Back to Journals » International Journal of Nanomedicine » Volume 9 » Issue 1
2'-(2-bromohexadecanoyl)-paclitaxel conjugate nanoparticles for the treatment of non-small cell lung cancer in an orthotopic xenograft mouse model
Authors Peng L, Schorzman A, Ma P, Madden A, Zamboni W, Benhabbour SR, Mumper R
Received 12 April 2014
Accepted for publication 15 May 2014
Published 30 July 2014 Volume 2014:9(1) Pages 3601—3610
DOI https://doi.org/10.2147/IJN.S66040
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Lei Peng,1 Allison N Schorzman,2 Ping Ma,1 Andrew J Madden,2 William C Zamboni,2–4 Soumya Rahima Benhabbour,1 Russell J Mumper1,4
1Center for Nanotechnology in Drug Delivery, Division of Molecular Pharmaceutics, UNC Eshelman School of Pharmacy, University of North Carolina at Chapel Hill, 2Division of Pharmacotherapy and Experimental Therapeutics, UNC Eshelman School of Pharmacy, University of North Carolina at Chapel Hill, 3Center for Pharmacogenomics and Individualized Therapy, University of North Carolina at Chapel Hill, 4UNC Lineberger Comprehensive Cancer Center, University of North Carolina at Chapel Hill, North Carolina, NC, USA
Abstract: A nanoparticle (NP) formulation with 2'-(2-bromohexadecanoyl)-paclitaxel (Br-16-PX) conjugate was developed in these studies for the treatment of non-small cell lung cancer (NSCLC). The lipophilic paclitaxel conjugate Br-C16-PX was synthesized and incorporated into lipid NPs where the 16-carbon chain enhanced drug entrapment in the drug delivery system and improved in vivo pharmacokinetics. The electron-withdrawing bromine group was used to facilitate the conversion of Br-C16-PX to paclitaxel at the tumor site. The developed system was evaluated in luciferase-expressing A549 cells in vitro and in an orthotopic NSCLC mouse model. The results demonstrated that the Br-C16-PX NPs had a higher maximum tolerated dose (75 mg/kg) than Taxol® (19 mg/kg) and provided significantly longer median survival (88 days versus 70 days, P<0.05) in the orthotopic NSCLC model. An improved pharmacokinetic profile was observed for the Br-C16-PX NPs at 75 mg/kg compared to Taxol at 19 mg/kg. The area under the concentration versus time curve (AUC)0–96 h of Br-C16-PX from the NPs was 91.7-fold and 49.6-fold greater than Taxol in plasma and tumor-bearing lungs, respectively, which provided sustained drug exposure and higher antitumor efficacy in the NP-treated group.
Keywords: tubulin polymerization, pharmacokinetic, maximum tolerated dose, efficacy, pleural fluid, bioluminescence imaging, A549 cell tumor model
Introduction
Lung cancer is the leading cause of cancer mortality worldwide. An estimated 224,210 new cases and 159,260 deaths due to lung cancer are expected in 2014, accounting for 14% of cancer diagnoses and 27% of all cancer deaths, respectively.1 Among all lung cancer cases, 84% are classified as non-small cell lung cancer (NSCLC) and 15% as small-cell lung cancer for the purpose of treatment. The microtubule stabilization agent paclitaxel (PX) is used either as a monotherapy or in combination with carboplatin for the treatment of NSCLC.2
Currently PX is commercially marketed as Taxol® (Bristol-Myers Squibb, New York City, USA) and Abraxane® (Celgene, Basking Ridge, USA). Taxol contains Cremophor EL and dehydrated ethanol as co-solvents to solubilize PX and was given approval by the US Food and Drug Administration (FDA) in 1992. However, the hydrophobic nature of PX requires the amount of co-solvent to be so high that serious side effects including anaphylaxis and severe hypersensitivity are unavoidable. Abraxane, an albumin-bound PX nanoparticle (mean diameter 130 nm), was approved by the FDA in October 2012 for the first-line treatment of NSCLC in combination with carboplatin. In the phase III trial CA-031, weekly albumin-bound paclitaxel (nab-PX) exhibited decreased neuropathy and neutropenia compared to solvent-based paclitaxel (sb-PX), likely due to the absence of toxic excipients.3 However, the 10% improvement of progression-free survival (P=0.21) and overall survival (P=0.27) was not statistically significant. Moreover, the half-life (P=0.48) and area under the concentration versus time curve (AUC, P=0.52) of Abraxane at 260 mg/m2 were not significantly increased compared to Taxol at 175 mg/m2.4 Therefore, a PX formulation with decreased toxicity, improved therapeutic index, and better pharmacokinetic (PK) properties is needed.
Nano-formulations of PX have been investigated for safer and more effective chemotherapy.5–8 Due to moderate drug solubility or rapid drug release of most PX nano-formulations, lipophilic PX conjugates or derivatives have been developed to increase the stability in hydrophobic carrier systems. Previously Ali et al synthesized a series of lipophilic PX conjugates bearing 6, 8, 12, 14, or 16-carbon chains at the 2′-position.9 The PX conjugate with a long 16-carbon chain exhibited decreased cytotoxicity compared to conjugates with shorter chain lengths, but was therapeutically more efficacious when loaded into liposomes for the treatment of mouse ovarian cancer. Moreover, the incorporation of an electron-withdrawing bromine (Br) atom was evaluated in the PX conjugates. The bromoacyl PX conjugates were 50 to 250-fold more cytotoxic than their counterparts without Br in the human MCF-7 breast carcinoma cell line, indicating Br-induced hydrolysis. However, the liposomal formulation with PX conjugate had a relatively low molar ratio of bromoacyl taxane and a broad size range between 90 nm and 140 nm. Furthermore, the in vivo behavior of the system was not further investigated except for survival analysis.
