The streamlined procedure requires only minimal processing steps while ensuring that an adequate amount of rtPA for thrombolysis is encapsulated both within the particle matrix and on its surface, thereby maintaining sufficient thrombolytic efficacy. The precision and reproducibility of this top-down fabrication method support its scalability for large-scale biopharmaceutical production. By reducing procedural complexity and eliminating purification steps, this approach enhances efficiency and clinical feasibility for cost-effective thrombolytic therapies.
An important characteristic of our system is that the fragmented exposure of rtPA on the matrix can contribute to its efficacy for systemic thrombolysis while reducing the inherent side effects of rtPA. Notably, systemic thrombolysis via bolus injection of rtPA has been largely restricted to patients at high risk of pulmonary thromboembolism (approximately 23–30% of cases) due to the potential risk of hemorrhagic transformation. This limitation constrains the use of rtPA in patients predisposed to bleeding complications.47,48 Furthermore, thrombolytic particles designed for direct exposure of rtPA to the bloodstream may still pose a risk of drug dissociation during systemic circulation, potentially leading to off-target effects and systemic hemorrhage.17 Consequently, the DDS with excessive thrombolytic agent loading may not be optimal for the treatment of thrombotic disorders, including pulmonary thromboembolism. Instead, controlled/targeted DDS is likely to be more suitable for these diseases.
As demonstrated in Figure 2a and b, despite their limited EE capacity, it was sufficient to induce effective thrombolysis while exhibiting a lower lytic potential during the early phase of investigation (ie, < 60 minutes) compared to free rtPA. The sustained thrombolytic activity, characterized by a stable dissolution rate over time, was maintained throughout the assay period (Figure 2c) and was primarily driven by the gradual hydrolysis of the polymer matrix. Notably, the number of particles investigated in this study was controlled such that the total rtPA dose within the suspension remained equivalent to 10 µg. Considering that approximately 37% of rtPA was freely dissociated from the carriers within 4 hours (Figure 1g), a significantly lower drug dose (below 10 µg) was sufficient to achieve effective thrombolysis in the initial phase. Under shaking conditions, the enhanced dissolution rate observed for FRDPPs between 60–120 minutes may be attributed to the interaction between fucoidan and P-selectin on the activated platelets, a phenomenon not prominently observed in the free rtPA and RDPP groups. This strong affinity-based binding of FRDPPs to in vitro thrombi likely contributed to the sustained thrombolytic activity over the assay duration. Therefore, the unique characteristics of FRDPPs, including their controlled drug exposure and electrostatic binding capability, may help mitigate the risk of hemorrhagic transformation while maintaining thrombolytic efficacy. Additionally, these microscale particles are less likely to accumulate in parenchymal tissues, thereby reducing the incidence of hemorrhagic events. However, further investigations should be warranted to assess the potential risks associated with the dissociation of rtPA from the carriers and its systemic effects, which are beyond the scope of this article.16,17
Repetitively, the optimization of thrombolytic therapy hinges on the precise and controlled delivery of pharmacological agents to the thromboembolic lesion while minimizing off-target effects and systemic dissemination. Traditional thrombolytic approaches are significantly impeded by rapid systemic clearance, nonspecific biodistribution, and an elevated propensity for hemorrhagic sequelae. Systemic administration of thrombolytics frequently results in subtherapeutic concentrations at the thrombus site, necessitating increased dosages that, in turn, exacerbate the risk of deleterious complications. In response to these limitations, DDS integrating targeted thrombolytic modalities have been extensively explored for enhanced thrombolysis, incorporating a range of stimulus-responsive platforms such as magnetically guided carriers,20,21,49,50 ultrasound-mediated carriers,51–56 and electrostatic-based strategies.34–36 For example, magnetic nanoparticles conjugated with thrombolytic agents can be precisely directed to thrombi using externally applied magnetic fields, facilitating enhanced local drug accumulation and superior clot degradation while concurrently mitigating systemic toxicity. Similarly, ultrasound-sensitive carriers enable spatially localized thrombolysis via insonation, leveraging cavitation-induced microstreaming and enhanced clot permeability to optimize drug release kinetics. Various ultrasound-responsive vehicles, including echogenic liposomes, microbubbles, and phase-change nanodroplets, have been developed