Back to Journals » Clinical, Cosmetic and Investigational Dermatology » Volume 16
A Complete Form of Pachydermoperiostosis Accompanied by a Pituitary Microadenoma
Received 13 September 2022
Accepted for publication 30 December 2022
Published 6 January 2023 Volume 2023:16 Pages 47—52
DOI https://doi.org/10.2147/CCID.S389766
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Dr Anne-Claire Fougerousse
Yan Jing Chen, Li Li
Department of Dermatology and Venereology, West China Hospital, Sichuan University, Chengdu, Sichuan, People’s Republic of China
Correspondence: Li Li, Department of dermatology and venereology, West China Hospital, Sichuan University, Guoxue Alley No. 37, Wuhou District, Chengdu, Sichuan, 610041, People’s Republic of China, Tel +86 18980601692, Email [email protected]
Abstract: Pachydermoperiostosis is a rare genetic disease that is associated with HPGD (15-hydroxyprostaglandin dehydrogenase) and SLCO2A1 (solute carrier organic anion transporter family member 2A1) gene mutations. It is characterized by three major phenotypes, namely, pachydermia, periostosis, and digital clubbing. Clinically, misdiagnoses such as acromegaly and thyroid acropachy are commonly confused with pachydermoperiostosis. Integral medical history, physical examination, endocrinological tests, and multiple disciplinary cooperation are extremely significant in the accurate diagnosis of pachydermoperiostosis. The co-existence of pachydermoperiostosis and pituitary adenoma is rarely recorded and discussed. In this case, we present a young male patient with a complete form of pachydermoperiostosis and a nonfunctional pituitary microadenoma, which has rarely been reported.
Keywords: pachydermoperiostosis, pituitary microadenoma, acromegaly, thyroid acropachy
Introduction
Pachydermoperiostosis (PDP), also known as primary hypertrophic osteoarthropathy, is an autosomal recessive or perhaps an X-linked inherited disorder that is associated with HPGD (15-hydroxyprostaglandin dehydrogenase) and SLCO2A1 (solute carrier organic anion transporter family member 2A1) gene mutations.1 The proteins encoded by the genes are responsible for the metabolism of prostaglandins. Increased levels of prostaglandin E2 resulting from gene mutations contribute to bone and soft tissue changes in PDP by simulating the activity of osteoblasts and osteoclasts as well as prolonging local vasodilatory effects.2 PDP is characterized by pachydermia, periostosis, digital clubbing, oily skin, hyperhidrosis and arthropathy.1 It occurs predominantly in adolescent males.2 Three clinical variants of PDP were defined in 1935: the complete form, with both pachydermia, periostosis and digital clubbing (as shown in this case); the incomplete form, with periostosis but lacking cutis verticis gyrata (CVG); and the forme fruste, with classical skin changes but with minimal periostosis.3
Pituitary adenomas are one of the most common brain tumours and display an array of hormonal and proliferative activities.4,5 They are classified into microadenomas (size < 10 mm) and macroadenomas (size ≥ 10 mm). According to a previous literature report, the prevalence of pituitary adenomas was estimated at 16.7% (14.4% in autopsy studies and 22.5% in radiologic studies).5 Nonfunctional pituitary adenomas account for 14–54% of pituitary adenomas, lack clinical or biochemical evidence of hormone excess and can be referred to as clinically silent adenomas.6 Most pituitary tumours occur sporadically without known genetic predisposition, but in a significant minority of cases, somatic mutations and germline mutations can be identified in patients with pituitary adenomas.7
Pituitary adenomas are fairly common in the general population. However, the incidence of pituitary adenoma is scarcer in PDP patients. To the best of our knowledge, only a 58-year-old male with a pituitary microadenoma and a 34-year-old male with a pituitary macroadenoma have been previously reported.8,9 The present case reports a young male patient with a complete form of PDP and a nonfunctional pituitary microadenoma that has not been previously described.
