Back to Journals » International Journal of Nanomedicine » Volume 11
A blue fluorescent labeling technique utilizing micro- and nanoparticles for tracking in LIVE/DEAD® stained pathogenic biofilms of Staphylococcus aureus and Burkholderia cepacia
Authors Klinger-Strobel M, Ernst J, Lautenschlaeger C, Pletz MW, Fischer D, Makarewicz O
Received 15 October 2015
Accepted for publication 18 November 2015
Published 5 February 2016 Volume 2016:11 Pages 575—583
DOI https://doi.org/10.2147/IJN.S98401
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Thomas Webster
Mareike Klinger-Strobel,1,2,* Julia Ernst,3,* Christian Lautenschläger,4 Mathias W Pletz,1,2 Dagmar Fischer,3,5 Oliwia Makarewicz1,2
1Center for Infectious Diseases and Infection’s Control, 2Center for Sepsis Control and Care, Jena University Hospital, 3Department of Pharmaceutical Technology, Friedrich Schiller University Jena, 4Department of Internal Medicine IV, Jena University Hospital, 5Jena Center for Soft Matter (JCSM), Friedrich Schiller University Jena, Jena, Germany
*These authors contributed equally to this work
Abstract: Strategies that target and treat biofilms are widely applied to bacterial cultures using popular live/dead staining techniques with mostly red or green fluorescent markers (eg, with SYTO® 9, propidium iodide, fluorescein). Therefore, visualizing drugs or micro- and nanoparticulate delivery systems to analyze their distribution and effects in biofilms requires a third fluorescent dye that does not interfere with the properties of the live/dead markers. The present study establishes and evaluates a model for tracking polymeric particles in fluorescently stained biological material. To this end, poly(D,L-lactide-co-glycolide) (PLGA)-based micro- and nanoparticles were used as well-established model systems, which, because of their favorable safety profiles, are expected to play important future roles with regard to drug delivery via inhalation. PLGA was covalently and stably labeled with 7-amino-4-methyl-3-coumarinylacetic acid (AMCA), after which blue fluorescent poly(ethylene glycol)-block-PLGA (PEG-PLGA) particles were prepared using a mixture of fluorescent AMCA-PLGA and PEG-PLGA. Because chitosan is known to reduce negative surface charge, blue fluorescent PEG-PLGA-particles with chitosan were also prepared. These micro- and nanoparticles were physicochemically characterized and could be clearly distinguished from live/dead stained bacteria in biofilms using confocal laser scanning microscopy.
Keywords: 7-amino-4-methyl-3-coumarinylacetic acid, PLGA, PEG, confocal laser scanning microscopy, cystic fibrosis, chitosan, hydrodynamic diameter
Introduction
Most bacterial species prefer life in matrix-enclosed communities referred to as biofilms. Biofilms are highly complex structures of sometimes mixed species embedded in a highly variable hydrogel (matrix) containing proteins, complex carbohydrates, nucleic acids, and other polymers.1
Biofilms play a major role in infectious diseases because they are formed on artificial medical implants (eg, intracardiac devices, prosthetic joints) and indwelling catheters as well as natural surfaces, such as heart valves, wounds, and the mucosa of the respiratory tract, particularly in patients with cystic fibrosis (CF).2 According to the National Institutes of Health, biofilms are present in more than 80% of all nosocomial bacterial infections.3 Due to the rising number of medical implants and patients treated in intensive care units, this form of bacterial infection is of high therapeutic relevance. Accrued biofilms are almost impossible to eradicate using common antibiotics because the microbes embedded in a biofilm benefit from the protective biofilm environment and, thus, exhibit strongly reduced susceptibility to antibiotics.2 The phenotypic antibiotic resistance of biofilms is attributed to the properties of the matrix, including the following: 1) decreased penetration due to limited diffusion, electrostatic repulsion, or sequestration by matrix polymers, 2) specific microenvironment conditions such as low pH or accumulation of enzymes that hydrolyze or modify the drug, and 3) reduced metabolic activity of the biofilm-embedded bacteria.4 In consequence, the best strategy currently for treating patients with life-threatening biofilm-associated infections is to surgically remove the affected implants or infected body parts (eg, heart valves), which imposes risks of complications for the patients as well as increasing health care costs.
