Back to Journals » Clinical Interventions in Aging » Volume 9
A case of atypical progressive supranuclear palsy
Authors Spaccavento S, Del Prete M, Craca A, Loverre A
Received 17 July 2013
Accepted for publication 27 August 2013
Published 16 December 2013 Volume 2014:9 Pages 31—39
DOI https://doi.org/10.2147/CIA.S51640
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Simona Spaccavento, Marina Del Prete, Angela Craca, Anna Loverre
IRCCS Salvatore Maugeri Foundation, Cassano Murge, Bari, Italy
Background: Progressive supranuclear palsy (PSP) is a neurodegenerative extrapyramidal syndrome. Studies have demonstrated that PSP can present clinically as an atypical dementing syndrome dominated by a progressive apraxia of speech (AOS) and aphasia.
Aim: We aimed to investigate the clinical presentation of PSP, using a comprehensive multidimensional evaluation, and the disease response to various pharmacological treatments.
Methods: A 72-year-old right-handed male, with 17 years education, who first presented with aphasia, AOS, depression, apathy, and postural instability at 69 years; a complete neuropsychological evaluation, tapping the different cognitive domains, was performed.
Results: Testing revealed a moderate global cognitive deficit (Mini-Mental State Examination test score =20), low memory test scores (story recall, Rey’s 15-word Immediate and Delayed Recall), and poor phonemic and semantic fluency. The patient’s language was characterized by AOS, with slow speech rate, prolonged intervals between syllables and words, decreased articulatory accuracy, sound distortions, and anomia. Behavioral changes, such as depression, anxiety, apathy, and irritability, were reported. The neurological examination revealed supranuclear vertical gaze palsy, poor face miming, and a mild balance deficit. Magnetic resonance imaging showed only widespread cortical atrophy. Single photon emission computed tomography demonstrated left > right frontotemporal cortical abnormalities. After 6 months, a further neuropsychological assessment showed a progression in cognitive deficits, with additional attention deficits. The patient reported frequent falls, but the neurological deficits remained unchanged. Neuroimaging tests showed the same brain involvement.
Conclusion: Our case highlights the heterogeneity of the clinical features in this syndrome, demonstrating that atypical PSP can present as AOS and aphasia, without the classical features or involvement of the subcortical gray and brainstem region, commonly affected in typical PSP.
Keywords: pharmacological treatments, neuropsychological deficits
Introduction
Progressive supranuclear palsy (PSP) is a neurodegenerative extrapyramidal syndrome, characterized by motor symptoms, such as postural instability, rigidity, akinesia, and behavioral and cognitive symptoms. The disease is progressive, and patients have a median survival of around 6 years after onset of symptomatology, which usually occurs between 55 and 70 years of age. Both sexes are nearly equally affected, with an annual incidence of 5.3 per 100,000 inhabitants in Europe.1–3 The most common problem of PSP is postural instability and frequent falls, followed by dysarthria as the second most common symptom, and bradykinesia as the third. Visual disturbances are also early symptoms. Supranuclear gaze deficits involve either downward or upward gaze and later, horizontal gaze.4
The pathological changes are characterized by neuronal loss, with gliosis and neurofibrillary tangles in the subcortical and brainstem nuclei and the cerebellar dentate nucleus.5
As regards the neuropsychological features, many cases of PSP with subcortical dementia have been described in literature.6,7 Patients typically have cognitive deficits in executive functions, attention, and memory.8,9 General slowness of information processing, deficits in focused and divided attention and reduced verbal fluency are all characteristics related to impaired frontal-executive functions. However, many studies have also shown the involvement of other aspects of cognition, such as language and visuospatial skills.8,10,11 Nonfluent aphasia and progressive apraxia of speech (AOS) can characterize the clinical symptomatology of PSP, as recently described in repeated studies.12–14
As to the neuropsychiatric aspects, apathy has been described as the most frequent behavioral abnormality in these patients, followed by disinhibition, and depression.15 These symptoms appear in the early stages of the disease, advancing independently with disease duration, and are mediated by abnormalities of the frontal lobe or frontal–subcortical connections.
The alterations of the neurotransmitter system in PSP mainly involve the dopaminergic and cholinergic systems.16 Degeneration of the dopaminergic system could be related to some of the parkinsonian symptoms seen with PSP, whereas the cholinergic deficits seem to be associated with cognitive impairment. Also, the γ-aminobutyric acid (GABA)ergic system may be involved in the pathogenesis of PSP.5
The aim of this paper was to present the case of a patient with atypical PSP, presenting aphasia and AOS without the classical involvement of either the subcortical gray or brainstem region. We focused both on the neuropsychological profile and on the response of the disease to the various pharmacological treatments.
Materials and methods
Case description
The patient was a 72-year-old, right-handed male, with 17 years of education and no family history of a neurodegenerative disorder. He was a retired university office manager. The patient only suffered from high blood pressure and was in good health until approximately 2 years before presentation, when he was affected by a depression of mood state.