Case Report
A 26-year-old male presented to our outpatient department with a 6-year history of digital clubbing of the fingers and a 5-year facial appearance of CVG. He had excessive excretion of facial sebum. Hyperhidrosis and arthralgia were denied. Visual symptoms were absent. He denied any similar symptoms in his family. His parents were in a nonconsanguineous marriage. On physical examination, thickening and furrowing of the facial skin, biliteral eyelid ptosis (Figure 1A), digital clubbing (Figure 1B) and enlargement of the wrist, elbow, knee, and ankle joints were observed. Laboratory tests revealed normal levels of thyroid stimulating hormone (2.910 mU/L, reference range (RR) 0.27–4.2 mU/L), free triiodothyronine (5.10 pmol/L, RR 3.60–7.50 pmol/L), free thyroxine (16.90 pmol/L, RR 12.0–22.0 pmol/L), growth hormone (GH) (0.09 ng/mL, RR 0.030–2.47 ng/mL), adrenocorticotropic hormone (48.72 ng/L, RR 5.00–78.00 ng/L), and plasma total cortisol at 8.00 h (507.00 nmol/L, RR 133.0–537.0 nmol/L) and a decreased level of insulin-like growth factor-1 (IGF-1) (64.83 ng/mL, RR 137–278 ng/mL). Computed tomography of his chest and echocardiography were unremarkable. Radiographic examination of the long bones showed cortical thickening (Figure 1C). Contrast-enhanced magnetic resonance imaging (MRI) of the pituitary revealed a hypointense signal with a size of 3 mm×5 mm on coronal T2-weighted images (Figure 1D). The patient received diagnoses of a complete form of PDP and a nonfunctional pituitary microadenoma. He was referred to receive further gene analysis and a treatment modality with oral etoricoxib, but he refused.
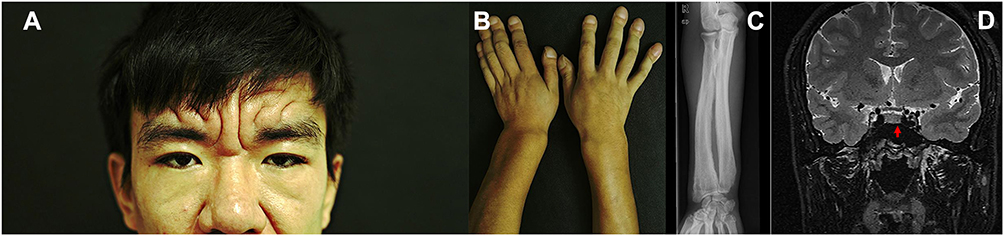 |
Figure 1 Skin furrowing and thickening, biliteral eyelids ptosis (A), digital clubbing of the fingers (B), radiographic examination showed cortical thickening in the long bones (C), contrast-enhanced magnetic resonance imaging of the pituitary revealed a hypointense signal on T2-weighted images (3×5mm) (D). |
Discussion
PDP is rare and is characterized by pachydermia, periostosis, and digital clubbing, as noted. Pachydermia, which usually presents as an appearance of CVG on the scalp, forehead, cheek, or chin, is the most frequent skin symptom. Periostosis of PDP might manifest as swelling of periarticular tissue and arthritis and radiographically display as periosteal thickening or reaction, commonly involving the tibia, fibula, radius and ulna.10,11 Digital clubbing, which often results in a drumstick appearance on the distal fingers or toes, can be determined by measuring the hyponychial angle, Lovibond angle, phalangeal depth ratio or Schamroth sign.10 Notably, diarrhoea, gastric ulcer, anaemia, hypoalbuminemia and even visual field disturbances, and blindness were also reported in PDP patients along with skin and arthrological symptoms.12,13 Therefore, regular follow-up monitoring is recommended for the possible association with gastrointestinal and ophthalmological involvement.
Before the final diagnosis of PDP is made, other possible diseases should be ruled out, such as acromegaly and thyroid acropachy (TA) (Table 1). Acromegaly is a rare and underdiagnosed disease that results from the overproduction of GH and IGF-1. The most common signs and symptoms are acral growth, deformity of facial features, soft tissue swelling and increased sweating.14 The systemic comorbidities related to acromegaly are variable. Digital clubbing and periostosis are not seen in acromegaly.11,15 Endocrine testing and pituitary MRI examination are important to confirm the diagnosis of acromegaly. Measurement of serum IGF-1 to rule out acromegaly has been recommended in any patients harbouring a pituitary adenoma, and a normal serum IGF-1 concentration is strong evidence that the patient does not have acromegaly.11,16 Both serum GH concentrations and IGF-1 concentrations are increased in virtually all patients with acromegaly.15 TA is a rare extrathyroid manifestation of autoimmune thyroid disease (AITD), which manifests as soft tissue swelling with digital clubbing and always associated with thyroid ophthalmopathy and dermopathy. For the chronological sequence of extrathyroidal manifestations of AITD, thyroid dysfunction develops first, followed by ophthalmopathy, dermopathy, and finally, acropachy.17,18 TA can occur in hyperthyroid, euthyroid or hypothyroid patients.17,18 Radiological study reveals periosteal reactions predominantly in metacarpals and phalanges of the fingers, and rarely in long bones.10,19 In this case, the patient was diagnosed with PDP due to facial disfigurement, digital clubbing and the periostosis of the long bones, although a pituitary microadenoma was found.