In the current studies, the 2′-(2-bromohexadecanoyl)-paclitaxel (Br-C16-PX) conjugate was synthesized and incoporated into lipid-based NPs for the treatment of NSCLC. The 16-carbon chain helps to increase drug entrapment and stability in the NPs, and the electron-withdrawing Br group at the 2-position of the fatty acid chain enables faster hydrolysis to release PX. Tubulin polymerization activity and cytotoxicity of the conjugate was investigated. In vivo performance of the Br-C16-PX NPs was evaluated in an orthotopic NSCLC mouse model previously established and characterized in our laboratory.10 Lung cancer survival is largely related to cancer stage at the time of diagnosis.11 To mimic the therapy of advanced NSCLC, the treatment in the current studies was started later than 50% of the median survival. The Br-C16-PX NPs were shown to have superior therapeutic efficacy and improved PK properties compared to Taxol in the orthotopic xenograft model.
Materials and methods
Materials
Polyoxyl 20-stearyl ether (Brij 78) was purchased from Uniqema (Wilmington, DE, USA). D-alpha-tocopheryl polyethylene glycol-1000 succinate (Vitamin E-TPGS) was purchased from Eastman Chemicals (Kingsport, TN, USA). Miglyol 812 was purchased from Sasol (Witten, Germany). PX powder was bought from LC Laboratories (Woburn, MA, USA). Taxol was purchased from Hospira Inc. (Lake Forest, IL, USA). Sodium fluoride, 2-bromohexadecanoic acid and 4-dimethylaminopyridine (DMAP) were purchased from Sigma-Aldrich Co. (St Louis, MO, USA). Sepharose CL-4B was acquired from GE Healthcare Life Sciences (Uppsala, Sweden). The tubulin polymerization assay kit was purchased from Cytoskeleton Inc. (Denver, CO, USA). D-luciferin potassium salt, A549-luc-c8 cell line and Caliper IVIS Lumina II were from Caliper (Hopkinton, MA, USA). Roswell Park Memorial Institute (RPMI) 1640 medium and Dulbecco’s phosphate-buffered saline (DPBS) were purchased from Thermo Fisher Scientific (Waltham, MA, USA). Fetal bovine serum (FBS) was purchased from ATCC (Manassas, VA, USA). Matrigel was obtained from BD Biosciences (San Jose, CA, USA).
Optimization and characterization of Br-C16-PX NPs
Br-C16-PX was synthesized as previously reported.9 Briefly, PX was conjugated with 2-bromohexadecanoic acid via a one-step esterification reaction with DMAP (Sigma-Aldrich Co.) as the catalyst. The product was purified by preparative thin layer chromatography. The structure and molecular mass were confirmed by nuclear magnetic resonance (NMR) and mass spectrometry.9
Our laboratory has previously prepared PX BTM NPs from a warm oil-in-water microemulsion precursor with Brij 78 and Vitamin E-TPGS as surfactants and Miglyol 812 as the oil phase (abbreviated as “BTM” NPs).12,13 For the current formulation with Br-C16-PX, the system was optimized by decreasing the amount of surfactants while enhancing drug loading and drug entrapment. The particle diameter of the NPs was measured using a Coulter N5 Plus Submicron Particle Sizer (Beckman Coulter, FL, USA). The drug entrapment efficiency was determined by gel permeation chromatography (GPC) with Sepharose CL-4B. Briefly, the NPs were loaded onto a 15 cm GPC column, and fifteen fractions were collected (1 mL per fraction) with PBS as the eluent. Light scattering intensity of each fraction was determined with the Beckman Coulter N5 Submicron Particle Size Analyzer. Br-C16-PX concentration was determined by High Performance Liquid Chromatography (HPLC) (γ=230 nm) with an Inertsil ODS-3 column (4.6×150 mm, 4 μm particle size, GL Sciences Inc., Torrance, CA, USA) and a mobile phase consisting of acetonitrile/water/isopropanol (55:10:35, v/v/v) with a flow rate of 1 mL/min.
For in vivo studies in nude mice, the Br-C16-PX NPs were pegylated by adding 0.2 mg/mL Brij 700 (Uniqema) during the preparation.
Tubulin polymerization activity of Br-C16-PX
Tubulin polymerization activity of Br-C16-PX was evaluated using a tubulin polymerization assay kit. The standard (control) polymerization condition includes 3 mg/mL tubulin in 80 mM piperazine-N,N′-bis(2-ethanesulfonic acid) (PIPES) buffer at pH 6.9 in the presence of glycerol. PX at 10 μM was used as the positive control, and Br-C16-PX was evaluated at 10 μM and 34 μM. The polymerization reaction was conducted at 37°C and the degree of tubulin polymerization was quantified by the change in absorbance at 340 nm. The OD340 nm (optical density) was monitored with a Synergy 2 Multi-Detection Microplate Reader (BioTek, Winooski, VT, USA) over a 60 minute period with one reading per minute. The maximum rate of tubulin polymerization (Vmax, mOD/min) was plotted for the control, and the PX at 10 μM, Br-C16-PX at 10 μM, and Br-C16-PX at 34 μM groups.
Cell culture
Luciferase-expressing human lung adenocarcinoma cell line A549-luc-c8 was maintained in RPMI 1640 supplemented with 10% FBS and antibiotic. Cells were cultured in a humidity controlled incubator with 5% CO2 at 37°C.
Cytotoxicity of Br-C16-PX
The cytotoxicity of Br-C16-PX in A549-luc-c8 human lung adenocarcinoma cells was determined using a 3-(4, 5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay and compared to PX. Briefly, A549-luc-c8 cells were cultured as described above and seeded in a 96-well plate with 5,000 cells/100 μL growth medium per well. PX, Br-C16-PX NPs, or blank NPs without Br-C16-PX were added to the plate at a series of different concentrations. After 48-hour incubation at 37°C, the cells were incubated with MTT solution for another 3 hours. Lastly, dimethyl sulfoxide (DMSO) was added to solubilize the converted dye, and the absorbance at 570 nm was measured with a Synergy 2 Multi-Detection Microplate Reader (BioTek). The IC50 of the drug (the concentration that leads to 50% cell survival inhibition) was determined from survival plots.