Consequently, there has been increasing scientific interest in compounds or strategies that enhance antibiofilm activity for the purpose of preventing biofilm formation or eradicating matured biofilms. Inhaled polymeric nanoparticles (NP) made of biodegradable organic compounds have been shown to penetrate mucus plugs in the respiratory tract and biofilm matrices and, thus, may provide applicable methodologies for delivering antibiotics into respiratory biofilms, particularly in patients with CF.5
To analyze the penetration properties and efficacies of embedded compounds, we looked for a means of obtaining stable and convenient labeling of the particles so that we could visualize both the particles and the viable and dead bacteria using confocal laser scanning microscopy. A common method for selectively staining viable and dead bacteria is the LIVE/DEAD® Bacterial Viability Kit (BacLight™), which contains the following two nucleic acid dyes: SYTO® 9, a green fluorescent dye that penetrates all cell membranes; and propidium iodide, a red fluorescent dye that penetrates only damaged cell membranes. Problematically, the most popular fluorescent dyes for analyzing NP properties in biofilms, fluorescein (green) or rhodamine (red),6,7 both interfere with LIVE/DEAD® staining. To facilitate visualization of NP and microparticles (MP) in red/green fluorescently stained biofilms, we covalently labeled the polymer poly(D,L-lactide-co-glycolide) (PLGA) with 7-amino-4-methyl-3-coumarinylacetic acid (AMCA) to prepare blue fluorescent NP and MP. These were investigated in biofilms of Burkholderia cepacia (a Gram-negative pathogen) and Staphylococcus aureus (a Gram-positive pathogen). Both species cause recurrent and difficult-to-treat biofilm-associated respiratory tract infections in patients with CF,8 but B. cepacia infections are usually associated with rapid clinical deterioration.
Materials and methods
Materials for particle preparation
To prepare the MP and NP, the following compounds were used: AMCA (~90%, Sigma-Aldrich, Steinheim, Germany); N-ethyl-N′-(3-dimethylaminopropyl) carbodiimide hydrochloride (EDC) (≥95%, Sigma-Aldrich); acetonitrile (≥99.5%, pharmaceutical quality, Sigma-Aldrich); ethanol (≥95%, Rotipuran, Carl Roth GmbH, Karlsruhe, Germany); acetone (ROTISOLV® ≥99.9%, GC Ultra Grade, Carl Roth GmbH); PLGA 50:50 (RESOMER® RG 502H, MW 7,000–17,000 g/mol, pharmaceutical quality, kindly provided by Boehringer Ingelheim, Ingelheim am Rhein, Germany); poly(ethylene glycol)-block-PLGA (PEG-PLGA) 50:50 (RESOMER® RGP d 50155, 5 kDa PEG, pharmaceutical quality, Evonik, Essen, Germany); polyvinyl alcohol (Mowiol 4–88, 31,000 g/mol, Kuraray Specialities Europe, Frankfurt, Germany); ultrapure chitosan chloride (30–400 kDa, pharmaceutical quality, Heppe Medical Chitosan, Halle, Germany); and ethyl acetate (≥99.5%, p.a., ISO, Carl Roth GmbH).
Fluorescent labeling of PLGA with AMCA
AMCA-PLGA was synthesized according to the method described by Horisawa et al9 and modified by the methodology of Lautenschläger et al.10 For fluorescence labeling, 1.5 g PLGA and 28 mg AMCA were mixed with 20 mg EDC in 5.0 mL acetonitrile. After incubation under stirring (150 U/min) at room temperature for 16 hours in the dark, the AMCA-PLGA was precipitated by adding deionized water followed by rinsing (to remove excessive reagents) via repeated dissolution/precipitation in acetone and 70% ethanol. The AMCA-PLGA was separated via centrifugation at 4,640× g (ROTANTA 460, Hettich, Beverly, MA, USA) for 30 minutes, after which it was vacuum-dried for 2 days in the dark.
