")
Back to Journals » International Journal of Nanomedicine » Volume 19
Nicorandil-Pretreated Mesenchymal Stem Cell-Derived Exosomes Facilitate Cardiac Repair After Myocardial Infarction via Promoting Macrophage M2 Polarization by Targeting miR-125a-5p/TRAF6/IRF5 Signaling Pathway
Authors Gong ZT , Xiong YY, Ning Y, Tang RJ, Xu JY, Jiang WY, Li XS, Zhang LL, Chen C, Pan Q, Hu MJ, Xu J, Yang YJ
Received 20 September 2023
Accepted for publication 18 February 2024
Published 29 February 2024 Volume 2024:19 Pages 2005—2024
DOI https://doi.org/10.2147/IJN.S441307
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Yan Shen
Zhao-Ting Gong,1 Yu-Yan Xiong,1,2 Yu Ning,1 Rui-Jie Tang,1 Jun-Yan Xu,3 Wen-Yang Jiang,1 Xiao-Song Li,1 Li-Li Zhang,1 Cheng Chen,1 Qi Pan,1 Meng-Jin Hu,1 Jing Xu,1 Yue-Jin Yang1
1State Key Laboratory of Cardiovascular Disease, Department of Cardiology, Fuwai Hospital, National Center for Cardiovascular Diseases, Chinese Academy of Medical Science and Peking Union Medical College, Beijing, 100037, People’s Republic of China; 2Department of Cardiology, Union Hospital, Tongji Medical College, Huazhong University of Science and Technology, Wuhan, 430022, People’s Republic of China; 3Department of Guangdong Provincial Key Laboratory of Coronary Heart Disease Prevention, Guangdong Cardiovascular Institute, Guangdong Provincial People’s Hospital, Guangdong Academy of Medical Sciences, Guangzhou, 510080, People’s Republic of China
Correspondence: Yue-Jin Yang, Email [email protected]
Background: Exosomes derived from bone marrow mesenchymal stem cells (MSC-exo) have been considered as a promising cell-free therapeutic strategy for ischemic heart disease. Cardioprotective drug pretreatment could be an effective approach to improve the efficacy of MSC-exo. Nicorandil has long been used in clinical practice for cardioprotection. This study aimed to investigate whether the effects of exosomes derived from nicorandil pretreated MSC (MSCNIC-exo) could be enhanced in facilitating cardiac repair after acute myocardial infarction (AMI).
Methods: MSCNIC-exo and MSC-exo were collected and injected into the border zone of infarcted hearts 30 minutes after coronary ligation in rats. Macrophage polarization was detected 3 days post-infarction, cardiac function as well as histological pathology were measured on the 28th day after AMI. Macrophages were separated from the bone marrow of rats for in vitro model. Exosomal miRNA sequencing was conducted to identify differentially expressed miRNAs between MSCNIC-exo and MSC-exo. MiRNA mimics and inhibitors were transfected to MSCs or macrophages to explore the specific mechanism.
Results: Compared to MSC-exo, MSCNIC-exo showed superior therapeutic effects on cardiac functional and structural recovery after AMI and markedly elevated the ratio of CD68+ CD206+/ CD68+cells in infarcted hearts 3 days post-infarction. The notable ability of MSCNIC-exo to promote macrophage M2 polarization was also confirmed in vitro. Exosomal miRNA sequencing and both in vivo and in vitro experiments identified and verified that miR-125a-5p was an effector of the roles of MSCNIC-exo in vivo and in vitro. Furthermore, we found miR-125a-5p promoted macrophage M2 polarization by inhibiting TRAF6/IRF5 signaling pathway.
Conclusion: This study suggested that MSCNIC-exo could markedly facilitate cardiac repair post-infarction by promoting macrophage M2 polarization by upregulating miR-125a-5p targeting TRAF6/IRF5 signaling pathway, which has great potential for clinical translation.
Keywords: mesenchymal stem cell, exosomes, nicorandil, macrophage polarization, myocardial infarction
Graphical Abstract:

Background
Cardiovascular disease is the leading cause of mortality worldwide, and nearly half of deaths are due to ischemic heart disease.1,2 Acute myocardial infarction (AMI) is a common disease caused by coronary acute closure, resulting in myocardial damage and functional impairment as consequences. Myocardium necrosis can occur as early as 1h after AMI with inflammatory signaling pathways activated and immune reactions induced. Macrophage is one of the most vigorous cells participating in all stages after AMI, including pro-inflammatory, cardioprotective, and tissue repair phases.3 In the early stage, macrophages adopt a pro-inflammatory phenotype which can produce pro-inflammatory mediators to recruit lymphocytes and also phagocytize necrotic cells, usually peaking between day 3 and day 5 after AMI. After the infarct area is cleared of cell debris, pro-inflammatory macrophages begin to undergo apoptosis, and newly arrived and differentiated macrophages polarize to anti-inflammatory M2 phenotype which facilitates the resolution of inflammation and tissue repair, peaking on day 7 post-AMI.4–6 These unique characteristics of macrophages can be a promising target in facilitating cardiac repair for AMI.
Mesenchymal stem cell (MSC)-based therapy is regarded as one of the most promising strategies for ischemic heart disease because of easy isolation and expansion, as well as its unique properties of regulating immune response and lack of immunogenicity.7–9 However, the beneficial effects of MSC observed in animal models have not been translated to AMI patients mainly attribute to low retention and survival of transplanted MSC in a local stressful microenvironment post-myocardial infarction (MI).10,11 The cardio-protection property of MSC exerts paracrine effects, mainly through exosomes.12 Exosomes are endosomal-originated extracellular vesicles with 30–150 nm in diameter that can be secreted by diverse cell types.13–15 MSC-derived exosomes (MSC-exos) not only had been testified to exhibit similar effects in cardiac repair as MSC for AMI,16 but also showed several strengths including low immunogenicity, high product stability and feasibility in clinical use.17 Although these advantages of exosomes have sparked great research interest in the academic community, the low efficacy of exosomes as MSC in cardiac repair still requires to be improved.18,19 In previous studies, when parent MSC were under different environmental stimuli or treatment with drugs, the bioactive contents of exosomes including mRNAs, non-coding RNAs, proteins and lipids, etc., would subsequently be changed and correspondingly conferred the new biological effects,20–22 thereby achieving powerful therapeutic effects.
Nicorandil has two pharmacological effects, which can be used as a mitochondrial KATP+ channel opener and a nitric oxide (NO) donor.23 Its pharmacological characteristics make it widely used in the treatment of ischemic heart disease and heart failure.24,25 Our previous studies found that both treatment with cardioprotective drugs (atorvastatin and Tongxinluo) and the two drug-pretreated MSC could enhance the effects of cardiac repair post-AMI via altering the mRNA or non-coding RNA profiles of their exosomes.18,26,27 Similar to statins and Tongxinluo, the nicorandil treatment also showed superior cardioprotective effect to MSC alone in heart failure rats28–31 and nicorandil-pretreatment could protect MSC from hypoxia/serum-deprivation injury in vitro as well.31,32 Therefore, we hypothesized that nicorandil-pretreated MSC-derived exosomes (MSCNIC-exo) could have distinct non-coding RNA profiles and also bettertherapeutic effects than MSC-derived exosomes (MSC-exo) in treating AMI.
In this study, we used a rat AMI model and in vitro experiments to investigate the efficacy of MSCNIC-exo in myocardial repair and M2 macrophage polarization. We found that miR-125a-5p was upregulated in MSCNIC-exo and then transferred to macrophage which modulated the TRAF6/IRF5 pathway to facilitate M2 macrophage polarization, resulting in a remarkably improvement in the efficacy of cardiac repair in AMI.
Methods
Fuwai Hospital’s Animal Department provided all the animals for this study. The study was conducted with the consent of the Experimental Animals Ethics Committee of Fuwai Hospital (FW-2022-0032), and all protocols adhered to the guidelines set by the National Institutes of Health. More detailed descriptions of experimental procedures are available in Additional File 1.
Rat Bone Marrow MSC Isolation and Pretreatment
Male Sprague-Dawley rats (3 weeks old, weighing 60–80g) were used to extract bone marrow. The bone marrow from femurs and tibias was flushed into a 10cm dish with IMDM and then cultured in complete medium (IMDM, 10% fetal bovine serum, 100 U/mL penicillin, and 100 μg/mL streptomycin) at 37°C in a 5% CO2 environment. The medium was changed after the first 24h culture to remove the non-adherent cells and after that fresh complete medium was changed every 48h. MSC were passaged when reached 80–90% confluence. MSC at passage 4 appeared spindle-shaped and were identified as CD11−, CD45−, CD29+, and CD90+ were used for further study. For nicorandil pretreatment, MSC at passage 4 were treated with 200 μmol/L nicorandil for 24h. Dimethyl sulfoxide (DMSO) was administered in equal volumes to the control groups.
Rat Bone Marrow-Derived Macrophage Isolation and Culture
Bone marrow was obtained by the same method as bone marrow MSC. Then bone marrow was centrifuged at 500g for 5min and cells were suspended in complete medium (DMEM, 10% fetal bovine serum, 100 U/mL penicillin, 100μg/mL streptomycin, 20ng/mL M-CSF) and cultured for another 6 days. Fresh complete medium was changed every 72h. We established M1 macrophage model in vitro by treating bone marrow derived macrophage (BMDM) with 100ng/mL lipopolysaccharide (LPS) and 50ng/mL interferon-γ (IFN-γ) for 12h, M2 macrophage in vitro model was established by treating BMDM with 10ng/mL interleukin-4 (IL-4) and 10ng/mL IL-13 for 48h.33,34 For exosome incubation experiments, we stimulated BMDM with 100 ng/mL LPS and 50ng/mL IFN-γ for 6 h and then incubated the cells with exosomes for another 48 h.
