Back to Journals » Clinical, Cosmetic and Investigational Dermatology » Volume 19
A Case of Angioimmunoblastic T-Cell Lymphoma Misdiagnosed as Eczema-Like Dermatitis in an Elderly Patient
Authors Xie L, Huang Z ![]() , Zhang R
, Zhang R
Received 13 October 2025
Accepted for publication 17 January 2026
Published 3 February 2026 Volume 2026:19 573866
DOI https://doi.org/10.2147/CCID.S573866
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Rungsima Wanitphakdeedecha
Lanqing Xie,1,* Zeyu Huang,2,* Ruzhi Zhang1
1Department of Dermatology, The Second Affiliated Hospital of Wannan Medical College, Wuhu, People’s Republic of China; 2Department of Dermatology, The Third Affiliated Hospital of Soochow University, Changzhou, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Ruzhi Zhang, Department of Dermatology, The Second Affiliated Hospital of Wannan Medical College, No. 10 Rehabilitation Road, Jinghu District, Wuhu, 241101, People’s Republic of China, Email [email protected]
Abstract: Angioimmunoblastic T-cell lymphoma (AITL) is a clinically heterogeneous subtype of peripheral T-cell lymphoma that frequently presents with nonspecific cutaneous manifestations. Approximately 40%– 50% of patients initially develop diffuse erythema or papular eruptions that may mimic benign inflammatory or allergic dermatoses, posing a diagnostic challenge in early disease stages. We report the case of an 81-year-old man who presented with generalized pruritic papules and was initially diagnosed with an eczema-like inflammatory dermatitis. Although the skin lesions showed transient improvement with symptomatic treatment, they recurred repeatedly. The subsequent onset of chills and cervical lymphadenopathy prompted further evaluation, including skin and lymph node biopsies, which established the diagnosis of AITL. Bone marrow involvement was identified by morphologic examination and flow cytometry. Immunohistochemical analysis demonstrated neoplastic T cells expressing CD3, CD4, and CD5, along with follicular helper T-cell–associated markers including CD10, BCL-6, PD-1, and CXCL13, with a Ki-67 proliferation index of approximately 70%. This case underscores the diagnostic complexity of AITL presenting with predominant allergic-like skin lesions and highlights the importance of considering underlying lymphoproliferative disorders in elderly patients with persistent, recurrent, or atypical dermatoses. Timely histopathological and immunophenotypic evaluation is essential for accurate diagnosis and appropriate management.
Keywords: angioimmunoblastic T-cell lymphoma, eczema, misdiagnosed
Introduction
Angioimmunoblastic T-cell lymphoma (AITL) is an aggressive subtype of non-Hodgkin lymphoma derived from follicular helper T (TFH) cells. In recent years, its incidence has increased among elderly individuals, accounting for approximately 1%–2% of all non-Hodgkin lymphomas and 15%–20% of mature T-cell lymphomas.1,2 Clinically, AITL typically presents with systemic manifestations such as fever, night sweats, weight loss, generalized lymphadenopathy and hepatosplenomegaly. Notably, cutaneous involvement is common and may precede systemic findings, with nearly half of patients initially presenting with skin manifestations. These include diffuse erythema, papules, or maculopapular eruptions that often resemble allergic or inflammatory dermatoses. In the absence of overt systemic symptoms, such nonspecific cutaneous presentations may mimic eczema-like or other hypersensitivity reactions, posing a significant diagnostic challenge in early disease stages.3–5
More broadly, various hematologic proliferative disorders can manifest with paraneoplastic or immune-mediated skin changes that resemble benign allergic conditions. This clinical overlap may obscure the recognition of underlying malignancy, particularly when routine dermatologic findings dominate the initial presentation. Consequently, early cutaneous manifestations of AITL and related disorders are frequently difficult to distinguish from common inflammatory dermatoses based on clinical features alone.
