Back to Journals » International Medical Case Reports Journal » Volume 14
A Case of Extensive Bilateral Idiopathic Sclerochoroidal Calcification and Review of Literature
Authors Thomson AC ![]() , Brown GT
, Brown GT ![]() , Dolores-Rodriguez A, Hunter AA
, Dolores-Rodriguez A, Hunter AA
Received 2 September 2021
Accepted for publication 7 October 2021
Published 28 October 2021 Volume 2021:14 Pages 749—755
DOI https://doi.org/10.2147/IMCRJ.S336237
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Scott Fraser
Andrew C Thomson,1 Gordon T Brown,2 Angel Dolores-Rodriguez,3 Allan A Hunter2
1McGovern Medical School at The University of Texas Health Science Center at Houston, Houston, TX, USA; 2Oregon Eye Consultants LLC, Eugene, OR, USA; 3Alameda Health System-Highland Hospital, Oakland, CA, USA
Correspondence: Andrew C Thomson 3783 International Ct Suite 290, Springfield, OR, 97477, USA
Tel +1 541 687 1927 ext 5765
Fax +1 541 683 8779
Email [email protected]
Abstract: A 62-year-old Caucasian male was referred to retina for choroiditis and uveitis. Multiple areas of yellow irregularities were noted on fundus exam throughout the periphery of both eyes, corresponding to lesions at the sclerochoroidal junction on OCT. A diagnosis of sclerochoroidal calcifications (SCC) was confirmed by B-ultrasonography, fundus photography, OCT imaging, and fluorescein and indocyanine green angiography. Systemic metabolic studies were performed, which showed reduced renal function with increased serum calcium; however, SCC lesions in this case were most likely idiopathic. In this work, we report the clinical findings, appearance on multimodal imaging, and systemic associations of sclerochoroidal calcification. Sclerochoroidal calcifications are an unusual clinical finding that tends to be idiopathic, but a focused workup and specialist referral may be warranted to exclude systemic conditions associated with abnormal calcium-phosphate metabolism or hypokalemic metabolic alkalosis syndromes.
Keywords: case report, literature review, multimodal imaging, sclerochoroidal calcification, systemic associations
Introduction
Sclerochoroidal calcification (SCC) is an uncommon, benign ophthalmic finding occurring in predominantly elderly, Caucasian patients of either gender.1 Histologically, calcium pyrophosphate is deposited within the sclera, choroid, or both. The peripheral retina has been defined by Duke-Elder and Rutnin, respectively, as either outside the macula or anterior to the vortex veins entrances.2 In this work, the former definition will be used. SCC is characterized by yellow-white sub-retinal lesions, classically located in the superotemporal or superonasal quadrants and mid-peripheral region of the fundus, often between the equator and retinal arcades or alongside the arcades.1,3 Despite well-known features, this condition is often misdiagnosed as a choroidal metastasis, nevus, melanoma, osteoma, or lymphoma and has resulted in unnecessary intervention.4 In the setting of concomitant cancer, differentiating the presence of incidental SCC from metastasis is especially important.5 SCC is most commonly idiopathic, around 79% of cases,6 but it may be associated with recurrent episodes of inflammation or disorders of calcium metabolism or the renal tubulopathies, Bartter and Gitelman syndromes.4
Case Report
A 62-year-old Caucasian male with a history of hypertension, hypercholesterolemia, type 2 diabetes without ocular complications, and arthritis was referred to retina for choroiditis and uveitis. The patient had no ocular complaints besides an occasional “eye itch.” Systemic medications included metformin, allopurinol, losartan, atorvastatin, and Vitamin D. Social and family history were noncontributory. At presentation, corrected visual acuity by Snellen chart was 20/60-2 right eye and 20/60 left eye. On exam, bilateral mild nuclear sclerotic and trace cortical cataracts were noted with rare flare in the anterior chamber. Trace cells and 1+ flare was noted in the left eye at a 3-week follow-up. Fundus examination revealed yellow-white lesions predominantly in the superotemporal near-peripheral and mid-peripheral regions of the fundus bilaterally. The exam was otherwise normal. Fundus photos, fluorescein angiography (FA), Indocyanine green angiography (ICGA), macula OCT, and B-scan ultrasonography were obtained. Fundus photography demonstrated extensive bilateral lesions (Figure 1A and B). B-scan ultrasonography revealed multiple hyperechoic lesions at the sclerochoroidal junction (Figure 1C and D). Fundus autofluorescence showed hyperautofluorescence of the lesions (Figure 2). Late-phase FA revealed peripheral staining without obvious leakage, and ICGA revealed multifocal, scattered hypocyanescent lesions in the peripheral retina of both eyes in all phases with some hypervascularity of the macular area (Figure 3). The FA also appear to demonstrate hypofluorescent lesions corresponding to areas of choroidal compression. Macular OCT showed scleral thickening with a “rocky-rolling” contour and compression of overlying choroid at and surrounding lesions (Figure 4). Calcium metabolism and renal function labs were obtained, revealing reduced renal function with increased serum calcium (Table 1). The diagnosis of sclerochoroidal calcification was made and additional hematologic, infectious, and autoimmune (HLA-A29) testing was negative for causes of uveitis. Upon 6 month follow-up, the examination and OCT imaging results were unchanged.
