Back to Journals » International Medical Case Reports Journal » Volume 13
A Concurrent Ischemic Stroke, Myocardial Infarction, and Aortic Thrombi in a Young Patient with Hyperhomocysteinemia: A Case Report
Authors Rawashdeh SI ![]() , Al-Mistarehi AH
, Al-Mistarehi AH ![]() , Yassin A
, Yassin A ![]() , Rabab'ah W, Skaff H, Ibdah R
, Rabab'ah W, Skaff H, Ibdah R ![]()
Received 2 September 2020
Accepted for publication 21 October 2020
Published 5 November 2020 Volume 2020:13 Pages 581—590
DOI https://doi.org/10.2147/IMCRJ.S279603
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Ronald Prineas
Sukaina I Rawashdeh,1 Abdel-Hameed Al-Mistarehi,2 Ahmed Yassin,3 Walaa Rabab’ah,1 Hussam Skaff,4 Rasheed Ibdah1
1Department of Internal Medicine, Faculty of Medicine, Jordan University of Science and Technology, Irbid, Jordan; 2Department of Public Health and Family Medicine, Faculty of Medicine, Jordan University of Science and Technology, Irbid, Jordan; 3Division of Neurology, Department of Neurosciences, Faculty of Medicine, Jordan University of Science and Technology, Irbid, Jordan; 4Department of Diagnostic Radiology, Faculty of Medicine, Jordan University of Science and Technology, Irbid, Jordan
Correspondence: Sukaina I Rawashdeh
Department of Internal Medicine, Faculty of Medicine, Jordan University of Science and Technology, P.O. Box: 630001, Irbid 22110, Jordan
Tel +962 7 9885 9054
Email [email protected]
Abdel-Hameed Al-Mistarehi
Department of Public Health and Family Medicine, Faculty of Medicine, Jordan University of Science and Technology, P.O.Box: 630001, Irbid 22110, Jordan
Tel +962 7 9828 4360
Email [email protected]
Abstract: We are presenting a case report of a previously healthy 39-year-old man who was found to have acute inferior ST-elevation myocardial infarction (STEMI) and acute large right middle cerebral artery (MCA) ischemic stroke with hemorrhagic transformation. Transesophageal echocardiogram and chest CT angiogram revealed two thrombi; one attached to the wall of the ascending aorta just above the right coronary artery sinus, and one at the origin of the brachiocephalic trunk. The occlusion of the coronary artery and right MCA most likely could be because of embolization from these thrombi. Extensive workup looking for underlying etiology and risk factors for these concurrent vascular events in this young man revealed hyperhomocysteinemia along with unfavorable lipid profile, and family history of premature coronary artery disease which increased the suspicion of familial hypercholesterolemia. Besides, the presence of vitamin B12 and folate deficiencies. The elevated serum homocysteine is likely a major risk factor for thromboembolism in this patient. The patient received antithrombotics and vitamin supplementations and gradually improved without any worsening of the stroke’s hemorrhagic transformation. We suggest that hyperhomocysteinemia needs to be considered in the differential etiology of vascular events in young people or those with no significant history of major vascular risk factors. Besides, vitamin supplementation could be a cost-effective, safe, and efficient way to decrease elevated serum homocysteine levels and prevent vascular complications. As well as this case report demonstrates that antithrombotics can safely be used after stroke’s hemorrhagic transformation without neurological deterioration or aggravation of hemorrhagic transformation.