Analysis of the covalent binding of AMCA-PLGA
The AMCA-PLGA preparation was analyzed by size exclusion chromatography (SEC) using a Shimadzu system equipped with an LC-10AD pump, a degasser DGU-14A, a CTO-10A column oven, a RID-10A refractive index detector, and a SPD-10AD VP ultraviolet detector. A 5% lithium chloride solution (ACROS Organic, Morris Plains, NJ, USA) in N,N-dimethylacetamide (Sigma-Aldrich) was used as eluent at a flow rate of 1 mL/min and a column oven temperature of 40°C. Polystyrene (Polymer kit) was used to calibrate the GRAM guard/1000/30 Å column (10 μm particle size) (PPS Polymer Standards Service GmbH, Mainz, Germany). The absorbance of AMCA-PLGA was determined at 350 nm.
For thin layer chromatography (TLC), AMCA (0.1 mg/mL) and AMCA-PLGA (2 mg/mL) were diluted in methanol (Merck KGaA, Darmstadt, Germany) and 1 μL aliquots were spotted on silica thin-layer plates (TLC Silica gel 60 F254, Merck KGaA). A methanol/chloroform (both Merck KGaA) 1:9 (v/v) mixture was used as the mobile phase. After drying the plates at room temperature, spots were identified by exposure to ultraviolet light at 366 nm.
Stability of AMCA-PLGA
A 4 mg/mL solution of AMCA-PLGA was prepared in dimethyl sulfoxide (DMSO) (ROTIPURAN®, ≥99.9%, p.a., Carl Roth GmbH), and 200 μL aliquots were transferred to 96-well plates (Greiner Bio-One, Frickenhausen, Germany). Fluorescence intensities at 450 nm (emission) were measured, using 350 nm as the excitation wavelength, at predetermined time intervals up to 120 hours using a Fluostar OPTIMA microplate reader (BMG LABTECH, Ortenberg, Germany). DMSO was used as the blank control. The 96-well plates were stored during the experiment under light protection at different temperatures (4°C, room temperature, 37°C). Measurements were performed in triplicate.
Preparation of AMCA-labeled NP and MP
Preparations of AMCA-NP and AMCA-MP were based on a modified solvent evaporation method described previously.11,12 The organic phase (O) comprised PEG-PLGA (30 mg) and AMCA-PLGA (20 mg) dissolved in 5 mL ethyl acetate. The aqueous phase (W) was prepared by dissolving 1% polyvinyl alcohol in deionized water, with 0.2% chitosan hydrochloride added on an optional basis. The organic phase was added drop-wise to 15 mL of the aqueous phase under stirring at 400× g. Afterwards, this O/W emulsion was stirred at 200× g for 3 hours to evaporate the organic solvent. The resultant O/W emulsion was homogenized using an Ultra-Turrax T25 (IKA-Werke, Staufen, Germany) and applying 5,500× g for 15 minutes (for the preparation of AMCA-PLGA MP) or 13,500× g for 15 minutes (for the preparation of AMCA-PLGA NP). The particles were precipitated by adding deionized water while stirring at 200× g to obtain a final volume of 50 mL. The residual organic solvent was evaporated via continuous stirring (200× g) for 24 hours. The MP and NP obtained were washed three times in deionized water, via resuspension in 10 mL deionized water and centrifugation (1 hour, 4,640× g, 20°C) (ROTANTA 460). The particles were vacuum-dried (ModulyoD Freeze Dryer; Thermo Fisher Scientific, Braunschweig; Germany) for 2 days in the dark.
Characterization of MP and NP
The mean hydrodynamic diameters and the zeta potentials (ZPs) of the particles were determined using a Zetasizer Nano ZS instrument (Malvern Instruments, Herrenberg, Germany). All experiments were performed in at least three independent replicates (five runs per sample), and mean values were calculated. ZP measurements were performed in deionized water by determining the electrophoretic mobility at 25°C in DTS1060 capillary cells (Malvern). The hydrodynamic diameter and the polydispersity index were measured via photon correlation spectroscopy in deionized water using low-volume cuvettes (ZEN 0112, Brand, Wertheim, Germany), a temperature of 25°C, and a scattering angle of 173°. Data were analyzed using Zetasizer v7.11 software (Malvern Instruments Ltd, Worcestershire, UK) and the refractive index (1.33) and viscosity (0.88 mPa*s) of distilled water at 25°C.