Orthotopic lung tumor implantation
Four to six week old female nude mice (Harlan Sprague Dawley) were housed in a pathogen-free room. Orthotopic lung tumors were established as previously reported.10 Briefly, after mice were anesthetized, a 5 mm incision was made on the dorsal side over the left lung. Fat and muscles were separated to visualize lung movement. Five million A549-luc-c8 cells suspended in 40 μL PBS/matrigel were injected directly into the left lung parenchyma at the depth of 3 mm. All mouse studies were conducted according to a protocol approved by the University of North Carolina Institutional Animal Care and Use Committee.
Whole-body bioluminescence imaging
Weekly whole-body bioluminescence imaging was performed in mouse studies to monitor orthotopic lung tumor growth. D-luciferin potassium salt reconstituted in DPBS was injected intraperitoneally at 150 mg/kg. Mice were anesthetized with 2% isoflurane and imaged by Caliper IVIS Lumina II (Caliper) 15 minutes after luciferin injection.
In vivo antitumor efficacy studies
A549-luc-c8 cells were implanted into the left lung of nude mice on day 0 as described above. Mice were randomized into four groups (9 mice/group): control group (untreated), Taxol at 19 mg/kg (established maximum tolerated dose [MTD]), Br-C16-PX NPs at 60 mg/kg (“NP 60 group”), and Br-C16-PX NPs at 75 mg/kg (“NP 75 group”). Treatments were started as intravenous (IV) injections on day 16 with the treatment schedule of Q7d ×6. Mice were sacrificed when Body Condition Scoring (BCS) was 2 or less, or when a total weight loss of 20% was reached at any time for any mouse in the study.14
Histology staining
The efficacy study was terminated on day 154 post tumor cell implantation, with one mouse living in the NP 60 group and two mice living in the NP 75 group. Lungs of the surviving mice were harvested. After formalin fixing and paraffin embedding, hematoxylin and eosin staining was performed for 4 μm sections of the lungs. Histological staining was performed by the Animal Histopathology Core at UNC-Chapel Hill. Microscopic observations were carried out with Olympus Bx-61 (Olympus Corporation, Tokyo, Japan) in Microscopy Services Laboratory at UNC-Chapel Hill.
Quantification of PX and Br-C16-PX by Liquid Chromatography Tandem Mass Spectrometry (LC-MS/MS)
PX, Br-C16-PX and docetaxel (internal standard) were separated on a Waters XSelect CSH Phenyl-Hexyl column (2.1×50 mm, 130 Å pore size, 5 μm particle size; Waters Corporation, Milford, MA, USA) using a gradient mobile phase consisting of 0.1% formic acid in water (mobile phase A) and 0.1% formic acid and 10% isopropanol in acetonitrile (mobile phase B) on a Shimadzu LC-20AD (Shimadzu, Columbia, MD) liquid chromatography. The flow rate was 0.33 mL/min and the total run time was 6 minutes. The compounds were measured using a Thermo TSQ Ultra Triple Quadrupole Mass Spectrometer (Thermo Fisher Scientific, Inc, Waltham, MA) equipped with a heated electrospray ionization source in the positive ion mode. The discharge current was held at 3.7 kV and the vaporizer temperature at 225°C. PX, Br-C16-PX, and docetaxel were detected by selected-reaction monitoring using the transitions 854 → 286, 1172 → 604, and 808 → 527, respectively.
PK and biodistribution studies
A549-luc-c8 cells were injected into the left lung parenchyma on day 0 as described above. On day 16 post implantation, either Taxol at 19 mg/kg or Br-C16-PX NPs at 75 mg/kg of the Br-C16-PX conjugate were administered as a single IV bolus. Mice were sacrificed at predetermined time points (3 mice/time point). Plasma and tissues including tumor-containing lung, liver, spleen, and kidneys were harvested. Discernible lung tumor nodules were dissected for analysis when possible. Since pleural effusion was often observed at the time of sacrifice, the excess pleural fluid was collected when present. Plasma was harvested in sodium fluoride/potassium oxalate blood collection tubes and tissues were homogenized in the presence of 2.5 mg/mL sodium fluoride to serve as an esterase inhibitor to prevent ex vivo conversion. The drugs were extracted from the matrix by acetonitrile with 0.1% formic acid. The concentrations of PX from Taxol, Br-C16-PX from the NPs, and PX converted from Br-C16-PX were determined by LC-MS/MS as described above. Non-compartmental PK analysis was performed using WinNonlin version 6.2.0 (Pharsight Corp., Cary, NC, USA). The AUC from 0 to 96 hr (AUC0–96 h), AUC from 48 to 96 hr (AUC48–96 h), half-life (T1/2), volume of distribution (Vd), clearance (CL), and mean residence time (MRT) were reported.
Statistical analysis
Statistical analysis was performed with Prism (GraphPad Software Inc., La Jolla, CA, USA). One-way ANOVA was applied for multiple-group comparison, with Tukey’s Multiple Comparison Test as a post-test to compare any two groups. Log-rank Test was performed for survival comparison. Differences were considered statistically significant when P<0.05.
Results
Optimization and characterization of Br-C16-PX NPs
Br-C16-PX was synthesized through the conjugation of PX and 2-bromohexadecanoic acid as previously reported (Figure 1).9 Br-C16-PX NPs were formulated using a microemulsion precursor method with Miglyol 812 as the oil phase, and Brij 78 and Vitamin E-TPGS as surfactants.12,13 To further optimize the Br-C16-PX NPs in terms of drug loading, drug entrapment and particle size, four different formulations (Formulations I–IV) of the conjugate were investigated with various concentrations of surfactants and Br-C16-PX (Table 1). Drug entrapment of each formulation was determined using GPC with PBS elution. Out of 15 fractions for each formulation, NPs of 200 nm size eluted in fractions 3–5 as monitored by light scattering intensity and particle size measurement. For Formulation I and Formulation II, an additional population of smaller particles (~30 nm) was identified in fractions 6–9. These Br-C16-PX-containing smaller particles were likely to be micelles generated due to excess amount of surfactants.