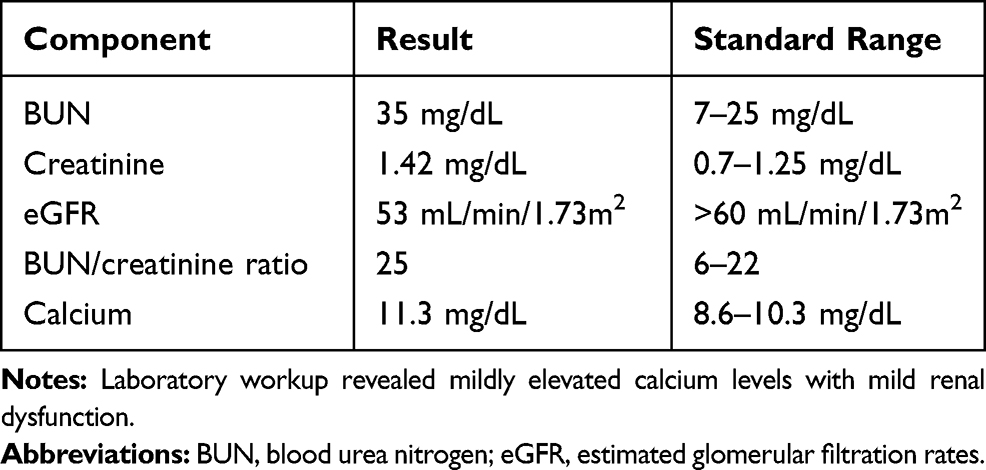 |
Table 1 Laboratory Workup |
 |
Figure 1 Fundus photography and B-scan ultrasonography of sclerochoroidal calcification lesions. (A) There are dense yellow-white lesions in the superotemporal region near the superior retinal arcades in the right eye. (B) Fundus photography shows scattered calcifications in the superotemporal region of the fundus in the left eye. (C and D) Ultrasonography in both eyes shows hyperechoic lesions at the level of the choroid-scleral junction with posterior shadowing. |
 |
Figure 2 Blue-light autofluorescence of the right eye (left eye not shown) displaying an autofluorescent lesion adjacent to the superior and inferior retinal arcades. |
 |
Figure 3 Fluorescein angiography of right (A) and left (B) eyes displays mild-moderate late phase staining with no obvious leakage. Indocyanine green angiography (ICGA) of right (C) and left (D) eyes shows extensive, multifocal hypofluorescent lesions in the near-peripheral and mid-peripheral fundus with choroidal vascular hyperpermeability of the macular area. |
 |
Figure 4 OCT imaging of representative SCC lesions in the right eye (left eye not shown) near the (A) superior and (B) inferior retinal arcades. The sclera appears to have a “rocky-rolling” surface with thinning of overlying choroid. |
Discussion
SCC is characterized by calcium deposition within the sclera, eventually invading the choroid and manifesting as a yellow-white choroidal mass. Lesions are bilateral in approximately 52% of cases.1 Patients with SCC are typically Caucasian with a mean age of 69 years-old and have a slight female predominance (60%).1 SCC is most often asymptomatic; however, a relative scotoma in areas of corresponding chorioretinal atrophy overlying SCC lesions may be seen.7 Rarely, visual deterioration is seen in SCC due to the development of choroidal neovascularization (CNV) with exudation extending to the macula.8,9 The differential includes choroidal metastasis, melanoma, nevus, osteoma, chorioretinitis, and intraocular lymphoma which may be differentiated upon exam, imaging, and angiography.4 SCC findings on multimodal imaging are summarized in Table 2.
