Back to Journals » International Medical Case Reports Journal » Volume 7
18F-fluorodeoxyglucose positron emission tomography might be useful for diagnosis of hepatic amyloidosis
Authors Tawada A, Kanda T, Oide T, Tsuyuguchi T, Imazeki F, Nakatani Y, Yokosuka O
Received 1 March 2014
Accepted for publication 3 May 2014
Published 20 June 2014 Volume 2014:7 Pages 103—109
DOI https://doi.org/10.2147/IMCRJ.S63296
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Akinobu Tawada,1 Tatsuo Kanda,1 Takashi Oide,2 Toshio Tsuyuguchi,1 Fumio Imazeki,1,3 Yukio Nakatani,2 Osamu Yokosuka1
1Department of Gastroenterology, 2Department of Diagnostic Pathology, Chiba University Hospital, Chuo-ku, Chiba, Japan; 3Safety and Health Organization, Chiba University, Inage-ku, Chiba, Japan
Abstract: We report on a woman with hepatic involvement of primary systemic (immunoglobulin light chain, AL) amyloidosis. Her diagnosis was confirmed by liver biopsy. Clinical symptoms of hepatic amyloidosis are generally mild at its first stage, with most frequent findings being hepatomegaly and alkaline phosphatase elevation. Recent advances in the understanding of the pathophysiology of systemic amyloidosis have made several treatments available. However, its prognosis is occasionally poor. Because liver biopsy is not always safe, other modalities for the diagnosis are needed. Of interest was that fluorodeoxyglucose (FDG) uptake into the liver was observed, compared with that into the spleen, in this patient, indicating that FDG positron emission tomography and computed tomography might be useful for the diagnosis of hepatic amyloidosis with mild liver dysfunction.
Keywords: amyloidosis, diagnosis, hepatic involvement, FDG PET
Introduction
Systemic amyloidosis has been recognized as one of the most serious incurable diseases. It is a relatively rare disease consisting of several subtypes according to amyloid origin, and it causes multiple organ dysfunctions by the deposition of insoluble amyloid protein in intercellular spaces of these organs.1 Hepatic involvement is one of its phenotypes, and it is reported to complicate 56%–70% cases of systemic amyloidosis.2,3 However, it is too difficult to diagnose in its earlier stages because its symptoms and laboratory abnormalities are usually milder – irrespective of the amounts of amyloid deposition. Understanding of the pathophysiology and treatment of the disease has been improving, but its prognosis is still poor in some patients. Other diagnostic modalities are urgently required.
We report a case of hepatic involvement of primary systemic (immunoglobulin light chain, AL) amyloidosis, with severe amyloid deposition in the liver. Of note in this patient, fluorodeoxyglucose (FDG) uptake into the liver was observed, compared with that into the spleen, indicating that FDG positron emission tomography (PET) might prove to be helpful for the diagnosis of hepatic amyloidosis with mild liver dysfunction. FDG PET is one of the safe and useful diagnostic methods for determining her hepatic involvement of primary systemic AL amyloidosis, compared to liver biopsy.
Patient
A 76-year-old Japanese woman was referred to our hospital due to malaise, itching, anorexia, and liver dysfunction for 6 months. At her first visit to our clinic, her alkaline phosphatase (ALP) and gamma-glutamyl transpeptidase (γ-GTP) were 678 IU/L and 330 IU/L, respectively. Alanine aminotransferase, aspartate aminotransferase, and whole blood cell counts were almost within normal range, although prothrombin time was slightly prolonged. Serum hepatitis viral markers and autoantibodies were negative (Table 1). The immunofixation electrophoresis of serum and urine did not demonstrate the presence of a monoclonal light chain (data not shown).