A definitive diagnosis of AITL relies on an integrated assessment including lymph node biopsy, histopathologic evaluation, immunophenotyping, and, when available, T-cell clonality analysis. Recent molecular studies have identified recurrent mutations in genes such as RHOA, TET2, IDH2 and DNMT3A that play a role in AITL development. These mutations could form the basis of molecular classification and targeted therapies.6 However, despite advances in diagnostic techniques, recognizing AITL at an early stage remains challenging when initial manifestations are limited to the skin.
Here, we report a case of AITL that initially presented with predominant allergic-like cutaneous manifestations. This case underscores the diagnostic complexity of AITL with atypical skin involvement and highlights the importance of maintaining clinical vigilance and considering systemic evaluation, including histopathological examination, in patients with recurrent, treatment-resistant, or atypical dermatoses.
Case Report
An 81-year-old man presented with a three-day history of generalized pruritic skin eruptions. Based on the clinical appearance of diffuse erythematous papules and the absence of systemic symptoms at that time, he was initially diagnosed with an eczema-like inflammatory dermatitis (Figure 1A and B). Treatment was initiated with oral desloratadine citrate, topical halometasone cream, and a urea–vitamin E emulsion. The skin lesions showed transient improvement but failed to resolve completely.
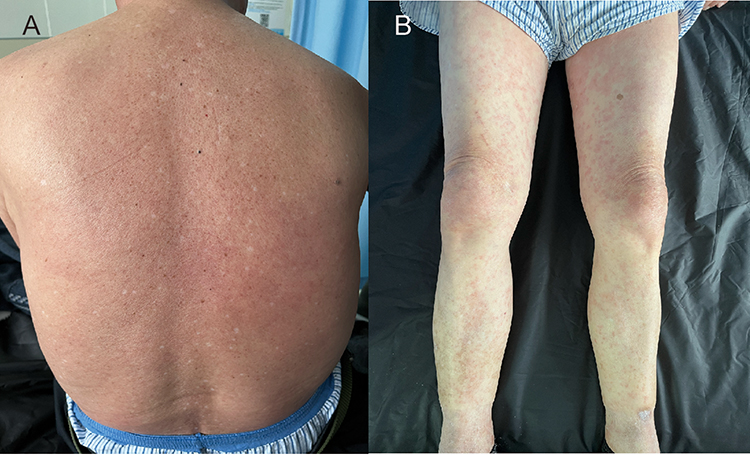 |
Figure 1 Cutaneous manifestations on the day of admission. Diffuse erythema with ill-defined borders on the back (A); scattered erythema and papules on both lower extremities (B). |
Approximately one month later, the patient returned with recurrent skin eruptions accompanied by chills. Physical examination revealed multiple enlarged superficial cervical lymph nodes, prompting further evaluation and biopsy. The patient had no personal or family history of atopic dermatitis, asthma, allergic rhinitis, or other chronic allergic diseases.
Specialist Examination
Scattered erythematous papules and maculopapular lesions ranging in size from millet to soybean were noted on the trunk and extremities. The lesions were mildly indurated and exhibited pitting on pressure. Enlarged, soft and mobile lymph nodes were palpable in the cervical, axillary and inguinal regions, and were mildly tender.
Ancillary Examinations
Imaging
Color Doppler ultrasonography demonstrated multiple enlarged lymph nodes in the bilateral cervical, axillary, and inguinal regions.
Laboratory Findings
White blood cell count: 12.5 × 109/L (reference range: 3.5–9.5); Neutrophils: 8.23 × 109/L (reference range: 1.8–6.3); Erythrocyte sedimentation rate (ESR): 93 mm/h (normal range:≤42); C-reactive protein (CRP): 74.4 mg/L (normal range: 0–6); Complement C4: mildly decreased; Immunoglobulins: IgG 28.6 g/L (normal range: 8–18), IgM 5.34 g/L (normal range: 0.4–2.3); Serology: positive for IgG for against rubella, cytomegalovirus and herpes simplex virus type 1; with detectable Epstein–Barr virus (EBV) DNA; Antinuclear antibody (ANA): weakly positive, cytoplasmic granular pattern (1:80).
