")
Back to Journals » Nature and Science of Sleep » Volume 16
Ferrostatin-1 Reversed Chronic Intermittent Hypoxia-Induced Ferroptosis in Aortic Endothelial Cells via Reprogramming Mitochondrial Function
Authors Chen J, Deng X, Lin T, Huang J , Yang Y, Lian N
Received 25 October 2023
Accepted for publication 10 April 2024
Published 23 April 2024 Volume 2024:16 Pages 401—411
DOI https://doi.org/10.2147/NSS.S442186
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Sarah L Appleton
Jia Chen,1– 4,* Xiaoyu Deng,1– 4,* Ting Lin,1– 4 Jiefeng Huang,1– 4 Yisong Yang,1– 4 Ningfang Lian1– 4
1Department of Respiratory and Critical Care Medicine, the First Affiliated Hospital of Fujian Medical University, Fuzhou, Fujian, People’s Republic of China; 2Fujian Provincial Sleep-Disordered Breathing Clinic Center, Fuzhou, Fujian, People’s Republic of China; 3Institute of Respiratory Disease, Fujian Medical University, Fuzhou, Fujian, People’s Republic of China; 4Department of Respiratory and Critical Care Medicine, National Regional Medical Center, Binhai Campus of the First Affiliated Hospital, Fujian Medical University, Fuzhou, Fujian, People’s Republic of China
*These two authors contributed equally to this work
Correspondence: Ningfang Lian, Department of Respiratory and Critical Care Medicine, the First Affiliated Hospital of Fujian Medical University, No. 20, Chazhong Road, Taijiang District, Fuzhou, Fujian Province, 350005, People’s Republic of China, Tel +86-591-87981698, Email [email protected]
Purpose: Chronic intermittent hypoxia (CIH) related arterial endothelium injury is a common cause of cardiovascular system injury. However, the mechanism still needs to be clarified. In this study, we aimed to clarify the role and mechanism of ferrostatin-1 (Fer-1) in CIH-related rat arterial endothelial cells (ROAEC) ferroptosis.
Methods: ROAEC was divided into control group, CIH group, and CIH+ Fer-1 group. Cell viability was detected by cell counting kit 8 kits (CCK8). The apoptotic rate, reactive oxygen species (ROS) levels, Fe2+ levels, and lipid ROS levels were detected by flow cytometry. Malondialdehyde (MDA) levels and nicotinamide adenine dinucleotide (NAD+)/NADH ratio were detected via Elisa kits. The mRNA and protein levels of cystine/glutamate antiporter solute carrier family 7 member 11 (SLC7A11) and glutathione peroxidase 4 (GPX4) were detected by qRT-PCR and Western blot. Mitochondrial structure and function were observed by transmission electron microscope (TEM) and mitochondrial membrane potential (MMP). Central carbon metabolism was measured to compare metabolites among each group.
Results: After the CIH exposure, ROAEC cell viability decreased; The levels of cell apoptosis, ROS, Fe2+, MDA, and lip ROS increased; The levels of NAD+/NADP ratio decreased; The mRNA and protein levels of GPX4 and SLC7A11 decreased (all p< 0.05). Co-cultured with Fer-1 reversed the levels of apoptosis rate, cell viability, ROS, Fe2+, MAD, lipid ROS, NAD+/NADH ratio and the mRNA and protein expression of GPX4 and SLC7A11 (all p< 0.05). The TEM results showed that damaged mitochondrial membrane and the matrix spillover in the CIH group. The results of the JC-1 assay showed decreased MMP in the CIH group. Fer-1 treatment ameliorated the mitochondrial injury. The results of central carbon metabolism found that CIH altered the metabolites in the TCA cycle, which were reversed by Fer-1 treatment.
Conclusion: CIH-induced ferroptosis in ROAEC, which were reversed by Fer-1 via reprogramming mitochondrial function.