The morphologies of the resulting MP and NP were examined via scanning electron microscopy (SEM). An aqueous particle suspension (4 μL) was incubated for 4 minutes on previously glow-discharged grids and blotted with filter paper; holey carbon grids R 1.2/1.3, mesh 300 (Quantifoil Micro Tools GmbH, Jena, Germany) were utilized for MP, and formvar-carbon coated grids S162, mesh 400 (Plano GmbH, Wetzlar, Germany) were used for NP. The specimens were frozen immediately by manually plunging them into liquid propane/ethane (1:1 v/v, Linde AG, Munich, Germany) cooled with liquid nitrogen. The frozen grids were transferred into freeze fracture devices (BAF 060, Bal-Tec AG, Balzers, Liechtenstein) and freeze-dried at −90°C and ~5×10−7 mbar.
The grids were then mounted onto SEM sample holders and sputter coated with 8 nm platinum at 60 mA using a Bal-Tec Med 020 Coating System (Bal-Tec AG). SEM images were obtained with a LEO 1530 Gemini microscope (Carl Zeiss AG, Oberkochen, Germany) operated at 4 kV using the in-lens-detector. The geometric mean diameters of the particle batches were determined using Makroaufmassprogramm version 0.9.2 software (open source software by Jens Rüdig, http://ruedig.de/tmp/messprogramm.htm).
Strains and culture conditions
Biofilm assays were performed using B. cepacia 3003/3 (a clinical CF-isolate provided by the Institute of Medical Microbiology, University Hospital Jena) and S. aureus (ATCC 43300). A single colony of each strain was transferred from Lysogeny broth agar plates into 10 mL Mueller-Hinton (MH) medium (Carl Roth GmbH) and grown overnight at 37°C under rotation (150× g). The bacteria suspensions were adjusted to an optical density of 0.5 McFarland (corresponding to 107 bacteria/mL).
Determination of biocompatibility with bacteria
The biocompatibility of AMCA was determined by mixing a 105 CFU/mL bacteria suspension with serially diluted AMCA or AMCA-PLGA in MH medium in 96-well microtiter plates (Greiner Bio-One GmbH, Frickenhausen, Germany). The final concentrations of AMCA ranged from 0.125 to 128 mg/L (double-dilution series). Growth was indicated by the visible turbidity of the cultures after incubation at 37°C for 24 hours. This testing was performed in four replicates.
Biofilm screening and analysis
For biofilm formation, the bacteria suspension (0.5 McFarland) was freshly inoculated into MH medium via 1:100 dilution, and 300 μL was applied into X-Well Tissue Culture Chambers (Sarstedt AG, Nümbrecht, Germany). Biofilms were grown without shaking at 37°C for 24 hours. Subsequently, the planktonic bacteria were carefully removed, and the biofilms were washed with 200 μL phosphate buffered saline. Thereafter, 100 μL of the particles, prepared as a 2 g/L water suspension, was added to the biofilms followed by incubation for 4, 24, 48, 72, 96, 120, and 144 hours.
The biofilms were stained using a LIVE/DEAD® BacLight™ Bacterial Viability Kit for microscopy (Life Technologies GmbH, Darmstadt, Germany) according to the manufacturer’s protocol. After staining, the bacterial supernatants were carefully removed, and the biofilms were washed carefully once with 300 μL 0.9% NaCl solution. Stained biofilms were analyzed under vital conditions using an inverse confocal laser scanning microscope LSM510 (Carl Zeiss Jena GmbH, Jena, Germany) and a 40× air objective and 10× ocular (Carl Zeiss Jena GmbH). An area of ~100 μm (X) ×100 μm (Y) was screened in 1 μm Z-intervals (Z-stack) via green (excitation 488 nm/emission filter 501–545 nm), red (excitation 488 nm/emission filter 570–670 nm), and blue (excitation 405 nm/emission filter 371–450 nm) channels, respectively. The pinhole was adjusted to 0.95 Airy Units (corresponding to 1 μm). The experiments were repeated independently at least four times. The biofilm data were visualized using ZEN 9.0 software (Carl Zeiss Jena GmbH, Jena, Germany).