Exosome Extraction, Identification, and Quantification
After washed with Phosphate-buffered saline (PBS, pH7.4) for 3 times, MSC or nicorandil pretreated MSC were cultured with exosome-free FBS contained IMDM for 48h. Then, the culture supernatants were collected. To eliminate cells and debris, the culture supernatants were initially subjected to centrifugation at 300g for 10 minutes and 2000g for 20 minutes, followed by an additional centrifugation at 16500g for 30 minutes to remove large extracellular vesicles. The rough exosome pellet was acquired through ultracentrifugation at a force of 120000g for a duration of 70 minutes. Subsequently, the sediment was cleansed in PBS and subjected to another round of ultracentrifugation at the identical speed for 70 minutes. After that, the exosome pellet was resuspended in a specific amount of PBS and kept at −80°C for future investigation. We used 40 μg/mL exosomes for in vitro exosome incubation experiments.
Exosomes were detected using a transmission electron microscope (Tecnai G2 Spirit BioTwin, FEI, USA) and Nanoparticle Tracking Analysis (ZetaVIEW S/N 17–310, PARTICLE METRIX, Germany). Western blotting with antibodies against Tsg101 and Alix was used to identify exosomes. The microBCA protein assay kit (23235, Thermo Fisher Scientific, USA) was used to determine the levels of proteins in exosomes.
Rat AMI Model Establishment and Exosome Delivery
The animals were randomly assigned to the following groups: the Sham group, the AMI group (treated with PBS), and the Exo group (treated with different exosomes). Before the surgical procedure, female SD rats weighing 200–220g were given an intraperitoneal injection of pentobarbital sodium (50 mg/kg) to induce anesthesia. AMI model was induced by ligation of the left anterior descending coronary artery following the previously mentioned method.27 The successful induction of AMI was confirmed by obvious pale color change of distal site below the ligation site. The rats in the Sham group went through identical procedures, with the exception of not having left anterior descending coronary artery ligation. For other groups, exosomes (105 μg, in 100 μL PBS) or 100 μL PBS were injected into the border zone of infarcted hearts at three sites using a 31-gauge Hamilton syringe 30 minutes after the left anterior descending ligation. After the operation, ibuprofen was used for postoperative analgesia for 1 week.35
Cardiac Function Measurements
After administering isoflurane anesthesia, rats underwent transthoracic two-dimensional M-mode echocardiography using the Vevo 2100 high-resolution micro imaging system (Visual Sonic, Canada) at both baseline (3 days post-surgery) and endpoint (28 days post-surgery). Rats with left ventricular ejection fraction (LVEF) above 60% by baseline echocardiography 3 days post-surgery were excluded from the study. Throughout the measurement, the rats’ body temperature remained at 37°C while ensuring the heart rate stayed above 350 beats per minute. Additionally, measurements were taken for the thickness of left ventricular anterior wall and left ventricular posterior wall (LVAW and LVPW) and left ventricular inner diameter (LVID) at end systole (LVAWs, LVIDs, LVPWs) and end diastole (LVAWd, LVIDd, LVPWd). Calculations of left ventricular end-diastolic volume (LVEDV), and left ventricular end-systolic volume (LVESV), left ventricular ejection fraction (LVEF) and left ventricular fractional shortening (LVFS) were conducted following equations:  ,
, 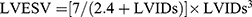 ,
,  , and
, and  . All procedures and analyses were conducted by a skilled researcher who was blinded to treatment.
. All procedures and analyses were conducted by a skilled researcher who was blinded to treatment.
Histological Analysis
Rats were sacrificed after endpoint echocardiography measurements were recorded. Hearts were collected and then fixed for 72 hours in 4% paraformaldehyde. Fixed hearts were then subjected to a dehydration process and embedded in paraffin, and subsequently sliced into serial sections that were 5 μm-thick. To measure the size of the infarct and the level of fibrosis in the left ventricle, Masson’s trichrome and Sirius red staining were used on the cardiac sections. The infarct size and collagen area were determined using ImageJ software. The infarct size was calculated as [(epicardial infarct ratio + endocardial infarct ratio)/2] × 100. The epicardial infarct ratio was obtained by dividing the epicardial infarct length by the epicardial circumferences. The endocardial infarct ratio was calculated similarly. Collagen area was calculated as the average ratio of collagen area to the total LV area (collagen area/total LV area × 100%). Hematoxylin–eosin (HE) staining was used to evaluate inflammatory cell infiltration. Angiogenesis was quantified by immunofluorescence of α-SMA and CD31. M2 macrophage polarization was determined by immunofluorescence of CD68 and CD206. The method of immunofluorescence staining is detailed described in Additional file 1. Corresponding primary antibodies were listed in Table S1.
RNA Extraction and Real-Time PCR Analysis
Trizol reagent (15596-018, Invitrogen, USA) was used to extract Total RNA (>200nt) from heart tissue, cultured cells, or exosomes following the instructions provided by the manufacturer. The RNA was converted into complementary DNA (cDNA) using a reagent kit (RR047B, Takara, Japan). The PowerUp™ SYBR™ Green Master Mix (A25742, Applied Biosystems, USA) was utilized for conducting real-time PCR on the QuantStudio 3 Real-Time PCR system (A28136, Applied Biosystems, USA). The miDETECT A Track miRNA qRT-PCR Starter Kit (C10712-2, RiboBio, China) was used to reverse transcribe and detect small RNA (20–200nt) following the manufacturer’s recommended procedure. After normalizing to GAPDH or U6, the expression levels of the genes and miRNAs were determined using the 2−ΔΔCT method. GenePharma synthesized the primers, and their sequences can be found in Table S2.
Exosomal miRNA Sequencing
RiboBio Company (Guangzhou, China) conducted miRNA sequencing on both MSC-exo and MSCNIC-exo. The manufacturer’s protocol was followed to extract exosomal RNA using MagZol (Magen). To evaluate the amount and quality of the exosomal RNA, the Qubit2.0 (Invitrogen, USA) and Agilent 2200 TapeStation (Agilent Technologies, USA) were utilized individually. In short, libraries of small RNA were prepared following the guidelines provided by NEBNext Multiplex Small RNA Library Prep Set for Illumina (Illumina, USA). The sequencing of the libraries was performed using HiSeq 2500 from Illumina, USA, with a single-end 50bp approach. Each group had three samples processed. The edgeR algorithm was used to identify miRNAs that were expressed differently based on the criteria of log2 (fold change) > 1 and P value < 0.05. The miRNA sequencing data are available in the Gene Expression Omnibus database (http://www.ncbi.nlm.nih.gov/gds) under accession number GSE229889.
MicroRNA and siRNA Transfection
Bone marrow MSC or BMDM cells were transfected with miR-125a-5p mimics (100 nM), miR-125a-5p inhibitors (100 nM), TRAF6 small interfering RNA (siRNA) (200 nM), or relative NC (100 nM) using Lipofectamine RNAiMAX (13778030, Invitrogen, USA) following the manufacturer’s instructions. After a 6-hour incubation, cell supernatants were discarded and replaced with fresh medium, cells were subsequently cultured for another 48 hours. The RiboBio (Guangzhou, China) synthesized microRNA mimics, inhibitors, and relative NCs. GenePharma (Shanghai, China) synthesized the siRNA and its corresponding NC.
Cell Line and Cell Culture
The human embryonic kidney cell line HEK293T were obtained from ATCC. Cells were cultured in complete medium (Dulbecco’s modified Eagle medium, 10% fetal bovine serum, 100 U/mL penicillin, and 100 μg/mL streptomycin) at 37°C in a 5% CO2 environment.
Statistical Analysis
Data are expressed as means ± standard error (SEM). GraphPad Prism 9.0 conducted all the analyses. For comparisons between two groups, the two-sided Student’s t-test was utilized, while for comparisons involving more than two groups, one-way ANOVA was conducted. P value < 0.05 is considered statistically significant.
Results
Characterization of Rat Bone Marrow MSC and MSC-Derived Exosomes
MSC were extracted from the bone marrow of 3-week-old SD rats. MSC passed to the fourth generations were spindle-shaped and were further identified as CD45−, CD11−, CD29+, and CD90+ by standard surface marker detection (Figure 1A). To evaluate the proper pretreatment concentration of nicorandil, MSC were treated with different concentrations of nicorandil for 24h. We found when the pretreatment concentration of nicorandil was higher than or equal to 300μM, the cell viability of MSC decreased, while at the concentration of 100 and 200μM the cell viability of MSC did not decrease, as shown by CCK-8 assay. And pretreatment concentration of 200μM significantly enhanced the effectiveness of exosomes derived from pretreated MSC in polarizing M2 macrophage compared with 100 μM. Therefore, we choose the concentration of 200μM for nicorandil pretreatment of MSC (Figure S1).
 |
Figure 1 Characterization of MSC-exo. (A) surface markers of bone marrow MSC detected by flow cytometry were shown as CD45−CD11−CD90+CD29+. (B) Representative Western blot images of exosomal protein markers (Alix and Tsg101) and MSC markers (CD73). (C) MSC-exo and MSCNIC-exo manifested cup-shaped morphology under TEM. Scale bar = 200 μm. (D) The particle size and concentration were analyzed by nanoparticle tracking analysis. No significant difference was found between MSC-exo and MSCNIC-exo. (E) Representative confocal images suggested that exosomes were endocytosed by BMDM. Scale bar = 10 μm. |
Furthermore, we verified the characteristics of exosomes derived from MSC. Cell supernatants from non-pretreated or nicorandil-pretreated MSC were collected, and exosomes were harvested by ultracentrifugation. Through transmission electron microscopy (TEM) imaging analysis, we confirmed that the exosomes derived from MSC (MSC-exo) and nicorandil pretreated MSC (MSCNIC-exo) both displayed a typical cup-shaped morphology (Figure 1C). Nanoparticle tracking analysis (NTA) was used to assess the concentration and size distribution of exosomes. Particle mean size and concentration did not differ significantly between MSC-exo and MSCNIC-exo (Figure 1D). By Western blot, we affirmed that typical exosome markers (Alix, Tsg101) and MSC-specific surface marker (CD73) were positive in MSC-exo (Figure 1B).