Keywords: homocysteine, hyperhomocysteinemia, myocardial infarction, stroke, hemorrhagic transformation, thrombus, vascular diseases, vitamin B12, folate, antithrombotics, anticoagulant, antiplatelet
Introduction
Homocysteine (Hcy) is an intermediary sulfur-containing nonproteinogenic amino acid derived from the metabolism of methionine via the one‐carbon metabolism (in the methionine cycle).1 After production of Hcy, is normally removed by two processes: 1) Transsulfuration pathway for producing cysteine and eventually glutathione (an antioxidant) or proteins, and this pathway is responsible for the metabolism of around 50% of Hcy; 2) The rest of Hcy can be metabolized through methionine cycle that synthesizes methionine from it.2,3 However, serum Hcy level is considered normal between 5 and 15 μmol/l, while hyperhomocysteinemia (HHcy) refers to an elevated blood Hcy level above 15 µmol/L, and is categorized into mild-moderate for values between 15 to 30 μmol/l, intermediate when ranging from 31 to 100 μmol/l, and severe for values exceeding 100 μmol/l.4 HHcy can be caused by genetic defects in the enzymes involved in Hcy metabolisms, such as cystathionine‐β synthase or methylenetetrahydrofolate reductase (MTHFR);5,6 nutritional deficiencies in vitamins that act as co-factors for Hcy metabolism (vitamins B12, B6, and folate);7 chronic medical conditions (eg chronic renal failure, hypothyroidism, and systemic lupus erythematosus);8,9 drugs (eg theophylline, bile acid resins, methotrexate, levodopa, fibrates, and nicotinic acid);10 and smoking.11
HHcy has atherogenic and prothrombotic effects. Several experimental studies implicated histopathologic hallmarks of vascular injury related to elevated plasma Hcy levels including smooth muscle hypertrophy, intimal thickening, elastic lamina disruption, platelet accumulation, and the formation of platelet-enriched occlusive thrombi.12–14 HHcy is an established independent risk factor for occlusive vascular diseases and it has been confirmed to be associated with vascular diseases including premature peripheral vascular, cerebrovascular and coronary artery diseases, in addition to venous thromboembolic diseases.15–19
Many epidemiological observations indicated the correlation between HHcy and the incidence of ischemic stroke.20–22 A prolonged elevated level of homocysteine is implicated in the pathogenesis of ischemic stroke by the initiation of complex processes include increased oxidative stress, reactive oxygen species production, oxidizing membrane lipids and proteins, protein homocysteinylation, Ca2+ dysregulation, and DNA methylation. These events with epigenetic risk factors can result in neuronal cell apoptosis, and blood-brain barrier dysregulation manifested as ischemic stroke.21–24
Several studies indicated a significant correlation of HHcy with the risk and severity of cardiovascular diseases and its complications such as heart attacks and strokes.25–27 Underlying mechanisms might include HHcy inducing endothelial cells damage, reduction in the flexibility of blood vessels, and altering the hemostasis process.25 In addition, HHcy could exacerbate the adverse effects of ischemic heart disease (IHD) risk factors such as high blood pressure, smoking, unfavorable lipid profile, lipoprotein metabolism, high creatinine, and faulty diet.25,26 Also, it is believed that HHcy could promote the process of inflammation.25 A previous meta-analysis, that was conducted on 120 genetic and prospective studies, reported significant associations between serum homocysteine levels and cardiovascular diseases, deep venous thrombosis, and strokes.28 Also, it strengthened the evidence that a raised serum homocysteine level is a cause of IHD.28
Homocystinuria is a rare inherited disorder characterized by the accumulation of homocysteine in the body and elevated its excretion in the urine.29 Clinical findings of homocystinuria include mental retardation, ocular abnormalities, bone fractures, thromboembolic disorders, and premature atherosclerosis.29
This case report is about a young man who developed acute myocardial infarction and ischemic stroke with hemorrhagic transformation most likely due to thromboembolism from clots in the ascending aorta and brachiocephalic trunk and was found to have hyperhomocysteinemia. Thrombi were resolved and the patient’s symptoms improved with vitamin supplementations, anticoagulants, and antiplatelet treatment.
Materials and Methods
Institutional approval was not required for a case report. Written Informed consent was obtained from the patient to have the case details published. We reviewed the electronic medical records to investigate this clinical case including hospital course, vital signs, laboratory findings, radiological images, and treatments. Serum homocysteine (Hcy), vitamin B12, and folate levels were measured by enzyme-linked immunosorbent assay (ELISA) kits (Roche COBAS INTEGRA® 6000). Genetic tests were conducted for MTHFR (A1298C, and C677T), and the prothrombin gene. All tests were conducted in the laboratories of Jordan University of Science and Technology (JUST), Irbid, Jordan.
Case Presentation
A 39-year-old male patient experienced an acute severe central non-radiating chest pain while asleep (2:00 am). As the patient woke up due to the pain, he also felt dizzy, had a headache, and vomited twice. The pain settled down on its own and the patient went back to sleep. He did not seek medical help until the morning (8:00 am) when he woke up from sleep and found that he already had developed left upper limb and facial weakness.
The patient works as a taxi driver, is an active smoker (20 pack-year), is not previously known to have diabetes mellitus, hypertension, or any other chronic medical diseases, is not on any medications, has no history of alcohol intake or illicit drug abuse, but has a positive family history for premature coronary artery diseases; his brother underwent coronary artery bypass graft at age of 50, and his mother had unprovoked deep vein thrombosis. Although he is not a strict vegetarian, he only eats meat a few times per year. He denied any history of appetite or weight loss.