Result and discussion
Synthesis and characterization of AMCA-linked PLGA
AMCA was covalently bound to the terminal carboxy groups of PLGA by EDC, a carbodiimide crosslinker that activates the carboxy group, which enables nucleophilic attack by primary amines as present in AMCA. The ratio of the compounds PLGA: EDC:AMCA was nearly equimolar.
The covalent linkage of AMCA to PLGA was indicated by SEC detection of the PLGA-specific refractive index unit and the AMCA-specific absorbance (mAU) at 350 nm (Figure 1A). Both, AMCA and PLGA signals were detected at the same elution volume of ~20 mL, indicating successful covalent binding. This observation was also confirmed by the TLC results, which indicated the absence of free AMCA in the AMCA-PLGA sample (data not shown). The peak corresponding to free AMCA (0.233 kDa, elution volume 24.1 mL) in the SEC elugram that did not match the low-molecular fraction (<24.5 mL) most likely represented compounds of the cross-linkage reaction and hydrolytically degraded PLGA.
 | Figure 1 (A) SEC elugram of AMCA-PLGA; the black line indicates the refractive index of the PLGA expressed as relative units (RIU), and the gray line indicates the ultraviolet absorption by AMCA at 350 nm expressed as relative milli-units (mAU). (B) Stability studies via fluorescence intensity measurements of AMCA-PLGA in DMSO for >120 hours at 4°C (solid line, crosses), 20°C (dotted line, triangles) and 37°C (broken line, circles). Data are expressed as ratios of I/I0 and shown as the mean ± SD of three independent experiments. |
AMCA-PLGA dissolved in DMSO demonstrated high and stable fluorescence over a 72 hour time period at three applied temperatures: 4°C (refrigerator), 20°C (room temperature), and 37°C (body temperature) (Figure 1B). The fluorescence intensity only slightly decreased, to ~70%, after 120 hours of storage at 37°C.
PLGA and PEG were already known to be biocompatible, but AMCA is known to be an irritant. Thus, the biocompatibility of free AMCA was tested using B. cepacia and S. aureus. Based on the mixture ratio, the maximal calculated amount of AMCA in the particles was 0.7% (w/w), meaning the estimated maximal concentration that might be released by degradation of 2 g/L particles (as used for the assays) was 28.6 mg/L. For concentrations ranging from 0.125 to 128 mg/L, no inhibition of bacteria growth was observed, indicating that AMCA has no visible toxic effects on the bacteria.
Preparation and physiochemical characterization of MP and NP
Blue fluorescent AMCA-PLGA MP and NP were prepared as mixtures of PEG-PLGA and AMCA-PLGA using a modified emulsion/solvent diffusion technique. PEG-PLGA has been incorporated into the particles to improve the penetration properties. PEG has been shown to prevent absorbance of the particles to matrix compounds when the PEG content is higher than 40%.13 We used a 60:40 mixture of PEG-PLGA:AMCA-PLGA for particle preparation. In another experiment, chitosan, a cationic polymer that modifies the charge of the particles from negative to neutral, was incorporated into the particles (Chi-AMCA-PLGA MP or Chi-AMCA-PLGA NP) to decrease electrostatic repulsion in biofilms. Table 1 provides an overview of the particles’ physicochemical characteristics obtained.
 | Table 1 Physicochemical characteristics of AMCA-labeled particles determined by dynamic laser light scattering techniques in water |
AMCA-PLGA MPs and AMCA-PLGA NPs without chitosan exhibited mean hydrodynamic diameters of 1,068 and 234 nm, respectively, with narrow size distributions, as indicated by the low polydispersity index and comparable ZPs (around -33 mV). The incorporation of cationic chitosan not only increased the ZPs to nearly neutral values (-4 to -10 mV) but also increased the hydrodynamic diameters of both MP and NP.