MSCNIC-Exo Improved Cardiac Function and Restrained Inflammation After Myocardial Infarction
To assess the therapeutic effect of MSCNIC-exo on cardiac function after AMI, we analyzed and compared echocardiographic cardiac function and pathological MI size among all groups. As shown in Figure 2 and Table S3, echocardiography revealed that MSCNIC-exo significantly improved LVEF and LVFS on the 28th day post-AMI compared with both MSC-exo and AMI group (LVEF: 42.00 ± 2.060% vs 33.13 ± 2.401% and 23.63 ± 2.554%; LVFS: 21.64 ± 1.205% vs 16.63 ± 1.326% and 11.51 ± 1.361%; p < 0.05–0.0001) (Figure 2A and B). MSCNIC-exo group had a trend to have smaller LVEDV and also have significant reduced LVESV compared to AMI group (Figure 2B). MSCNIC-exo was also demonstrated to markedly reduce both the infarct size (30.38 ± 2.089% vs 44.83 ± 1.100% and 53.88 ± 1.811%, p < 0.05–0.0001) and collagen area (19.49 ± 1.249% vs 29.33 ± 3.495% and 40.05 ± 2.348%, p < 0.05–0.0001) (Figure 2C–F, Table S3). Moreover, HE staining showed that MSCNIC-exo decreased the infiltration of inflammatory cells in MI border zone (Figure 2G, Table S3). Additionally, the levels of pro-inflammatory cytokines IL-6 and TNF-α in heart tissue were detected using ELISA and were significantly decreased in MSCNIC-exo group compared to AMI group (Figure 2H, Table S3 and Figure S2). Vessel densities of both arteriole (α-SMΑ staining) and capillary (CD31 staining) in border zone were also measured by immunofluorescence. Results showed that both arteriole and capillary density were notably increased in MSCNIC-exo group compared to MSC group and AMI group (p < 0.01–0.0001) (Figure 2I–L, Table S3). These results indicated that nicorandil pretreatment significantly improved the therapeutic efficacy of MSC-derived exosomes on the functional and structural repair of infarcted hearts, which presented as cardiac function improvement, infarct size and collagen area minimization, inflammation alleviation, and angiogenesis augmentation in the MI region.
 |
Figure 2 MSCNIC-exo strengthened cardiac repair in AMI rats. (A) Representative M-mode echocardiogram in different groups 4 weeks post-AMI. (B) Cardiac function including LVEF, LVFS, LVEDV, and LVESV were assessed 4 weeks post-AMI (n=9–10). (C and D) Representative images of heart cross-sections stained with Masson’s trichrome staining (C) and Sirius Red staining (D) 28 days after MI. Scale bar = 2000 μm. (E and F) Quantification of infarct size in C (E) and collagen area in D (F) (n=8–10). (G) Representative HE staining images of the border zone of the infarcted heart. Scale bar= 100 μm. (H) IL-6 expression levels in the infarct border zone tissue of rat hearts using ELISA (n=5). (I and J) Representative images of CD31 positively stained capillaries (I) and α-SMA positively stained arterioles (J) at the border zone. Scale bar= 50 μm. (K and L) Quantification of capillaries density in I (K) and arteriole density in J (L) (5 random fields per animal; n=4 for each group). All data are presented as mean ± SEM. Statistical analysis was performed by one-way ANOVA followed by Tukey’s test. *P<0.05, **P<0.01, ***P<0.001, ****P<0.0001. |
MSCNIC-Exo Promoted Polarization of Macrophage Toward M2 Phenotype Both in vivo and in vitro
Since a delayed transition of M1 toward M2 macrophage may interfere with the healing of the infarct heart, predisposing to the development of heart failure and adverse ventricular remodeling.36 We first use immunofluorescence to explore whether MSCNIC-exo treatment promotes the M2 polarization of macrophages in vivo. Infarcted hearts were collected on day 3 post-AMI and total macrophages were marked with CD68 and M2 macrophages were marked with CD206, and the ratio of double-positive cells (CD68+CD206+cells) to CD68+ cells in the border zone was used to evaluate M2 macrophage polarization. We found that MSCNIC-exo treatment significantly increased the ratio of (CD68+CD206+)/CD68+ cells in the border zone when compared to both MSC-exo and AMI groups (65.73 ± 3.280% vs 36.12 ± 2.371% and 7.303 ± 0.8796%, both p < 0.0001) (Figure 3A and B, Table S3). Meanwhile, we using qPCR and western blot to identify the mRNA and protein levels of M1 and M2 macrophage markers in infarct hearts. The results showed that MSCNIC-exo also significantly increased the mRNA expression of M2 macrophage markers of Arg1, Mrc1, IL-4R, and Tgfb1 (p < 0.05–0.001) compared to AMI groups (p < 0.05–0.001) with the former two level still higher than MSC-exo (both p < 0.05), while M1 macrophage markers of IL-1β, TNF-α, and IL-6 (p < 0.01–0.0001) decreased compared to both MSC-exo and AMI groups (Figure 3C–E, Table S3). Furthermore, the protein expression of Arg1 in myocardium tissue of the MSCNIC-exo group was markedly elevated compared with both MSC-exo and AMI groups as well (p < 0.01–0.001) (Figure 3F and G, Table S3).
 |
Figure 3 MSCNIC-exo polarized macrophage toward M2 phenotype both in vivo and in vitro. (A) M2 macrophage at the border zone on day 3 post-MI was identified by labeling with CD206 (green) and CD68 (red) and nuclei (blue). (B) Quantification of the CD206+CD68+/CD68+ ratio of each group in A (5 random fields per animal; n=5 for each group). (C and D) mRNA expression levels of M1 macrophage markers IL-1β, IL-6, and TNF-α in infarcted myocardium at day 3 after AMI (n=5). (E) mRNA expression levels of M2 macrophage markers Mrc1, Tgfb1, IL-4R, and Arg1 in infarcted hearts at day 3 after AMI (n=5). (F and G) Representative western blot image (F) and quantification of protein expressions (G) of Arg1 in infarcted hearts at day 3 after AMI (n=5). (H) IL-1β and TNF-α levels in the supernatants of different group using ELISA (n=3) (I) IL-4, Vegfa and IL-10 levels in the supernatants of different group using ELISA (n=3). (J) mRNA levels of M2 macrophage markers Mrc1, Tgfb1, IL-4R, IL-10, Vegfa, and Arg1 in BMDM of each group (n=6). (K) mRNA expression levels of M1 macrophage markers IL-1β, and TNF-α in BMDM (n=6). (L) Representative western blot image of Arg1 expression in BMDM. (M) Quantification of protein expressions of Arg1 in BMDM (n=4). All data are presented as mean ± SEM. Statistical analysis was performed with one-way ANOVA followed by Tukey’s test. *P<0.05, **P<0.01, ***P<0.001, ****P<0.0001. |
We further explored whether MSCNIC-exo exerted greater capacity in M2 macrophage polarization in vitro. BMDM labeled with Dil dye were co-cultured with PKH67 labeled MSC-exo or MSCNIC-exo for 12 hours, and the internalization of exosomes was further verified by fluorescence imaging. We observed that both the MSCNIC-exo and MSC-exo can be uptaken by BMDM (Figure 1E). Next, we pretreated BMDM with LPS (100 ng/mL) and IFN-γ (50 ng/mL) to simulate in vitro inflammatory micro-environment after myocardial infarction. After stimulation with LPS and IFN-γ for 6 hours, we added MSCNIC-exo, MSC-exo, or PBS to the stimulated BMDM for another 24h or 48h. Neither LPS and IFN-γ nor PBS/exosomes were added to the control group. Results of CCK8 showed similar cell viability of BMDM after treating with exosomes for 24 or 48 hours (Figure S3). We first collected culture supernatants of control group and BMDM treated with MSCNIC-exo, MSC-exo, or PBS for 24h or 48h, and detected the secreted proteins by ELISA. Compared with control group, the notably increased levels of IL-1β and TNF-α in PBS group (all P < 0.0001) represented the successful simulation of inflammatory micro-environment in vitro (Figures S3, 3H and L). We also found culture supernatants of BMDM treated with exosomes for 48 hours showed lower expression levels of M1 macrophage marker (IL-1β and TNF-α) as well as higher levels of M2 macrophage marker expression (IL-4, Vegfa and IL-10) (Figures S3, 3H and L). Therefore, we chose to treat BMDM with different exosomes for 48 hours in following in vitro experiments. ELISA results also revealed that MSCNIC-exo not only significantly increased the concentrations of IL-4, Vegfa, and IL-10 but also downregulated the concentrations of IL-1β and TNF-α in the supernatant compared with PBS and MSC-exo group (p < 0.05–0.0001) (Figure 3H and L). PCR results showed that MSCNIC-exo significantly upregulated the mRNA expression levels of M2 markers including Mrc1, Arg1, Tgfb1, IL-4R, Vegfa, and IL-10 (p < 0.05–0.0001), while significantly reducing M1 markers including IL-1β and TNF-α compared with both the MSC-exo and PBS groups (p < 0.05–0.0001) (Figure 3J and K, Table S4). Consistently, the MSCNIC-exo group also displayed significantly increased protein level of Arg1 (p < 0.01–0.0001) compared with the other two groups (Figure 3L and M, Table S4). These results indicated that MSCNIC-exo remarkably promoted macrophage M2 polarization both in vivo and in vitro.