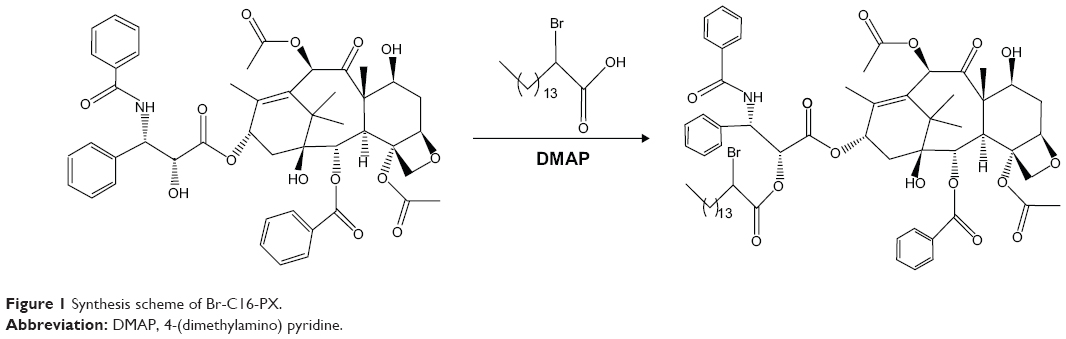 | Figure 1 Synthesis scheme of Br-C16-PX. |
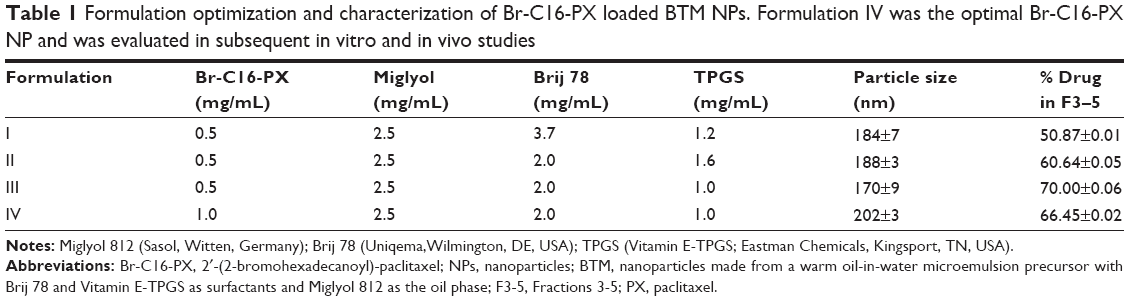 | Table 1 Formulation optimization and characterization of Br-C16-PX loaded BTM NPs. Formulation IV was the optimal Br-C16-PX NP and was evaluated in subsequent in vitro and in vivo studies |
With decreasing concentrations of the surfactants, a decreasing amount of Br-C16-PX was observed in fractions 6–9 of the GPC elution profile, leading to higher drug entrapment in the 200 nm oil-filled NPs in fractions 3–5. The drug entrapment of 0.5 mg/mL Br-C16-PX increased from 50.87%±0.01% to 70.00%±0.06% from Formulation I to Formulation III. In Formulation IV, Br-C16-PX concentration was increased to 1.0 mg/mL which yielded a drug entrapment of 66.45%±0.02%. Although Formulation IV appeared to have a slightly lower drug entrapment than Formulation III, it was considered to be the optimal formulation since the drug loading capacity was as high as 40% (w/w, drug/oil).
Tubulin polymerization activity of Br-C16-PX
The principle of the tubulin polymerization assay is that the extent of light being scattered by microtubules is proportional to the concentration of microtubule polymer. Under standard assay conditions at 37°C, the tubulin polymerization process is described by an increasing OD340 nm curve comprised of nucleation phase, growth phase, and equilibrium phase. The maximum polymerization rate (Vmax) obtained from the first derivative of the polymerization curve represents how fast the tubulin is polymerized under a certain condition. As shown in Figure 2A, a dose-dependent acceleration of tubulin polymerization was observed for Br-C16-PX. Br-C16-PX at 10 μM had a Vmax similar to the negative control (20.0±1.4 versus 17.5±2.1 mOD/min). When Br-C16-PX concentration was increased to 34 μM, the Vmax (30.5±2.1 mOD/min) was comparable to PX at 10 μM (29.0±0.0 mOD/min). It should be noted that the Vmax was reached later for Br-C16-PX (8–9 minutes) in the 60 minute time course than PX (0–3 minutes).
 | Figure 2 Toxicity study of Br-C16-PX. |
Cytotoxicity of Br-C16-PX NPs
An in vitro cytotoxicity study of free PX, blank NPs and the optimized Br-C16-PX NPs (Formulation IV) was carried out in the A549-luc-c8 cell line using the MTT assay. As shown in Figure 2B, the IC50 of Br-C16-PX NPs was 62.2-fold higher than free PX (5.3±3.2 nM). This was expected and consistent with other reported studies of 2′-PX conjugate since the 2′-OH group is crucial to the cytotoxicity and tubulin polymerization activity of PX.9,15–17 The blank NPs did not show significant cytotoxicity (drug equivalent IC50 3080.5±406.6 nM), indicating some cytotoxic effect of the Br-C16-PX conjugate.
In vivo antitumor efficacy studies
The therapeutic efficacy of the optimized Br-C16-PX NPs (Formulation IV) was evaluated in the orthotopic NSCLC mouse model previously established with A549-luc-c8 cells in our laboratory.10 Starting on day 16 post tumor cell implantation, Taxol at 19 mg/kg (predetermined MTD) and Br-C16-PX NPs at 60 mg/kg (“NP 60 group”) and 75 mg/kg of the conjugate (“NP 75 group”) were administered IV with the treatment schedule of Q7d ×6. As shown in Figure 3A, the control group had a median survival of 31 days, which was improved by Taxol treatment to 70 days. A dose response was observed for the treatment with Br-C16-PX NPs. Median survivals for the NP 60 group and the NP 75 group were 77 days and 88 days, respectively. The median survival was significantly improved by Br-C16-PX NPs at 75 mg/kg compared to the Taxol standard of care at MTD (P=0.03).