SEM results indicated spherical shapes for all particles as well as smooth surfaces and variable diameters (Figure 2A–D) indicating a slightly smaller particle size range than that indicated by hydrodynamic diameters (Table 1). Analogous to photon correlation spectroscopy measurements, the averaged geometric diameter of chitosan-coated particles determined by SEM increased (Figure 2D) compared to particles without chitosan (Figure 2C).
 | Figure 2 Morphological studies of (A) AMCA-PLGA MP, (B) Chi-AMCA-PLGA MP, (C) AMCA-PLGA NP, and (D) Chi-AMCA-PLGA NP by SEM. Aqueous suspensions of representative particles were freeze-dried on specific grids and sputter coated with 8 nm platinum (holey carbon grids for MP, formvar-carbon grids for NP). |
Visualization of particles in biofilms
To visualize the particles within the biofilms, we used 24-hour-old biofilms of B. cepacia and S. aureus. B. cepacia formed thicker and denser biofilms compared to S. aureus (Figures 3 and 4). The biofilms were treated with phosphate buffered saline (control), AMCA-PLGA or Chi-AMCA-PLGA MP and NP, respectively, and penetrations of the particles were visualized via confocal laser scanning microscopy from 4 to 144 hours posttreatment.
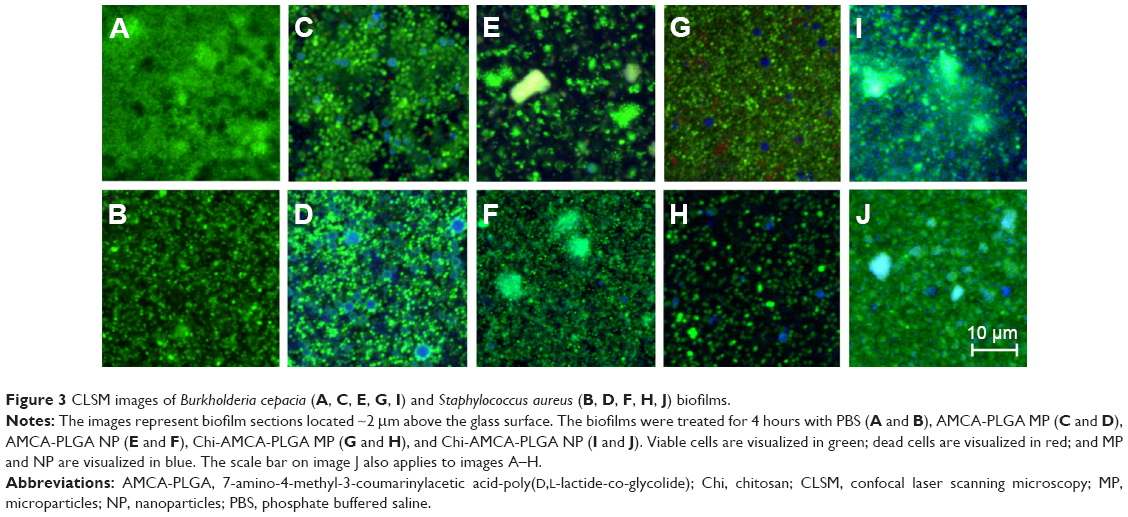 | Figure 3 CLSM images of Burkholderia cepacia (A, C, E, G, I) and Staphylococcus aureus (B, D, F, H, J) biofilms. |
 | Figure 4 Cross-sections through biofilms of Burkholderia cepacia (A, C, E, G, and I) and Staphylococcus aureus (B, D, F, H, and J), untreated (A, B) or treated with AMCA-PLGA MP (C, D), Chi-AMCA-PLGA MP (G, H), AMCA-PLGA NP (E, F) or Chi-AMCA-PLGA NP (I, J). Viable cells are visualized in green; dead cells are visualized in red; and MP and NP are visualized in blue. The glass slide surface is located at the bottom of each image. |
Both MP and NP were visible within deep layers of the biofilms after 4 hours, indicating their substantial penetration abilities (Figures 3C–J and 4C–J). MP were clearly visible as spherical blue structures of expected 1–2 μm diameters (Figure 3C, D, G, and H), and NP were found to be distributed via their diffuse blue fluorescence within the biofilm channels (Figures 3E, F, I, J and 4E, F, I, J). NP, in general, are insufficiently visible by light or fluorescent microscopy due the limited magnification (400-fold) and resolution (0:45 μm) of the 40× air lens.