A neuropsychological evaluation performed at that time, in 2009, highlighted only an impairment in episodic memory and reduced phonemic fluency and a pathological performance in the Frontal Assessment Battery.17
At the first visit in our clinic, in January 2011, a general physical examination did not reveal any known pathology. A neurological examination detected hypomimic facies; impaired balance; ataxic-type gait; pyramidal-extrapyramidal hypertonia in all four limbs; reduced strength, especially on the right side; wide-base standing position; and supranuclear upgaze paresis.
Magnetic resonance imaging (MRI) showed the ventricular–cisternal system in its normal axis, normal morphology and size, and no areas of altered signal in the brain parenchyma. Diffuse cortical atrophy was present.
Single photon emission computed tomography (SPECT) showed widespread hypoperfusion of the cerebral cortex, with thinning of cortical thickness. The hypoperfusion of both hemispheres appeared more pronounced in the posterior temporal cortex, with a prevalence in the left hemisphere. Perfusion of the basal ganglia and cerebellar hemispheres was preserved.
The patient was assessed by the same neuropsychologist for all follow-ups, using a wide battery of neuropsychological tests to investigate all cognitive functions.
Tests
Global cognitive impairment was evaluated using Mini–Mental State Examination (MMSE).18 This is a screening test for mental deterioration, assessing the following five areas: orientation to time (score 0–5) and place (score 0–5); immediate recall, ie, short-term verbal memory (score 0–3); attention and calculation (score 0–5); delayed recall (score 0–3), language (in the sections: naming, verbal fluency, comprehension, reading, and writing) (score 0–6); and constructional ability (score 0–3). The possible maximum total score is 30. A score of 24 or more in this test is considered diagnostic of normal cognitive status. The MMSE score must be corrected for age and education, in this case, according to procedures standardized for the Italian population.19
Mental Deterioration Battery
The Mental Deterioration Battery (MDB)20 is a standardized neuropsychological test battery of seven tests used to obtain eight performance scores, four for the elaboration of verbal stimuli and four for the elaboration of visuospatial material. The tests investigate different cognitive areas: language (Phonological Verbal Fluency21; Sentence Construction22), verbal memory (Rey’s 15-word Immediate and Delayed Recall23), visual memory (Immediate Visual Memory24), logical reasoning (Raven’s Progressive Matrices25), and constructional praxis (Copying Drawing and Landmark Drawing Copy Test26).
Individual performance can range between 0 and 8, according to the number of test scores above the cutoff; a score of below 4 characterizes demented subjects.
We added other cognitive tests to this test battery, with the aim of obtaining a more complete and exhaustive neuropsychological profile assessing all cognitive functions (memory, language, attention, and executive functions).
Memory
Three additional tests were administered in order to assess memory functions more comprehensively: the Corsi block-tapping test27 and digit span test,28 respectively, for visuospatial and verbal short-term memory, and the story recall test27 for the evaluation of episodic memory.
Language
To assess language functioning, we used the Aachener Aphasie Test,29 comprising six subtests: spontaneous speech, token test, repetition, written language, naming, and comprehension; and a semantic verbal fluency test.27
Attention
To assess attention deficits we used the Alertness test from the German Testbatterie zur Aufmerksamkeitsprufung or Test for Attentional Performance (TAP).30
This test measures reaction time (RT) with or without a warning signal (tone). In the test, a cross appears in the middle of the computer screen, and the subject has to press a button as rapidly as possible. The order of block presentation is ABBA, in which A is the block without a tone and B is the block with a warning signal. A total of 80 trials were presented. The three parameters evaluated are: reaction times, number of omissions, and the index of phasic alertness.
Executive functions
We assessed various aspects of executive abilities, such as shifting, interference inhibition, reasoning, and planning.
The Trail Making Test (TMT)31 was used to explore visual–conceptual and visual–motor tracking.
The Stroop test32 measured the ability to control and inhibit the automatic response.
Depression assessment
We used a short version (15 items) of the Geriatric Depression Scale (GDS)33 to evaluate the possible presence of depression. The GDS questions are answered by stating “yes” or “no.” This simplicity enables the scale to be used with individuals who are ill or moderately cognitively impaired. A score higher than 5 is indicative of depression.
Psychiatric evaluation
Neuropsychiatric symptoms were evaluated using the Neuropsychiatric Inventory (NPI).34 Twelve behavioral domains are evaluated in this test: delusions, hallucinations, agitation/aggression, dysphoria, anxiety, euphoria, apathy, disinhibition, irritability/lability, aberrant motor activity, nighttime behavioral disturbances, and appetite/eating disorders.
The NPI is comprised of screening questions used to ask the caregiver whether the patient’s behavior has changed since the onset of dementia and if so, whether the altered behavior was present during the last month.
The Italian version of the NPI was validated by Binetti et al35 in Alzheimer’s disease patients and has demonstrated comparable psychometric properties.