Keywords: obstructive sleep apnea, chronic intermittent hypoxia, arterial endothelium injury, ferroptosis, mitochondrial function
Introduction
Obstructive Sleep Apnea (OSA) is an independent risk factor for cardiovascular diseases such as hypertension, coronary heart disease, and heart failure.1 The characteristic of OSA is chronic intermittent hypoxia (CIH), which results from the repetitive collapse of the upper airway with oxygen desaturation;2 Growing evidences indicated that CIH-induced arterial endothelial injury was an important pathophysiological process of CIH-related vascular lesions, such as atherosclerosis.3,4 Activated local and systemic inflammatory responses, and mitochondria-dependent apoptotic pathways, were the early pathogenesis of CIH-associated atherosclerosis.5,
Figure S1). The pattern and time of chronic intermittent hypoxia were the same as CIH group.
Cell Viability Assay
We used a cell counting kit-8 assay (CCK-8) (Beyotime, C0037, China) to test cell viability. The ROAEC were added to the 96-well plate with 5 multiple wells in each group. After molding, 10μL CCK-8 solution was added to each well.
After incubating for 2 h at 37°C, the absorbance was detected by a microplate reader at 450nm. The results were calculated by the following formula: (Experimental group-blank control)/(negative control-blank control) × 100%.
Apoptosis Assay
After the CIH model, cells were collected in 1.5 mL ep tubes, washed with PBS thrice, and then resuspended in 200ul binding buffer with 10 µL PI and 5 µL Annexin V-FITC (Beyotime, C10625, China). After 10 minutes incubated at room temperature, the apoptosis rates were detected by C6 flow cytometry.
ROS Assay
ROS levels were measured via ROS assay (Beyotime, S00335, China). ROAEC were planted in the 6-well plate. After the CIH modeling, cells were collected into a 1.5 mL ep tube. 2’,7’-Dichlorodihydrofluorescein diacetate (DCFH-DA) was added to each ep tube. After being incubated for 20 min in a 37°C incubator, cells were washed by non-serum DMEM thrice, suspended with serum-free DMEM, and tested by flow cytometry.
Detection of the Intracellular Fe2+ Level
FerroOrange (F-374, Dojindo, China) was used to detect the Fe2+ level in different groups. Cells were collected into 1.5 mL ep tubes, washed with non-serum DMEM, then incubated at 37°C incubator with 1 µM FerroOrange for 30 min and tested by flow cytometry.
Malondialdehyde (MDA) Assay
To assess the levels of MDA, an MDA detection kit was utilized. The cells were seeded in a 10 cm dish, and collected in a 5 mL EP tube after the modeling process. According to the manufacturer’s instructions, the samples and reagent were prepared. The absorbance at 532 nm and 600 nm was then measured by a microplate reader. Subsequently, the MDA levels were calculated using the standard curve.
Measurement of Lipid Peroxidation
The cells were seeded in the 6-well-plate. After CIH modeling, the cells were collected in the 1.5EP tube, and washed by non-serum DMEM three times. Then incubated in the 1 µM of 4.4-difluoro-5-(4-phenyl-1,3-butadienyl)-4-bora-3a,4a-diazas-indacene-3-undecanoic acid (C11-BODIPY 581/591) (Thermo Fisher, USA) for 30 min in the 37°C incubator. Washed by non-serum DMEM three times, and tested by flow cytometry.
Detection of Nicotinamide Adenine Dinucleotide (NAD+)/NADH Ratio
The NAD+/NADH ratio was detected using a NADP+/NADH assay kit with WST-8 (Elabscience). Cells were seeded in a 6-well plate, washed with pre-cold PBS, and collected in the 1.5 EP tube. Follow the instructions to prepare the samples and reagents, and then add the sample and reagent to the labeled plate. For NADH detection, the samples should be incubated at 60°C for 30 minutes to remove NAD+. After incubating for 30 minutes at 37°C, the absorbance was detected by a microplate reader at 450nm. NADtotal and NADH were calculated using the standard curve. NAD+/NADH ratio= (NADtotal -NADH)/NADH*100%.