Compared to MP, NP exhibited large surface areas, which expose more polymers, and tended to aggregate to microparticle-like blue spheres or light-blue and green clusters, indicating attachment to SYTO9®. SYTO9® is a cationic cyanine dye that intercalates with helical polymers (such as nucleic acids) via a complex cooperative mechanism including van der Waal, ionic and electrostatic interactions.14 Therefore, SYTO9® might interact with the PEG and PLGA portion and promote aggregation. SYTO9® accumulation was also visible as a green corona on the AMCA-PLGA MP’s surface (Figure 3C and D); however, this effect generally did not interfere with live/dead staining because the MP were still clearly visible. Addition of chitosan to the Chi-AMCA-PLGA MP reduced this effect (Figure 3G and H) without visible differences in the MP’ distributions in biofilms, indicating that chitosan prevents attachment of SYTO9® to PEG-PLGA. However, chitosan increased light scattering of NP (visible as opacity in Figure 3I and J) and, therefore, remains the only suitable additive for microparticle applications such as the one reported here.
The amounts of visible MP and NP decreased over the treatment time. After 48 hours, neither MP nor NP were clearly visible within the biofilms (Figure S1), indicating that the organic particles were most likely hydrolytically degraded, as described previously for PEG-PLGA.15,16 Notably, AMCA fluorescence also faded, and, after 48 hours, no blue fluorescence was observed in the treated biofilms. AMCA inactivation due to specific milieu conditions, such as low pH, within each biofilm is unlikely because AMCA dyes are pH insensitive. Because AMCA and its derivatives have been shown to be relatively resistant to photobleaching,17 it is also unlikely that photobleaching caused the reduction in blue fluorescence. More likely, ester bond breakage released AMCA-derivatives with altered or lost fluorescent properties. This hypothesis is supported by the reduced fluorescence of AMCA-PLGA after longer storage periods (Figure 1B). However, contribution from enzymatic digestion within the biofilms, even if unknown, cannot be excluded.
Conclusion
Biodegradable polymers like PEG-PLGA exhibit great potential as nanocarrier systems for therapeutics and may therefore be suitable for encapsulating antibiotics to improve their deposition and efficacy in deeper biofilm layers. AMCA constitutes an excellent blue fluorescent label for visualization of PEG-PLGA-based NP and MP in biofilms stained by most commercially available live/dead protocols utilizing green and red fluorescent dyes. The most popular green fluorescent dye is SYTO9® that is permeant to cell membranes and nonselectively stains all cells upon binding to nucleic acids. To distinguish between live and dead cells, red fluorescent dyes are used, often propidium iodide that also interacts with the nucleic acids but is not permeant to membranes of living cells and stains dead cells due to membrane damages, or resazurin derivatives that are reduced to resurfin upon reduction being selective for viable cells. Therefore, the AMCA labeling methodology not only allows tracking of the particles during their penetration into live/dead stained biofilms but also enables the investigation of the efficacies of encapsulated antibiotics against biofilms.
Acknowledgments
The authors thank Dr Jana Thamm (Department of Pharmaceutical Technology) and Susanne Linde (Center for Electron Microscopy, Jena University Hospital) for the SEM analysis reported herein. We also gratefully acknowledge Meike Nicole Leiske (Laboratory of Organic and Macromolecular Chemistry, Friedrich Schiller University, Jena) for assistance with polymer characterization.