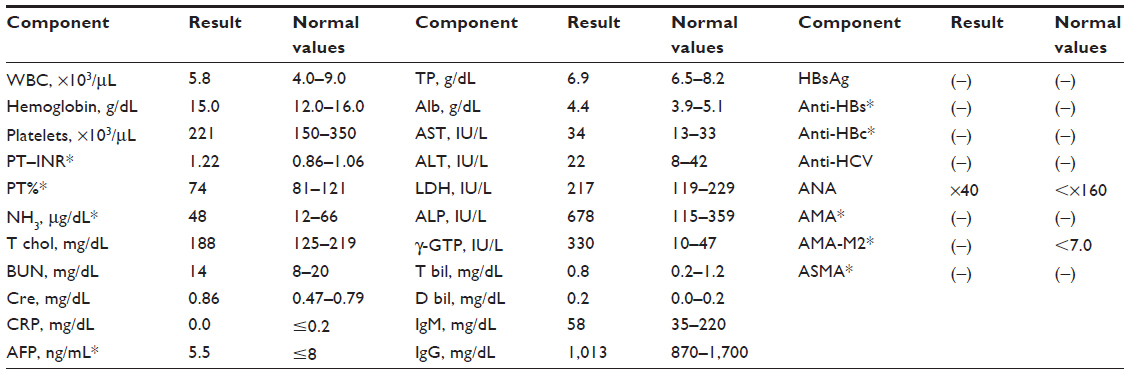 | Table 1 Laboratory data on first visit to Chiba University Hospital |
She had pulmonary tuberculosis at the age of 26. She took oral contraceptives, and she underwent surgery and blood transfusion for endometriosis at age 47. She had surgery without transfusion for osteoarthritis of the right knee and removal of colonic polyps, at 68 and 75 years old, respectively. Hypertension, hypercholesterolemia, and hypertrophic cardiomyopathy were diagnosed at age 56, 56, and 66, respectively, but no treatments were given. She did not have a past history of rheumatoid arthritis and had never received hemodialysis. She never smoked or drank alcohol. She was not a drug abuser, and no tattoo was found. At the time of admittance to our hospital, she was not taking any medications. Her father had esophageal cancer and alcoholic liver dysfunction, and her sister had skin cancer.
Although several clinical tests were performed at the outpatient clinic during a 6-month period, as the cause of her liver dysfunction remained unknown, she was then admitted to our hospital. Contrast-enhanced computed tomography (CT) revealed hepatomegaly with narrow portal vein, hepatic vein, and mild splenomegaly (Figure 1A–C). Although the association of liver and cardiac involvement is frequent, as her electrocardiogram and echocardiography showed normal cardiac movement (left ventricular ejection fraction, 75%) and no dilatation of the inferior vena