miRNA-125a-5p Enriched in MSCNIC-Exo and Stood Out as the Candidate Effector in Mediating Cardiac Reparative Effects
To explore whether MSCNIC-exo contain any key miRNAs that contribute to its strong cardiac repair and inflammation modulatory potential. Small RNA sequencing was conducted on MSCNIC-exo and MSC-exo, and analysis of differentially expressed miRNA was performed with a two-fold change and p < 0.05 threshold cutoff standard. We then identified 23 candidate miRNAs (Figure 4A and B). Based on 11 miRNAs enriched in MSCNIC-exo, we searched for miRNAs known to be involved in either immunomodulation (miR-148a-3p, miR-125a-5p, miR-100-5p, miR-23a-3p, miR-146b-5p), or cardiac repair facilitation (miR-125a-5p, miR-93-5p, miR-351-5p), miR-125a-5p emerged as a probable candidate effector for cardiac repair and macrophage polarization (Figure 4C). To confirm the results provided by miRNA sequencing, we detected the expression of miR-125a-5p in nicorandil-pretreated and untreated MSC (MSCNIC and MSC) by qPCR. Nicorandil pretreatment significantly increased the expression of miR-125a-5p in both MSCNIC and MSCNIC-exo compared with MSC and MSC-exo (both p < 0.01) (Figure 4D and E).
 |
Figure 4 Differential expressed exosomal miRNA expression and miR-125a-5p exerted as a main effector for cardiac repair capacity of MSCNIC-exo. (A) Heatmap of differentially expressed miRNAs between MSC-exo and MSCNIC-exo (n=3). (B) Volcano plot showing log2 (Fold change) on the x-axis and -log10 (P value) on the y-axis (n=3). (C) miRNAs upregulated in MSCNIC-exo which engaged in inflammation modulation or cardiac repair. (D and E) miR-125a-5p expression levels in MSC (D) (n =5) and exosome (E) (n =6). (F) miR-125a-5p expression levels in MSC or MSCNIC transfected with miR-125a-5p mimics, inhibitors, or its negative control (NC) (n = 4). (G) Representative images of echocardiograms at 28 days post-AMI. (H) Analysis of LVEF, LVFS, LVEDV, and LVESV 4 weeks post-AMI (n = 9–10). (I and J) Representative images of Masson’s trichrome staining (I) and Sirius red staining (J) 4 weeks post-AMI. Scale bar = 2000 μm. (K and L) Analysis of Masson’s trichrome staining (K) and Sirius red staining (L) at 4 weeks post-AMI (n=7). (M) Representative HE staining images of the border zone of the infarcted heart. Scale bar= 100 μm. (N and O) Representative images of CD31 positively stained capillaries (N) and α-SMA positively stained arterioles (O). Scale bar= 50 μm. (P and Q) Quantification of capillaries density in N (P) and arteriole density in O (Q) (5 random fields per animal; n=4 for each group). All data are presented as mean ± SEM. Statistical analysis was performed with one-way ANOVA followed by Tukey’s test. *P<0.05, **P<0.01, ***P<0.001, ****P<0.0001. |
To further clarify whether miR-125a-5p mediates the beneficial impact of MSCNIC-exo on improving post-infarct cardiac repair in vivo, we transfected miR-125a-5p inhibitors, mimics, and their corresponding negative controls (NC) into MSCNIC or MSC and collected exosomes from these MSC. The efficiency of miR-125a-5p overexpression in MSC or knockdown in MSCNIC was quantified (Figure 4F). Then, we delivered exosomes of MSC-exo(mimic NC), MSC-exo(mimics), MSCNIC-exo(inhibitor NC), MSCNIC-exo(inhibitors)) to the hearts of AMI rats. In comparison with the MSCNIC-exo(inhibitor NC) group, the MSCNIC-exo(inhibitors) group showed significantly lower LVEF (30.75 ± 1.213% vs 48.09 ± 1.486%, p < 0.0001) and LVFS (15.20 ± 0.6576% vs 25.24 ± 0.8648%, p < 0.01) with enlarged LVESV (p < 0.05) in the fourth week after AMI, while the MSC-exo(mimics) group showed significantly higher LVEF (41.15 ± 2.263% vs 33.29 ± 1.667%, p < 0.0001) and LVFS (21.13 ± 1.324% vs 16.59 ± 0.9273%, p < 0.01) compared to MSC-exo(mimic NC) group (Figure 4G and H, Table S5). Consistently, the MSCNIC-exo(inhibitors) extended both the infarct size and collagen area (p < 0.01–0.001), while the MSC-exo(mimics) minimized the infarct size and collagen area (p < 0.05–0.01) (Figure 4I–L, Table S5). HE staining showed that the degree of inflammation infiltration in the infarcted hearts of the MSC-exo(mimics) group were alleviated compared to AMI and MSC-exo(mimic NC) group (Figure 4M). Furthermore, MSCNIC-exo(inhibitors) significantly suppressed angiogenesis in the infarcted heart, while correspondingly MSC-exo(mimics) greatly promoted angiogenesis. (Figure 4N–Q, Table S5). These results verified that miR-125a-5p contributed to the superior effect of MSCNIC-exo on cardiac repair post-MI.
miRNA-125a-5p in MSCNIC-Exo Promoted Macrophage M2 Polarization Both in vivo and in vitro
We further clarified the role of miR-125a-5p in MSCNIC-exo in macrophage polarization both in vivo and in vitro. As shown in Figure 5 and Table S5, double immunofluorescence staining of heart tissues displayed that MSCNIC-exo(inhibitors) group had a significantly lower ratio of (CD68+ CD206+)/CD68+ cells in the border zone on the 3rd day after AMI that the MSCNIC-exo(inhibitors NC) group (47.64 ± 1.189% vs 62.44 ± 4.530%, p < 0.05), while the MSC-exo(mimics) group had a greatly increased ratio of (CD68+ CD206+)/CD68+ cells compared to MSC-exo(mimics NC) group (52.32 ± 4.922% vs 38.33 ± 2.138%, p < 0.05). (Figure 5A and B, Table S5). Next, we detected the mRNA levels of markers of M1 and M2 macrophage in the infarct hearts. MSCNIC-exo(inhibitors) also significantly downregulated the mRNA expression of M2 macrophage markers Arg1, Mrc1, IL-4R, and Tgfb1 compared to MSCNIC-exo(inhibitors NC) group (p < 0.01–0.0001), and all of them except Mrc1 were upregulated in MSC-exo(mimics) group compared with MSC-exo(mimics NC) group (p < 0.05–0.0001) (Figure 5C, Table S5). Correspondingly, MSCNIC-exo(inhibitors) resulted in upregulation of the expression of M1 macrophage markers IL-6 and TNF-α except IL-1β compared with MSCNIC-exo(inhibitors NC) group (p < 0.05–0.01), while MSC-exo(mimics) significantly decreased only TNF-α compared with MSC-exo(mimic NC) group (p<0.05) (Figure 5D, Table S5). Additionally, MSCNIC-exo(inhibitors) significantly repressed the protein expression of Arg1 in myocardial tissue in comparison to MSCNIC-exo(inhibitors NC) (p < 0.05) (Figure 5E and F, Table S5).