The patient was taken by the family to a nearby hospital where an electrocardiogram (ECG) and a brain computed tomography (CT) scan were done and showed acute inferior ST-elevation myocardial infarction (STEMI) with a lateral extension (Figure 1) and acute large right middle cerebral artery (MCA) ischemic stroke with the hemorrhagic transformation (Figure 2). The patient was later referred to our hospital for further evaluation. Within six hours from the patient’s presentation to the primary hospital emergency room, and 12 hours from the chest pain onset (at 2:00 pm) the patient arrived at our hospital, and he was immediately admitted to the coronary care unit. His chest pain was gone but he was tachycardic with slightly elevated blood pressure. His detailed exam revealed an overweight man with a low relative risk of mortality from excess weight following A Body Shape Index z-score. The patient was conscious, oriented with a left lower facial weakness and 1/5 left upper limb weakness. His exam was otherwise normal (Table 1).
 |
Table 1 The Main Anthropometric Indices and Vital Signs of Our Patient at Admission |
 |
Figure 1 ECG of the patient on presentation showing inferior ST-elevation myocardial infarction (STEMI) with a lateral extension. |
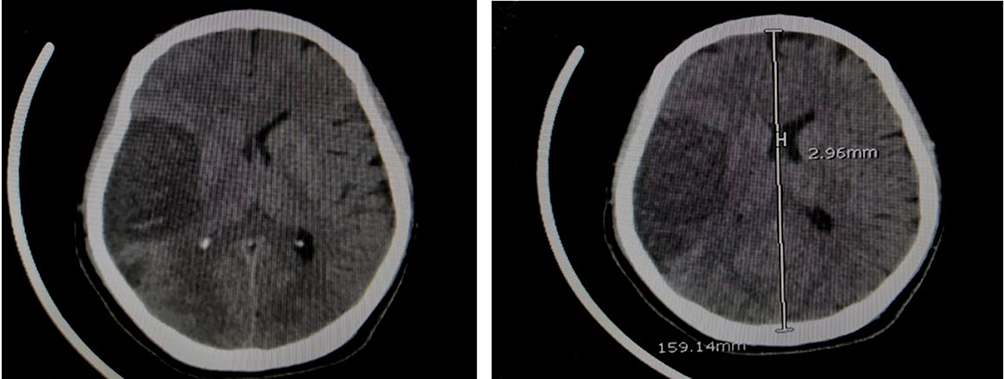 |
Figure 2 CT brain without contrast showing evidence of a large hypoattenuating area seen involving the right fronto-parietal region with loss of the gray-white matter differentiation containing few hyperdense foci associated with right cerebral hemispheric edema, effacement of sulci, compression effect on the ipsilateral lateral ventricle and midline shift to the left side measuring about 3 mm, overall findings representing acute right MCA territory infarction with hemorrhagic transformation. |
The ECG, and brain CT scan were repeated in our hospital and showed the same findings of previous hospital tests. Additional tests were conducted including a brain Magnetic Resonance Imaging (MRI) which showed the same above-mentioned findings on the CT scan in addition to a small area of chronic ischemic nature in the periventricular white matter of the left frontal lobe (Figure 3). Head CT angiography (CTA) was remarkable for occluded branches of the right MCA (Figure 4).
 |
Figure 3 Brain MRI: (A-C) T2, FLAIR and DWI showing right MCA territory infarction. A small chronic ischemic lesion seen in the white matter of the left frontal lobe. (D) SWI image showing blooming artifacts at the site of infarction representing blood component (hemorrhagic transformation). |
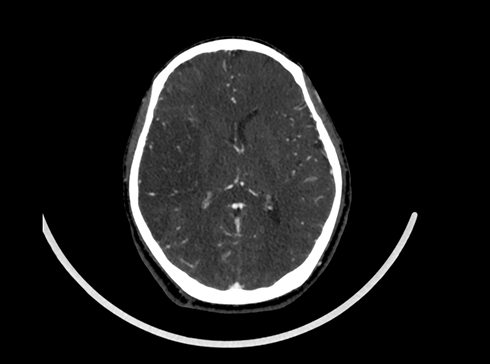 |
Figure 4 Brain CTA showing a Lesser extent of opacification of the cortical branches of right MCA compared to the left. |
Transesophageal echocardiogram (TEE) showed a fresh, freely oscillating, 1.6 *1.2 cm thrombus attached to the wall of the ascending aorta just above the right coronary artery sinus (Figure 5). Also, TEE and Transthoracic Echocardiogram showed a hypertrophied moderately impaired left ventricle with an ejection fraction of 43%, inferior akinesia, and lateral hypokinesia. Atherosclerotic changes in the aorta with visible small calcified plaques in the aortic arch were also noted. Chest CTA showed another thrombus at the origin of the brachiocephalic trunk (Figure 6).
 |
Figure 5 Transesophageal echocardiogram (TEE) images showing ascending aorta thrombus. |
 |
Figure 6 Chest CTA showing non-occlusive filling defects at the origin of the Brachiocephalic artery and in the proximal ascending aorta. |
Laboratory investigations at presentation showed elevated cardiac enzymes, unfavorable lipid profile, intermediately elevated serum Hcy level (65.91 μmol/L), low vitamin B12 (112.7 pg/mL), low folate (4.17 ng/mL), heterozygote MTHFR (A1298C, C677T), and heterozygote prothrombin gene. Other extensive hypercoagulability workups were negative (Table 2).