cava, we did not consider the possibility of cardiac involvement. Renal function was within normal range as late as 2 months before her death. Fluorine-18 (18F) labeled FDG PET/CT was performed. The FDG uptake into the liver but not into the spleen was observed (Figure 1D), although the mean standard uptake value was not so high (2.0 in the left lobe of the liver; 1.8–2.0 in the right lobe; and 1.3–1.4 in the spleen). Magnetic resonance cholangiopancreatography and endoscopic retrograde cholangiopancreatography revealed narrowed intrahepatic bile ducts with smooth walls (Figure 2).
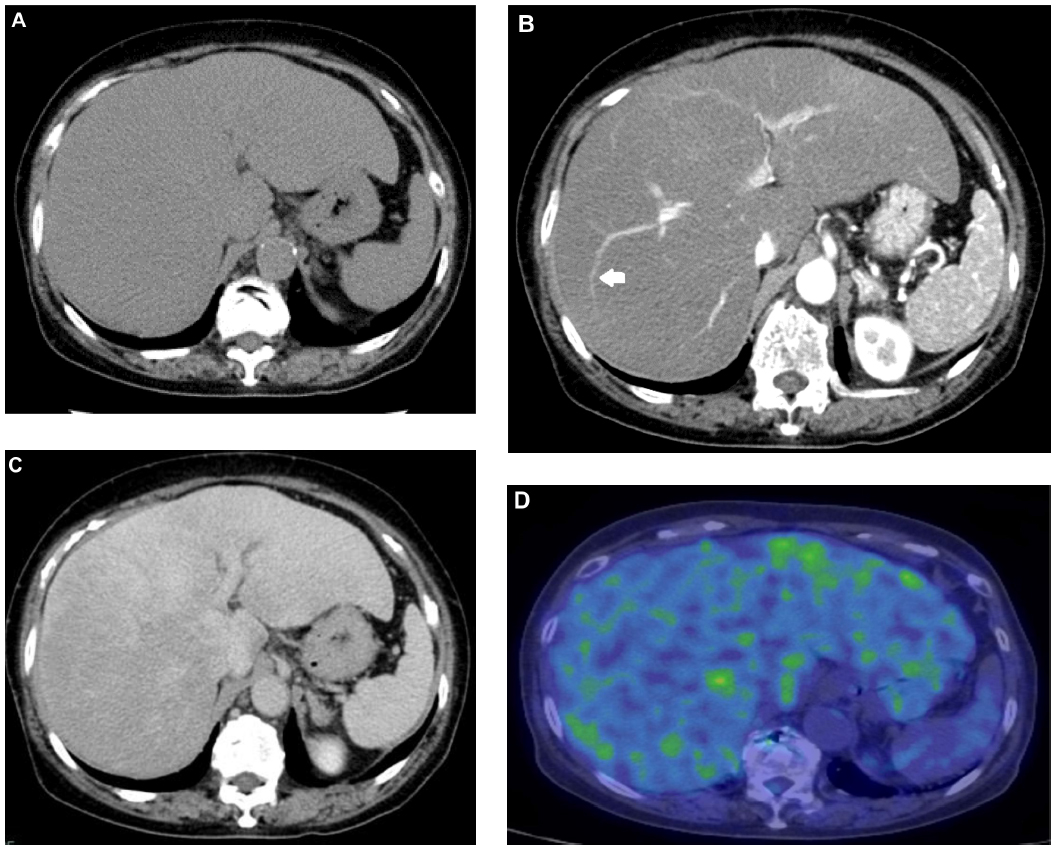 | Figure 1 Contrast-enhanced CT revealed hepatomegaly and heterogeneity of contrast staining. |
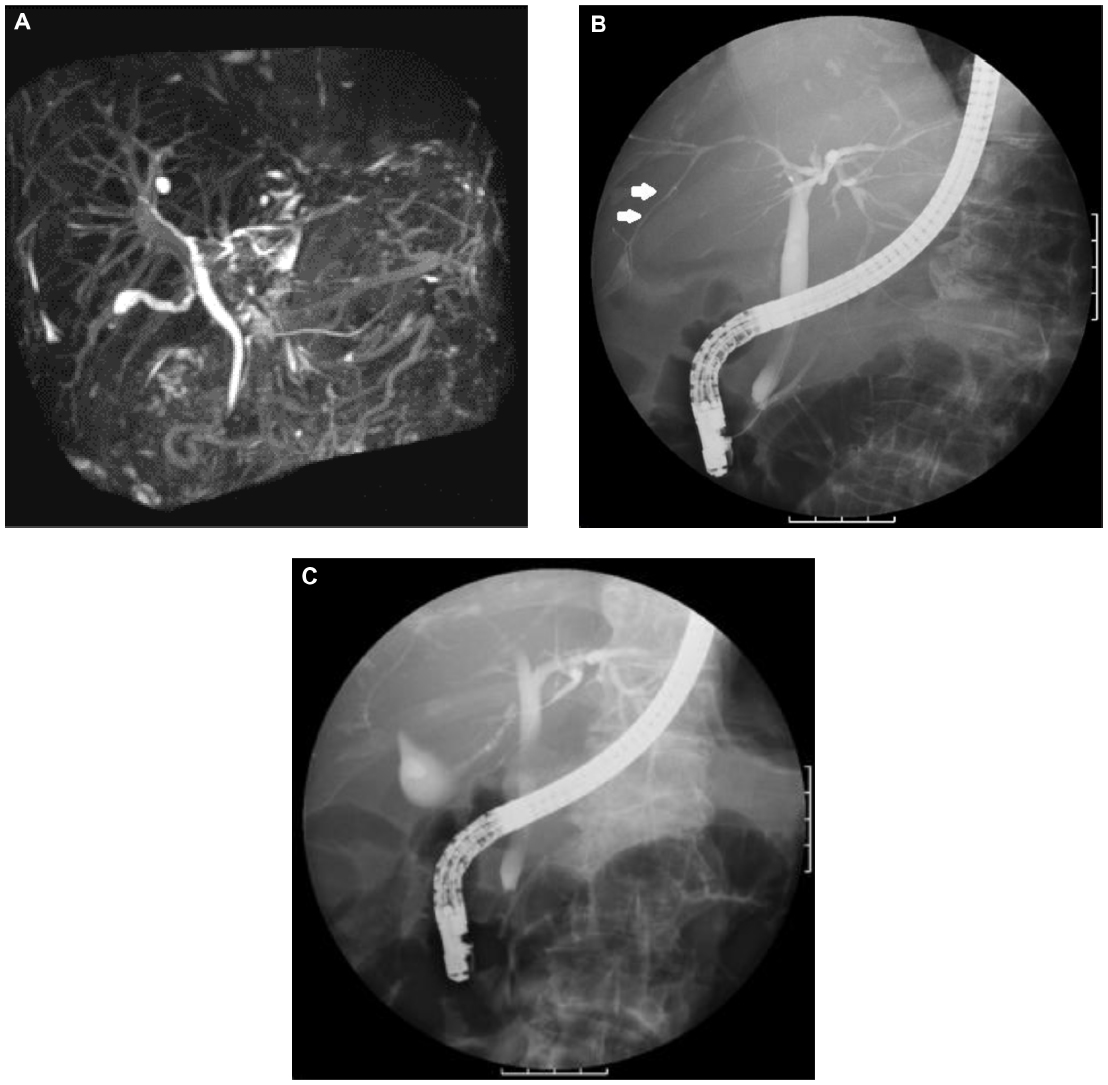 | Figure 2 MRCP (A) and ERCP (B and C) revealed narrowed whole intrahepatic bile ducts with smooth walls (white arrows). |
Upper gastrointestinal endoscopy showed no abnormal gross findings, but a biopsy specimen from the duodenal bulb mucosa exhibited amyloid deposition in the interspace of smooth muscles (not shown); macroglossia was observed. Eosinophilic amorphous depositions positive for Dylon staining4 that were resistant to potassium permanganate treatment5 and positive for amyloid P component were found.6 These results strongly suggested AL amyloidosis.