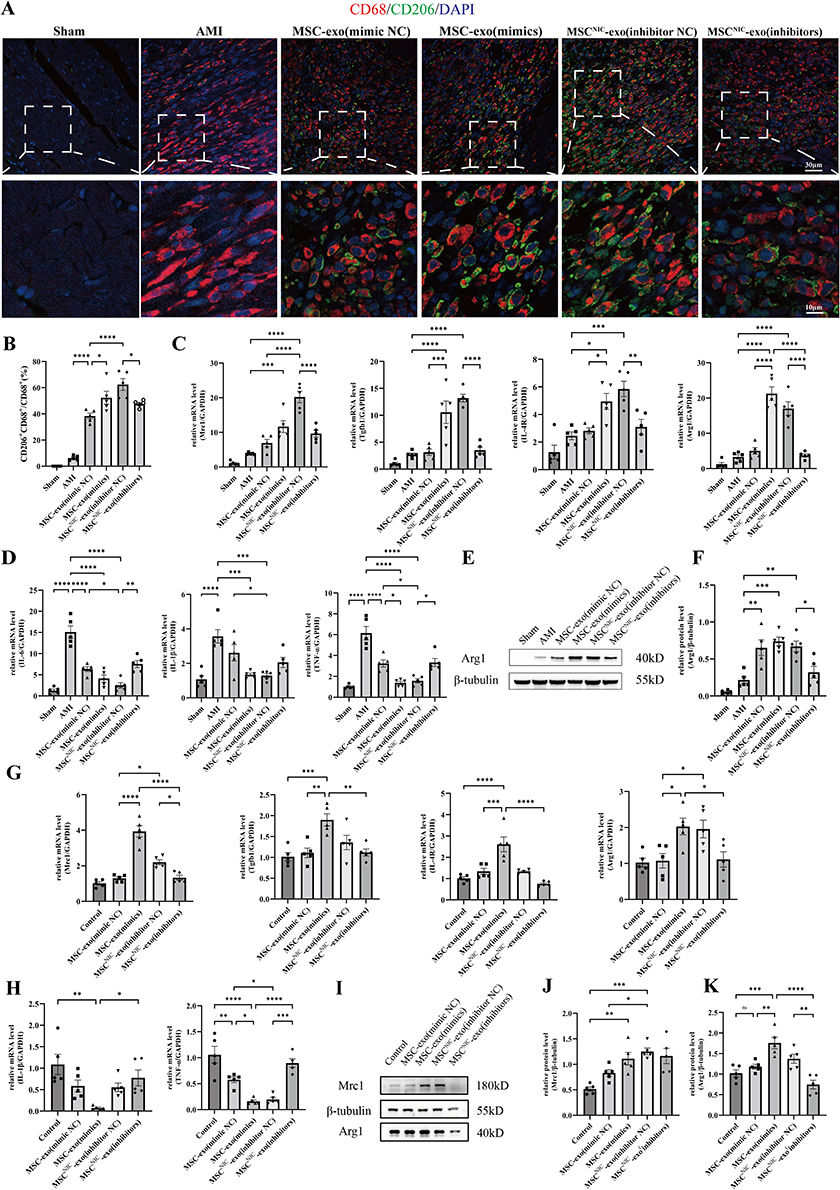 |
Figure 5 miR-125a-5p played a crucial role in MSCNIC-exo mediated M2 macrophage polarization both in vivo and in vitro. (A) M2 macrophage at the border zone 3 days post-AMI was identified by staining with CD206 (green) and CD68 (red) and nuclei (blue). (B) Quantification of the CD206+CD68+/CD68+ ratio of each group in (E)(5 random fields per animal; n=5 for each group). (C) mRNA expression of M2 macrophage markers Mrc1, Tgfb1, IL-4R, and Arg1 in infarcted hearts 3 days post-AMI (n=5). (D) mRNA expression of M1 macrophage markers IL-1β, IL-6, and TNF-α in infarcted hearts 3 days post-injury (n=5). (E) Representative western blot image of Arg1 expression in infarcted hearts. (F) Quantification of protein expressions of Arg1 in infarcted hearts 3 days post-AMI (n=5). (G) mRNA expression levels of M2 macrophage markers Mrc1, Tgfb1, IL-4R, and Arg1 in BMDM (n=5). (H) mRNA expression levels of M1 macrophage markers IL-1β and TNF-α in BMDM (n=5). (I) Representative western blot image of Arg1 and Mrc1 expression in BMDM. (J and K) Quantification of protein expression levels of Mrc1 (J) and Arg1 (K) in BMDM (n=5). All data are presented as mean ± SEM. Statistical analysis was performed with one-way ANOVA followed by Tukey’s test. *P<0.05, **P<0.01, ***P<0.001, ****P<0.0001. |
We next performed in vitro experiments by incubating LPS/IFN-γ-stimulated BMDM with differently transfected MSC-derived exosomes for 48h. As shown in Figure 5 and Table S6, MSC-exo(mimics) remarkably enhanced mRNA expression of M2 polarization-related genes including Mrc1, Arg1, Tgfb1, IL-4R (p < 0.05–0.0001), and restrained M1 markers expression of TNF-α (p<0.05) and IL-1β without reaching significance (p > 0.05) (Figure 5G and H, Table S6). Accordingly, MSCNIC-exo(inhibitors) downregulated M2 markers of Mrc1 (p < 0.01) although Arg1, Tgfb1 and IL-4R did not reach significance (P > 0.05) compared to MSCNIC-exo(inhibitors NC), and upregulated the M1 markers of TNF-α (p < 0.01) and IL-1β without reaching significance (p > 0.05) (Figure 5G and H, Table S6). Additionally, MSC-exo(mimics) also increased the protein level of Arg1 (p<0.01) and Mrc1 (p > 0.05), while MSCNIC-exo(inhibitors) conducting the opposite effect (Figure 5I–K, Table S6). These results indicated that miR-125a-5p is crucial in implementing the function of MSCNIC-exo in promoting cardiac repair post-AMI via facilitating the polarization of macrophage toward M2 phenotype.
miRNA‑125a‑5p Polarize Macrophage to M2 Phenotype by Inhibiting TRAF6/IRF5 Pathway
As shown in Figure 6, the expression of miR-125a-5p was significantly increased in M2 macrophages compared with M1 macrophages in vitro, indicating a possible correlation between the increase of miR-125a-5p and M2 polarization of macrophages (Figure 6A). We further embark on verifying the role of miR-125a-5p in macrophage polarization. Transfection of miR-125a-5p mimics or inhibitors was demonstrated to significantly upregulate or downregulate the miR-125a-5p expressions in BMDM compared with corresponding NC (both p < 0.01) (Figure 6B). The results of qPCR showed miR-125a-5p mimics markedly increased the expression of M2 macrophage markers (Mrc1, Tgfb1, IL-4R and Arg1, all p < 0.05–0.0001) and decreased the expression of M1 macrophage markers (IL-1β and TNF-α, p < 0.05 and 0.001) compared with the mimic NC (Figure 6C and D, Table S7). Consistently, miR-125a-5p inhibitors significantly downregulated Mrc1 and Arg1 (both p < 0.05) as well as upregulated IL-1β and TNF-α (both p < 0.001) when compared with the inhibitor NC (Figure 6C and D, Table S7). Furthermore, western blot analysis showed miR-125a-5p mimics significantly elevated the protein expression of Mrc1 and Arg1 (p < 0.05 and 0.0001), while miR-125a-5p inhibitors notably suppressed Arg1 protein level (p < 0.0001) (Figure 6E–G, Table S7), confirming the key regulatory role of miR-125a-5p in promoting macrophage M2 polarization.
 |
Figure 6 miR-125a-5p participated in the polarization of M2 macrophage by regulating TRAF6. (A) miR-125a-5p expression levels in M1 and M2 macrophage (n=6). (B) miR-125a-5p expression levels in BMDM transfected with miR-125a-5p mimics, inhibitors, or its negative control (NC) (n = 3). (C and D) mRNA expression of M1 macrophage markers (IL-1β and TNF-α) (C) and M2 markers (Mrc1, Tgfb1, IL-4R, and Arg1) (D) in BMDM after transfected with miR-125a-5p mimics, inhibitors, or corresponding NC (n=5). (E–G) Representative western blot image (E) and quantification of protein expression (F and G) of M2 markers (Mrc1 and Arg1) in BMDM of each group (n=5). (H) Relative luciferase activity of miR-125a-5p mimics-treated HEK293T cells which transfected with TRAF6-wild-type or TRAF6-mutant plasmids was determined. (I) TRAF6 mRNA expression level in BMDM (n=4). (J and K) Representative western blot image (J) and quantification of the protein expression level (K) of TRAF6 in BMDM (n=5). (L) TRAF6 mRNA expression level in BMDM after transfected with miR-125a-5p mimics, inhibitors or corresponding NC (n=5). (M–O) Representative western blot image (M) and quantification of protein expression of TRAF6 and IRF5 (N and O) in BMDM (n=4). All data are presented as mean ± SEM. Statistical analysis was performed with one-way ANOVA followed by Tukey’s test. *P<0.05, **P<0.01, ***P<0.001, ****P<0.0001. |
TargetScan and miRDB predicted that TRAF6 is a potential downstream target of miR-125a-5p (Table S8). To confirm the direct binding between miR-125a-5p and the 3′UTR target region of TRAF6, we conducted the luciferase reporter assay. The results indicated that the miR-125a-5p mimics specifically reduced luciferase activity in the TRAF6 wild type 3′UTR group, while it did not have any impact on the TRAF6 mutated 3′UTR group. This suggests that miR-125a-5p binds to the 3′-UTR of TRAF6 mRNA and thus resulting in the inhibition of TRAF6 (Figures 6H and S4). In vitro experiments showed that the mRNA and protein levels of TRAF6 in the MSC-exo (mimics) group were significantly lower than the MSC-exo (mimic NC) group (p < 0.01 and <0.05), and MSCNIC-exo (inhibitors) increased the levels of TRAF6 though not reaching significance (both p > 0.05) (Figure 6I–K, Table S6). Next, we validated the relationship between miR-125a-5p and TRAF6 by directly overexpressing or knocking down miR-125a-5p in BMDM as well. Results from qPCR showed that miR-125a-5p mimics significantly reduced TRAF6 mRNA expressions (p < 0.05), and miR-125a-5p inhibitors significantly increased TRAF6 levels (p < 0.0001) (Figure 6L, Table S7). Results from western blot confirmed that miR-125a-5p mimics transfection caused a notable downregulation of TRAF6 protein levels (p < 0.01), while inhibitors caused a markedly upregulation in TRAF6 protein levels (p < 0.05) (Figure 6M and N, Table S7). Hence, we affirmed that miR-125a-5p can downregulate TRAF6. We further found that miR-125a-5p mimics also inhibited IRF5 expression (p < 0.01 vs mimic NC), while miR-125a-5p inhibitors showed the opposite outcome (Figure 6M and O, Table S7), revealing that IRF5 might be a potential downstream molecule of miR-125a-5p and TRAF6.
To further verify the role of TRAF6 in M2 macrophage polarization and whether TRAF6 can regulate the expression of IRF5, we used siRNA to knock down TRAF6 in BMDM. We verified the knockdown efficiency of TRAF6 through qPCR and western blotting (Figure 7A–C, Table S9). Results showed that knocking down TRAF6 resulted in markedly lower expression of IRF5 in BMDM (p < 0.05 vs siRNA NC) (Figure 7B and D, Table S9), with increased expression of Mrc1 and Arg1 as well as repressed IL-1β and TNF-α expression (p < 0.05–0.0001 vs siRNA NC) (Figure 7E–H, Table S9). Correspondingly, TRAF6 knockdown resulted in downregulation of Arg1 protein expression (Figure 7I and J, Table S9). Therefore, we identified the pivotal role of TRAF6 in macrophage M2 polarization and confirmed that IRF5 was a downstream effector of TRAF6.
 |
Figure 7 IRF5 may be a down-stream effector of TRAF6 in M2 macrophage polarization. (A) TRAF6 mRNA expression level in BMDMs after si-TRAF6 or corresponding NC transfection (n=4). (B–D) Representative western blot image (B) and quantification of the protein expression level of TRAF6 (C) (n=4) and IRF5 (D) (n=6) in BMDM after transfected with si-TRAF6 or corresponding NC. (E and F) mRNA expression of M2 markers (Mrc1 and Arg1) in BMDM (n=6). (G and H) mRNA expression of M1 markers (IL-1β and TNF-α) in BMDM (n=5–6). (I and J) Representative western blot image (I) and quantification of protein expression levels of Arg1 (J) in BMDM (n=4). All data are presented as mean ± SEM. Statistical analysis was performed with one-way ANOVA followed by Tukey’s test. *P<0.05, **P<0.01, ***P<0.001, ****P<0.0001. |
Taken together, these results suggest that miR-125a-5p plays a crucial role in M2 macrophage polarization via TRAF6/IRF5 signaling pathway inhibition, which is a key downstream mechanism.