Metabolites Analysis
2*107 cells were collected in each group. After centrifugation, the cells were stored in liquid nitrogen for 30s and kept at −80°C. Metabolites were immediately extracted, and targeted metabolomics analysis of central carbon metabolism was performed. After adding 500 μL precooled MeOH/H2O (3/1, v/v), the samples were precooled in dry ice and repeated freeze-thaw in liquid nitrogen three times; a 400 μL aliquot of the clear supernatant was collected and dried by spin. Then, the samples were reconstituted with 200 μL of purified water. Reconstituted samples were vortexed before filtration through the centrifuge tube filter and were subsequently transferred to inserts in injection vials for HPIC-MS/MS analysis. The HPIC separation was carried out using a Thermo Scientific Dionex ICS-6000 HPIC System (Thermo Scientific), equipped with Dionex IonPac AS11-HC (2× 250 mm) and AG11-HC (2 mm×50 mm) columns. An AB SCIEX 6500 QTRAP+ triple quadrupole mass spectrometer (AB Sciex), equipped with an electrospray ionization (ESI) interface, was applied for assay development. Heat map R package (corrplot) (version 0.89) was used to analyze the heat map of central carbon metabolism.
qRT‑PCR
The Total RNA Isolation Kit V2 (Vazyme, China) was used to extract RNA. The HiScript III All-in-one RT SuperMix Perfect for qPCR (Vazyme, R333-01, China) was used to reverse transcribe cDNA. RT-qPCR was performed on an (ABI QuantStudio 5) ABI 7500 thermocycler (Applied Biosystems, Foster City, CA, USA) by using the SYBR Green PCR Master Mix (Vazyme, Q712, China). GPX4 and SLC7A11 were detected. Table 1 shows the appropriate primers. The relative expression levels of mRNA were calculated via the 2-Δ(CT) method.
 |
Table 1 PCR primer sequence |
Western Blotting
Radio Immunoprecipitation Assay (RIPA) lysis buffer (Beyotime, China) was used to extract proteins. A BCA protein concentration kit (Beyotime, China) was used to detect protein concentration. Then we made Sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) (Meilunbio, China) to separate the proteins. Electrophoresis transfers the protein molecules to the membrane (Millipore, 150 Billerica, MA, USA). Then, the membranes were soaked in 5% nonfat dry milk powder and dissolved in Tris Buffer Solution Tween (TBST) (servicebio, G0001-2L, China) for 2 h. The primary antibodies SLC7A11 (solute carrier family 7 member 11, Abcam, ab175186, 1:1000), GPX4 (glutathione peroxidase 4, Abcam, ab252833, 1:1000), and β-actin (Abcam, ab5694, 1:1000) were then incubated at 4 °C overnight. After being washed with TBST, the membranes were incubated with goat anti-rabbit IgG secondary antibody (Abcam, ab205718, 1:10,000) at room temperature for 1 h and then washed with TBST thrice. Some of the blots were divided before soaking in antibodies. With the help of an improved ECL kit (Meilunbio, MA0186-1, China), the images were taken and analyzed by Image J.
Transmission Electron Microscopy
After modeling, discarding the culture medium, and adding electron microscope fixation solution at 4°C for 2–4h, cells were collected after low-speed centrifugation. Samples were embedded in 1% agarose and rinsed with 0.1m phosphate buffer thrice, each 15 min. They were then fixed in the 1% osmic acid with 0.1m phosphate buffer mixture at room temperature. They were then rinsed with 0.1m phosphate buffer thrice, each 15min. The samples were successively dehydrated with 30–50-70-80-95-100-100% alcohol for 15 min each time, and 100% acetone twice for 15 min each time at room temperature. After embedding, ultrathin sections were cut into 70nm, stained with lead citrate, and photographed by a transmission electron microscope (Hitachi, HT7700, Japan).
Detection of Mitochondrial Membrane Potential
JC-1 assay (Beyotime, C2006, China) was used to test mitochondrial membrane potential (MMP). JC-1 working solution was prepared according to the instructions, then was added 500 μL into each group, mixed, and incubated at 37°C for 20 min. After centrifugation at 300g/min for 5 min, the supernatant was dropped and washed twice with precooled 1*JC-1 Assay Buffer. The cells were re-suspended by 1*JC-1 Assay Buffer and then detected by flow cytometry.