A liver biopsy was performed. The specimen was occupied by a diffuse eosinophilic amorphous structure, and any cellular component could not be found, suggesting hepatic amyloidosis (Figure 3). The biopsy specimen had the appearance of inappropriate material, but we later confirmed the hepatic involvement of primary systemic AL amyloidosis. Her general condition was too poor to allow aggressive treatment. She died 4 months after first admission to our hospital. Necropsy showed cardiac, renal, and hepatic involvement of amyloidosis.
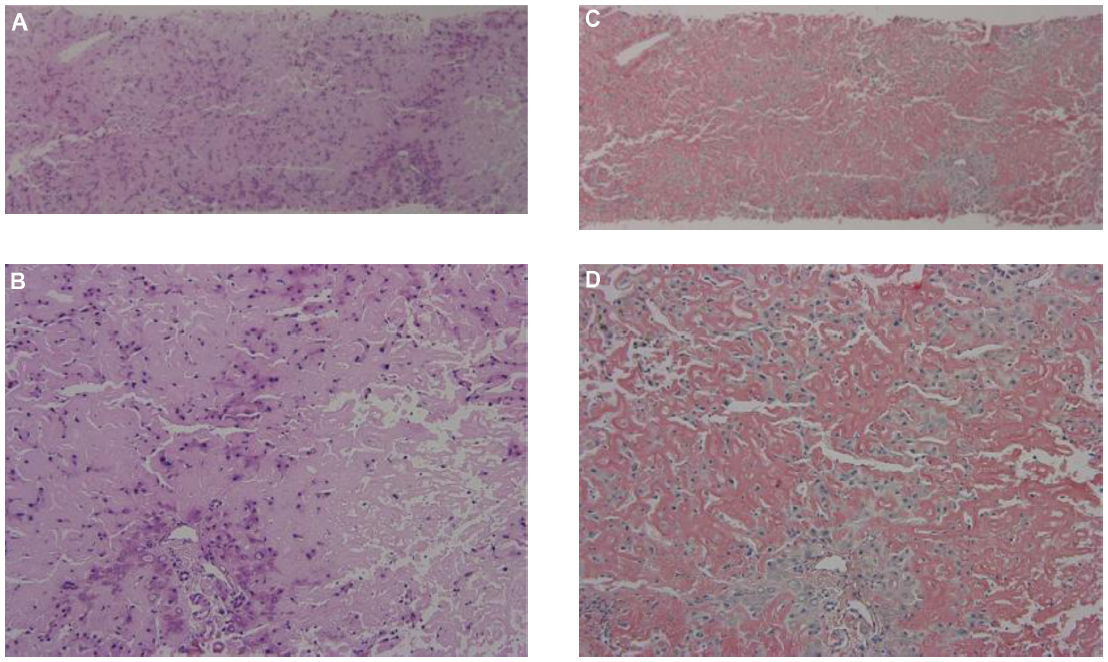 | Figure 3 Liver biopsy showing deposition of amyloid protein. |
Discussion
We present a case of hepatic amyloidosis with severe amyloid deposition in the liver. Deposit diseases, including amyloidosis, metabolic diseases, and nonalcoholic fatty liver disease, which are increasing in our country7 as well as the diffuse infiltration of various malignant diseases, such as malignant lymphoma, should be considered as differential diagnoses for hepatomegaly. It is reported that 56%–95% of autopsy cases of amyloidosis were complicated by hepatic amyloidosis.2,8 The prognosis of hepatic amyloidosis is very poor, with a reported mean survival time of about 12–17 months.9,10 Clinical symptoms of hepatic amyloidosis are generally mild at the early stage, and the most frequent findings are hepatomegaly and ALP elevation. In addition, 57%–83% of hepatic amyloidosis cases are reported to exhibit hepatomegaly,2,8 but the severity does not correspond to the amount of amyloid deposition. Jaundice is rare, only being observed in about 5% of the hepatic amyloidosis cases, but once jaundice appears, the remaining life duration will be very limited.11–13
Contrast-enhanced CT findings, such as hepatomegaly with ill-balanced inappropriate narrow intrahepatic vessels, and/or heterogeneitic stain pattern, are reported, but besides these, the disease is thought to lack definitive findings. Recently, the extraskeletal uptake of 99mTc-pyrophosphate in the liver was reported, and it was concluded to be specific for hepatic amyloidosis.14,15 Of interest, the sites of the liver with comparatively remaining hepatocytes become hypertrophic, compared with the sites with hepatocytes compressed by severe deposition of amyloid.16
An interesting, perhaps vital, observation was the uptake of FDG into the liver, compared with the spleen of this patient (Figure 1D), indicating the possible usefulness of FDG PET/CT for the diagnosis of hepatic amyloidosis with mild liver dysfunction. Son et al reported that FDG uptake was observed in hepatic amyloidosis or primary amyloidosis associated with multiple myeloma.17 The fixation of FDG was observed in certain localizations of amyloidosis,18 but the fixation of a satellite lymphoma cannot be excluded. No hepatic uptake was described in these two studies, which supported our observation. It is difficult to provide a pathophysiological mechanism to explain FDG uptake in extracellular sites. It is possible that the target cells might be macrophages that retain the amyloid deposits.