Functional assessment
Functional autonomy was evaluated using the basic Activities of Daily Living (ADL) scale36 and the Instrumental Activities of Daily Living (IADL) scale.37 These are two indices of functional dependence; the ADL scale measures the patient’s abilities in basic self-care tasks, such as feeding, bathing, walking and transferring, maintaining continence, etc; whereas the IADL scale explores instrumental activities, including telephoning, outdoor mobility and grocery shopping, travelling, taking medicines, managing money, housekeeping, preparing meals, and laundry.
Results
Tables 1–4 summarize the patient data for each test. The patient’s score and the cutoff in the standardization sample for each test are shown. For the attention test of the TAP battery, the medians of reaction times are shown.
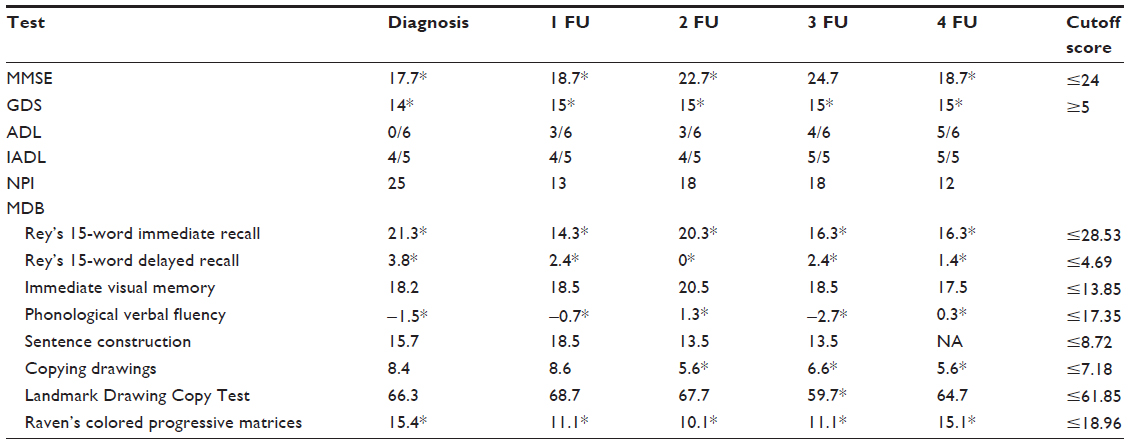 | Table 1 The patient’s scores and cutoff score of the cognitive tests |
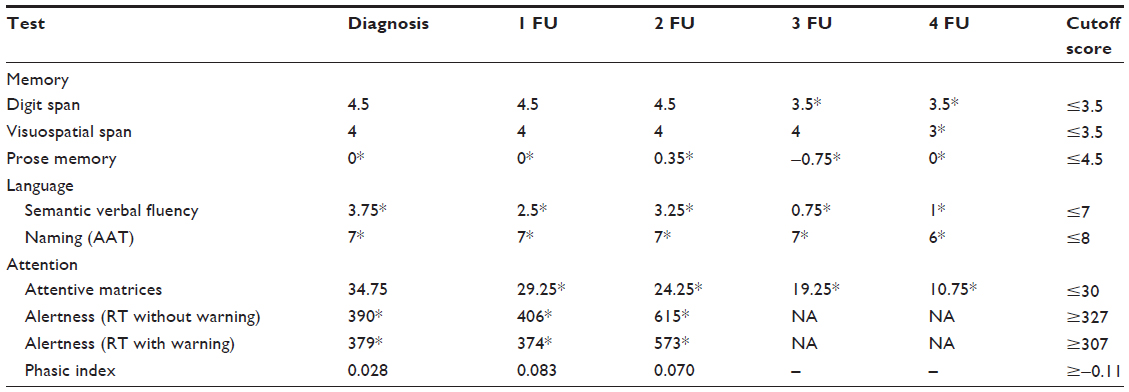 | Table 2 The patient’s scores and cutoff score of the other cognitive tests |
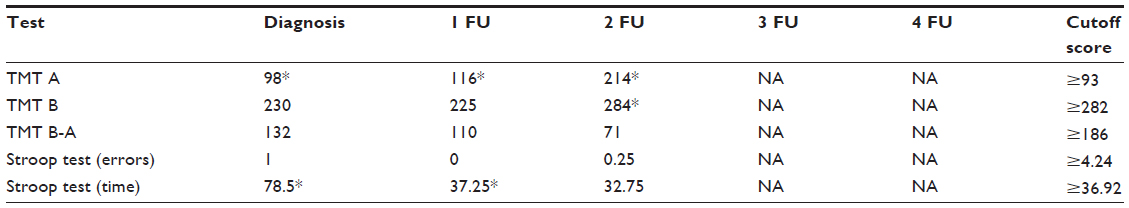 | Table 3 The patient’s scores and cutoff score of the executive tests |
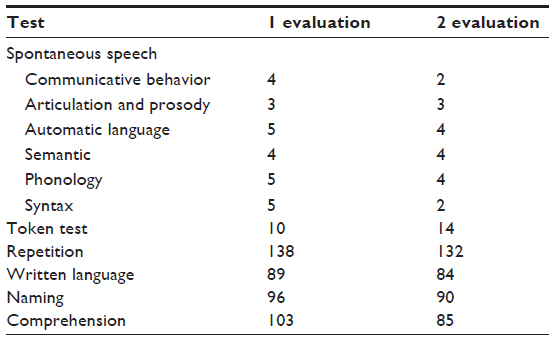 | Table 4 Patient’s scores and cutoff score of the language test |
At diagnosis, the patient showed a severe depression of mood state, with dysarthric language and an impairment of cognitive functioning characterized by memory deficits, reduced phonemic and semantic fluency, and deficits in abstract reasoning. The reaction times were mildly slow.