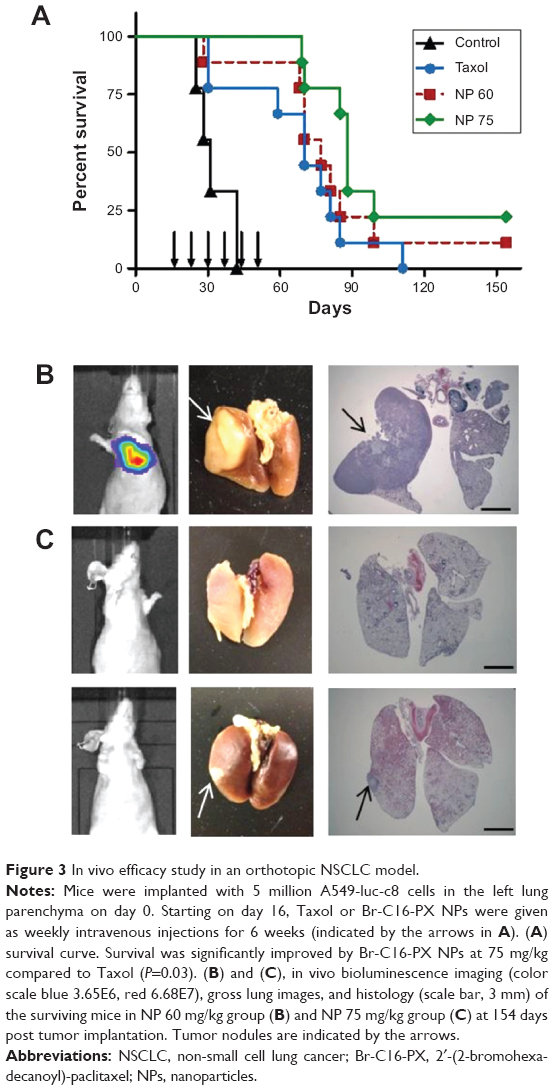 | Figure 3 In vivo efficacy study in an orthotopic NSCLC model. |
The efficacy study was terminated on day 154 post tumor cell implantation, when one mouse in the NP 60 group and two mice in the NP 75 group were still alive. Stronger bioluminescence signals were observed in the lungs of the surviving mouse in the NP 60 group (Figure 3B). The two mice in the NP 75 group had very low bioluminescence signals, indicating minimal tumor growth (Figure 3C). Gross images and hematoxylin and eosin staining of the lungs harvested from the surviving mice agreed with the bioluminescence imaging observations. The lungs of the mice in the NP 75 group had either no solid tumor nodule or a very small nodule in the left lung. While in the surviving mouse in the NP 60 group, an apparent tumor nodule occupied almost the entire lobe of the left lung.
PK and biodistribution studies
PK and biodistribution studies of the Br-C16-PX NPs and Taxol were carried out in the orthotopic NSCLC model at 75 mg/kg and 19 mg/kg, respectively. Tumors were distributed throughout the lung in the form of multiple nodules and possibly diffuse cells at the time of tissue collection, which made it difficult to dissect all the tumors from the lung in a time-efficient manner. Therefore, tumor-containing lungs were analyzed as a whole in the PK/biodistribution studies. However, a number of clearly discernible lung tumors were successfully dissected at several time points including 5 minutes, 15 minutes, 48 hours, 72 hours, and 96 hours. This dissection is believed to have minimal impact on quantified drug concentrations in the whole tumor-bearing lungs due to the small size of the dissected tumors and relatively low drug concentration. Excess pleural fluid was generated due to tumor growth and was collected for analysis when present.
As shown in Figure 4A and Table 2, plasma concentration of PX from Taxol decreased rapidly and was below the LC-MS/MS quantification range after 8 hours, while the concentration of Br-C16-PX was detectable up to 96 hours. The clearance and volume of distribution of Br-C16-PX in plasma were significantly decreased resulting in increased AUC and drug exposure. The clearance of Br-C16-PX from the NPs was only 4.3% of the PX clearance from Taxol, and the AUC0–96 h was 91.7-fold greater, suggesting prolonged systemic circulation of the NPs. The T1/2 (27.2 hr versus 0.8 hr) and MRT (14.5 hr versus 0.7 hr) of PX converted from Br-C16-PX were much longer compared to Taxol, indicating sustained conversion of PX from the Br-C16-PX NPs.