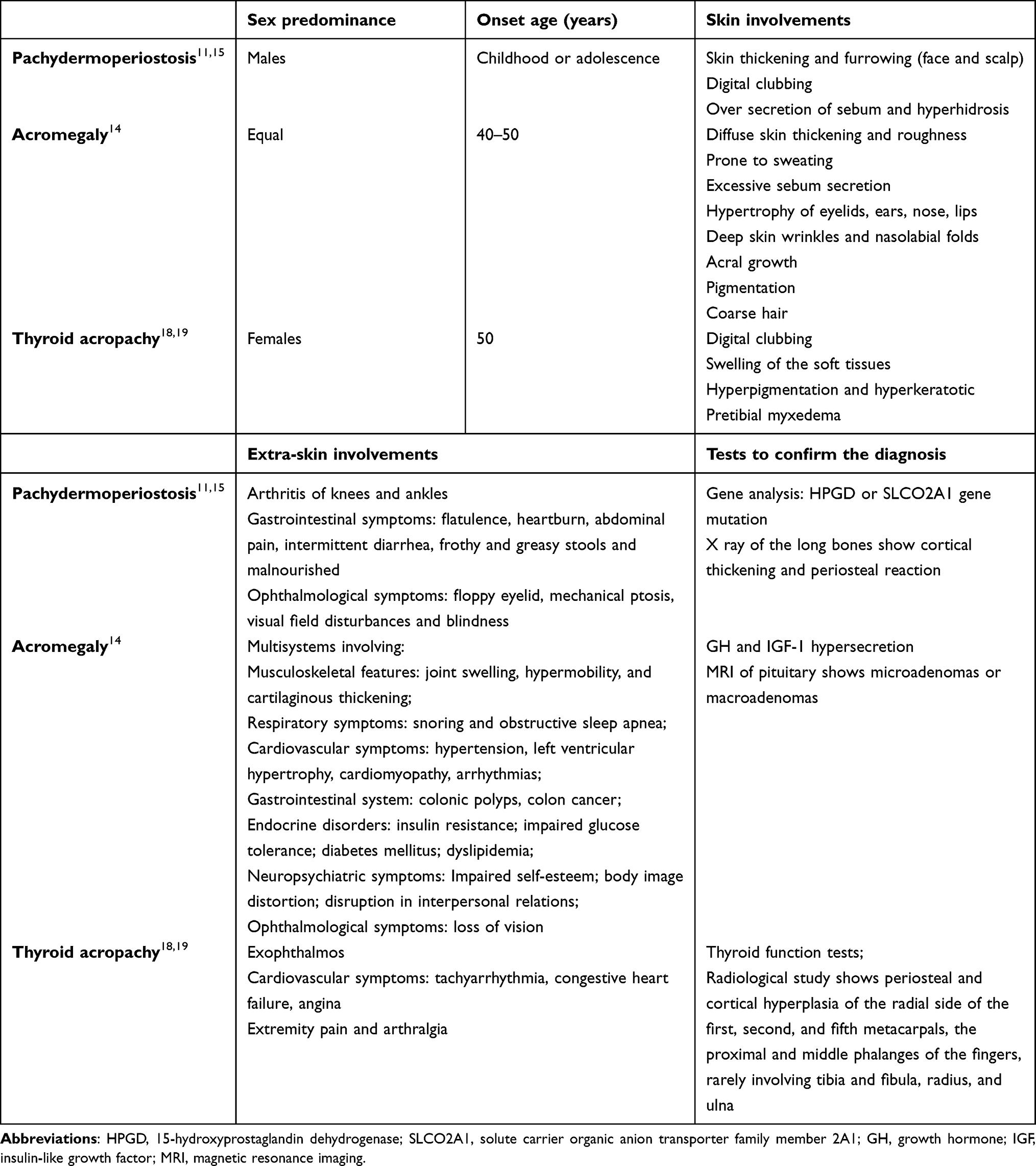 |
Table 1 The Main Differential Diagnoses Compared with Pachydermoperiostosis |
According to the previous literature, only one case reported a nonsecreting pituitary macroadenoma in a male PDP patient, and another reported a functional pituitary microadenoma in an aged Japanese male (Table 2).8,9 A nonfunctional pituitary microadenoma in a young male patient with PDP has never been described before. All the reported patients were male with characteristic features of PDP, but no family history was noted. Endocrinological examinations found an increased GH level in a patient with a pituitary microadenoma, but the diagnosis was acromegaly associated with PDP. Generally, microadenomas are more likely to be asymptomatic or lack hormone excess than macroadenomas, corresponding with the condition in our patient. It is intriguing that the level of IGF-1 was low in our patient; conditions that can reduce IGF-1 levels, such as malnutrition, sepsis, liver disease, uncontrolled diabetes, hypothyroidism, and obesity, were not present.20 We suggest that the possible causes may be related to GH deficiency, which could be confirmed by further determination of GH secretion provocative tests or GH insensitivity, which may be associated with internal medical conditions, as mentioned above, and characterized by low serum IGF-1, GH binding protein and increased serum GH.21 In our case, the cause was not ultimately identified.