to capitalize on acoustic cavitation, thereby achieving enhanced intrathrombus penetration while maintaining a high therapeutic index. Beyond externally controlled DDS, endogenous targeting mechanisms, similar to the fucoidan binding, present an autonomous and clinically translatable alternative, circumventing the necessity for external activation devices. In the present study, we developed FRDPPs designed for molecularly targeted thrombolysis. The targeting efficacy of FRDPPs was systematically evaluated under both static and physiologically relevant dynamic flow conditions, as demonstrated in Figure 3. Consistent with prior literature,34,35 FRDPPs exhibited enhanced targeting efficiency under both conditions. Given the hemodynamic parameters of pulmonary circulation (v of 44–70 cm⋅s−1 in venous conduits57 and 60–100 cm⋅s−1 in arterial networks58), active targeting remains a viable therapeutic approach for venous thromboembolism. The high binding affinity of fucoidan for P-selectin (Kd = 1.2 nM) surpasses that of conventional polysaccharides such as heparin and dextran sulfate, a property attributed to the structural homology between sulfated fucose moieties in fucoidan and sLex motifs in PSGL-1. This molecular mimicry facilitates high-affinity interaction with P-selectin, thereby enhancing thrombus localization and optimizing therapeutic outcomes.29
An essential characteristic of systemically circulating thrombolytic carriers is their ability to directly interact with thromboembolic deposits under physiological flow conditions, independent of immune-mediated clearance. Conventional lung-targeted therapeutic strategies have predominantly relied on either inhalation or intravenous administration to enhance drug bioavailability and efficacy.59,60 As emphasized in this study, the physicochemical properties of discoidal particles confer a unique advantage for lung-targeted thrombolysis, increasing their propensity to engage with thromboembolic obstructions upon intravenous administration. Specifically, oblate discoidal particles with tailored aspect ratios (ARs) exhibit preferential localization within the RES, particularly in the pulmonary vasculature, kidneys, and spleen. The biodistribution profile of these particles is strongly dictated by their dimensional characteristics, with optimal accumulation occurring within the 150 nm to 7 µm range. Particles smaller than 20 nm are rapidly eliminated via renal clearance, limiting their efficacy for sustained therapeutic action.59,61 Therefore, the size of FRDPPs with 3 µm is suitable for targeting the lungs’ vasculature. As demonstrated in Figure 4c, approximately 50% of the injected particles accumulated in the lungs within the first 15 minutes post-injection, with a significant fraction persisting for at least 60 minutes. Consistent with prior literature, these physicochemical attributes of FRDPPs substantiate their potential as an effective platform for targeted thrombolytic intervention in pulmonary vasculature.24
Despite the promise of the developed thrombolytic system, several critical limitations remain. For example, the intervention did not reduce mortality in models of severe pulmonary thromboembolism. In murine subjects administered a lethal dose of thromboplastin (≥ 50 µg), bolus injection of rtPA or FRDPPs failed to prevent fatality following the onset of acute thrombotic symptoms (data were not shown). Given that FRDPPs demonstrated a dose-dependent reduction in the severity of pulmonary thromboembolism, further investigations are warranted to optimize the dosing strategy and therapeutic regimen to maximize thrombolytic efficacy. This dual-targeted approach may be better suited for patients presenting with mild to moderate thromboembolic events. Furthermore, our results underscore the necessity of adjunctive pharmacological interventions, such as anticoagulants, to improve therapeutic outcomes. The strategic integration of thrombolytic and anticoagulant therapies, coupled with continuous monitoring, may significantly enhance patient prognosis and therapeutic success.
Conclusion
This study introduces fucoidan-functionalized, rtPA-loaded discoidal polymeric particles as a dual-targeted platform for pulmonary thromboembolism, integrating physicochemical targeting by disc-shaped microparticles (~3 µm) with biological affinity via fucoidan–P-selectin interactions. Our results demonstrated rapid and preferential pulmonary accumulation, with approximately 50% of the injected particles localizing in the lungs within 15 minutes and a substantial fraction persisting for at least 60 minutes. Fucoidan-mediated binding was systematically assessed under both static and dynamic flow conditions, confirming superior performance of fucoidan-functionalized particles. Based on these effects, this dual-targeting strategy produced a more sustained thrombolytic effect between 60 and 120 minutes in in vitro thrombolysis assays compared to free rtPA.