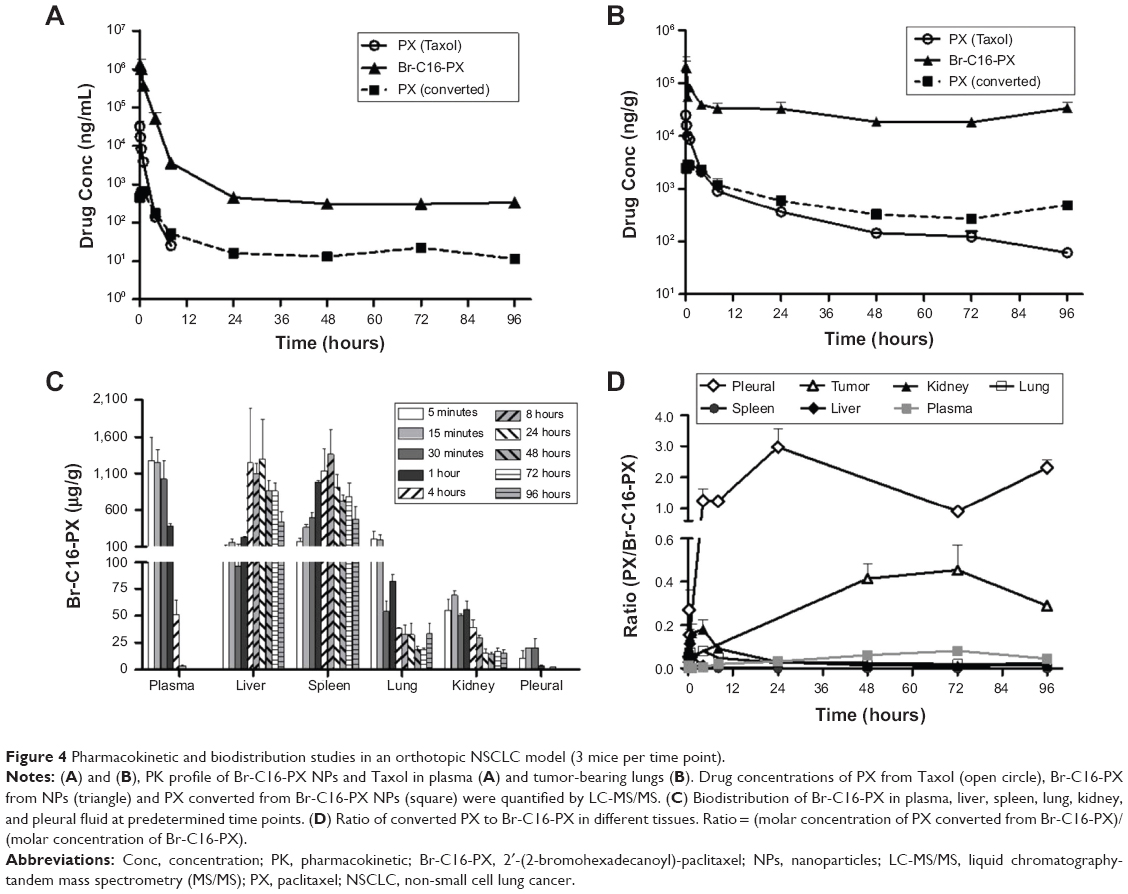 | Figure 4 Pharmacokinetic and biodistribution studies in an orthotopic NSCLC model (3 mice per time point). |
 | Table 2 Summary of important pharmacokinetic parameters of PX from Taxol, converted PX and Br-C16-PX from Br-C16-PX NPs. These parameters were calculated from the pharmacokinetic data by non-compartmental analysis |
Drug concentrations and PK parameters of the tumor-bearing lungs are illustrated in Figure 4B and Table 2. Br-C16-PX had a 49.6-fold greater AUC0–96 h and longer T1/2 (80.9 hr versus 30.3 hr) compared to Taxol. The concentration of PX converted from Br-C16-PX NPs reached higher numbers than Taxol at 4 hours and remained higher up to 96 hours with a much longer MRT (32.2 hr versus 14.7 hr). The AUC0–96 h and AUC48–96 h of PX from the NPs were 1.1-fold and 2.7-fold higher than Taxol, respectively. The significantly increased and prolonged exposures of tumor-bearing lungs to both Br-C16-PX and converted PX contributed to the superior therapeutic efficacy of the Br-C16-PX NPs over Taxol observed in the efficacy studies. The biodistribution of Br-C16-PX in different specimens is summarized in Figure 4C. Br-C16-PX was distributed primarily in the plasma from 5 minutes to 4 hours, and then mainly in the liver and spleen after 8 hours. The pegylation with 0.2 mg/mL Brij 700 may not be dense enough to shield the NPs from the reticuloendothelial (RES) system. Denser pegylation is currently being investigated to further decrease RES uptake and prolong circulation and ultimately increase tumor accumulation.
Importantly, the ratio of converted PX over the conjugate for the Br-C16-PX NPs was higher in pleural fluid and lung tumors compared to other tissues (Figure 4D). PX/Br-C16-PX from 48 hours to 96 hours was between 0.29 and 0.45 in dissected lung tumors, and between 1.24 and 2.97 in pleural fluid containing pleural tumors, likely indicating favorable conversion of the conjugate to PX in these compartments. Considering that the tubulin stabilization activity and cytotoxicity of the conjugate was lower than PX at the same concentration (Figure 2), the higher conversion in tumor tissue enhances the in vivo efficacy of the NPs. The higher conversion in pleural fluid may provide a better opportunity for the converted PX to diffuse into adjacent tumor sites in the lung, and enable a therapeutic benefit against pleural tumors. On the other hand, minimal conversion in non-tumor tissues and organs could help protect healthy tissues from systemic toxicity.
Discussion
In current studies, a lipophilic PX conjugate 2′-(2-bromohexadecanoyl)-paclitaxel (Br-C16-PX) was synthesized and incorporated into lipid-based NPs. The conjugate was evaluated for tubulin stabilization activity and cytotoxicity. In the tubulin polymerization assay (Figure 2), Br-C16-PX at 10 μM exhibited a lower tubulin stabilization effect compared to PX at the same concentration presumably due to the modification of the 2′-OH group.15–17 However, when the Br-C16-PX was increased to 34 μM, accelerated microtubule polymerization was observed similar to PX at 10 μM, indicating a tubulin stabilization effect of the conjugate.
Similar to compromised tubulin polymerization activity, free Br-C16-PX was expected to be less cytotoxic than the parent drug PX due to the conjugation of the 2′ hydroxyl group.15 Due to increased lipophilicity of Br-C16-PX after conjugation of the 16-carbon chain, its solubility in DMSO was not high enough to enable the IC50 analysis of free Br-C16-PX via MTT assay. Cytotoxicity of the Br-C16-PX NPs was investigated instead and results showed an IC50 that was 62.2-fold higher than free PX, which is consistent with other reported 2′-PX conjugates.9,15–17
The lipid-based NPs with Br-C16-PX were optimized to have a drug loading as high as 40% (w/w, drug/oil) and a drug entrapment of 66.45%±0.02%. In contrast, previously reported PX BTM NPs and C22-PX BTM NPs had a drug loading of only 7.5% and 7.7% by weight, respectively,18,19 likely due to relatively low affinity of PX to the Miglyol oil core in the PX BTM NPs or excess amount of surfactant in the C22-PX BTM NPs. From Formulation I to Formulation IV (Table I), Brij 78 concentration was decreased from 3.7 mg/mL to 2.0 mg/mL and Vitamin E-TPGS concentration was decreased from 1.2 mg/mL to 1.0 mg/mL. Higher drug entrapment was achieved with decreasing concentrations of the surfactants.