Bone Marrow Analysis
Morphology: Marked myeloid hyperplasia (65.5%), erythroid lineage 17%, lymphocytes 9.6%, plasma cells 6%, blasts 0.5%. Flow cytometry: Granulocytes: 71.5%, myeloid cells: 76.5%, lymphocytes: 8.5%; CD38^bright^ population ~3%, without light chain restriction.
Histopathology and Immunohistochemistry
A skin biopsy obtained from the left thigh revealed focal infiltration of atypical tumour cells involving the epidermis and superficial dermis (Figure 2A and B). A cervical lymph node biopsy demonstrated histopathological features characteristic of angioimmunoblastic T-cell lymphoma, including diffuse lymphoid infiltration, prominent vascular proliferation, and dilated high endothelial venules within a polymorphous inflammatory background (Figure 2C and D). Immunohistochemical analysis of the lymph node showed tumor cells positive for leukocyte common antigen (LCA) with diffuse expression of CD3, CD4, and CD5. Partial expression of CD8 and CD10 was observed. Tumor cells expressed TFH-associated markers, including BCL-6 and CXCL13, supporting TFH lineage derivation. Scattered positivity for BCL-2 and MUM-1 was noted. The Ki-67 proliferation index was approximately 70%. The follicular dendritic cell (FDC) meshwork, as highlighted by CD21 and CD23 staining, was markedly expanded. CD20 positivity was confined to scattered background B cells, consistent with reactive B-cell proliferation (Figure 3).