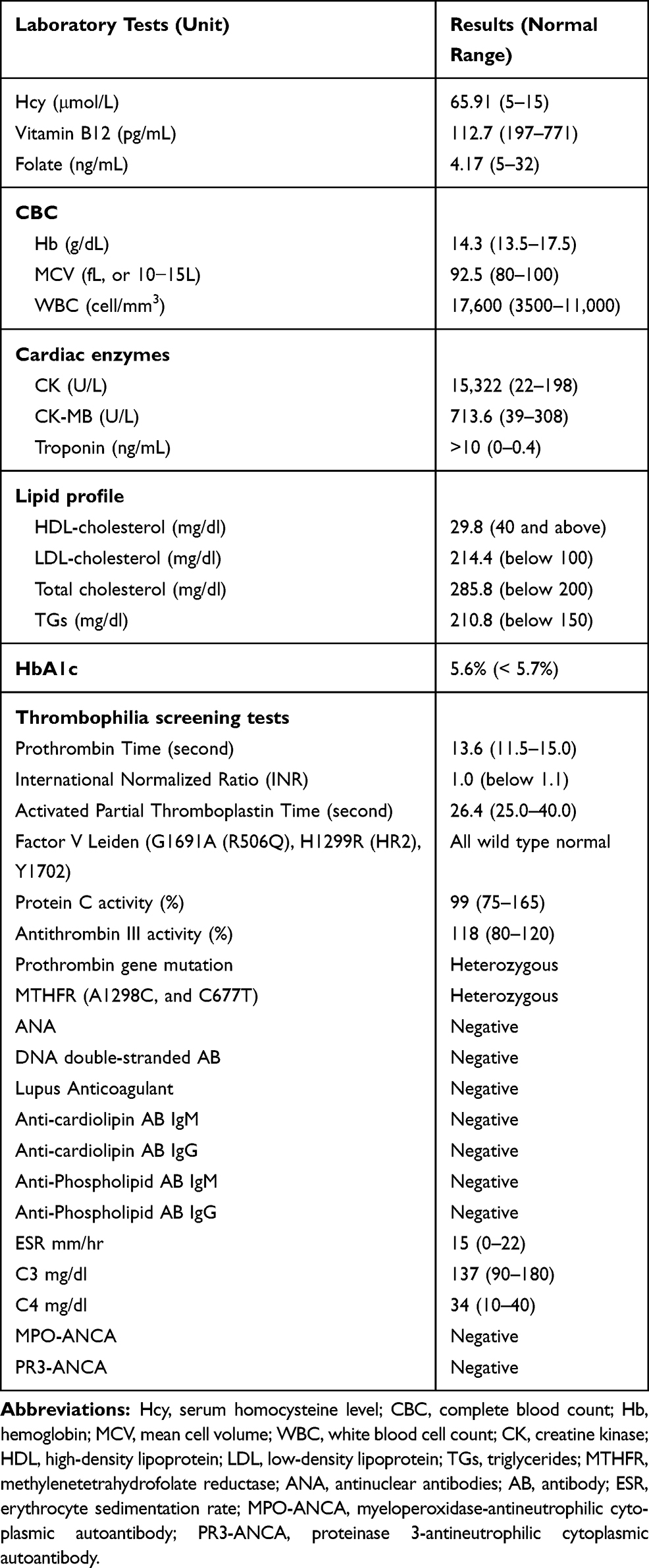 |
Table 2 Summary of Patient’ Laboratory Findings at Admission |
Since there is a high risk of further embolization from the freely oscillating thrombi in the ascending aorta and the brachiocephalic trunk, and after agreement with the patient and his family, heparin infusion was started without a loading dose. Besides, cardiac catheterization was postponed for several reasons, including the high risk of aortic thrombus rupture and dislodgment during catheterization, the patient’s late presentation to our hospital when the chest pain was resolved and ECG showed established Q waves in the inferior leads, and the worry that we needed full anticoagulation therapy (if stent to be placed) before comprehensively evaluating the case the benefit-risk ratio in the presence of ischemic stroke with hemorrhagic transformation.
Frequent neurological assessments and daily brain CT follow up were done to quickly detect any worsening of the stroke’s hemorrhagic transformation. Aspirin, high dose statin, ACE inhibitor, and beta-blocker were also started. The patient’s hospital clinical course was smooth. Subsequent follow-up CT brain images initially showed an increase in the midline shift to the left side to 7 mm and a right-sided uncal herniation which gradually improved without any worsening of the stroke’s hemorrhagic transformation. The patient was discharged on warfarin; vitamin B 12, vitamin B complex and folic acid supplements; aspirin; high dose statin; ACE inhibitor; and a beta-blocker.