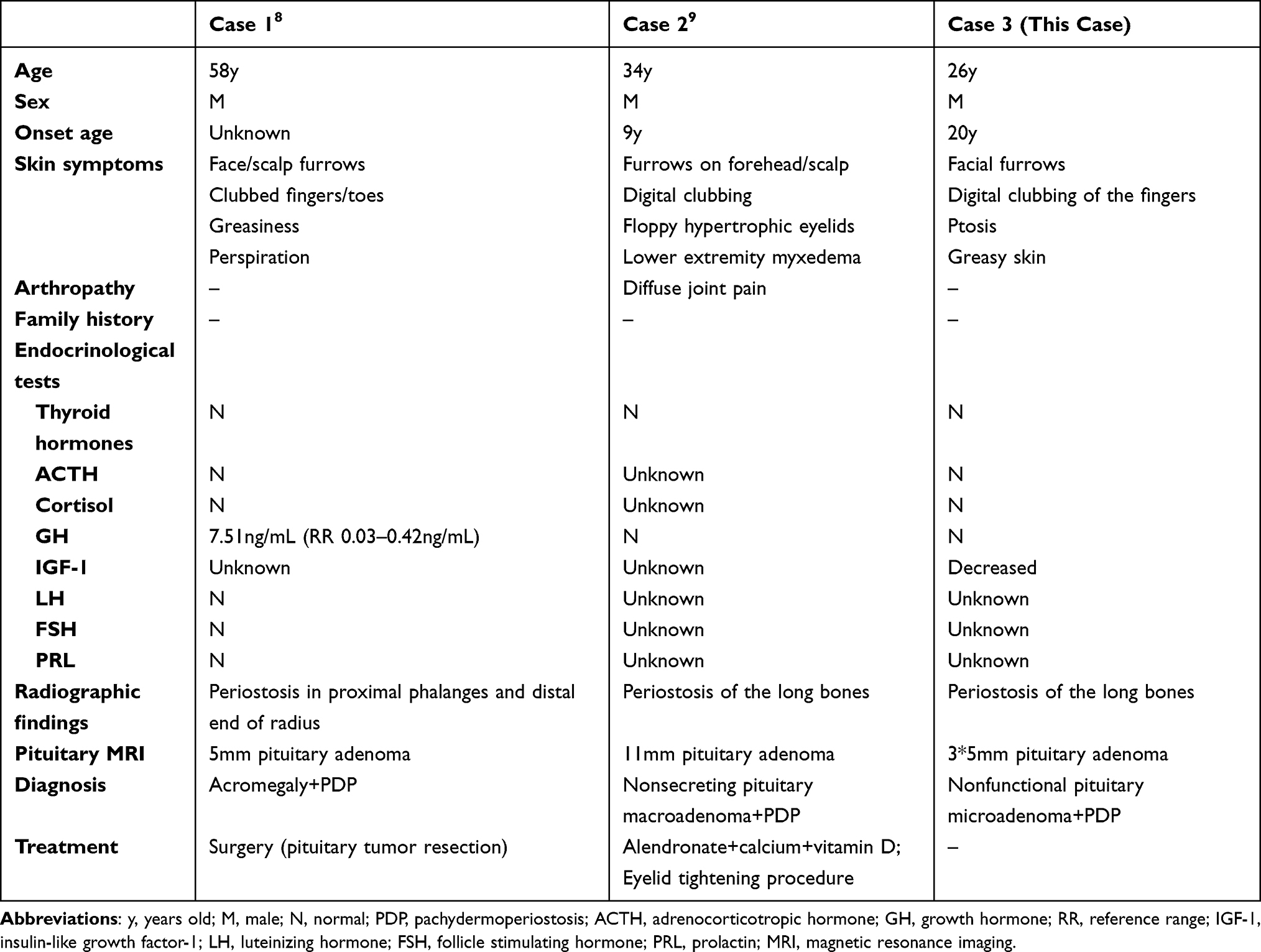 |
Table 2 Comparison Among PDP Patients with Pituitary Adenomas |
In the absence of studies on this disease, treatment options are limited and largely based on case reports. Administration of oral nonsteroidal anti-inflammatory drugs could relieve joint pain and swelling. Etoricoxib (60 mg per day) and hydroxychloroquine (400 mg per day) were proven to be effective in the control of both skin and joint conditions.22,23 Plastic surgery and injection of botulinum toxin (BTX) may improve skin syndrome.24 However, BTX is an off-label use, and both procedures may cause a severe financial burden on patients. None of the treatment strategies were attempted in this patient, as he refused to take the risks of medication complications or any surgery-related adverse effects. Microadenoma growth occurs in only 10.6% of patients. Imaging follow-up of the pituitary MRI is recommended every 1, 2, and 5 years, and repeat scanning should be performed less frequently in these patients. Surgical resection is generally not indicated.25
In general, this case report presents a young male patient with complete PDP and a nonfunctional pituitary microadenoma. Age and sexuality predilection, clinical features, radiographic changes and endocrinological tests should be addressed in the diagnosis of PDP. Genetic study is of significance. Regretfully, it was unable to be performed in this case. Multidisciplinary cooperation should be recommended for periodic follow-ups in PDP patients. Despite the absence of clinical symptoms, the association between PDP and pituitary microadenoma or macroadenoma needs to be mentioned.
Ethics Statement
Ethical committee approval was not required for this case report.
Patient Consent
Informed consent for this case and associated images were obtained from the patient for publication.
Acknowledgment
We thank Dr. Yu Jiang from the radiological department for her kind help in analyzing the contrast-enhanced MRI of the pituitary.
Funding
This work was supported by 1·3·5 project for disciplines of excellence, West China Hospital, Sichuan University.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Xiao J, Zhang DD, Zhang L. A novel mutation in the SLCO2A1 gene in a Chinese family with pachydermoperiostosis. Australas J Dermatol. 2019;60(4):e348–e350. doi:10.1111/ajd.13041