 |
Table 2 Sclerochoroidal Calcification Findings on Multimodal Imaging |
Diagnosis of SCC can be made with fundoscopic examination. Ancillary testing may confirm the diagnosis.4 However, extensive testing and imaging for incidental findings of SCC is unnecessary. Hasanreisoglu et al6 have classified these calcium deposits based on enhanced depth imaging optical coherence tomography (EDI-OCT) appearance. They described four types of lesions (flat, rolling, rocky-rolling, or table mountain) based on scleral contour, and the “rocky-rolling” appearance may be unique to SCC, helping differentiate it from more concerning lesions. However, the exact utility of classifying these lesions in this way requires further research. On OCT imaging, the choroid is thin above these lesions, and there may be thinning of the retina with an absence of the external limiting membrane and disruption of the ellipsoid zone.3 The RPE may be thin, normal, or thickened above the lesion.3,6 “Rocky-rolling” type lesions have a significantly higher rate of RPE and ellipsoid zone abnormalities compared to the other types.6 B-Scan ultrasonography will demonstrate a dense echogenic mass with posterior shadowing.6 If the calcium deposits are located within the outer sclera, ultrasonography may uncover hyperechoic plaques in eyes with mild RPE alterations or an otherwise normal appearance.4
On FAF, lesions may be hyperautofluorescent, but hypoautofluorescent lesions with a hyperautofluorescent halo have also been observed.3,10,11 Fung et al3 hypothesized the hyperautofluorescence was due to thinning of the choroid, revealing hyperautofluorescent sclera or calcific tissue. Another explanation has been proposed by Caminal-Mitjana et al10 based on correlating FAF and spectral domain-OCT findings. The authors assert that choriocapillaris compression by scleral lesions leads to RPE dysfunction and accumulation of lipofuscin, a fluorophore, within the RPE. As RPE-choriocapillaris outpouching develops on SD-OCT, there is diminished autofluorescence likely corresponding to RPE atrophy and lipofuscin loss. Where the RPE is healthy, the FAF is isoautofluorescent. FA findings over lesions may demonstrate hypofluorescence in the arterial phase followed by progressively intensifying hyperfluorescence, possibly due to increased scleral staining around the lesions.10 In areas of RPE atrophy, the transmission of choroidal vasculature through window defects may be seen. FA may also reveal CNV, a rare association with SCC.4
ICGA findings vary and are somewhat lacking in the literature. It may show hypocyanescence in all phases12 or hypocyanescence during early phases with mild and progressive hypercyanescence in later phases.5 Mitamura et al11 described a case of bilateral SCC in the setting of secondary hyperparathyroidism with ICGA findings similar to our patient’s. Their patient displayed choroidal vascular hyperpermeability in the macular area with scattered hypocyanescent lesions in the peripheral fundus on ICGA with dilated Haller layer veins on OCT. They hypothesize this was due to multiple points of choroidal circulation impairment due to scleral lesions causing compression and subsequent vascular congestion. However, their patient had dialysis-dependent chronic kidney disease, which also causes systemic vascular dysfunction. Lastly, orbital computer tomography will show a radiodense scleral lesion, confirming the presence of calcification.3
The pathogenesis of SCC may be either dystrophic, idiopathic, or metastatic.4,12 Dystrophic calcification occurs after chronic tissue damage or necrosis, such as senile degeneration, chronic inflammation, or severe trauma in the setting of normal calcium-phosphorus metabolism.4 Idiopathic cases of SCC typically seen in elderly patients may be related to the dystrophic changes caused by chronic movement of the oblique muscles, which has its insertion near a common location for SCC, the temporal arcades. Evidencing this hypothesis is that the calcified plaques near insertion sites of horizontal recti muscles are histologically identical to SCC.4 Metastatic calcification develops in normal tissue due to an abnormal calcium-phosphorus metabolism seen in hyperparathyroidism, pseudohypoparathyroidism, sarcoidosis, hypervitaminosis D, and chronic renal failure.