TEE and chest CTA were repeated one month following discharge and the previously described thrombi resolved. INR was in therapeutic range, the patient had improvement in his left upper limb and facial weakness, and the Modified Rankin Score was 2. Besides, after two months following discharge, significant improvements in serum levels of Hcy, vitamin B12, and folate were observed with values of 42.1 μmol/L; 236 pg/mL; and 11 ng/mL respectively. Till the time of writing this report, approximately 1 year and 3 months, the patient had not developed any thromboembolic events with preserved vitamin therapy.
Discussion
HHcy may present with vascular thromboembolic events, in the presence or absence of the traditional vascular risk factors.15–19 Our patient was a smoker male with dyslipidemia, that was diagnosed after admission, along with intermediate HHcy which increased his risk profile. He had an unusual presentation of concurrent myocardial infarction and stroke caused by thromboembolism from ascending aorta and brachiocephalic trunk thrombi. There was a positive family history for premature coronary artery diseases, and heterozygote prothrombin and MTHFR genes mutations. This case sheds light on the importance of testing for HHcy as part of a workup for atherothrombotic diseases, especially in patients of young age or those without other significant risk factors. Also, the patient clinically improved on vitamin supplementations and antithrombotics therapy without any worsening of the stroke’s hemorrhagic transformation. Therefore, anticoagulants and antiplatelet drugs may be safely used in patients with stroke hemorrhagic transformation especially when the risk of recurrent embolism is high.
Our patient had a serum Hcy level of 66 μmol/L which reflects an intermediate HHcy. Previous studies, which were conducted on the Jordanian population, showed that the mean serum Hcy level ranged from 10.3 to 17.7 as the highest reported mean among a subsample of healthy individuals in Jordan.30,31 Thus, our patient had a higher serum Hcy level than the general population in Jordan. Given the family history of premature coronary heart disease, and elevated LDL-cholesterol and cholesterol levels increase the suspicion of familial hypercholesterolemia (FH).32 However, a genetic confirmation test and serum apolipoprotein B level were not conducted. The concordant occurrence of acute myocardial infarction and ischemic stroke and the presence of two large oscillating thrombi increased the possibility of an embolic phenomenon from the thrombi as the main cause of these events. However, we could not exclude a heavy atheromatic burden in the arteries as a cause since it is likely that the patient had FH.
Although the results from many studies conducted over the last 30 years have shown consistent evidence that suggests a strong relationship between HHcy and various atherothrombotic diseases including cerebrovascular and coronary artery diseases, in addition to venous thromboembolic diseases,15–19 several clinical trials have failed to achieve improvement in vascular diseases’ risks and outcomes with administering Hcy lowering agents like folic acid and vitamin-B supplementations.33–35 In contrast to those studies which included patients with mild-moderate elevations in serum Hcy levels, our patient had an intermediately elevated Hcy level. On the other hand, a recent meta-analysis of observational studies found that a 25% reduction in the serum Hcy level is associated with an 11% lower IHD risk with an odds ratio (OR) of 0.89 (95% CI, 0.83 to 0.96) and a 19% lower stroke risk with OR of 0.81 (95% CI, 0.69 to 0.95).27 Another systemic review and meta-analysis by Wald David et al reported that lowering Hcy levels by 3 μmol/l (achievable by increasing folic acid intake) would reduce the risk of IHD by 16% (11% to 20%), and stroke by 24% (15% to 33%).28 Thus, It has been suggested that the relationship is complex and needs further testing and stratification for possible outliers and effect modifiers.
Veeranna et al study found that the addition of a serum Hcy measurement to the Framingham risk scores significantly improved coronary heart disease risk prediction. A significant number of individuals were reclassified to an intermediate risk level of coronary heart disease events.36 Therefore, HHcy needs to be considered in the work-up of acute myocardial infarction not explained by traditional risk factors.
One of the reported factors in this patient is double heterozygous for MTHFR (A1298C, C677T). The MTHFR is an important enzyme involved in the Hcy metabolism by stimulating the conversion of 5,10-methylenetetrahydrofolate into 5-methyltetrahydrofolate which is a co-substrate for Hcy remethylation back to methionine.1–3 However, the MTHFR gene has at least two functional polymorphisms, A1298C, and C677T. Heterozygosity for MTHFR C677T is quite common in the general population and ranged from 22% up to 78% in some ethnic groups.37,38 A previous study on the Arab populations showed that the prevalence of heterozygous C677T genotype was 25.8% while 51.5% for heterozygous A1298C genotype among healthy individuals.39 Besides, the prevalence of A1298C, C677T compound heterozygous was 9.6% for the healthy group.39 Although double heterozygosity status for MTHFR, particularly C677T, can result in a lower MTHFR activity and thus decreased levels of circulating folate and methionine,40,41 it is rarely associated with moderately elevated homocysteine levels.42,43 However, previous studies have failed to establish a link between these 2 MTHFR polymorphisms and coronary artery disease.39,43 Thus, we could not recommend the MTHFR test for patients unless elevated serum Hcy levels are investigated.