Pathologically, hepatic amyloidosis is divided into subtypes, according to the pattern of amyloid deposition.19 Of the three subtypes, the intralobular type features amyloid deposition in the space of Disse in the liver and is most common in AL amyloidosis. The portal type is defined with amyloid deposition in the vessel walls of the portal areas. The mixed type is defined with amyloid deposition in both hepatic lobules and portal areas. The present case was assumed to be the intralobular type, which can present massive amyloid deposition and hepatocyte atrophy (Figure 3).20 Among the five major subtypes of systemic amyloidosis, the amyloid A protein (AA) amyloidosis was negative in the liver because the amyloid resisted the potassium permanganate treatment by direct fast scarlet staining; it was also negative by immunostaining, although amyloid P component was positive in biopsy specimens from the duodenum. Familial or senile transthyretin-related amyloidosis was also thought to be negative because there was no family history of amyloidosis, and the amyloid was negative by immunostaining. The beta 2-microglobulin-related amyloidosis was also negative since this patient had not undergone dialysis. The CD138-positive plasma cells were observed to proliferate in the bone marrow (data not shown). These accumulating plasma cells were accompanied by atypia, such as the disparity of nucleus, irregularity of karyotype, and some with two or three nuclei. The bone marrow analysis from autopsy samples showed that immunoglobulin (lambda) light chain-positive plasma cells were dominant (22% of total nucleated cell counts) over immunoglobulin (kappa) light chain-positive plasma cells, suggesting monoclonal proliferation of plasma cells. These results strongly suggested primary AL amyloidosis, A (lambda) type, associated with plasma cell dyscrasia. Multiple myeloma was negative because the proliferation of the plasma cells was confined to bone marrow, and there was no evidence of extramedullary plasmacytoma, obvious lymph node enlargement, or plasma cell infiltration other than in bone marrow. The amyloid itself was negative for both anti-immunoglobulin (lambda) light chain antibody and anti-immunoglobulin (kappa) light chain antibody immunostaining. Such results are apparently often observed in AL amyloidosis because of the three-dimensional structural change and the regional diversity of the immunoglobulin light chain.21
This patient presented negative serum and urine immunofixations, although Lachmann et al reported that a monoclonal excess of circulating free immunoglobulin light chain was identified at diagnosis in 98% of patients with primary AL amyloidosis.22
Variant forms of soluble proteins, such as fibrinogen, gelsolin, lysozyme, and apolipoprotein (apo) A-I have been associated with familial amyloidosis. As apo A-I is the main protein in high-density lipoprotein (HDL), most apo A-I variants are associated with decreased plasma HDL (hypoalphalipoproteinemia), hyperglyceridemia, and/or defective lecithin cholesterol acyltransferase activation.23 Because the HDL was within normal limits, apo A-I-related amyloidosis could also be excluded in our case.
Liver biopsy has been thought to be contraindicative for hepatic amyloidosis because of the risks of hepatic bleeding and rupture. There are two reports concerning this issue.24,25 One reported that among the 98 hepatic amyloidosis patients undergoing liver biopsy, four were complicated by bleeding, with two of them needing blood transfusion. They recovered, and there was no hepatic rupture or biopsy-related death. The other reported that in 80 hepatic amyloidosis patients having liver biopsy, two were complicated by bleeding but recovered with conservative treatment.24 Liver biopsy is an effective procedure for patients suspected of hepatic amyloidosis. Still, contrary opinions exist concerning liver biopsy for patients with hepatic amyloidosis.25 Szturz et al26 reported a case of AL amyloidosis presenting with spontaneous liver hemorrhage and hemoperitoneum. Because liver stiffness is greater in patients with hepatic involvement of amyloidosis, liver stiffness measurement may also be a simple method with diagnostic value in such patients.27 Testing of the abdominal fat aspirates might also help to eliminate the risky invasive procedure of liver biopsy.28 Thus, in terms of risk/benefit, a combination of these noninvasive modalities, together with FDG PET/CT, should be considered before a liver biopsy, as FDG PET/CT is not histologically specific, and – of course – hepatic/systemic amyloidosis cannot be diagnosed with FDG PET/CT alone. However, we noticed splenic hypoperfusion using FDG PET/CT, and the radiological finding of splenic hypoperfusion has been well-described as a marker of systemic amyloidosis.29,30 Splenic hypoperfusion might be a useful clue when clinical findings fail to lead to an accurate diagnosis. If one PET image is insufficient, it might be necessary to use a maximum intensity projection image. Overall, there have been very few case reports on the possible usefulness of FDG/PET in the diagnosis of hepatic amyloidosis.31 Recent advances in the understanding of the pathophysiology of the disease have made several treatments possible, such as dexamethasone,11 melphalan with prednisolone,31 melphalan with dexamethasone,32 thalidomide with dexamethasone,33 lenalidomide with dexamethasone,34 bortezomib with dexamethasone,35 and autologous hematopoietic stem cell transplantation.36 Survival benefits have been reported. But once the disease stage has progressed beyond a certain extent, these treatments can no longer be performed due to the serious symptoms and complications. Stem cell transplant is a preferred therapeutic modality, but only a minority of patients are eligible, due to either advanced disease stage at presentation or advanced age.