Disorders of abnormal calcium-phosphorus metabolism are commonly found in patients with SCC (Table 3). A review of 179 eyes from 118 patients by Shields et al1 found 27% of patients had hyperparathyroidism, and 15% had a parathyroid adenoma. SCC is also seen in secondary hyperparathyroidism due to chronic renal failure, wherein phosphate retention and decreased vitamin D activation lead to hypocalcemia and elevated parathyroid compensation.1 Increased serum calcium, phosphate, and calcium-phosphate products inducing soft tissue calcification, and SCC has been observed in hypervitaminosis D-related disorders such as sarcoidosis producing 1,25-dihydroxy-cholecalciferol and vitamin D intoxication from ingesting large doses of vitamin D.12,13 Two cases of SCC associated with hypovitaminosis D have also been reported. Sierra-Rodriguez et al14 and Ale & David15 describe a case of SCC associated with isolated vitamin D deficiency of 4 ng/mL and 8.9 ng/mL, respectively. As with hypervitaminosis D, the exact mechanism of calcification development is unclear, but hypovitaminosis D may lead to calcification by disinhibiting PTH secretion or causing dysfunction of inflammatory pathways.13
 |
Table 3 Diseases Associated with Sclerochoroidal Calcification |
Pseudohypoparathyroidism (PHP) includes several related disorders involving resistance to parathyroid hormone and subsequent hypocalcemia and hyperphosphatemia. Extensive SCC has been reported in several cases of type 1a PHP, known as Albright hereditary osteodystrophy. This disease is due to the inactivation of the GNAS1 gene, and it presents with characteristic facies, brachydactyly, renal dysfunction, electrolyte abnormalities, subcutaneous calcifications, and developmental delay.16,17
In 118 patients with SCC, Shields et al1 found primary Bartter syndrome was present in 2% and Gitelman syndrome secondary to diuretic use in 11%. SCC in primary Gitelman syndrome has also been described.18 It is important to determine the presence of these diseases because electrolyte abnormalities, and potential cardiac arrhythmias, can be fatal without appropriate management.19 Although Bartter and Gitelman syndromes have different etiologies, the clinical presentation—normotensive hypokalemic metabolic alkalosis—is often similar.
There are five variants of Bartter syndrome with defects of different channels located in the thick ascending limb of the nephron. Most variants present antenatally with polyhydramnios and postnatally with characteristic facies, salt wasting, polyuria, polydipsia, marked hypokalemia, metabolic alkalosis, hypercalciuria, and failure to thrive.19 Classic Bartter’s syndrome, type III, may display some or all of these features, but it is historically considered a milder variant and may present in adulthood with or without developmental delay.20 With its milder features, classic Bartter syndrome is often confused with Gitelman syndrome. The presence of normal or high urine calcium found in Bartter syndrome will differentiate it from Gitelman syndrome, where urine calcium is always low. Gitelman syndrome is caused by an autosomal recessively inherited mutation of the thiazide-sensitive sodium chloride cotransporter in the distal convoluted tubule. It usually presents after the age of 6 years old with metabolic alkalosis, hypomagnesemia, hypokalemia, hypocalciuria, and chondrocalcinosis. Hypomagnesemia may be present in Bartter syndrome but is more characteristic of Gitelman syndrome.20
Bartter and Gitelman syndromes and other causes of hypomagnesemia have been associated with calcium pyrophosphate dihydrate deposition disease (CPPD), also called pseudogout.21 CPPD, particularly familial disease,22 has been associated with SCC as well.23 Although the mechanisms behind these associations are unclear, hypomagnesemia has been implicated in the development of CPPD and may also contribute to SCC development via the formation of calcium pyrophosphate dihydrate crystal.20,21 Magnesium inhibits crystal formation by several mechanisms; it increases crystal solubility, decreases inorganic pyrophosphate production, and acts as a cofactor of alkaline phosphatase, which hydrolyzes inorganic pyrophosphate.12,21
After finding incidental SCC, further laboratory screening for calcium-phosphate metabolism dysfunction and renal tubulopathies is warranted with a prompt nephrology referral if concerning abnormalities are detected. A list of possible testing includes renal function tests; serum levels of parathyroid hormone, calcitonin, vitamin D, and alkaline phosphatase; and blood and urine levels of calcium, potassium, phosphate, and magnesium.1,9 In our case, workup revealed mild hypercalcemia and mildly impaired renal function. Our patient was also taking vitamin D. However, our patient’s overall clinical gestalt suggests idiopathic SCC.