Our patient improved clinically and follow-up CTs showed no worsening of the stroke’s hemorrhage despite the use of antithrombotics Up to date, there have been no appropriate guidelines about the use of antithrombotics after hemorrhagic transformation. However, previous retrospective studies showed that the use of antithrombotics did not worsen or increase the risk for hemorrhagic transformation in stroke patients and was not associated with neurological deterioration after hemorrhagic transformation.44–46 However, many physicians are hesitant to use antithrombotics after hemorrhagic transformation detected by imaging even when there is an indication for anticoagulation therapy. Since our patient had STEMI and MCA strokes and two large oscillating thrombi, the consensus between caring physicians was to give antithrombotics. Therefore, we suggest that the use of antithrombotics following stroke hemorrhagic transformation, after balancing the risk of hemorrhage worsening against the benefit of preventing further thromboembolism, can be safe and not necessarily be associated with neurological deterioration and worsening of hemorrhagic transformation.
Conclusion
This case demonstrates that hyperhomocysteinemia, even if it is not severe, can be a risk factor for thromboembolic events. We conclude that hyperhomocysteinemia needs to be considered in the work-up of acute myocardial infarction or stroke not sufficiently explained by the traditional vascular risk factors. Although several studies have shown no vascular benefits from correcting mildly elevated homocysteine levels by vitamin supplementations, we suggest that intermediate or severe elevations in serum homocysteine levels should be corrected with appropriate vitamin therapy to potentially prevent vascular complications. Also, we suggest that antithrombotics can safely be used in ischemic stroke after hemorrhagic transformation especially when the risk of recurrent embolism is high and can not be associated with worsening of hemorrhagic transformation. Further studies on large cohorts should be conducted to look more carefully at antithrombotics use after hemorrhagic transformation, hyperhomocysteinemia as a vascular hazard, and the clinical impact of its correction.
Clinical Implications
The workup of stroke-in-the-young should include a measurement of serum homocysteine level, and hyperhomocysteinemia should be corrected with vitamin therapy that may alleviate the risk of further vascular events. Although using anticoagulants may increase the risk of bleeding in ischemic stroke with hemorrhagic transformation, the benefits of using them, in cases where thrombi are concurrently present, can outweigh the risk. Individualized decisions in complicated cases should be sought.
Ethics and Consent
Institutional approval was not required for a case report. Written informed consent has been provided by the patient to have the case details published.
Funding
No Funding was received for this case report.
Disclosure
The authors declare that they have no competing interests.
References
1. Selhub J. Homocysteine metabolism. Annu Rev Nutr. 1999;19:217–246. doi:10.1146/annurev.nutr.19.1.217
2. Veeranki S, Tyagi SC. Defective homocysteine metabolism: potential implications for skeletal muscle malfunction. Int J Mol Sci. 2013;14(7):15074–15091. doi:10.3390/ijms140715074
3. Majumder A, Singh M, George AK, et al. Remote ischemic conditioning as a cytoprotective strategy in vasculopathies during hyperhomocysteinemia: an emerging research perspective. J Cell Biochem. 2019;120(1):77–92. doi:10.1002/jcb.27603
4. Kang SS, Wong PWK, Malinow MR. Hyperhomocyst(e)inemia as a risk factor for occlusive vascular disease. Annu Rev Nutr. 1992;12:279–298. doi:10.1146/annurev.nu.12.070192.001431
5. Brustolin S, Giugliani R, Félix TM. Genetics of homocysteine metabolism and associated disorders. Braz J Med Biol Res. 2010;43(1):1–7. doi:10.1590/s0100-879x2009007500021
6. Majumder A, Singh M, George AK, Behera J, Tyagi N, Tyagi SC. Hydrogen sulfide improves postischemic neoangiogenesis in the hind limb of cystathionine-β-synthase mutant mice via PPAR-γ/VEGF axis. Physiol Rep. 2018;6(17):e13858. doi:10.14814/phy2.13858.