Neurologically, gait ataxic disturbances, balance deficits, and supranuclear upgaze paresis were already present. No falls were reported.
As regards behavioral symptoms, an interview with relatives revealed depression, anxiety, apathy, irritability, and sleep disorders.
As to functional independence, the patient was autonomous in activities of daily living, such as basic self-care tasks but needed help with instrumental activities.
After the evaluation the patient began pharmacological treatment with rivastigmine 6 mg daily for 6 months and paroxetine 20 mg daily.
At the first follow-up visit, after 6 months from diagnosis, he showed a further impairment of cognitive deficits and of balance disturbances, with bradykinesia. The patient appeared to relatives to be less anxious, depressed, and apathetic but also more dependent on others for simple daily living activities, such as feeding and bathing. However, the patient himself complained of sadness and depression, although he still showed interest in his life.
The patient’s relatives reported the onset of great difficulty in his building management, a task that he had previously carried out without any problems.
A further SPECT showed widespread hypoperfusion of the cerebral cortex, with thinning of the cortical thickness. The hypoperfusion of both hemispheres in the frontal and temporal cortexes appeared more pronounced than earlier. Perfusion of the basal ganglia and cerebellar hemispheres was preserved.
The results of neuropsychological evaluation showed a further worsening of cognitive performance, above all of memory, attention, and language, with the patient demonstrating anomia, and semantic and verbal paraphasic errors.
The language examination using Aachener Aphasie Test (AAT) showed, at that time, a mild nonfluent, Broca-like aphasia with language deficits, which were most evident in expression and were characterized by word retrieval difficulties and anomia, and some semantic paraphasias. His conversational/narrative speech was characterized by a slow speech rate, prolonged intervals between syllables and words, and decreased articulatory accuracy with increased speaking rate. These are all characteristics of AOS. His prosody was also abnormal. He also displayed word repetition, characterized by phonemic substitution; his comprehension was better than expression, but the patient had more difficulty with the syntactically complex sentences. He printed dictated words and sentences adequately, such as words and phrases read aloud.
The early clinical and neuropsychological manifestations in this case were consistent with PSP syndrome, even if the neuroradiological data were unusual in regard to the usual involvement of the subcortical gray and brainstem region reported in the literature.
It was decided to change the pharmacological therapy, substituting paroxetine with 50 mg of sertraline, daily. Sertraline hydrochloride is an antidepressant of the selective serotonin reuptake inhibitor (SSRI) class and is also a dopamine reuptake inhibitor. Moreover, a combination of melevodopa (l-dopa methylester, a highly soluble prodrug of l-dopa) plus carbidopa in an effervescent tablet formulation (CHF 1512; Sirio Pharma Co, Ltd, Shantou, People’s Republic of China) and zolpidem, a nonbenzodiazepine hypnotic drug (with an imidazopyridine structure that binds to the GABA-benzodiazepine receptor complex found in high density in basal ganglia), were prescribed.
After 6 months, a new multidimensional evaluation showed a rather stable situation, both in cognitive and functional aspects. The pharmacological therapy with rivastigmine, sertraline, levodopa, and zolpidem was confirmed.
The onset of a spastic smile and psychomotor slowing led to the patient requesting a return visit. The assessment identified an impairment of the patient’s functional autonomy, both in simple and instrumental daily activities, and in executive functions, as demonstrated by the phonological and semantic verbal fluency tests and the patient’s inability to perform the TMT and the Stroop Test. A new drug, memantine, at a daily dose of 20 mg, was added to the therapy.
After 6 months of treatment with memantine, the neuropsychiatric symptomatology was reduced in our patient, although moderate depression and mild irritability continued. As regards cognitive functions, the patient’s spontaneous speech had become poor, telegraphic, and echolalic; compared with the previous language assessment, we observed a further impairment of sentence comprehension and repetition, with more severe difficulty with phrases and syntagma.
The patient’s wife had noticed a frequent cough when he drank and sometimes during a meal. A videofluoroscopic examination showed moderate dysphagia.