The optimized Br-C16-PX NPs (Formulation IV) were evaluated for the treatment of NSCLC. About 15% of lung cancer cases are diagnosed at early stage with a 5 year survival of 52.6%. For the majority of patients who are diagnosed with distant tumors, the 5 year survival rate is only 3.5%.11 More efficacious therapy options for advanced lung cancer are in great demand. However, treatments are started relatively early in the majority of preclinical studies in orthotopic lung cancer mouse models. Bao et al treated orthotopic A549 tumor with a heat shock protein 90 inhibitor starting on day 4 after tumor implantation, which is 10.5% of the 38 day median survival in control group.20 Jin et al started the treatment of orthotopic H460 tumor on day 10, which is 23.5% of the 42.5 day median survival.21 Furthermore, treatment of advanced lung tumors is more challenging due to the well-developed and likely more aggressive primary tumor as well as potential metastasis. Doki et al treated orthotopic Lewis lung carcinoma with cisplatin either starting on day 1 or day 10, which is 4.8% and 47.6% of the 21 day median survival, respectively.22 Cisplatin suppressed primary lung tumor growth and lymph node metastasis only in the day 1 treatment group, and failed to show any significant therapeutic effect in the day 10 treatment group.
In our in vivo efficacy studies in the orthotopic NSCLC mouse model, the Br-C16-PX NP treatment was started on day 16 post tumor cell implantation (51.5% of the 31 day median survival) instead of day 5 in the previously published studies with Br-C16-DX NPs10 to mimic the therapy of advanced NSCLC. The MTD of the Br-C16-PX NPs in non-tumor-bearing mice in the previously published studies was determined to be between 90 mg/kg and 120 mg/kg of the conjugate. However, the chemotherapeutic tolerance of orthotopic lung tumor-bearing mice was decreased due to compromised lung function and was related to the extent of tumor progression. Therefore, the drug dose in the current studies was decreased proportionately based on the MTD in healthy mice. Two doses (60 mg/kg and 75 mg/kg) of the Br-C16-PX NPs were chosen and evaluated for in vivo efficacy. The median survival was slightly improved by the Br-C16-PX NPs at the lower dose and further improved at the high dose (P=0.03) compared to Taxol standard of care (Figure 3). After the treatment was started in the NP 75 group, a minor weight loss was observed from which the mice recovered quickly, so it is believed that 75 mg/kg is very close to the actual MTD in the tumor-bearing mice.
There are several possible reasons for the superior therapeutic efficacy of the Br-C16-PX NPs over Taxol. First, the MTD of the NPs was greatly increased (4-fold higher than Taxol) likely due to the fact that the NP formulation consists of generally regarded as safe excipients instead of toxic Cremophor and ethanol in Taxol, allowing for higher delivered dose. Moreover, the decreased clearance and prolonged circulation provided more time for the NPs to circulate and accumulate into the orthotopic tumors via the enhanced permeability and retention (EPR) effect. Indeed, in the PK studies (Figure 4 and Table 2), the plasma T1/2 of Br-C16-PX from the NPs was increased to 7.9 hours compared to PX from Taxol at 0.8 hours. AUC0–96 h of Br-C16-PX in plasma and tumor-bearing lung was 91.6-fold and 49.6-fold higher than Taxol, respectively. In tumor-bearing lungs, the MRT of converted PX (32.2 hr) was 2.2-fold longer than Taxol (14.7 hr). The AUC0–96 h and AUC48–96 h of converted PX from the NPs were 1.1-fold and 2.7-fold greater than Taxol, respectively.
In addition to increased drug exposure, higher ratio of converted PX/Br-C16-PX was observed primarily in tumor (ratio up to 0.45) and pleural fluid (ratio up to 2.97). The higher ratio likely indicated favorable conversion of the conjugate to PX in these compartments which would enable more effective tumor treatment. The improved conversion was attributable to the electron-withdrawing Br group at the 2-position of the 16-carbon chain. The selective conversion in pleural fluid and tumor was likely caused by tissue-specific enzymatic activity and/or pH environment. Shaffer et al found that the metabolism of a paclitaxel poliglumex is at least partially mediated by the lysosomal enzyme cathepsin B, whose expression is up-regulated in many tumor types including lung tumors.23 Additionally, lower pH in the tumor interstitial fluid and also late endosomes and lysosomes could facilitate chemical hydrolysis of the Br-C16-PX conjugate especially with the presence of Br.24,25 Pleural effusion generated in the orthotopic mouse model was a thick fluid containing tumor cells, red blood cells, and some immune cells. The higher ratio of converted PX/Br-C16-PX in pleural fluid as compared to lung tumors may be caused by sequestration of the NPs in pleural cavity, and the absence of tumor stroma in the fluid environment thus easier access of the NPs to pleural tumors compared to solid lung tumors.
Conclusion
In the current studies, lipid-based NPs with Br-C16-PX were developed and evaluated for the treatment of NSCLC. The optimized Br-C16-PX NPs had a drug entrapment of 66.45% and exhibited moderate cytotoxicity in A549-luc-c8 cells. The Br-C16-PX NPs had superior antitumor efficacy as compared to Taxol for the treatment of advanced NSCLC in a novel orthotopic mouse model (P=0.03). Prolonged circulation, enhanced drug exposure, and selective conversion of Br-C16-PX to release PX contributed to the improved therapeutic efficacy of the Br-C16-PX NPs. Denser pegylation and CD44 targeted NPs are under investigation to further enhance the therapeutic index of the developed system.8
Acknowledgments
This work was supported by NIH-NCI U54 CA151652. The authors are very grateful to the Animal Studies Core at UNC-Chapel Hill especially Charlene M Santos for their skill and expertise with animal studies. The authors would also like to acknowledge Animal Histopathology Core at Lineberger Comprehensive Cancer Center for histology staining and the Microscopy Services Laboratory at UNC-Chapel Hill for assistance with microscopic observations.