7. Sánchez-Moreno C, Jiménez-Escrig A, Martín A. Stroke: roles of B vitamins, homocysteine and antioxidants. Nutr Res Rev. 2009;22(1):49–67. doi:10.1017/S0954422409990023
8. Williams RH, Maggiore JA. Hyperhomocysteinemia: pathogenesis, clinical significance, laboratory assessment, and treatment. Lab Med. 1999;30(7):468–475. doi:10.1093/labmed/30.7.468
9. Timlin H, Manno R, Douglas H. Hyperhomocysteinemia and lupus nephritis. Cureus. 2019;11(7):e5065. doi:10.7759/cureus.5065
10. Desouza C, Keebler M, McNamara DB, Fonseca V. Drugs affecting homocysteine metabolism: impact on cardiovascular risk. Drugs. 2002;62(4):605–616. doi:10.2165/00003495-200262040-00005
11. Bazzano LA, He J, Muntner P, Vupputuri S, Whelton PK. Relationship between cigarette smoking and novel risk factors for cardiovascular disease in the United States. Ann Intern Med. 2003;138(11):891–897. doi:10.7326/0003-4819-138-11-200306030-00010
12. Tsai JC, Perrella MA, Yoshizumi M, et al. Promotion of vascular smooth muscle cell growth by homocysteine: a link to atherosclerosis. Proc Natl Acad Sci U S A. 1994;91:6369–6373. doi:10.1073/pnas.91.14.6369
13. McCully KS. Vascular pathology of homocysteinemia: implications for the pathogenesis of arteriosclerosis. Am J Pathol. 1969;56:111.
14. Harker LA, Ross R, Slichter SJ, Scott CR. Homocystine-induced arteriosclerosis. The role of endothelial cell injury and platelet response in its genesis. J Clin Invest. 1976;58:731.
15. Chrysant GS, Chrysant GS. The current status of homocysteine as a risk factor for cardiovascular disease: a mini review. Expert Rev Cardiovasc Ther. 2018;16(8):559–565. doi:10.1080/14779072.2018.1497974
16. Liu Y, Song JH, Hou XH, et al. Elevated homocysteine as an independent risk for intracranial atherosclerotic stenosis. Aging (Albany NY). 2019;11(11):3824–3831. doi:10.18632/aging.102019
17. Khandanpour N, Loke YK, Meyer FJ, Jennings B, Armon MP. Homocysteine and peripheral arterial disease: systematic review and meta-analysis. Eur J Vasc Endovasc Surg. 2009;38(3):316–322. doi:10.1016/j.ejvs.2009.05.007
18. Ray JG. Meta-analysis of hyperhomocysteinemia as a risk factor for venous thromboembolic disease. Arch Intern Med. 1998;158:2101. doi:10.1001/archinte.158.19.2101
19. Clarke R, Daly L, Robinson K, et al. Hyper-homocysteinemia: an independent risk factor for vascular disease. NEJM. 1991;324:1149–1155.
20. Herrmann W, Obeid R. Homocysteine: a biomarker in neurodegenerative diseases. Clin Chem Lab Med. 2011;49(3):435–441. doi:10.1515/CCLM.2011.084
21. Petras M, Tatarkova Z, Kovalska M, et al. Hyperhomocysteinemia as a risk factor for the neuronal system disorders. J Physiol Pharmacol. 2014;65(1):15–23.
22. Modi M, Prabhakar S, Majumdar S, Khullar M, Lal V, Das CP. Hyperhomocysteinemia as a risk factor for ischemic stroke: an Indian scenario. Neurol India. 2005;53(3):297–302. doi:10.4103/0028-3886.16927
23. Lehotsky J, Petras M, Kovalska M, Tothova B, Drgova A, Kaplan P. Mechanisms involved in the ischemic tolerance in brain: effect of the homocysteine. Cell Mol Neurobiol. 2015;35(1):7–15. doi:10.1007/s10571-014-0112-3
24. Lehotský J, Tothová B, Kovalská M, et al. Role of homocysteine in the ischemic stroke and development of ischemic tolerance. Front Neurosci. 2016;10:538. doi:10.3389/fnins.2016.00538
25. Baszczuk A, Kopczyński Z. Hiperhomocysteinemia u chorych na schorzenia układu krążenia [Hyperhomocysteinemia in patients with cardiovascular disease]. Postepy Hig Med Dosw (Online). 2014;68:579–589. doi:10.5604/17322693.1102340
26. Shenoy V, Mehendale V, Prabhu K, Shetty R, Rao P. Correlation of serum homocysteine levels with the severity of coronary artery disease. Indian J Clin Biochem. 2014;29(3):339–344. doi:10.1007/s12291-013-0373-5
27. Homocysteine Studies Collaboration. Homocysteine and risk of ischemic heart disease and stroke: a meta-analysis. JAMA. 2002;288(16):2015–2022. doi:10.1001/jama.288.16.2015.