2. Honório MLP, Bezerra GH, Costa V. Complete form of pachydermoperiostosis. An Bras Dermatol. 2020;95(1):98–101. doi:10.1016/j.abd.2019.04.009
3. Prasad A, Shahi P, Sehgal A, Bhagirathi Mallikarjunaswamy M. Incomplete primary hypertrophic osteoarthropathy. BMJ Case Rep. 2020;13(5):e236034. doi:10.1136/bcr-2020-236034
4. Mehta GU, Lonser RR. Management of hormone-secreting pituitary adenomas. Neuro Oncol. 2017;19(6):762–773. doi:10.1093/neuonc/now130
5. Ezzat S, Asa SL, Couldwell WT, et al. The prevalence of pituitary adenomas: a systematic review. Cancer. 2004;101(3):613–619. doi:10.1002/cncr.20412
6. Ntali G, Wass JA. Epidemiology, clinical presentation and diagnosis of non-functioning pituitary adenomas. Pituitary. 2018;21(2):111–118. doi:10.1007/s11102-018-0869-3
7. Lim CT, Korbonits M. Update On The Clinicopathology Of Pituitary Adenomas. Endocr Pract. 2018;24(5):473–488. doi:10.4158/ep-2018-0034
8. Shimizu C, Kubo M, Kijima H, et al. A rare case of acromegaly associated with pachydermoperiostosis. J Endocrinol Invest. 1999;22(5):386–389. doi:10.1007/bf03343577
9. Berdia J, Tsai FF, Liang J, Shinder R. Pachydermoperiostosis: a rare cause of marked blepharoptosis and floppy eyelid syndrome. Orbit. 2013;32(4):266–269. doi:10.3109/01676830.2013.788672
10. Yap FY, Skalski MR, Patel DB, et al. Hypertrophic Osteoarthropathy: clinical and Imaging Features. Radiographics. 2017;37(1):157–195. doi:10.1148/rg.2017160052
11. Abdullah NRA, Jason WLC, Nasruddin AB. Pachydermoperiostosis: a rare mimicker of acromegaly. Endocrinol Diabetes Metab Case Rep. 2017;2017. doi:10.1530/edm-17-0029
12. Cheng SK, Michael A, Rizzuti AE. Pachydermoperiostosis presenting with vision loss secondary to severe phlyctenular keratoconjunctivitis. Cornea. 2022;41(1):113–115. doi:10.1097/ico.0000000000002700
13. Wang Q, Li YH, Lin GL, et al. Primary hypertrophic osteoarthropathy related Gastrointestinal complication has distinctive clinical and pathological characteristics: two cases report and review of the literature. Orphanet J Rare Dis. 2019;14(1):297. doi:10.1186/s13023-019-1264-5
14. Vilar L, Vilar CF, Lyra R, Lyra R, Naves LA. Acromegaly: clinical features at diagnosis. Pituitary. 2017;20(1):22–32. doi:10.1007/s11102-016-0772-8