In conclusion, the clinical diagnosis of systemic amyloidosis at an earlier stage is a major challenge. We presented a case of hepatic involvement of systemic amyloidosis in which we observed FDG uptake into the liver, compared with that into the spleen. Our case suggests that FDG PET/CT might be of potential interest in the search for a diagnosis. Further studies will be needed.
Acknowledgment
The authors thank all medical staff at the liver unit of Chiba University Hospital.
Disclosure
The authors report no conflicts of interest in this work.
References
Park MA, Mueller PS, Kyle RA, Larson DR, Plevak MF, Gertz MA. Primary (AL) hepatic amyloidosis: clinical features and natural history in 98 patients. Medicine (Baltimore). 2003;82(5):291–298. | |
Levine RA. Amyloid disease of the liver. Correlation of clinical, functional and morphologic features in forty-seven patients. Am J Med. 1962;33:349–357. | |
Buck FS, Koss MN. Hepatic amyloidosis: morphologic differences between systemic AL and AA types. Hum Pathol. 1991;22(9):904–907. | |
Uenishi T, Teramura K, Kitamura M, et al. Hyperkeratotic variant of porokeratosis Mibelli with dermal amyloid deposits. J Dermatol. 2010;37(5):475–479. | |
Pambuccian SE, Horyd ID, Cawte T, Huvos AG. Amyloidoma of bone, a plasma cell/plasmacytoid neoplasm. Report of three cases and review of the literature. Am J Surg Pathol. 1997;21(2):179–186. | |
Hachulla E, Janin A, Flipo RM, et al. Labial salivary gland biopsy is a reliable test for the diagnosis of primary and secondary amyloidosis. A prospective clinical and immunohistologic study in 59 patients. Arthritis Rheum. 1993;36(5):691–697. | |
Kawaguchi T, Sumida Y, Umemura A, et al; Takeshi Okanoue. Genetic polymorphisms of the human PNPLA3 gene are strongly associated with severity of non-alcoholic fatty liver disease in Japanese. PLoS One. 2012;7(6):e38322. | |
Ebert EC, Nagar M. Gastrointestinal manifestations of amyloidosis. Am J Gastroenterol. 2008;103(3):776–787. | |
Kyle RA, Gertz MA. Primary systemic amyloidosis: clinical and laboratory features in 474 cases. Semin Hematol. 1995;32(1):45–59. | |
Kyle RA, Gertz MA, Greipp PR, et al. A trial of three regimens for primary amyloidosis: colchicine alone, melphalan and prednisone, and melphalan, prednisone, and colchicine. N Engl J Med. 1997;336(17):1202–1207. | |
Levy M, Fryd CH, Eliakim M. Intrahepatic obstructive jaundice due to amyloidosis of the liver. A case report and review of the literature. Gastroenterology. 1971;61(2):234–238. | |
Peters RA, Koukoulis G, Gimson A, Portmann B, Westaby D, Williams R. Primary amyloidosis and severe intrahepatic cholestatic jaundice. Gut. 1994;35(9):1322–1325. | |
Goenka MK, Bhasin DK, Vasisth RK, Dhawan S. Hepatic amyloidosis presenting with severe intrahepatic cholestasis. J Clin Gastroenterol. 1996;23(2):134–136. | |
Ferraro EM, Alfelor FR, Lee M, Poon TT. Hepatic amyloidosis in an iv drug abuser detected by bone scintigraphy. Clin Nucl Med. 1987;12(4):274–276. | |
Lomeña F, Roselló R, Pons F, et al. Abnormal scintigraphic evolution in AA hepatic amyloidosis. Clin Nucl Med. 1988;13(3):194–196. | |
Suzuki K, Okuda K, Yoshida T, Kanda Y. False-positive liver scan in a patient with hepatic amyloidosis: case report. J Nucl Med. 1976;17(1):31–32. | |
Son YM, Choi JY, Bak CH, et al. 18F-FDG PET/CT in primary AL hepatic amyloidosis associated with multiple myeloma. Korean J Radiol. 2011;12(5):634–637. | |
Mekinian A, Jaccard A, Soussan M, et al; Centre de Référence des Amyloses immunoglobulinémiques et autres maladies liées aux dépots des immunoglobulines monoclonales. 18F-FDG PET/CT in patients with amyloid light-chain amyloidosis: case-series and literature review. Amyloid. 2012;19(2):94–98. | |