Treatment for SCC is conservative with the management of any underlying disease. SCC lesions have been shown in some cases to slowly enlarge over the span of years with, as of yet, unknown consequences.22,24 Given the rare yet possible development of CNV, some authors recommend regular visits every couple of years with imaging.24 In cases of CNV requiring treatment, anti-VEGF intravitreal injections, argon laser photocoagulation, and photodynamic therapy have shown success.9 Clinical prognosis will vary depending on systemic associations, but visual prognosis, especially in idiopathic cases, is generally good as parafoveal or foveal involvement or CNV development is rare.4
Conclusion
Although SCC association with underlying systemic conditions is rare, excluding systemic disease with a detailed examination, laboratory workup, and ancillary multimodal imaging may be warranted. Imaging such as OCT (preferably EDI-OCT), FA, ICGA, or B-scan ultrasonography and laboratory testing such as parathyroid, renal function, calcium, and vitamin D testing along with assessing diuretic use, may elucidate this rare, incidental finding and rule out concerning disease or prevent unnecessary intervention.
Informed Consent/Institutional Approval
Informed consent to publish their case details and images was obtained from the patient. This case report did not require institutional approval for publication.
Acknowledgments
We would like to thank Olivia Crowley for her contribution.
Disclosure
All authors report no conflicts of interest in this work.
References
1. Shields CL, Hasanreisoglu M, Saktanasate J, Shields PW, Seibel I, Shields JA. Sclerochoroidal calcification: clinical features, outcomes, and relationship with hypercalcemia and parathyroid adenoma in 179 eyes. Retina. 2015;35(3):547–554. doi:10.1097/IAE.0000000000000450
2. Quinn N, Csincsik L, Flynn E, et al. The clinical relevance of visualising the peripheral retina. Prog Retin Eye Res. 2019;68(June 2018):83–109. doi:10.1016/j.preteyeres.2018.10.001
3. Fung AT, Arias JD, Shields CL, Shields JA. Sclerochoroidal calcification is primarily a scleral condition based on enhanced depth imaging optical coherence tomography. JAMA Ophthalmol. 2013;131(7):960–963. doi:10.1001/jamaophthalmol.2013.67
4. Shields JA, Shields CL. CME review: sclerochoroidal calcification. The 2001 Harold Gifford lecture. Retina. 2002;22(3):251–261. doi:10.1097/00006982-200206000-00001
5. Lassandro NV, Danieli L, Nicolai M, Pirani V, Pelliccioni P, Mariotti C. Sclerochoroidal calcification as an incidental finding during oncological staging of a patient with parotid malignancy: a case report. Eur J Ophthalmol. 2021;112067212199962. doi:10.1177/1120672121999629
6. Hasanreisoglu M, Saktanasate J, Shields PW, Shields CL. Classification of sclerochoroidal calcification based on enhanced depth imaging optical coherence tomography mountain-like features. Retina. 2015;35(7):1407–1414. doi:10.1097/IAE.0000000000000468
7. Hara K, Tanito M, Kodama T, Ohira A. A case of chorioretinal atrophy due to sclerochoroidal calcification. Acta Ophthalmol. 2013;91(2):e167–e168. doi:10.1111/j.1755-3768.2012.02470.x
8. Bessette AP, Singh AD. Multimodal imaging of choroidal neovascularization associated with sclerochoroidal calcification. Ocul Oncol Pathol. 2016;2(4):234–238. doi:10.1159/000446214