Discussion
A spectrum of clinical syndromes and neurodegenerative disorders must be considered in a patient who presents with these neuropsychological and clinical symptoms. Frontotemporal dementia, progressive nonfluent aphasia, and corticobasal degeneration (CBD) all represent syndromes reflecting asymmetric cortical dysfunction.38 PSP has been associated with CBD syndrome.39 Dementia with Lewy bodies rarely presents as an asymmetrical cortical degeneration syndrome.40
We tested a wide range of cognitive functions in this patient and the response of his disease to all the drugs available for this pathology.
The early clinical and neuropsychological manifestations were the most consistent with PSP syndrome. He exhibited gait impairment and supranuclear gaze palsy, typical of PSP. The overlap of PSP with CBD has been increasingly recognized lately. Our patient did not present limb apraxia, asymmetric extrapyramidal syndrome, alien limb behavior, myoclonus, or cortical sensory loss, all typical findings of CBD.
Our patient showed depression of mood state as the first symptom of the pathology. Moreover, this was resistant to all pharmacological treatment as the patient continued to be depressed at all follow-up visits, as shown by the GDS scores.
Most published reports41,42 on behavior in PSP patients have been single case studies and have emphasized symptoms related to depression, psychosis, or obsessive features. Borroni et al,8 in a sample of 24 PSP patients, found a high frequency of depression, anxiety, sleep, and eating disorders. More recently, Fukui et al42 studied 74 PSP patients, observing a relatively high incidence (24%) of obsessive–compulsive symptoms. These are frequent but under recognized neuropsychiatric symptoms of dementia in PSP, probably due to a dysfunction of the fronto–caudate–thalamus–cerebellum circuit.
Unlike the results of Litvan et al,15 which indicated apathy as the dominant behavioral change in PSP, the main behavioral symptom in our case was depression. The apathy that the patient showed in the early stage of disease was replaced by an unmistakable depression.
Depressive symptoms can appear as an initial manifestation of an incipient cognitive change or can be interpreted as a risk factor for cognitive decline, or can be a concomitant disease, possibly the result of an awareness of cognitive difficulties, as suggested by previous authors.43,44
With regard to cognitive functions, only a mild executive deficit was found in the patient at the first neuropsychological examination performed following the onset of the depression.
Functional imaging studies of PSP patients show a deafferentation of the frontal lobes. The frontal lobes have reduced perfusion despite limited local pathological changes. The metabolic deficits are found in areas disconnected from the prominent subcortical pathology of PSP.
There are five frontal subcortical circuits, originating in the supplementary motor area, frontal eye fields, dorsolateral prefrontal cortex, orbitofrontal cortex, and anterior cingulated cortex. These circuits unite regions of the frontal lobe with the striatum, globus pallidus, and thalamus in functional systems that mediate motor activities, eye movements, cognition, and behavior. In PSP, all five circuits are impaired.15,45
MRI of our patient showed no involvement of either the frontal areas or of the subcortical structures. Moreover, the first SPECT imaging performed showed a hypoperfusion in both temporal posterior hemispheres, although moderate depression and mild cognitive symptoms were evident. Only the second SPECT scan showed a reduced perfusion of both frontal lobes.
As suggested in the literature,46 there is a worsening of executive functions only in the later stages of disease when the frontal structures are more involved, while the onset of neuropsychiatric disorders are independent of cognitive deficits, due to the dysfunction of different subcortical circuits.
The peculiarity of our case is the lack of an obvious involvement of subcortical structures, as the MRI and SPECT images showed, even in the phase of disease during which the patient showed obvious motor deficits.
With regard to neuropsychological characteristics, our results are in accordance with the literature. At the first stage of the disease, our patient showed a mild deficit in verbal phonemic fluency and in long-term memory, and slow reaction times. In fact, one study has reported that letter fluency tends to be more severely affected than category fluency.47 The short-term memory is considered to be relatively well preserved in PSP.48 Other authors49,50 have demonstrated the prolonged reaction times and a cognitive slowing independent of the motor function. As to language, repeated studies12–14 have demonstrated the nonfluent aphasia in this neurodegenerative disorder, such as an impairment of oral and written comprehension, also found by Podoll et al51 in five patients with PSP.
Certainly the disorder of speech and communication in PSP can take different forms. Dysarthria is the most common, but dysprosody, aphonia, stuttering, palilalia, and echolalia, as well as abnormal loquacity have also been described.52,53
To date, there is no approved treatment for PSP. Treatment with levodopa54 may alleviate some motor symptoms, such as bradykinesia and rigidity, but only 20%–40% of PSP patients respond to it. Levodopa can be combined with other therapies, such as serotonergic drugs, which improve depressive symptoms but which are ineffective in the treatment of cognitive and other symptoms.