Discussion
The main findings of this study were as follows: (1). Nicorandil pretreatment enhanced the cardiac function and structural repair ability of MSC-exo in AMI. (2). MSCNIC-exo can promote the polarization of macrophages toward M2 phenotype both in vivo and in vitro. (3). MiR-125a-5p enriched in MSCNIC-exo acted as the main effector molecule facilitating macrophage M2 polarization in enhancing cardiac repair after MI. (4). The effect of miR-125a-5p on macrophage M2 polarization was mainly achieved through inhibiting the TRAF6/IRF5 signaling pathway.
Over the past decade, MSC transplantation has been found to have beneficial effects on heart repair in animal models or humans with AMI,37 and the cardioprotective effects of MSC are mainly achieved through their paracrine effects and exosome transfer.27,38 However, the low survival rate of MSC in infarct region limits its therapeutic effect.39 Cell-free therapy with MSC-derived exosome has similar therapeutic effects on AMI with more advantages, such as minimal immunogenicity, product stability, and systemic delivery efficacious.17,18 Hence, cell-free therapy with the exosomes derived from MSC has attracted increasing attention. Exosomes are nanosized (30–150 nm) extracellular vesicles with double-layer lipid membrane and play a vital role in cell-to-cell communication.14,15 Despite the advantages that the cell-free therapy may have, the limited therapeutic efficacy is the same shortcoming as MSC and needs to be further enhanced.18 Although transgenic methods are the most effective strategy to improve the efficacy of bone marrow MSC and the exosomes, these methods are currently difficult for clinical translation.18,40 In contrast, we reported that pretreatment of MSC with cardioprotective drug is a clinically promising method to enhance the effect of MSC and exosomes on cardiac repair for AMI.18,26,27 Since Nicorandil has been proven to have significant effects in protecting cardiac cells, reducing cardiovascular events and directly protecting MSC from apoptosis under H/SD conditions in vitro,25,31,32 we used nicorandil as pretreatment drug for MSC in the present study and found that intramyocardial injection of MSCNIC-exo had superior therapeutic effects in cardiac repair post-MI with cardiac function enhanced, infarct size and collagen area minimized, inflammatory cells and cytokines infiltration suppressed, angiogenesispromoted, as well as macrophage M2 polarization facilitated in the infarct region.
After AMI, injury-related molecules are released by necrotic myocardium, and cardiac inflammation is initiated41,42 as the process of myocardial repair following AMI which can be divided into three stages.3,43 The initial inflammatory phase begins with the removal of dead cells and debris, gradually develops into the proliferative and reparative phase with wound healing and scar formation. The excessive inflammatory cells and cytokines during the inflammatory phase not only clear necrotic myocardium, but also cause further cellular damage and adverse tissue remodeling.44–46 Therefore, in the process of myocardial repair following AMI, a balanced inflammatory process mediated by various immune cells and inflammatory factors is decisive.47 Macrophages, in particular, are active during all phases after AMI and play an important role in infarcted myocardium healing.3,48 Generally, there are two subpopulations of macrophages: pro-inflammatory macrophages (classically activated M1 macrophages) and constructive macrophages (alternatively activated M2 macrophages).49 M2 macrophages dominate in infarct myocardium repair by producing proangiogenic and reparative factors such as Tgfb1, IL-10, and Vegfa.50 It has been reported that the transition of M1 toward M2 macrophage was associated with the healing process of the infarcted heart, and delayed M2 polarization in infarcted heart lay a higher risk of adverse ventricular remodeling and heart failure.36 Therefore, macrophage polarization is considered a promising therapeutic target for AMI.51–53 Our results showed that MSCNIC-exo can effectively shift M1 macrophage toward M2 phenotype both in vivo and vitro, thus verifying that MSCNIC-exo indeed has a strong ability in facilitating M2 macrophage polarization. Furthermore, we found that M2 macrophages polarized by MSCNIC-exo exhibited elevated expression of Tgfb1 which is a proangiogenic and reparative factor, corresponding to a smaller infarct size and enhanced angiogenesis in the MSCNIC-exo treated rat infarcted hearts. Hence, the ability in polarizing M2 macrophage underlies the superior cardiac repair efficacy of MSCNIC-exo for AMI.
The influence of exosomes on recipient cells is largely determined by miRNA, an important component of exosomes.54 By miRNA sequencing of exosomes, we found that miR-125a-5p was highly expressed in MSCNIC-exo. MiR-125a-5p is correlated to cardiovascular diseases55 and is highly expressed in M2 macrophages,56 suggesting that it could be a key candidate effector for the cardio-protection and immunomodulation ability of MSCNIC-exo. MiR-125a-5p was reported to be associated with cardioprotection in the ischemia-reperfusion model of mice and swine.57 MiR-125a-5p in the serum of patients with advanced heart failure was downregulated and negatively correlated with the biomarkers of heart failure, suggesting that miR-125a-5p is a protective effector of heart failure.58 However, the role of miR-125a-5p in acute myocardial infarction has not been elucidated. In the present study, we verified the key role of miR-125a-5p in promoting cardiac repair by overexpressing or knocking down the level of miR-125a-5p in MSC-exo or MSCNIC-exo. We found that MSCNIC-exo (inhibitors) with downregulated miR-125a-5p expression worsened the powerful cardiac repair effects of MSCNIC-exo in AMI, resulting in cardiac function deterioration, infarct size extension, angiogenesis suppression and M2 macrophage reduction. Accordingly, MSC-exo (mimics) with higher level of miR-125a-5p showed stronger cardiac repair effects and macrophage polarization toward M2 phenotype. The key effect of miR-125a-5p in cardiac repair after AMI was then revealed.
In the present study, we confirmed that miR-125a-5p could downregulate downstream molecular TRAF6. TRAF6 is crucial in the induction of inflammatory response,59,60 and is also significantly associated with proinflammatory cytokine production and neutrophil infiltration in the myocardium after ischemic/reperfusion injury.61,62 TRAF6-mediated signals play a crucial role in the immune system by significantly impacting the development, stability, and stimulation of innate immune cells including macrophages,63 particularly regulating macrophage phenotypic switching.64,65 It was also reported that negative regulation of TRAF6 contributes to attenuation of postinfarct cardiac dysfunction and remodeling,66 indicating that TRAF6 may be a key downstream molecule of the effect of miR-125a-5p on the macrophage M2 polarization and cardiac repair. Our in vitro data identified that not only M2 polarized macrophages have lower TRAF6 levels but also macrophages with TRAF6 knockdown can polarize toward M2 phenotype, thus confirming the vital role of TRAF6 downregulation in M2 macrophage polarization.
We also found that downregulation of TRAF6 inhibited the expression of IRF5, indicating that IRF5 is a downstream molecule of TRAF6. IRF5 is a member of the interferon-regulatory factor family, which directly regulates TNF-α and IL-1β and participates in the regulation of a range of other inflammatory genes.53,67 IRF5 is a master regulator of macrophage activation and phenotype transformation, enhanced expression of IRF5 drives macrophage toward M1 phenotype and the ectopic expression of IRF5 in M2 macrophages leads to a decrease in the expression of many M2 phenotypic markers, such as Mrc1 and IL-10 after LPS stimulation.67–69 Our in vitro data demonstrated that M2 polarized macrophages have lower levels of IRF5, confirming that IRF5 is an important effector of macrophage polarization. IRF5 is also a promising therapeutic target in inflammation-associated cardiovascular diseases, such as atherosclerosis, ischemic/reperfusion injury and MI.53,70–72 Silencing IRF5 promotes cardiac repair and mitigates heart failure by facilitating myocardial inflammation resolution after infarction.53 Our results in the present study verified that the TRAF6/IRF5 signaling pathway is a crucial downstream pathway for exosomal miR-125a-5p taking effect in cardiac repair and macrophage polarization after AMI, and could be a promising therapeutic target for cardiac repair.
There are still several limitations that need to be noted. Firstly, exosomes can also be up-taken by other types of cells in infarcted hearts, such as cardiomyocytes or fibroblasts, and further research is needed on the unknown effects. Secondly, there may be some other differentially expressed miRNAs in MSCNIC-exo that may also have cardioprotective effects or regulate macrophage polarization, and miR-125a-5p may have other target molecules in macrophages, so further exploration is needed. Finally, this study adopted intramyocardial injection to deliver exosomes to the infarcted myocardium. Whether other clinically feasible delivery methods such as coronary or intravenous injection are still effective has not been verified.
Conclusions
We found that nicorandil-pretreated MSC-derived exosomes had markedly enhanced cardiac repair effect after AMI by facilitating M2 macrophage polarization, which can be partly attributed to upregulated miRNA-125a-5p and subsequently suppressed TRAF6/IRF5 signaling pathway. Our study provides a novel and clinically feasible strategy for enhancing the therapeutic efficacy of MSC-exo for cardiac repair in AMI with high clinical translational potential.