However, despite these promising findings, the proposed drug delivery system did not reduce mortality in a severe thromboembolism model with a lethal thromboplastin dose, suggesting that its therapeutic potential may be more suitable for mild-to-moderate events. Taken together, this approach provides dose-responsive thrombolysis with sustained activity and target engagement, although its superiority over native rtPA remains inconclusive. Future investigations should focus on optimizing dosing strategies, elucidating particles–thrombus interactions in vivo, and exploring combination therapies with adjunct anticoagulants to enhance clinical translatability.
Abbreviations
DDS, drug delivery system; rtPA, recombinant tissue plasminogen activator; RES, reticuloendothelial system; Sialyl Lewis X, sLex; DPP, discoidal polymeric particles; FDPP, fucoidan-functionalized discoidal polymeric particles; RDPP, rtPA-loaded discoidal polymeric particles; FRDPP, fucoidan-functionalized and rtPA-loaded discoidal polymeric particles.
Funding
This research was supported by the Smart HealthCare Program through the Korea Institutes of Police Technology (KIPoT) funded by the Korean National Police Agency (RS-2022-PT000188), the Technology Innovation Program (20014477, Development of non-contact AI health monitoring system based on multimodal sensors) funded by the Ministry of Trade, Industry and Energy (MOTIE, Korea), KIST Intramural Grants (2E33771), the Regional Innovation System & Education (RISE) program through the Gangwon RISE Center, funded by the Ministry of Education (MOE) and the Gangwon State (G.S.), Republic of Korea (2025-RISE-10-006), and the Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Education (2022R1F1A1069516 and RS-2024-00460405).
Disclosure
The author(s) report no conflicts of interest in this work. Wonseok Choi and Hyeyoun Cho are co–first authors.
References
1. Goldhaber SZ, Bounameaux H. Pulmonary embolism and deep vein thrombosis. Lancet. 2012;379(9828):1835–1846. doi:10.1016/S0140-6736(11)61904-1
2. Gotzinger F, Lauder L, Sharp ASP, et al. Interventional therapies for pulmonary embolism. Nat Rev Cardiol. 2023;20(10):670–684. doi:10.1038/s41569-023-00876-0
3. Martin KA, Molsberry R, Cuttica MJ, Desai KR, Schimmel DR, Khan SS. Time trends in pulmonary embolism mortality rates in the United States, 1999 to 2018. J Am Heart Assoc. 2020;9(17):e016784. doi:10.1161/JAHA.120.016784
4. Schulman S, Hu Y, Konstantinides S. Venous thromboembolism in COVID-19. Thromb Haemost. 2020;120(12):1642–1653. doi:10.1055/s-0040-1718532
5. Tang N, Li D, Wang X, Sun Z. Abnormal coagulation parameters are associated with poor prognosis in patients with novel coronavirus pneumonia. J Thromb Haemost. 2020;18(4):844–847. doi:10.1111/jth.14768
6. Wichmann D, Sperhake JP, Lutgehetmann M, et al. Autopsy findings and venous thromboembolism in patients with COVID-19: a prospective cohort study. Ann Intern Med. 2020;173(4):268–277. doi:10.7326/M20-2003
7. Sharifi M, Bay C, Skrocki L, Rahimi F, Mehdipour M, Investigators M. Moderate pulmonary embolism treated with thrombolysis (from the “MOPETT” Trial). Am J Cardiol. 2013;111(2):273–277. doi:10.1016/j.amjcard.2012.09.027
8. Wang C, Zhai Z, Yang Y, et al. Efficacy and safety of low dose recombinant tissue-type plasminogen activator for the treatment of acute pulmonary thromboembolism: a randomized, multicenter, controlled trial. Chest. 2010;137(2):254–262. doi:10.1378/chest.09-0765
9. Connors JM. Testing and monitoring direct oral anticoagulants. Blood. 2018;132(19):2009–2015. doi:10.1182/blood-2018-04-791541