9. Goerlitz-Jessen M, Ali MH, Grewal DS. Rare Complication of Sclerochoroidal Calcifications. JAMA Ophthalmol. 2019;137(1):111–112. doi:10.1001/jamaophthalmol.2018.2457
10. Caminal-Mitjana JM, Padrón-Pérez N, Arias-Barquet L, Rubio-Caso MJ, Catala-Mora J. Correlation between spectral-domain optical coherence tomography and autofluorescence findings in sclerochoroidal calcification. Can J Ophthalmol. 2013;48(4):331–334. doi:10.1016/j.jcjo.2013.01.024
11. Mitamura M, Kase S, Ishida S. Multimodal imaging in sclerochoroidal calcification: a case report and literature review. BMC Ophthalmol. 2020;20(1). doi:10.1186/s12886-020-01520-y
12. Honavar SG, Shields CL, Demirci H, Shields JA. Sclerochoroidal calcification: clinical manifestations and systemic associations. Arch Ophthalmol. 2001;119(6):833–840. doi:10.1001/archopht.119.6.833
13. Wang J, Zhou JJ, Robertson GR, Lee VW. Vitamin D in vascular calcification: a double-edged sword? Nutrients. 2018;10(5):652. doi:10.3390/nu10050652
14. Sierra-Rodríguez MA, Bailez Fidalgo C, Sáenz-Francés F, Gonzalez Romero JC, Muñoz Bellido L. Calcificaciones esclerocoroideas asociadas a hipovitaminosis D. Arch Soc Esp Oftalmol. 2014;89(7):290–292. doi:10.1016/j.oftal.2012.12.016
15. Ali ZC, David VP. Sclerochoroidal calcification associated with hypovitaminosis D. Can J Ophthalmol. 2017;52(4):e121–e122. doi:10.1016/j.jcjo.2017.02.001
16. Lee H, Kumar P, Deane J. Sclerochoroidal calcification associated with Albright’s hereditary osteodystrophy. BMJ Case Rep. 2012;2012:bcr0320126022–bcr0320126022. doi:10.1136/bcr-03-2012-6022
17. Wong S, Zakov ZN, Albert DM. Scleral and choroidal calcifications in a patient with pseudohypoparathyroidism. Br J Ophthalmol. 1979;63(3):177–180. doi:10.1136/bjo.63.3.177
18. Gupta R, Hu V, Reynolds T, Harrison R. Sclerochoroidal calcification associated with Gitelman syndrome and calcium pyrophosphate dihydrate deposition. J Clin Pathol. 2005;58(12):1334–1335. doi:10.1136/jcp.2005.027300
19. Fulchiero R, Seo-Mayer P. Bartter Syndrome and Gitelman Syndrome. Pediatr Clin North Am. 2019;66(1):121–134. doi:10.1016/j.pcl.2018.08.010
20. Rodríguez-Soriano J. Bartter and related syndromes: the puzzle is almost solved. Pediatr Nephrol. 1998;12(4):315–327. doi:10.1007/s004670050461
21. Joshi A, Chokkalingam S. Magnesium disorders can cause calcium pyrophosphate deposition disease: a case report and literature review. Eur J Rheumatol. 2018;5(1):53–57. doi:10.5152/eurjrheum.2017.16116
22. Boutboul S, Bourcier T, Heligon JP, et al. Familial pseudotumoral sclerochoroidal calcification associated with chondrocalcinosis. Br J Ophthalmol. 2004;88(8):1094–1095. doi:10.1136/bjo.2003.039925
23. Ciaffi J, Borlandelli E, Mancarella L, Brusi V, Meliconi R, Ursini F. Sclerochoroidal calcifications associated with early-onset calcium pyrophosphate deposition disease. Clin Rheumatol. 2020;39(9):2825–2826. doi:10.1007/s10067-020-05148-7
24. Slean GR, Kalevar A, Chen J, Johnson R. Enlargement of sclerochoroidal calcifications: multimodal imaging update. Retin Cases Brief Rep. 2018;12(1):S122–S124. doi:10.1097/ICB.0000000000000644
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.