Abbreviations
MSC, Mesenchymal stem cell; SD, Sprague–Dawley; NIC, Nicorandil; AMI, Acute myocardial infarction; BMDM, Bone marrow-derived macrophage; CSF, Colony stimulating factor; LPS, Lipopolysaccharide; IFN-γ, Interferon—γ; IL, Interleukin; Mrc1, Mannose Receptor C Type 1; Tgfb1, Transforming growth factor beta 1; Vegfa, Vascular Endothelial Growth Factor A; Arg1, Arginase 1; TRAF6, TNF receptor associated factor 6; IRF5, Interferon regulatory factor 5; miRNA, MicroRNA; PBS, Phosphate-buffered saline; TEM, Transmission electron microscope; NTA, Nanoparticle tracking analysis; LVEF, Left ventricular ejection fraction; LVFS, Left ventricular fractional shortening; LVEDV, Left ventricular end-diastolic volume; LVESV, Left ventricular end-systolic volume; HE, Hemoxylin-eosin; NC, Negative control; qRT-PCR, Quantitative reverse transcription-polymerase chain reaction; IL-4R, IL-4 receptor.
Data Sharing Statement
The datasets generated and analyzed in the present study are available via the Gene Expression Omnibus under accession code GSE229889, https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE229889.
Ethics Approval and Consent to Participate
Animal protocols were performed with approval of the Experimental Animals Ethics Committee of Fuwai Hospital (FW-2022-0032).
Funding
This project was supported by the grants from the National Key Research and Development Program of China (No. 2017YFC1700503), CAMS Innovation Fund for Medical Sciences (CIFMS) (2016-12M-1-009), the National Natural Science Foundation of China (Nos. 81573957, 81774292, 81874461, 82100313, 82300332 and 82070307), and NSFC Incubation Project of Guangdong Provincial People’s Hospital (KY0120220041).
Disclosure
The authors declare that they have no competing interests in this work.
References
1. Benjamin EJ, Muntner P, Alonso A, et al. Heart disease and stroke statistics-2019 update: a report from the American Heart Association. Circulation. 2019;139(10):e56–e528.
2. Bergmark BA, Mathenge N, Merlini PA, et al. Acute coronary syndromes. Lancet. 2022;399(10332):1347–1358. doi:10.1016/S0140-6736(21)02391-6
3. Yap J, Irei J, Lozano-Gerona J, et al. Macrophages in cardiac remodelling after myocardial infarction. Nat Rev Cardiol. 2023;20(6):373–385. doi:10.1038/s41569-022-00823-5
4. Weissman D, Maack C. Mitochondrial function in macrophages controls cardiac repair after myocardial infarction. J Clin Invest. 2023;133(4). doi:10.1172/JCI167079
5. Frantz S, Nahrendorf M. Cardiac macrophages and their role in ischaemic heart disease. Cardiovasc Res. 2014;102(2):240–248. doi:10.1093/cvr/cvu025
6. Niu XH, Liu R-H, Lv X, et al. Activating α7nAChR helps post-myocardial infarction healing by regulating macrophage polarization via the STAT3 signaling pathway. Inflamm Res. 2023;72(4):879–892. doi:10.1007/s00011-023-01714-2
7. Karantalis V, Hare JM. Use of mesenchymal stem cells for therapy of cardiac disease. Circ Res. 2015;116(8):1413–1430. doi:10.1161/CIRCRESAHA.116.303614
8. Wang Q, Zhang L, Sun Z, et al. HIF-1α overexpression in mesenchymal stem cell-derived exosome-encapsulated arginine-glycine-aspartate (RGD) hydrogels boost therapeutic efficacy of cardiac repair after myocardial infarction. Mater Today Bio. 2021;12:100171. doi:10.1016/j.mtbio.2021.100171
9. Karp JM, Leng Teo GS. Mesenchymal stem cell homing: the devil is in the details. Cell Stem Cell. 2009;4(3):206–216. doi:10.1016/j.stem.2009.02.001
10. Tu C, Mezynski R, Wu JC. Improving the engraftment and integration of cell transplantation for cardiac regeneration. Cardiovasc Res. 2020;116(3):473–475. doi:10.1093/cvr/cvz237
11. Yan W, Guo Y, Tao L, et al. C1q/tumor necrosis factor-related protein-9 regulates the fate of implanted mesenchymal stem cells and mobilizes their protective effects against ischemic heart injury via multiple novel signaling pathways. Circulation. 2017;136(22):2162–2177. doi:10.1161/CIRCULATIONAHA.117.029557
12. Rani S, Ritter T. The exosome - a naturally secreted nanoparticle and its application to wound healing. Adv Mater. 2016;28(27):5542–5552. doi:10.1002/adma.201504009
13. Kalluri R. The biology and function of exosomes in cancer. J Clin Invest. 2016;126(4):1208–1215. doi:10.1172/JCI81135
14. Tkach M, Théry C. Communication by extracellular vesicles: where we are and where we need to go. Cell. 2016;164(6):1226–1232. doi:10.1016/j.cell.2016.01.043
15. Wang Y, Liu J, Ma J, et al. Exosomal circRNAs: biogenesis, effect and application in human diseases. Mol Cancer. 2019;18(1):116. doi:10.1186/s12943-019-1041-z
16. Lin KC, Yip H-K, Shao P-L, et al. Combination of adipose-derived mesenchymal stem cells (ADMSC) and ADMSC-derived exosomes for protecting kidney from acute ischemia-reperfusion injury. Int J Cardiol. 2016;216:173–185. doi:10.1016/j.ijcard.2016.04.061
17. Marbán E. The secret life of exosomes: what bees can teach us about next-generation therapeutics. J Am Coll Cardiol. 2018;71(2):193–200. doi:10.1016/j.jacc.2017.11.013
18. Huang P, Wang L, Li Q, et al. Atorvastatin enhances the therapeutic efficacy of mesenchymal stem cells-derived exosomes in acute myocardial infarction via up-regulating long non-coding RNA H19. Cardiovasc Res. 2020;116(2):353–367. doi:10.1093/cvr/cvz139
19. Chien KR, Frisén J, Fritsche-Danielson R, et al. Regenerating the field of cardiovascular cell therapy. Nat Biotechnol. 2019;37(3):232–237. doi:10.1038/s41587-019-0042-1
20. Barile L, Moccetti T, Marbán E, et al. Roles of exosomes in cardioprotection. Eur Heart J. 2017;38(18):1372–1379. doi:10.1093/eurheartj/ehw304
21. Zhu J, Lu K, Zhang N, et al. Myocardial reparative functions of exosomes from mesenchymal stem cells are enhanced by hypoxia treatment of the cells via transferring microRNA-210 in an nSMase2-dependent way. Artif Cells Nanomed Biotechnol. 2018;46(8):1659–1670. doi:10.1080/21691401.2017.1388249
22. Gray WD, French KM, Ghosh-Choudhary S, et al. Identification of therapeutic covariant microRNA clusters in hypoxia-treated cardiac progenitor cell exosomes using systems biology. Circ Res. 2015;116(2):255–263. doi:10.1161/CIRCRESAHA.116.304360
23. Lee FY, Lu H-I, Zhen -Y-Y, et al. Benefit of combined therapy with nicorandil and colchicine in preventing monocrotaline-induced rat pulmonary arterial hypertension. Eur J Pharm Sci. 2013;50(3–4):372–384. doi:10.1016/j.ejps.2013.08.004
24. Lefer DJ, Lefer AM. Studies on the mechanism of the vasodilator action of nicorandil. Life Sci. 1988;42(19):1907–1914. doi:10.1016/0024-3205(88)90031-8
25. Horinaka S. Use of nicorandil in cardiovascular disease and its optimization. Drugs. 2011;71(9):1105–1119. doi:10.2165/11592300-000000000-00000
26. Ning Y, Huang P, Chen G, et al. Atorvastatin-pretreated mesenchymal stem cell-derived extracellular vesicles promote cardiac repair after myocardial infarction via shifting macrophage polarization by targeting microRNA-139-3p/Stat1 pathway. BMC Med. 2023;21(1):96. doi:10.1186/s12916-023-02778-x
27. Xiong Y, Tang R, Xu J, et al. Tongxinluo-pretreated mesenchymal stem cells facilitate cardiac repair via exosomal transfer of miR-146a-5p targeting IRAK1/NF-κB p65 pathway. Stem Cell Res Ther. 2022;13(1):289. doi:10.1186/s13287-022-02969-y
28. Qian H-Y, Yang Y-J, Huang J, et al. Effects of Tongxinluo-facilitated cellular cardiomyoplasty with autologous bone marrow-mesenchymal stem cells on postinfarct swine hearts. Chin Med J. 2007;120(16):1416–1425. doi:10.1097/00029330-200708020-00008
29. Mohamed SS, Ahmed LA, Attia WA, et al. Nicorandil enhances the efficacy of mesenchymal stem cell therapy in isoproterenol-induced heart failure in rats. Biochem Pharmacol. 2015;98(3):403–411. doi:10.1016/j.bcp.2015.10.004
30. Yang YJ, Qian H-Y, Huang J, et al. Atorvastatin treatment improves survival and effects of implanted mesenchymal stem cells in post-infarct swine hearts. Eur Heart J. 2008;29(12):1578–1590. doi:10.1093/eurheartj/ehn167
31. ShamsEldeen AM, El-Aal SAA, Aboulhoda BE, et al. Combined systemic intake of K-ATP opener (Nicorandil) and mesenchymal stem cells preconditioned with nicorandil alleviates pancreatic insufficiency in a model of bilateral renal ischemia/reperfusion injury. Front Physiol. 2022;13:934597. doi:10.3389/fphys.2022.934597
32. Zhang F, Cui J, Lv BO, et al. Nicorandil protects mesenchymal stem cells against hypoxia and serum deprivation-induced apoptosis. Int J Mol Med. 2015;36(2):415–423. doi:10.3892/ijmm.2015.2229
33. de Couto G, Gallet R, Cambier L, et al. Exosomal MicroRNA transfer into macrophages mediates cellular postconditioning. Circulation. 2017;136(2):200–214. doi:10.1161/CIRCULATIONAHA.116.024590
34. Murray PJ, Allen J, Biswas S, et al. Macrophage activation and polarization: nomenclature and experimental guidelines. Immunity. 2014;41(1):14–20. doi:10.1016/j.immuni.2014.06.008
35. Tang R, Wang K, Xiong Y, Meng J, Yang Y. A fluorescence assay for evaluating the permeability of a cardiac microvascular endothelial barrier in a rat model of ischemia/reperfusion. J Vis Exp. 2021;172:e62746.