15. Kartal Baykan E, Türkyılmaz A. Differential diagnosis of acromegaly: pachydermoperiostosis two new cases from Turkey. J Clin Res Pediatr Endocrinol. 2022;14(3):350–355. doi:10.4274/jcrpe.galenos.2021.2020.0301
16. Katznelson L, Laws ER, Melmed S, et al. Acromegaly: an endocrine society clinical practice guideline. J Clin Endocrinol Metab. 2014;99(11):3933–3951. doi:10.1210/jc.2014-2700
17. Jadidi J, Sigari M, Efendizade A, Grigorian A, Lehto SA, Kolla S. Thyroid acropachy: a rare skeletal manifestation of autoimmune thyroid disease. Radiol Case Rep. 2019;14(8):917–919. doi:10.1016/j.radcr.2019.04.021
18. Fatourechi V, Ahmed DD, Schwartz KM. Thyroid acropachy: report of 40 patients treated at a single institution in a 26-year period. J Clin Endocrinol Metab. 2002;87(12):5435–5441. doi:10.1210/jc.2002-020746
19. Perini N, Santos RB, Romaldini JH, Villagelin D. Thyroid acropachy: a rare manifestation of graves disease in joints. AACE Clin Case Rep. 2019;5(6):e369–e371. doi:10.4158/accr-2018-0591
20. Aimaretti G, Boschetti M, Corneli G, et al. Normal age-dependent values of serum insulin growth factor-I: results from a healthy Italian population. J Endocrinol Invest. 2008;31(5):445–449. doi:10.1007/bf03346389
21. Kato Y, Murakami Y, Sohmiya M, Nishiki M. Regulation of human growth hormone secretion and its disorders. Intern Med. 2002;41(1):7–13. doi:10.2169/internalmedicine.41.7
22. Li Z, Yang Q, Yang Y, Wang D, Wang S. Successful treatment of pachydermoperiostosis with etoricoxib in a patient with a homozygous splice‐site mutation in the SLCO2A1 gene. Br J Dermatol. 2018;180(3):682–684. doi:10.1111/bjd.14480
23. Alessandrella A, Della Casa R, Alessio M, Puente Prieto J, Strisciuglio P, Melis D. A novel homozygous mutation in the SLCO2A1 gene causing pachydermoperiostosis: efficacy of hydroxychloroquine treatment. Am J Med Genet A. 2018;176(5):1253–1257. doi:10.1002/ajmg.a.38677
24. Li X, Hao D, Li-Ling J, Jiang X. Complete form of pachydermoperiostosis with cutis verticis gyrata resulting from the SLCO2A1 gene mutation. Indian J Dermatol Venereol Leprol. 2019;85(6):681. doi:10.4103/ijdvl.IJDVL_911_17
25. Molitch ME. Management of incidentally found nonfunctional pituitary tumors. Neurosurg Clin N Am. 2012;23(4):543–553. doi:10.1016/j.nec.2012.06.003
 © 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.