28. Wald David S, Malcolm L, Morris Joan K. Homocysteine and cardiovascular disease: evidence on causality from a meta-analysis. BMJ. 2002;325:1202. doi:10.1136/bmj.325.7374.1202
29. Finkelstein JD, Mudd SH, Irreverre F, Laster L. Homocystinuria due to cystathionine synthetase deficiency: the mode of inheritance. Science. 1964;146(3645):785–787. doi:10.1126/science.146.3645.785
30. Gharaibeh MY, Gahtan RA, Khabour OF, Alomari MA. Hyperhomocysteinemia, low folate, and Vitamin B12 deficiency in elderly living at home and care residences: a comparative study. Lab Med. 2010;41(7):410–414. doi:10.1309/LM1P78OFXACYYHPQ
31. Mashal RH, Odeh A (2004). Plasma homocysteine levels and coronary heart disease risk in jordanian subjects.
32. Mortality in treated heterozygous familial hypercholesterolaemia: implications for clinical management. Scientific Steering Committee on behalf of the Simon Broome Register Group. Atherosclerosis. 1999;142(1):105–112. doi:10.1016/S0021-9150(98)00200-7
33. Li J, Li B, Qi J, Shen B. [Meta-analysis of clinical trials of folic acid, vitamin B12 and B6 supplementation on plasma homocysteine level and risk of cardiovascular disease]. Zhonghua Xin Xue Guan Bing Za Zhi. 2015;43(6):554–561.
34. Bazzano LA, Reynolds K, Holder KN, He J. Effect of folic acid supplementation on risk of cardiovascular diseases: a meta-analysis of rendomized controlled trials. JAMA. 2006;296(22):2720–2726. doi:10.1001/jama.296.22.2720
35. The Heart Outcomes Prevention Evaluation (HOPE). 2 Investigators: homocysteine lowering with folic acid and B vitamins in vascular disease. N Engl J Med. 2006;354:1567–1577. doi:10.1056/NEJMoa060900
36. Veeranna V, Zalawadiya SK, Niraj A, et al. Homocysteine and reclassification of cardiovascular disease risk. J Am Coll Cardiol. 2011;58(10):1025–1033. doi:10.1016/j.jacc.2011.05.028.
37. Botto LD, Yang Q. 5,10-Methylenetetrahydrofolate reductase gene variants and congenital anomalies: a HuGE review. Am J Epidemiol. 2000;151(9):862–877. doi:10.1093/oxfordjournals.aje.a010290
38. Ruiz-Argüelles GJ, Garcés-Eisele J, Reyes-Núñez V, Ramírez-Cisneros FJ. Primary thrombophilia in Mexico. II. Factor V G1691A (Leiden), prothrombin G20210A, and methylenetetrahydrofolate reductase C677T polymorphism in thrombophilic Mexican mestizos. Am J Hematol. 2001;66(1):28–31. doi:10.1002/1096-8652(200101)66:1<28::aid-ajh1003>3.0.CO;2-3
39. Abu-Amero KK, Wyngaard CA, Dzimiri N. Prevalence and role of methylenetetrahydrofolate reductase 677 C–>T and 1298 A–>C polymorphisms in coronary artery disease in Arabs. Arch Pathol Lab Med. 2003;127(10):1349–1352. doi:10.1043/1543-2165(2003)127<349:paromr>2.0.CO;2
40. Frosst P, Blom HJ, Milos R, et al. A candidate genetic risk factor for vascular disease: a common mutation in methylenetetrahydrofolate reductase. Nat Genet. 1995;10(1):111–113. doi:10.1038/ng0595-111
41. George AK, Majumder A, Ice H, et al. Genes and genetics in hyperhomocysteinemia and the “1-carbon metabolism”: implications for retinal structure and eye functions. Can J Physiol Pharmacol. 2020;98(2):51–60. doi:10.1139/cjpp-2019-0236
42. Raslová K, Bederová A, Gasparovic J, Blazícek P, Smolková B. Effect of diet and 677 C–>T 5, 10-methylenetetrahydrofolate reductase genotypes on plasma homocyst(e)ine concentrations in slovak adolescent population. Physiol Res. 2000;49(6):651–658.
43. Hanson NQ, Aras O, Yang F, Tsai MY. C677T and A1298C polymorphisms of the methylenetetrahydrofolate reductase gene: incidence and effect of combined genotypes on plasma fasting and post-methionine load homocysteine in vascular disease. Clin Chem. 2001;47(4):661–666. doi:10.1093/clinchem/47.4.661
44. Pessin MS, Estol CJ, Lafranchise F, Caplan LR. Safety of anticoagulation after hemorrhagic infarction. Neurology. 1993;43(7):1298–1303. doi:10.1212/wnl.43.7.1298.
45. Kim JT, Heo SH, Park MS, Chang J, Choi KH, Cho KH. Use of antithrombotics after hemorrhagic transformation in acute ischemic stroke. PLoS One. 2014;9(2):e89798. doi:10.1371/journal.pone.0089798
46. Marsh EB, Llinas RH, Hillis AE, Gottesman RF. Hemorrhagic transformation in patients with acute ischaemic stroke and an indication for anticoagulation. Eur J Neurol. 2013;20(6):962–967. doi:10.1111/ene.12126.
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.