36. Swirski FK, Nahrendorf M. Leukocyte behavior in atherosclerosis, myocardial infarction, and heart failure. Science. 2013;339(6116):161–166. doi:10.1126/science.1230719
37. Steinhoff G, Nesteruk J, Wolfien M, et al. Stem cells and heart disease - Brake or accelerator? Adv Drug Deliv Rev. 2017;120:2–24. doi:10.1016/j.addr.2017.10.007
38. Golpanian S, Wolf A, Hatzistergos KE, et al. Rebuilding the damaged heart: mesenchymal stem cells, cell-based therapy, and engineered heart tissue. Physiol Rev. 2016;96(3):1127–1168. doi:10.1152/physrev.00019.2015
39. Guo Y, Yu Y, Hu S, et al. The therapeutic potential of mesenchymal stem cells for cardiovascular diseases. Cell Death Dis. 2020;11(5):349. doi:10.1038/s41419-020-2542-9
40. Xu J, Xiong -Y-Y, Li Q, et al. Optimization of timing and times for administration of atorvastatin-pretreated mesenchymal stem cells in a preclinical model of acute myocardial infarction. Stem Cells Transl Med. 2019;8(10):1068–1083. doi:10.1002/sctm.19-0013
41. Arslan F, de Kleijn DP, Pasterkamp G. Innate immune signaling in cardiac ischemia. Nat Rev Cardiol. 2011;8(5):292–300. doi:10.1038/nrcardio.2011.38
42. Ghigo A, Franco I, Morello F, et al. Myocyte signalling in leucocyte recruitment to the heart. Cardiovasc Res. 2014;102(2):270–280. doi:10.1093/cvr/cvu030
43. Frangogiannis NG. The mechanistic basis of infarct healing. Antioxid Redox Signal. 2006;8(11–12):1907–1939. doi:10.1089/ars.2006.8.1907
44. Frangogiannis NG, Rosenzweig A. Regulation of the inflammatory response in cardiac repair. Circ Res. 2012;110(1):159–173. doi:10.1161/CIRCRESAHA.111.243162
45. Prabhu SD, Frangogiannis NG. The biological basis for cardiac repair after myocardial infarction: from inflammation to fibrosis. Circ Res. 2016;119(1):91–112. doi:10.1161/CIRCRESAHA.116.303577
46. Liehn EA, Postea O, Curaj A, et al. Repair after myocardial infarction, between fantasy and reality: the role of chemokines. J Am Coll Cardiol. 2011;58(23):2357–2362. doi:10.1016/j.jacc.2011.08.034
47. Wen H, Peng L, Chen Y. The effect of immune cell-derived exosomes in the cardiac tissue repair after myocardial infarction: molecular mechanisms and pre-clinical evidence. J Cell Mol Med. 2021;25(14):6500–6510. doi:10.1111/jcmm.16686
48. Minami E, Castellani C, Malchodi L, et al. The role of macrophage-derived urokinase plasminogen activator in myocardial infarct repair: urokinase attenuates ventricular remodeling. J Mol Cell Cardiol. 2010;49(3):516–524. doi:10.1016/j.yjmcc.2010.03.022
49. Aurora AB, Porrello ER, Tan W, et al. Macrophages are required for neonatal heart regeneration. J Clin Invest. 2014;124(3):1382–1392. doi:10.1172/JCI72181
50. Ma Y, Mouton AJ, Lindsey ML. Cardiac macrophage biology in the steady-state heart, the aging heart, and following myocardial infarction. Transl Res. 2018;191:15–28. doi:10.1016/j.trsl.2017.10.001
51. Liu M, Yin L, Li W, et al. C1q/TNF-related protein-9 promotes macrophage polarization and improves cardiac dysfunction after myocardial infarction. J Cell Physiol. 2019;234(10):18731–18747. doi:10.1002/jcp.28513
52. Weirather J, Hofmann UDW, Beyersdorf N, et al. Foxp3 + CD4 + T cells improve healing after myocardial infarction by modulating monocyte/macrophage differentiation. Circ Res. 2014;115(1):55–67. doi:10.1161/CIRCRESAHA.115.303895
53. Courties G, Heidt T, Sebas M, et al. In vivo silencing of the transcription factor IRF5 reprograms the macrophage phenotype and improves infarct healing. J Am Coll Cardiol. 2014;63(15):1556–1566. doi:10.1016/j.jacc.2013.11.023
54. Zhao J, Li X, Hu J, et al. Mesenchymal stromal cell-derived exosomes attenuate myocardial ischaemia-reperfusion injury through miR-182-regulated macrophage polarization. Cardiovasc Res. 2019;115(7):1205–1216. doi:10.1093/cvr/cvz040
55. Wang Y, Tan J, Wang L, et al. MiR-125 family in cardiovascular and cerebrovascular diseases. Front Cell Dev Biol. 2021;9:799049. doi:10.3389/fcell.2021.799049
56. Banerjee S, Cui H, Xie N, et al. miR-125a-5p regulates differential activation of macrophages and inflammation. J Biol Chem. 2013;288(49):35428–35436. doi:10.1074/jbc.M112.426866
57. Gao L, Qiu F, Cao H, et al. Therapeutic delivery of microRNA-125a-5p oligonucleotides improves recovery from myocardial ischemia/reperfusion injury in mice and swine. Theranostics. 2023;13(2):685–703. doi:10.7150/thno.73568
58. Galluzzo A, Gallo S, Pardini B, et al. Identification of novel circulating microRNAs in advanced heart failure by next-generation sequencing. ESC Heart Failure. 2021;8(4):2907–2919. doi:10.1002/ehf2.13371
59. Skaug B, Jiang X, Chen ZJ. The role of ubiquitin in NF-kappaB regulatory pathways. Annu Rev Biochem. 2009;78(1):769–796. doi:10.1146/annurev.biochem.78.070907.102750
60. Ock S, Ahn J, Lee SH, et al. Receptor activator of nuclear factor-κB ligand is a novel inducer of myocardial inflammation. Cardiovasc Res. 2012;94(1):105–114. doi:10.1093/cvr/cvs078
61. Wang X, Ha T, Liu L, et al. Increased expression of microRNA-146a decreases myocardial ischaemia/reperfusion injury. Cardiovasc Res. 2013;97(3):432–442. doi:10.1093/cvr/cvs356
62. Wang X, Ha T, Zou J, et al. MicroRNA-125b protects against myocardial ischaemia/reperfusion injury via targeting p53-mediated apoptotic signalling and TRAF6. Cardiovasc Res. 2014;102(3):385–395. doi:10.1093/cvr/cvu044
63. Walsh MC, Lee J, Choi Y. Tumor necrosis factor receptor- associated factor 6 (TRAF6) regulation of development, function, and homeostasis of the immune system. Immunol Rev. 2015;266(1):72–92. doi:10.1111/imr.12302
64. Zhang Y, Le X, Zheng S, et al. MicroRNA-146a-5p-modified human umbilical cord mesenchymal stem cells enhance protection against diabetic nephropathy in rats through facilitating M2 macrophage polarization. Stem Cell Res Ther. 2022;13(1):171. doi:10.1186/s13287-022-02855-7
65. Sun J, Liao Z, Li Z, et al. Down-regulation miR-146a-5p in Schwann cell-derived exosomes induced macrophage M1 polarization by impairing the inhibition on TRAF6/NF-κB pathway after peripheral nerve injury. Exp Neurol. 2023;362:114295. doi:10.1016/j.expneurol.2022.114295
66. Wu W, Hu Y, Li J, et al. Silencing of Pellino1 improves post-infarct cardiac dysfunction and attenuates left ventricular remodelling in mice. Cardiovasc Res. 2014;102(1):46–55. doi:10.1093/cvr/cvu007
67. Krausgruber T, Blazek K, Smallie T, et al. IRF5 promotes inflammatory macrophage polarization and TH1-TH17 responses. Nat Immunol. 2011;12(3):231–238. doi:10.1038/ni.1990
68. Crea F. New therapeutic targets to reduce inflammation-associated cardiovascular risk: the CCL2-CCR2 axis, LOX-1, and IRF5. Eur Heart J. 2022;43(19):1777–1781. doi:10.1093/eurheartj/ehac233
69. Saliba DG, Heger A, Eames H, et al. IRF5:RelA interaction targets inflammatory genes in macrophages. Cell Rep. 2014;8(5):1308–1317. doi:10.1016/j.celrep.2014.07.034
70. Edsfeldt A, Swart M, Singh P, et al. Interferon regulatory factor-5-dependent CD11c+ macrophages contribute to the formation of rupture-prone atherosclerotic plaques. Eur Heart J. 2022;43(19):1864–1877. doi:10.1093/eurheartj/ehab920
71. Liu Z, Liu Z, Zhou H, et al. Increased sympathetic outflow induced by emotional stress aggravates myocardial ischemia-reperfusion injury via activation of TLR7/MyD88/IRF5 signaling pathway. Inflamm Res. 2023;72(5):901–913.
72. Chen H, Hou Y, Zhai Y, et al. Peli1 deletion in macrophages attenuates myocardial ischemia/reperfusion injury by suppressing M1 polarization. J Leukoc Biol. 2023;113(2):95–108. doi:10.1093/jleuko/qiac012
 © 2024 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2024 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.