Disclosure
The authors report no commercial association or conflict of interest for the research in this article.
References
Siegel R, Ma J, Zou Z, Jemal A. Cancer statistics, 2014. CA Cancer J Clin. 2014;64(1):9–29. | ||
Azzoli CG, Baker S, Temin S, et al. American society of clinical oncology clinical practice guideline update on chemotherapy for stage IV non-small-cell lung cancer. J Clin Oncol. 2009;27(36):6251–6266. | ||
Socinski MA, Bondarenko I, Karaseva NA, et al. Weekly nab-paclitaxel in combination with carboplatin versus solvent-based paclitaxel plus carboplatin as first-line therapy in patients with advanced non-small-cell lung cancer: final results of a phase III trial. J Clin Oncol. 2012;30(17):2055–2062. | ||
Sparreboom A, Scripture CD, Trieu V, et al. Comparative preclinical and clinical pharmacokinetics of a cremophor-free, nanoparticle albumin-bound paclitaxel (ABI-007) and paclitaxel formulated in Cremophor (Taxol). Clin Cancer Res. 2005;11(11):4136–4143. | ||
Ma P, Mumper RJ. Paclitaxel nano-delivery systems: a comprehensive review. J Nanomed Nanotechnol. 2013;4(2):1000164. | ||
Önyüksel H, Jeon E, Rubinstein I. Nanomicellar paclitaxel increases cytotoxicity of multidrug resistant breast cancer cells. Cancer Lett. 2009; 274(2):327–330. | ||
Blanco E, Sangai T, Hsiao A, et al. Multistage delivery of chemotherapeutic nanoparticles for breast cancer treatment. Cancer Lett. 2013;334(2):245–252. | ||
Yang X, Li Y, Li M, Zhang L, Feng L, Zhang N. Hyaluronic acid-coated nanostructured lipid carriers for targeting paclitaxel to cancer. Cancer Lett. 2013;334(2):338–345. | ||
Ali S, Ahmad I, Peters A, et al. Hydrolyzable hydrophobic taxanes: synthesis and anti-cancer activities. Anticancer Drugs. 2001;12(2): 117–128. | ||
Peng L, Feng L, Yuan H, Benhabbour SR, Mumper RJ. Development of a novel orthotopic non-small cell lung cancer model and therapeutic benefit of 2′-(2-bromohexadecanoyl)-docetaxel conjugate nanoparticles. Nanomedicine. Epub 2014 Apr 4. | ||
Siegel R, Naishadham D, Jemal A. Cancer statistics, 2012. CA Cancer J Clin. 2012;62(1):10–29. | ||
Dong X, Mattingly CA, Tseng M, Cho M, Adams VR, Mumper RJ. Development of new lipid-based paclitaxel nanoparticles using sequential simplex optimization. Euro J of Pharm Biopharm. 2009;72(1): 9–17. | ||
Ma P, Dong X, Swadley CL, et al. Development of idarubicin and doxorubicin solid lipid nanoparticles to overcome Pgp-mediated multiple drug resistance in leukemia. J Biomed Nanotechnol. 2009;5(2): 151–161. | ||
Foltz CJ, Ullman-Cullere M. Guidelines for assessing the health and condition of mice. Lab Animal. 1999;28(4):28–32. | ||
Parness J, Kingston DGI, Powell RG, Harracksingh C, Horwitz SB. Structure-activity study of cytotoxicity and microtubule assembly in vitro by taxol and related taxanes. Biochem Biophys Res Commun. 1982;105(3):1082–1089. | ||
Lundberg BB, Risovic V, Ramaswamy M, Wasan KM. A lipophilic paclitaxel derivative incorporated in a lipid emulsion for parenteral administration. J Control Release. 2003;86(1):93–100. | ||
Perkins WR, Ahmad I, Li X, et al. Novel therapeutic nano-particles (lipocores): trapping poorly water soluble compounds. Int J Pharm. 2000;200(1):27–39. | ||
Dong X, Mattingly CA, Tseng M, Cho M, Adams VR, Mumper RJ. Doxorubicin and paclitaxel-loaded lipid-based nanoparticles overcome multidrug resistance by inhibiting P-glycoprotein and depleting ATP. Cancer Res. 2009;69(9):3918–3926. | ||
Ma P, Benhabbour SR, Feng L, Mumper RJ. 2′-Behenoyl-paclitaxel conjugate containing lipid nanoparticles for the treatment of metastatic breast cancer. Cancer Lett. 2013;334(2):253–262. | ||
Bao R, Lai CJ, Wang DG, et al. Targeting heat shock protein 90 with CUDC-305 overcomes erlotinib resistance in non–small cell lung cancer. Mol Cancer Ther. 2009;8(12):3296–3306. | ||
Jin H, Yang R, Ross J, et al. Cooperation of the agonistic DR5 antibody apomab with chemotherapy to inhibit orthotopic lung tumor growth and improve survival. Clin Cancer Res. 2008;14(23):7733–7740. | ||
Doki Y, Murakami K, Sugiyama S, Misaki T, Saiki I. Mediastinal lymph node metastasis model by orthotopic intrapulmonary implantation of Lewis lung carcinoma cells in mice. Br J Cancer. 1999;79(7–8): 1121–1126. | ||
Shaffer SA, Baker-Lee C, Kennedy J, et al. In vitro and in vivo metabolism of paclitaxel poliglumex: identification of metabolites and active proteases. Cancer Chemother Pharmacol. 2007;59(4):537–548. | ||
Wike-Hooley JL, Haveman J, Reinhold HS. The relevance of tumour pH to the treatment of malignant disease. Radiother Oncol. 1984;2(4): 343–366. | ||
Paradise RK, Lauffenburger DA, Van Vliet KJ. Acidic extracellular pH promotes activation of integrin α(v)β(3). PLoS ONE. 2011;6(1): e15746. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.