Levy M, Polliack A, Lender M, Eliakim M. The liver in amyloidosis. Digestion. 1974;10(1):40–51. | |
Kitamura Y, Yokomori H. Hepatic amyloidosis manifesting severe intrahepatic cholestasis. Intern Med. 2005;44(6):675–676. | |
Hoshii Y, Takahashi M, Ishihara T, Uchino F. Immunohistochemical classification of 140 autopsy cases with systemic amyloidosis. Pathol Int. 1994;44(5):352–358. | |
Lachmann HJ, Gallimore R, Gillmore JD, et al. Outcome in systemic AL amyloidosis in relation to changes in concentration of circulating free immunoglobulin light chains following chemotherapy. Br J Haematol. 2003;122(1):78–84. | |
Joy T, Wang J, Hahn A, Hegele RA. APOA1 related amyloidosis: a case report and literature review. Clin Biochem. 2003;36(8):641–645. | |
Gertz MA, Kyle RA. Hepatic amyloidosis (primary [AL], immunoglobulin light chain): the natural history in 80 patients. Am J Med. 1988;85(1):73–80. | |
Gertz MA, Comenzo R, Falk RH, et al. Definition of organ involvement and treatment response in immunoglobulin light chain amyloidosis (AL): a consensus opinion from the 10th International Symposium on Amyloid and Amyloidosis, Tours, France, April 18–22, 2004. Am J Hematol. 2005;79(4):319–328. | |
Szturz P, Kyclová J, Moulis M, et al. Extensive Al amyloidosis presenting with recurrent liver hemorrhage and hemoperitoneum: case report and literature review. Klin Onkol. 2013;26(1):49–52. | |
Loustaud-Ratti VR, Cypierre A, Rousseau A, et al. Non-invasive detection of hepatic amyloidosis: FibroScan, a new tool. Amyloid. 2011;18(1):19–24. | |
Duston MA, Skinner M, Meenan RF, Cohen AS. Sensitivity, specificity, and predictive value of abdominal fat aspiration for the diagnosis of amyloidosis. Arthritis Rheum. 1989;32(1):82–85. | |
Mainenti PP, Cantalupo T, Nicotra S, et al. Systemic amyloidosis: the CT sign of splenic hypoperfusion. Amyloid. 2004;11(4):281–282. | |
Mainenti PP, Camera L, Nicotra S, et al. Splenic hypoperfusion as a sign of systemic amyloidosis. Abdom Imaging. 2005;30(6):768–772. | |
Dhodapkar MV, Hussein MA, Rasmussen E, et al; United States Intergroup Trial Southwest Oncology Group. Clinical efficacy of high-dose dexamethasone with maintenance dexamethasone/alpha interferon in patients with primary systemic amyloidosis: results of United States Intergroup Trial Southwest Oncology Group (SWOG) S9628. Blood. 2004;104(12):3520–3526. | |
Palladini G, Perfetti V, Obici L, et al. Association of melphalan and high-dose dexamethasone is effective and well tolerated in patients with AL (primary) amyloidosis who are ineligible for stem cell transplantation. Blood. 2004;103(8):2936–2938. | |
Palladini G, Perfetti V, Perlini S, et al. The combination of thalidomide and intermediate-dose dexamethasone is an effective but toxic treatment for patients with primary amyloidosis (AL). Blood. 2005;105(7):2949–2951. | |
Dispenzieri A, Lacy MQ, Zeldenrust SR, et al. The activity of lenalidomide with or without dexamethasone in patients with primary systemic amyloidosis. Blood. 2007;109(2):465–470. | |
Kastritis E, Anagnostopoulos A, Roussou M, et al. Treatment of light chain (AL) amyloidosis with the combination of bortezomib and dexamethasone. Haematologica. 2007;92(10):1351–1358. | |
Dispenzieri A, Kyle RA, Lacy MQ, et al. Superior survival in primary systemic amyloidosis patients undergoing peripheral blood stem cell transplantation: a case-control study. Blood. 2004;103(10):3960–3963. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.