10. NINDS t-PA Stroke Study Group T. Intracerebral hemorrhage after intravenous t-PA therapy for ischemic stroke. The NINDS t-PA stroke study group. Stroke. 1997;28(11):2109–2118. doi:10.1161/01.str.28.11.2109
11. Powers WJ, Rabinstein AA, Ackerson T, et al. Guidelines for the early management of patients with acute ischemic stroke: 2019 update to the 2018 guidelines for the early management of acute ischemic stroke: a guideline for healthcare professionals from the American Heart Association/American Stroke Association. Stroke. 2019;50(12):e344–e418. doi:10.1161/STR.0000000000000211
12. Becattini C, Agnelli G. Acute treatment of venous thromboembolism. Blood. 2020;135(5):305–316. doi:10.1182/blood.2019001881
13. Allen TM, Cullis PR. Drug delivery systems: entering the mainstream. Science. 2004;303(5665):1818–1822. doi:10.1126/science.1095833
14. Jain KK. An Overview of Drug Delivery Systems. Methods Mol Biol. 2020;2059:1–54. doi:10.1007/978-1-4939-9798-5_1
15. Choi W, Key J, Youn I, Lee H, Han S. Cavitation-assisted sonothrombolysis by asymmetrical nanostars for accelerated thrombolysis. J Control Release. 2022;350:870–885. doi:10.1016/j.jconrel.2022.09.008
16. Choi W, Cho H, Kim G, Youn I, Key J, Han S. Targeted thrombolysis by magnetoacoustic particles in photothrombotic stroke model. Biomater Res. 2022;26(1):58. doi:10.1186/s40824-022-00298-y
17. Wang S, Guo X, Xiu W, et al. Accelerating thrombolysis using a precision and clot-penetrating drug delivery strategy by nanoparticle-shelled microbubbles. Sci Adv. 2020;6(31):eaaz8204. doi:10.1126/sciadv.aaz8204
18. Lin L, Ba Z, Tian H, et al. Ultrasound-responsive theranostic platform for the timely monitoring and efficient thrombolysis in thrombi of tPA resistance. Nat Commun. 2024;15(1):6610. doi:10.1038/s41467-024-50741-y
19. Huang Y, Gu B, Salles C II, et al. Fibrinogen-mimicking, multiarm nanovesicles for human thrombus-specific delivery of tissue plasminogen activator and targeted thrombolytic therapy. Sci Adv. 2021;7(23). doi:10.1126/sciadv.abf9033
20. Ma YH, Wu SY, Wu T, Chang YJ, Hua MY, Chen JP. Magnetically targeted thrombolysis with recombinant tissue plasminogen activator bound to polyacrylic acid-coated nanoparticles. Biomaterials. 2009;30(19):3343–3351. doi:10.1016/j.biomaterials.2009.02.034
21. Yang HW, Hua MY, Lin KJ, et al. Bioconjugation of recombinant tissue plasminogen activator to magnetic nanocarriers for targeted thrombolysis. Int J Nanomedicine. 2012;7:5159–5173. doi:10.2147/IJN.S32939
22. Migliavacca M, Correa-Paz C, Perez-Mato M, et al. Thrombolytic therapy based on lyophilized platelet-derived nanocarriers for ischemic stroke. J Nanobiotechnology. 2024;22(1):10. doi:10.1186/s12951-023-02206-5
23. Kohane DS. Microparticles and nanoparticles for drug delivery. Biotechnol Bioeng. 2007;96(2):203–209. doi:10.1002/bit.21301
24. Park JY, Park S, Lee TS, et al. Biodegradable micro-sized discoidal polymeric particles for lung-targeted delivery system. Biomaterials. 2019;218:119331. doi:10.1016/j.biomaterials.2019.119331
25. Key J, Palange AL, Gentile F, et al. Soft discoidal polymeric nanoconstructs resist macrophage uptake and enhance vascular targeting in tumors. ACS Nano. 2015;9(12):11628–11641. doi:10.1021/acsnano.5b04866
26. Decuzzi P, Godin B, Tanaka T, et al. Size and shape effects in the biodistribution of intravascularly injected particles. J Control Release. 2010;141(3):320–327. doi:10.1016/j.jconrel.2009.10.014
27. Gentile F, Curcio A, Indolfi C, Ferrari M, Decuzzi P. The margination propensity of spherical particles for vascular targeting in the microcirculation. J Nanobiotechnology. 2008;6:9. doi:10.1186/1477-3155-6-9