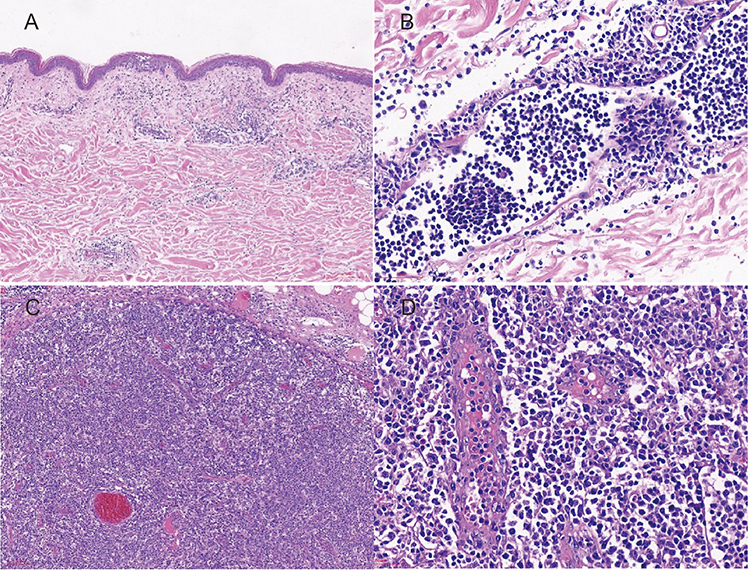 |
Figure 2 Histopathological findings of skin and cervical lymph node biopsies. Focal epidermal infiltration by atypical tumour cells in the skin biopsy ((A), H&E, ×100) and infiltration of atypical lymphoid cells in the superficial dermis ((B), H&E, ×400). Cervical lymph node biopsy showing diffuse tumour cell infiltration with prominent vascular proliferation and loss of follicular architecture ((C), H&E, ×100), and dilated high endothelial venules with irregular morphology surrounded by a polymorphous infiltrate composed of atypical lymphocytes, eosinophils, histiocytes, and plasma cells ((D), H&E, ×400). |
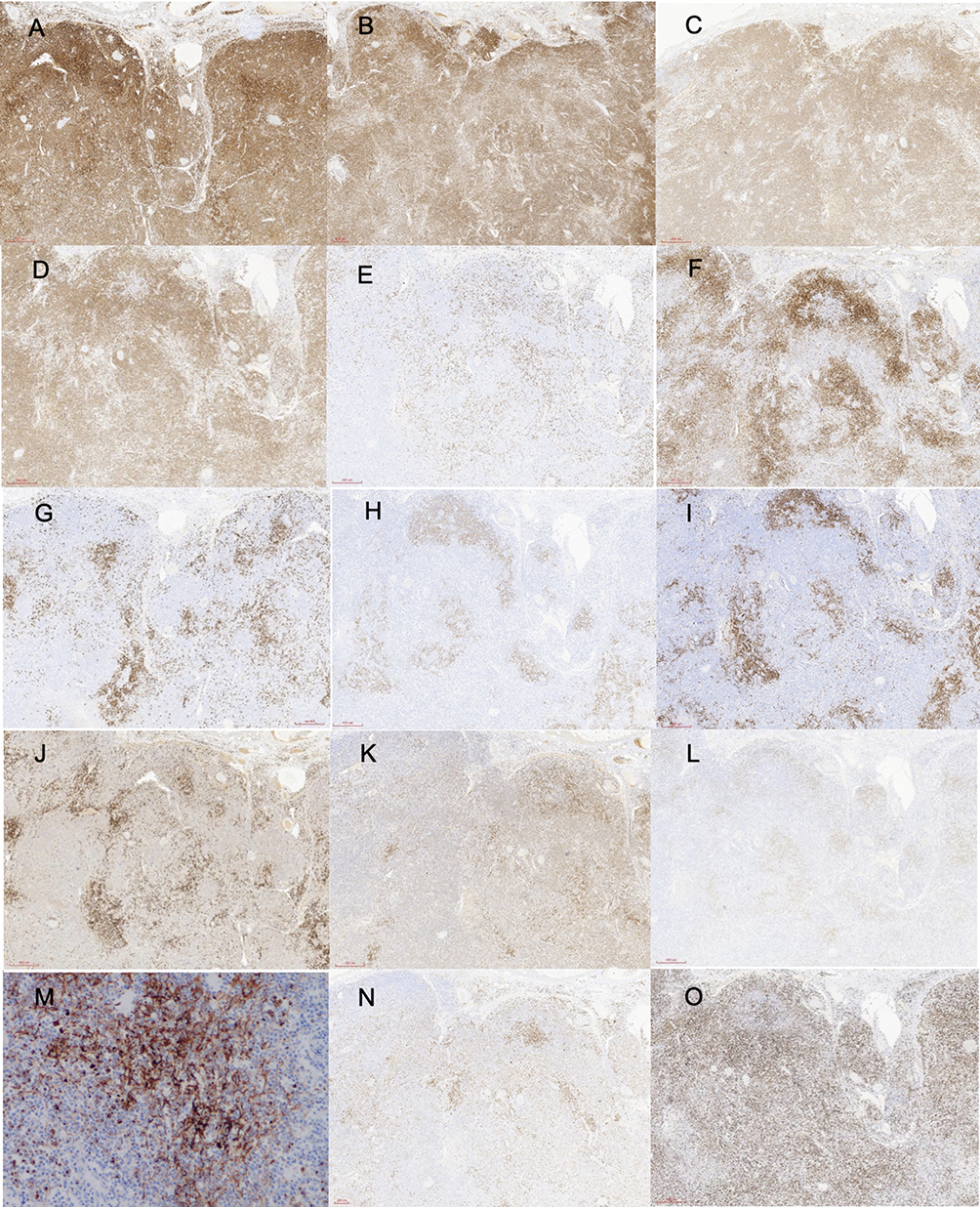 |
Figure 3 Immunohistochemical staining of the left cervical lymph node. The following markers were observed ((A–O), H&E ×100): LCA (+), CD3 (diffuse +), CD4 (diffuse +), CD5 (diffuse +), CD8 (partial +), CD10 (partial +), CD20 (scattered +), CD21 (expanded FDC network), CD23 (expanded FDC network), CD79α (scattered +), BCL-2 (scattered +), BCL-6 (scattered +), CXCL13(+), MUM-1 (scattered +). Ki-67 was observed in approximately 70% of cells. |
In the skin lesion, tumour cells were positive for CD3, CD4, CD5 and CD8, with a Ki-67 hotspot index of 60% (Figure 4).