The cholinergic system is impaired in PSP, as evidenced by a decrease in choline acetyltransferase activity in the basal ganglia and by a loss of mesencephalic cholinergic nuclei.55 This cholinergic deficit may explain the cognitive dysfunction. Donepezil is a centrally acting cholinesterase inhibitor that prolongs the action of acetylcholine in residual cholinergic neurons. Fabbrini et al55 investigated the efficacy and tolerability of donepezil in six PSP patients and found no significant changes in cognitive deficits or in autonomy in daily living activities.
In contrast to other cholinesterase inhibitors, rivastigmine inhibits both acetylcholinesterase and butyrylcholinesterase and, in a small number of patients, produces a slight improvement in cognitive symptoms.16
In 12 patients with PSP, Foster et al56 found a loss of interneurons containing benzodiazepine/GABA receptors, primarily in the anterior cingulated cortex, together with a deafferentation of the cerebral cortex from distant brain regions. Treatment with GABA agonists could be useful in PSP patients. Zolpidem is a GABA agonist of the benzodiazepine BZ1 subtype receptor and has been used in a small number of patients with PSP. Cotter et al5 found a sustained improvement in motor and ocular symptoms in a patient with PSP who was treated with controlled-release zolpidem over a period of 6 months.
Memantine is a new medication that acts on the glutamatergic system by blocking the N-methyl-D-aspartate (NMDA)-type glutamate receptors.
This medicine is used for the treatment of moderate to severe dementia of the Alzheimer type. Recent evidence57,58 has suggested that memantine is safe and might confer some cognitive improvement for individuals with dementia in Parkinson’s disease and dementia with Lewy bodies. There are no studies on memantine treatment in patients with PSP.
Recently, Gold et al1 investigated the use of davunetide, an advanced product in PSP. This is a novel neuroprotective peptide that is thought to impact neuronal integrity and cell survival through the stabilization of microtubules.
The studies on the efficacy and tolerability of pharmacological treatments in PSP patients have not provided any positive results. Our data also confirmed the inefficacy of available drugs, even in combination. Perhaps the aspect of associations between various therapies was not considered in the literature. This is a further point provided by our case. However, we investigated the efficacy of memantine in our patient with PSP and concluded that there was no significant slowing of disease progression, probably due to the stage of pathology.
A limitation of this study is that it involved a single case, and there was no control group. Moreover, we did not confirm the diagnosis, considering the lack of neuropathological findings. This weakness did not allow the achievement of any statistically significant data.
It could be interesting to investigate the use of memantine in a large group of patients and in the early stages of disease. Future strategies to treat PSP will require larger and prospective studies with multicenter trials.
Conclusion
In conclusion, in PSP patients, depression can represent a first symptom before an overt onset of disease. Moreover, the lack of involvement of the subcortical structures, as shown with MRI, even when the patient had a severe motor impairment, was an important and particular aspect of our case and is necessary to consider when diagnosing this neurodegenerative disease in other patients.
Disclosure
The authors report no conflicts of interest in this work.
References
Gold M, Lorenzl S, Stewart AJ, Morimoto BH, Williams DR, Gozes I. Critical appraisal of the role of davunetide in the treatment of progressive supranuclear palsy. Neuropsychiatr Dis Treat. 2012;8:85–93. | |
Schrag A, Ben-Shlomo Y, Quinn NP. Prevalence of progressive supranuclear palsy and multiple system atrophy: a cross-sectional study. Lancet. 1999;354(9192):1771–1775. | |
Winter Y, Bezdolnyy Y, Katunina E, et al. Incidence of Parkinson’s disease and atypical parkinsonism: Russian population-based study. Mov Disord. 2010;25(3):349–356. | |
Litvan I, Agid Y, Calne D, et al. Clinical research criteria for the diagnosis of progressive supranuclear palsy (Steele-Richardson-Olszewski syndrome): report of the NINDS-SPSP international workshop. Neurology. 1996;47(1):1–9. | |
Cotter C, Armytage T, Crimmins D. The use of zolpidem in the treatment of progressive supranuclear palsy. J Clin Neurosci. 2010;17(3):385–386. | |