28. Lee SY, Ferrari M, Decuzzi P. Shaping nano-/micro-particles for enhanced vascular interaction in laminar flows. Nanotechnology. 2009;20(49):495101. doi:10.1088/0957-4484/20/49/495101
29. Bachelet L, Bertholon I, Lavigne D, et al. Affinity of low molecular weight fucoidan for P-selectin triggers its binding to activated human platelets. Biochim Biophys Acta. 2009;1790(2):141–146. doi:10.1016/j.bbagen.2008.10.008
30. Chollet L, Saboural P, Chauvierre C, Villemin JN, Letourneur D, Chaubet F. Fucoidans in Nanomedicine. Mar Drugs. 2016;14(8):145. doi:10.3390/md14080145
31. Pouyani T, Seed B. PSGL-1 recognition of P-selectin is controlled by a tyrosine sulfation consensus at the PSGL-1 amino terminus. Cell. 1995;83(2):333–343. doi:10.1016/0092-8674(95)90174-4
32. Varki A. Selectin ligands. Proc Natl Acad Sci U S A. 1994;91(16):7390–7397. doi:10.1073/pnas.91.16.7390
33. Pochechueva TV, Galanina OE, Ushakova NA, et al. Uncharged P-selectin blockers. Glycoconj J. 2004;20(2):91–97. doi:10.1023/B:GLYC.0000018583.63140.91
34. Juenet M, Aid-Launais R, Li B, et al. Thrombolytic therapy based on fucoidan-functionalized polymer nanoparticles targeting P-selectin. Biomaterials. 2018;156:204–216. doi:10.1016/j.biomaterials.2017.11.047
35. Zenych A, Jacqmarcq C, Aid R, et al. Fucoidan-functionalized polysaccharide submicroparticles loaded with alteplase for efficient targeted thrombolytic therapy. Biomaterials. 2021;277:121102. doi:10.1016/j.biomaterials.2021.121102
36. Zhang H, Qu H, He Q, et al. Thrombus-targeted nanoparticles for thrombin-triggered thrombolysis and local inflammatory microenvironment regulation. J Control Release. 2021;339:195–207. doi:10.1016/j.jconrel.2021.06.043
37. Schneider CA, Rasband WS, Eliceiri KW. NIH image to imageJ: 25 years of image analysis. Nat Methods. 2012;9(7):671–675. doi:10.1038/nmeth.2089
38. Weiss EJ, Hamilton JR, Lease KE, Coughlin SR. Protection against thrombosis in mice lacking PAR3. Blood. 2002;100(9):3240–3244. doi:10.1182/blood-2002-05-1470
39. Page MJ, Lourenco AL, David T, et al. Non-invasive imaging and cellular tracking of pulmonary emboli by near-infrared fluorescence and positron-emission tomography. Nat Commun. 2015;6:8448. doi:10.1038/ncomms9448
40. Pircher J, Czermak T, Ehrlich A, et al. Cathelicidins prime platelets to mediate arterial thrombosis and tissue inflammation. Nat Commun. 2018;9(1):1523. doi:10.1038/s41467-018-03925-2
41. He H, Adili R, Liu L, Hong K, Holinstat M, Schwendeman A. Synthetic high-density lipoproteins loaded with an antiplatelet drug for efficient inhibition of thrombosis in mice. Sci Adv. 2020;6(49). doi:10.1126/sciadv.abd0130
42. Makadia HK, Siegel SJ. Poly Lactic-co-Glycolic Acid (PLGA) as Biodegradable Controlled Drug Delivery Carrier. Polymers. 2011;3(3):1377–1397. doi:10.3390/polym3031377
43. Park TG. Degradation of poly(lactic-co-glycolic acid) microspheres: effect of copolymer composition. Biomaterials. 1995;16(15):1123–1130. doi:10.1016/0142-9612(95)93575-x
44. Chung TW, Wang SS, Tsai WJ. Accelerating thrombolysis with chitosan-coated plasminogen activators encapsulated in poly-(lactide-co-glycolide) (PLGA) nanoparticles. Biomaterials. 2008;29(2):228–237. doi:10.1016/j.biomaterials.2007.09.027
45. Zamanlu M, Eskandani M, Barar J, Jaymand M, Pakchin PS, Farhoudi M. Enhanced thrombolysis using tissue plasminogen activator (tPA)-loaded PEGylated PLGA nanoparticles for ischemic stroke. J Drug Deliv Sci Tec. 2019;53. doi:10.1016/j.jddst.2019.101165
46. Colasuonno M, Palange AL, Aid R, et al. Erythrocyte-inspired discoidal polymeric nanoconstructs carrying tissue plasminogen activator for the enhanced lysis of blood clots. ACS Nano. 2018;12(12):12224–12237. doi:10.1021/acsnano.8b06021