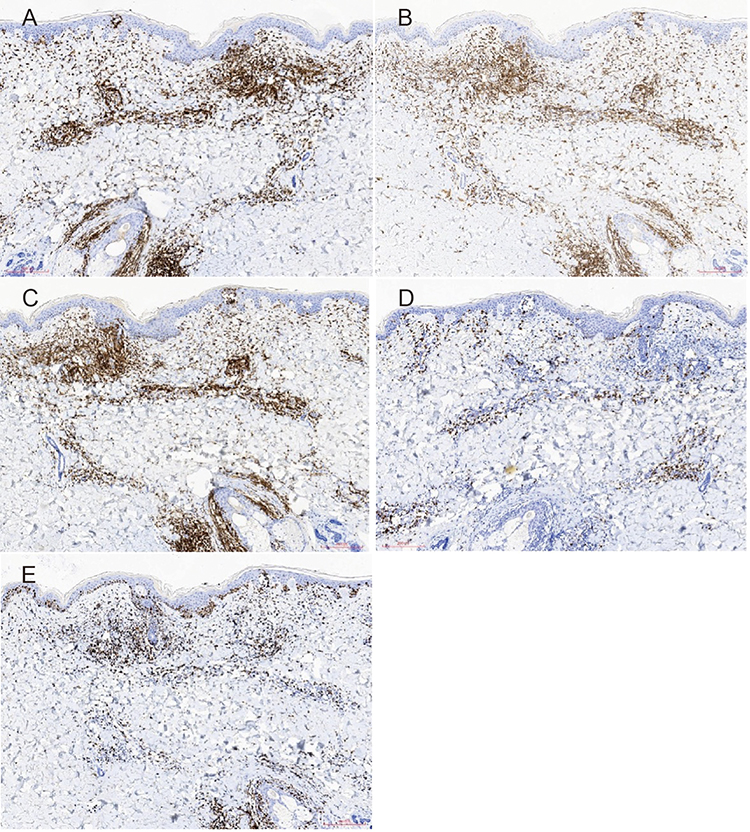 |
Figure 4 Immunohistochemical staining of skin lesion on the left thigh ((A–E), H&E ×100). CD3, CD4 and CD5 are all positive, while CD8 is partially positive and Ki-67 is positive in a hotspot region covering approximately 60%+). |
Discussion
AITL primarily affects middle-aged and elderly individuals and is characterized by an aggressive clinical course and an overall poor prognosis, with limited response to conventional therapies. The disease frequently involves multiple organ systems, manifesting as generalized lymphadenopathy, hepatosplenomegaly, ascites, and gastrointestinal dysfunction, and is often accompanied by non-specific systemic symptoms such as fever, night sweats, and weight loss.6 Cutaneous involvement is observed in more than half of patients and may precede systemic symptoms, often presenting a diagnostic challenge.7–9
The cutaneous manifestations of AITL are highly heterogeneous and frequently resemble benign inflammatory or allergic dermatoses, including eczema-like or psoriasiform eruptions. Pruritus is a common accompanying symptom. In the absence of early systemic findings, these nonspecific skin changes may obscure the underlying hematologic disorder, particularly in routine dermatologic practice.10 As a result, initial clinical evaluation may reasonably favor common inflammatory conditions, underscoring the inherent difficulty of early diagnosis rather than a true diagnostic delay.11
In the present case, the patient initially presented with generalized pruritic eruptions without lymphadenopathy or other systemic features, leading to a provisional diagnosis of an eczema-like inflammatory dermatitis. Such a clinical course is not uncommon and reflects the diagnostic complexity of AITL with predominant skin involvement. The subsequent development of lymphadenopathy appropriately prompted further investigation, ultimately leading to the correct diagnosis. This case therefore emphasizes the importance of maintaining a high index of suspicion for lymphoproliferative disorders in elderly patients with persistent, recurrent, or atypical dermatoses, and highlights the value of timely biopsy when clinical evolution raises concern.