Albert ML, Feldman RG, Willis AL. The “subcortical dementia” of progressive supranulcear palsy. J Neurol Neurosurg Psychiatry. 1974;37(2):121–130. | |
Cummings JL. Subcortical dementia. Neuropsychology, neuropsychiatry, and pathophysiology. Br J Psychiatry. 1986;149:682–697. | |
Borroni B, Turla M, Bertasi V, Agosti C, Gilberti N, Padovani A. Cognitive and behavioral assessment in the early stages of neurodegenerative extrapyramidal syndromes. Arch Gerontol Geriatr. 2008;47(1):53–61. | |
Kertesz A, McMonagle P. Behavior and cognition in corticobasal degeneration and progressive supranuclear palsy. J Neurol Sci. 2010;289(1–2):138–143. | |
Bak TH, Hodges JR. The neuropsychology of progressive supranuclear palsy. Neurocase. 1998;4(2):89–94. | |
Soliveri P, Monza D, Paridi D, et al. Neuropsychological follow up in patients with Parkinson’s disease, striatonigral degeneration-type multisystem atrophy, and progressive supranuclear palsy. J Neurol Neurosurg Psychiatry. 2000;69(3):313–318. | |
Boeve B, Dickson D, Duffy J, Bartleson J, Trenerry M, Petersen R. Progressive nonfluent aphasia and subsequent aphasic dementia associated with atypical progressive supranuclear palsy pathology. Eur Neurol. 2003;49(2):72–78. | |
Joseph KA, Boeve BF, Duffy JR, et al. Atypical progressive supranuclear palsy underlying progressive apraxia of speech and nonfluent aphasia. Neurocase. 2005;11(4):283–296. | |
Josephs KA, Duffy JR. Apraxia of speech and nonfluent aphasia: a new clinical marker for corticobasal degeneration and progressive supranuclear palsy. Curr Opin Neurol. 2008;21(6):688–692. | |
Litvan I, Mega MS, Cummings JL, Fairbanks L. Neuropsychiatric aspects of progressive supranuclear palsy. Neurology. 1996;47(5):1184–1189. | |
Liepelt I, Gaenslen A, Godau J, et al. Rivastigmine for the treatment of dementia in patients with progressive supranuclear palsy: Clinical observations as a basis for power calculations and safety analysis. Alzheimers Dement. 2010;6(1):70–74. | |
Dubois B, Slachevsky A, Litvan I, Pillon B. The FAB: a Frontal Assessment Battery at bedside. Neurology. 2000;55(11):1621–1626. | |
Folstein MF, Folstein SE, McHugh PR. “Mini-mental state”. A practical method for grading the cognitive state of patients for the clinician. J Psychiatr Res. 1975;12(3):189–198. | |
Measso G, Cavarzeran F, Zappalà G, et al. The mini-mental state examination: Normative study of an Italian random sample. Develop Neuropsychol. 1993;9(2):77–85. | |
Carlesimo GA, Caltagirone C, Gainotti G et al. The Mental Deterioration Battery: normative data, diagnostic reliability and qualitative analyses of cognitive impairment. The Group for the Standardization of the Mental Deterioration Battery. Eur Neurol. 1996;36:378–384. | |
Borkowsky JG, Benton AL, Spreen O. Word fluency and brain-damage. Neuropsychologia. 1967;5:135–140. | |
Gainotti G, Caltagirone C, Miceli G. Sui rapporti fra alcune prove di intelligenza verbale e lesioni focali monoemisferiche. Acta Neurologica. 1976;31:370–381. Italian. | |
Rey A. Memorisation d’une serie de 15 mots en 5 repetitions. In: Rey A, ed. L’examen Clinique en Psychologie. Paris, France: Presses Universitaires de France, 1958. | |
Caltagirone C, Gainotti G, Masullo C et al. Validity of some neuropsychological tests in assessment of mental deterioration. Acta Psychiatr Scand. 1979;60:50–56. | |
Raven JC. Progressive Matrices. Set A, Ab, B: Board and Book Forms. London, England: Lewis, 1947. | |
Gainotti G, Miceli G, Caltagirone C. Constructional apraxia in left brain-damaged patients: A planning disorder? Cortex. 1977;13:109–118. | |
Spinnler H, Tognoni G. Standardizzazione e taratura di test neuropsicologici [Italian normative values and standardization of neuropsychological tests]. Ital J Neurol Sci. 1987;6(Suppl 8):S1–S120. Italian. | |
Orsini A, Grossi D, Capitani E, Laiacona M, Papagno C, Vallar G. Verbal and spatial immediate memory span: normative data from 1355 adults and 1112 children. Ital J Neurol Sci. 1987;8(6):539–548. | |
Luzzatti C, Willmes K, De Bleser R. L‘Aachener Aphasie Test (AAT), Versione Italiana. Manuale e Dati Normativi [The Aachener Aphasie Test {AAT}, Italian Version. Manual and Normative Data]. 2nd ed. Firenze: Organizzazione Speciali; 1996. Italian. | |
Zimmermann P, Fimm B. Testbatterie zur Aufmerksamkeitsprufung [Tests of Attentional Performance{TAP}]. Wurselen: Psytest; 1992. German. | |
Giovagnoli AR, Del Pesce M, Mascheroni S, Simoncelli M, Laiacona M, Capitani E. Trail making test: normative values from 287 normal adult controls. Ital J Neurol Sci. 1996;17(4):305–309. | |