47. Keller K, Hobohm L, Ebner M, et al. Trends in thrombolytic treatment and outcomes of acute pulmonary embolism in Germany. Eur Heart J. 2020;41(4):522–529. doi:10.1093/eurheartj/ehz236
48. Stein PD, Matta F. Thrombolytic therapy in unstable patients with acute pulmonary embolism: saves lives but underused. Am J Med. 2012;125(5):465–470. doi:10.1016/j.amjmed.2011.10.015
49. Chen JP, Yang PC, Ma YH, Tu SJ, Lu YJ. Targeted delivery of tissue plasminogen activator by binding to silica-coated magnetic nanoparticle. Int J Nanomedicine. 2012;7:5137–5149. doi:10.2147/IJN.S36197
50. Tadayon A, Jamshidi R, Esmaeili A. Delivery of tissue plasminogen activator and streptokinase magnetic nanoparticles to target vascular diseases. Int J Pharm. 2015;495(1):428–438. doi:10.1016/j.ijpharm.2015.09.008
51. Tiukinhoy-Laing SD, Huang S, Klegerman M, Holland CK, McPherson DD. Ultrasound-facilitated thrombolysis using tissue-plasminogen activator-loaded echogenic liposomes. Thromb Res. 2007;119(6):777–784. doi:10.1016/j.thromres.2006.06.009
52. Ren X, Wang Y, Wang Y, et al. Thrombolytic therapy with rt-PA and transcranial color Doppler ultrasound (TCCS) combined with microbubbles for embolic thrombus. Thromb Res. 2015;136(5):1027–1032. doi:10.1016/j.thromres.2015.08.027
53. Hu B, Jiang N, Zhou Q, et al. Stable cavitation using acoustic phase-change dodecafluoropentane nanoparticles for coronary micro-circulation thrombolysis. Int J Cardiol. 2018;272:1–6. doi:10.1016/j.ijcard.2018.06.027
54. Uesugi Y, Kawata H, Jo J, Saito Y, Tabata Y. An ultrasound-responsive nano delivery system of tissue-type plasminogen activator for thrombolytic therapy. J Control Release. 2010;147(2):269–277. doi:10.1016/j.jconrel.2010.07.127
55. Kawata H, Uesugi Y, Soeda T, et al. A new drug delivery system for intravenous coronary thrombolysis with thrombus targeting and stealth activity recoverable by ultrasound. J Am Coll Cardiol. 2012;60(24):2550–2557. doi:10.1016/j.jacc.2012.08.1008
56. Ren ST, Kang XN, Liao YR, et al. The ultrasound contrast imaging properties of lipid microbubbles loaded with urokinase in dog livers and their thrombolytic effects when combined with low-frequency ultrasound in vitro. J Thromb Thrombolysis. 2014;37(3):303–309. doi:10.1007/s11239-013-0950-8
57. Meijburg HW, Visser CA, Westerhof PW, Kasteleyn I, van der Tweel I, Robles de Medina EO. Normal pulmonary venous flow characteristics as assessed by transesophageal pulsed Doppler echocardiography. J Am Soc Echocardiogr. 1992;5(6):588–597. doi:10.1016/s0894-7317(14)80324-6
58. Bouhemad B, Ferrari F, Leleu K, Arbelot C, Lu Q, Rouby JJ. Echocardiographic Doppler estimation of pulmonary artery pressure in critically ill patients with severe hypoxemia. Anesthesiology. 2008;108(1):55–62. doi:10.1097/01.anes.0000296067.02462.34
59. Dhand C, Prabhakaran MP, Beuerman RW, Lakshminarayanan R, Dwivedi N, Ramakrishna S. Role of size of drug delivery carriers for pulmonary and intravenous administration with emphasis on cancer therapeutics and lung-targeted drug delivery. Rsc Adv. 2014;4(62):32673–32689. doi:10.1039/c4ra02861a
60. Aryal S, Park S, Park H, et al. Clinical trials for oral, inhaled and intravenous drug delivery system for lung cancer and emerging nanomedicine-based approaches. Int J Nanomedicine. 2023;18:7865–7888. doi:10.2147/IJN.S432839
61. Blanco E, Shen H, Ferrari M. Principles of nanoparticle design for overcoming biological barriers to drug delivery. Nat Biotechnol. 2015;33(9):941–951. doi:10.1038/nbt.3330
 © 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.