Histopathological examination remains the cornerstone of AITL diagnosis. Lymph node biopsy typically demonstrates architectural effacement, proliferation of high endothelial venules, and expansion of follicular dendritic cell networks. However, diagnosis may be complicated by prominent reactive components, including EBV-positive B immunoblasts, plasma cells, and occasionally Reed–Sternberg–like cells, which can mask the underlying neoplastic T-cell population.12,13 In this context, immunohistochemical characterization is essential. The expression of pan–T-cell markers in conjunction with TFH-associated markers such as CD10, BCL-6, PD-1, and CXCL13 provides strong support for TFH-cell lineage and facilitates diagnostic accuracy.11,14,15
Cutaneous involvement in AITL typically manifests as dermal infiltrates, while epidermal infiltration is relatively uncommon. In this case, the presence of focal epidermal involvement represents an unusual but informative feature that broadens the histopathologic spectrum of AITL skin manifestations. Importantly, this finding necessitates careful differential diagnosis with epidermotropic cutaneous T-cell lymphomas, particularly mycosis fungoides. Integration of clinical context, systemic involvement, nodal histopathology, and TFH immunophenotype was critical in establishing the diagnosis of AITL and excluding primary cutaneous T-cell lymphoma.
AITL is also associated with an increased risk of secondary B-cell lymphomas, most commonly diffuse large B-cell lymphoma, a process thought to be driven by EBV-mediated B-cell proliferation, immune dysregulation, and a TFH-rich microenvironment that promotes B-cell activation.16 The presence of peripheral eosinophilia and reduced serum C4 levels in this case suggests systemic inflammation and autoimmune dysregulation, which are consistent with the immunological profile of AITL.17 Given the potential for histologic transformation and disease progression, repeat biopsy should be considered promptly if new lesions or atypical clinical features emerge during follow-up.6,18
Molecular testing for recurrent AITL-associated mutations, EBER in situ hybridization, and T-cell receptor gene rearrangement analysis were not performed in this case. Nevertheless, the diagnosis was established based on characteristic clinical features, typical histopathological findings, and a supportive immunophenotypic profile consistent with follicular helper T-cell differentiation.
In conclusion, when common dermatological diagnoses fail to explain persistent or evolving clinical features, particularly in elderly patients, clinicians should consider the possibility of AITL. Early histopathological and molecular evaluation is essential to avoid misdiagnosis or delayed diagnosis, enabling timely intervention and improving clinical outcomes.
Ethical Approval and Consent
Approval was obtained from the ethics committee of the Second Affiliated Hospital of Wannan Medical college. Signed consent was obtained from the patient for the publication of the case details, including publication of the images.
Acknowledgment
We thank the patient for providing consent and supporting this publication. This study was supported by the Project Fund for Excellent Research and Innovation Teams in Universities of Anhui Province (2024AH010032).
Disclosure
There is no conflict of interest.