Caffarra P, Vezzadini G, Dieci F, Zonato F, Venneri A. Una versione abbreviata del test di Stroop: dati normativi nella popolazione italiana [An abbreviated version of the Stroop Test: normative data in the Italian population]. Rivista di Neurologia. 2002;12(4):111–115. Italian. | |
Yesavage JA. Geriatric Depression Scale. Psychopharmacol Bull. 1988;24(4):709–711. | |
Cummings JL, Mega M, Gray K, Rosenberg-Thompson S, Carusi DA, Gornbein J. The Neuropsychiatric Inventory: comprehensive assessment of psychopathology in dementia. Neurology. 1994;44(12):2308–2314. | |
Binetti G, Mega MS, Magni E, et al. Behavioral disorders in Alzheimer’s disease: a transcultural perspective. Arch Neurol. 1998;55(4):539–544. | |
Katz S, Ford AB, Moskowitz RW, Jackson BA, Jaffe MW. Studies of illness in the aged. The index of ADL: a standardized measure of biological and psychosocial function. JAMA. 1963;185:914–919. | |
Lawton MP, Brody EM. Assessment of older people: self-maintaining and instrumental activities of daily living. Gerontologist. 1969;9(3):179–186. | |
Feldman RG, McKee AC. Case records of the Massachusetts General Hospital. Weekly clinicopathological exercises. Case 46–1993. A 75-year-old man with right-sided rigidity, dysarthria, and abnormal gait. N Engl J Med. 1993;329(21):1560–1567. | |
Sadler JZ, Kurtz NM, Rush AJ. Progressive supranuclear palsy presenting as pseudodementia. Psychosomatics. 1984;25(9):713–714. | |
Caselli R. Focal and asymmetric cortical degeneration syndromes. The Neurologist. 1995;1:1–19. | |
Schneider LS, Chui HC. Progressive supranuclear palsy manifesting with depressive features. J Am Geriatr Soc. 1986;34(9):663–665. | |
Fukui T, Lee E, Hosoda H, Okita K. Obsessive-compulsive behavior as a symptom of dementia in progressive supranuclear palsy. Dement Geriatr Cogn Disord. 2010;30(2):179–188. | |
Wilson RS, Barnes LL, Mendes de Leon CF, et al. Depressive symptoms, cognitive decline, and risk of AD in older persons. Neurology. 2002;59(3):364–370. | |
Bassuk SS, Berkman LF, Wypij D. Depressive symptomatology and incident cognitive decline in an elderly community sample. Arch Gen Psychiatry. 1998;55(12):1073–1081. | |
Blin J, Horwitz B, Baron JC, Agid Y. Does frontal cortex hypometabolism in progressive supranuclear palsy result from subcortical dysfunction? Eur J Neurol. 1995;1(3):221–228. | |
Pillon B, Dubois B, Agid Y. Severity and specificity of cognitive impairment in Alzheimer’s, Huntington’s, and Parkinson’s diseases and progressive supranuclear palsy. Neurology. 1991;41:634–643. | |
Rosser A, Hodges JR. Initial letter and semantic category fluency in Alzheimer’s disease, Huntington’s disease, and progressive supranuclear palsy. J Neurol Neurosurg Psychiatry. 1994;57(11):1389–1394. | |
Litvan I, Grafman J, Gomez C, Chase TN. Memory impairment in patients with progressive supranuclear palsy. Arch Neurol. 1989;46(7):765–767. | |
Dubois B, Pillon B, Legault F, Agid Y, Lhermitte F. Slowing of cognitive processing in progressive supranuclear palsy. A comparison with Parkinson’s disease. Arch Neurol. 1988;45(11):1194–1199. | |
Grafman J, Litvan I, Stark M. Neuropsychological features of progressive supranuclear palsy. Brain Cogn. 1995;28(3):311–320. | |
Podoll K, Schwarz M, Noth J. Language functions in progressive supranuclear palsy. Brain. 1991;114(Pt 3):1457–1472. | |
Pillon B, Dubois B. Cognitive and behavioral impairments. In: Litvan I, Agid Y, editors. Progressive Supranuclear Palsy: Clinical and Research Approaches. New York, NY: Oxford University Press; 1992:223–239. | |
Della Sala S, Spinnler H. Echolalia in a case of progressive supranuclear palsy. Neurocase.1998;4:155–165. | |
Lang AE. Treatment of progressive supranuclear palsy and corticobasal degeneration. Mov Disord. 2005;Suppl 12:S83–S91. | |
Fabbrini G, Barbanti P, Bonifati V, et al. Donepezil in the treatment of progressive supranuclear palsy. Acta Neurol Scand. 2001;103(2):123–125. | |
Foster NL, Minoshima S, Johanns J, et al. PET measures of benzodiazepine receptors in progressive supranuclear palsy. Neurology. 2000;54(9):1768–1773. | |
Aarsland D, Ballard C, Walker Z, et al. Memantine in patients with Parkinson’s disease dementia or dementia with Lewy bodies: a double-blind, placebo-controlled, multicentre trial. Lancet Neurol. 2009;8(7):613–618. | |
Boxer AL, Lipton AM, Womack K, et al. An open-label study of memantine treatment in 3 subtypes of frontotemporal lobar degeneration. Alzheimer Dis Assoc Disord. 2009;23(3):211–217. |
 © 2013 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2013 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.