This work was supported by Deutsche Forschungsgemeinschaft (DFG), grants PL 320/3-1 and FI 899/4-1.
Disclosure
The authors report no conflicts of interest in this work.
References
Bester E, Kroukamp O, Wolfaardt GM, Boonzaaier L, Liss SN. Metabolic differentiation in biofilms as indicated by carbon dioxide production rates. Appl Environ Microbiol. 2010;76(4):1189–1197. | ||
Macia MD, Rojo-Molinero E, Oliver A. Antimicrobial susceptibility testing in biofilm-growing bacteria. Clin Microbiol Infect. 2014;20(10):981–990. | ||
Römling U, Balsalobre C. Biofilm infections, their resilience to therapy and innovative treatment strategies. J Intern Med. 2012;272(6):541–561. | ||
Lewis K. Riddle of biofilm resistance. Antimicrob Agents Chemother. 2001;45(4):999–1007. | ||
Klinger-Strobel M, Lautenschläger C, Fischer D, et al. Aspects of pulmonary drug delivery strategies for infections in cystic fibrosis – where do we stand? Expert Opin Drug Deliv. 2015;12(8):1351–1374. | ||
Torché AM, Jouan H, Le Corre P, et al. Ex vivo and in situ PLGA microspheres uptake by pig ileal Peyer’s patch segment. Int J Pharm. 2000;201(1):15–27. | ||
White NS, Errington RJ. Fluorescence techniques for drug delivery research: theory and practice. Adv Drug Deliv Rev. 2005;57(1):17–42. | ||
Schechter MS, Fink AK, Homa K, Goss CH. The Cystic Fibrosis Foundation Patient Registry as a tool for use in quality improvement. BMJ Qual Saf. 2014;23 Suppl 1:i9–i14. | ||
Horisawa E, Kubota K, Tuboi I, et al. Size-dependency of DL-lactide/glycolide copolymer particulates for intra-articular delivery system on phagocytosis in rat synovium. Pharm Res. 2002;19(2):132–139. | ||
Lautenschläger C, Schmidt C, Lehr CM, Fischer D, Stallmach A. PEG-functionalized microparticles selectively target inflamed mucosa in inflammatory bowel disease. Eur J Pharm Biopharm. 2013;85(3 Part A):578–586. | ||
Sah H. Microencapsulation techniques using ethyl acetate as a dispersed solvent: effects of its extraction rate on the characteristics of PLGA microspheres. J Control Release. 1997;47(3):233–245. | ||
Nafee N, Taetz S, Schneider M, Schaefer UF, Lehr CM. Chitosan-coated PLGA nanoparticles for DNA/RNA delivery: effect of the formulation parameters on complexation and transfection of antisense oligonucleotides. Nanomedicine. 2007;3(3):173–183. | ||
Lai SK, Wang YY, Hanes J. Mucus-penetrating nanoparticles for drug and gene delivery to mucosal tissues. Adv Drug Deliv Rev. 2009;61(2):158–171. | ||
Hannah KC, Armitage BA. DNA-templated assembly of helical cyanine dye aggregates: a supramolecular chain polymerization. Acc Chem Res. 2004;37(11):845–853. | ||
Bhatia M, Girdhar A, Chandrakar B, Tiwari A. Implicating nanoparticles as potential biodegradation enhancers: a review. J Nanomed Nanotechol. 2013;4:175. | ||
Bala I, Hariharan S, Kumar MR. PLGA nanoparticles in drug delivery: the state of the art. Crit Rev Ther Drug Carrier Syst. 2004;21(5):387–422. | ||
Khalfan H, Abuknesha R, Rand-Weaver M, Price RG, Robinson D. Aminomethyl coumarin acetic acid: a new fluorescent labelling agent for proteins. Histochem J. 1986;18(9):497–499. |
Supplementary material
 | Figure S1 Burkholderia cepacia biofilms treated with AMCA-PLGA MP for 3 hours (A), 24 hours (B), 48 hours (C), 72 hours (D), and 96 hours (E). |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.