References
1. Mangana J, Guenova E, Kerl K, et al. Angioimmunoblastic T-cell lymphoma mimicking drug reaction with eosinophilia and systemic symptoms (DRESS syndrome). Case Rep Dermatol. 2017;9(1):74–7. doi:10.1159/000458752
2. Xing Y, Huang J, Zhang Y, et al. Advancing the understanding and management of angioimmunoblastic T-cell lymphoma: insights into its pathogenesis, clinical features, and emerging therapeutic strategies. Front Oncol. 2025;15:1479179. doi:10.3389/fonc.2025.1479179
3. Charest G, McBride M, Thomas AK, et al. The first sign of recurrent angioimmunoblastic T-cell lymphoma: a cutaneous presentation. Cureus. 2023;15(9):e44805. doi:10.7759/cureus.44805
4. Xu L, Dong H, Xue D, et al. Delayed diagnosis of angioimmunoblast T-cell lymphoma presenting with immunoglobulin a vasculitis. Ren Fail. 2024;46(2):2416935. doi:10.1080/0886022X.2024.2416935
5. Kumar S, Batra A, Budania A, et al. Generalised pruritus with prurigo-like lesions and generalised lymphadenopathy: could be an early diagnostic clue for angioimmunoblastic T-cell lymphoma (AITL) and or histoplasmosis. Indian J Dermatol. 2023;68(6):726. doi:10.4103/ijd.ijd_974_22
6. Matsumoto NP, Xu ML. Angioimmunoblastic T-cell lymphoma: current diagnostic insights and advances. Hum Pathol. 2025;156:105696. doi:10.1016/j.humpath.2024.105696
7. Mathieu M, de Vicq de Cumptich M, Kul A, et al. Cutaneous peri-ocular involvement in angioimmunoblastic T-cell lymphoma. J Eur Acad Dermatol Venereol. 2025;39(3):e234–e236. doi:10.1111/jdv.20190
8. Hsueh YH, Huang Y, Chen CB. Angioimmunoblastic T-cell lymphoma presenting as extensive pseudovesicular papuloplaques. Dermatologica Sinica. 2024;42(1):52–53. doi:10.4103/ds.DS-D-23-00040
9. Basnet A. Angioimmunoblastic T-cell lymphoma: an immunological masquerade. QJM. 2023;116(7):577–578. doi:10.1093/qjmed/hcad057
10. Choo ZY, Akinyemi AA, Cibull T, et al. Angioimmunoblastic T-cell lymphoma unmasked by treatment with dupilumab. JAAD Case Rep. 2023;33:87–90. doi:10.1016/j.jdcr.2023.01.008
11. Xie Y, Jaffe ES. How I diagnose angioimmunoblastic T-cell lymphoma. Am J Clin Pathol. 2021;156(1):1–14. doi:10.1093/ajcp/aqab090
12. Yabe M, Dogan A, Horwitz SM, et al. Angioimmunoblastic T-cell lymphoma. Cancer Treat Res. 2019;176:99–126.
13. Pesqué D, Marcantonio O, Vázquez I, et al. Cutaneous involvement of angioimmunoblastic T-cell lymphoma masquerading as B-cell reactive lymphoid hyperplasia. Am J Dermatopathol. 2022;44(4):e41–e45. doi:10.1097/DAD.0000000000002110
14. Yoon SE, Cho J, Kim YJ, et al. Comprehensive analysis of clinical, pathological, and genomic characteristics of follicular helper T-cell derived lymphomas. Exp Hematol Oncol. 2021;10(1):33. doi:10.1186/s40164-021-00224-3
15. Advani RH, Skrypets T, Civallero M, et al. Outcomes and prognostic factors in angioimmunoblastic T-cell lymphoma: final report from the international T-cell Project. Blood. 2021;138(3):213–220. doi:10.1182/blood.2020010387
16. Grossman M, Ruan J, Magro C. Epstein-Barr virus-positive, CD30-positive, diffuse large B-cell lymphoma in a patient with angioimmunoblastic T-cell lymphoma. JAAD Case Rep. 2022;25:58–62. doi:10.1016/j.jdcr.2022.04.029
17. Jin X, Liu H, Li J, et al. Composite B-cell and T-cell lymphomas: clinical, pathological, and molecular features of three cases and literature review. J Zhejiang Univ Sci B. 2023;24(8):711–722. doi:10.1631/jzus.B2300181
18. Vega F, Medeiros LJ. A suggested immunohistochemical algorithm for the classification of T-cell lymphomas involving lymph nodes. Hum Pathol. 2020;102:104–116. doi:10.1016/j.humpath.2020.05.006
 © 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.