Statistical Analysis
Results were all shown as mean ± standard deviation. Statistical analysis was performed using GraphPad Prism 7.0 (GraphPad Software Inc., USA). Student-t-test was used for statistical analysis between the two groups, and P < 0.05 was considered statistically significant.
Results
CIH‑induced ROAEC Injury and Ferroptosis
After being exposed to the CIH condition for 36 h, the apoptotic rate, the levels of ROS, the levels of Fe2+, the levels of lipid ROS, and the levels of MDA (Figure 1A-D, Figure 1H) increased remarkably comparing with the control group (all p < 0.05). The cell viability of ROAEC and the NAD+/NADH ratio reduced significantly after 36 h CIH exposure (Figure 1E and I) (p < 0.05). The mRNA and protein levels of GPX4 and SLC7A11 were significantly declined in the CIH group comparing with the levels in the control group, both p < 0.05, as shown in Figure 1F–G. The original Western blotting images are shown in Figure S2.
 |
Figure 1 CIH-induced ROAEC injury and ferroptosis. (A), the apoptosis rate in the CIH group and control group; (B), the ROS levels in CIH group and control group; (C), the Fe2+ levels in CIH group and control group; (D), the lipid ROS levels in CIH group and control group; (E), the cell viability of ROAEC decreased in CIH group and control group; (F), The mRNA levels of GPX4 and SLC7A11 in CIH group and control group; (G), the protein levels of GPX4 and SLC7A11 in CIH group and control group; (H), the MDA levels in CIH group and control group; (I), the NAD+/NADH ratio in CIH group and control group. Data were repeated at least three times and shown as the mean ± SD. *p < 0.05; **p < 0. 01; ***p < 0. 001; ****p < 0. 0001. |
Fer‑1 Alleviated CIH‑related ROAEC Injury and Ferroptosis
After being co-treated with CIH and Fer-1, the apoptosis rate, the levels of ROS, the levels of Fe2+, the levels of lipid ROS, and the levels of MDA (Figure 2A-D, Figure 2H) decreased; while the cell viability and the NAD+/NADH ratio increased comparing with the CIH group (Figure 2E, Figure 2I) (all p < 0.05). In the CIH + Fer-1 group, the mRNA and protein levels of GPX4 and SLC7A11 increased, comparing with the levels in the CIH group (Figure 2F-G) (both p < 0.05). The original Western blotting images are shown in Figure S3.
 |
Figure 2 Fer-1 alleviated CIH-related ROAEC ferroptosis. The ROAEC were divided into control group, CIH group, and CIH+Fer-1 group. (A), co-treatment with Fer-1 reversed the CIH-induced apoptosis; (B), co-treatment with Fer-1 reversed the CIH-induced ROS accumulation; (C), co-treatment with Fer-1 reversed the increase of Fe2+ levels induced by CIH; (D), co-treatment with Fer-1 reversed the lipid ROS levels induced by CIH; (E), co-treatment with Fer-1 reversed the decline in cell viability induced by CIH; (F), the mRNA levels of SLC7A11 and GPX4 increased due to Fer-1 co-treatment; (G), the protein levels of SLC7A11 and GPX4 increased after Fer-1 co-treatment; (H), co-treatment with Fer-1 reversed the increase of MDA levels induced by CIH; (I), co-treatment with Fer-1 reversed the decline in NAD+/NADH ratio induced by CIH. Data are shown as the mean ± SD. Compared with the control group, *p < 0.05, **p < 0. 01, ***p < 0. 001, ****p < 0. 0001; Compared with the CIH group, # p < 0.05, ## p < 0. 01, ### p < 0. 001, ####p < 0. 0001. |
Fer-1 Alleviated CIH-Induced Mitochondrial Structure and Function Injury
To observe the mitochondrial structure and function, we performed TEM and MMP detection. TEM results showed that comparing with the control group, the mitochondrial membrane was damaged, and matrix spillover was observed in the CIH group (Figure 3A). The result of the JC-1 assay showed that MMP in the CIH group was lower than that in the control group (Figure 3B). After co-treatment with Fer-1, the damage of mitochondrial structure and MMP induced by CIH were reversed (Figure 3C-D).
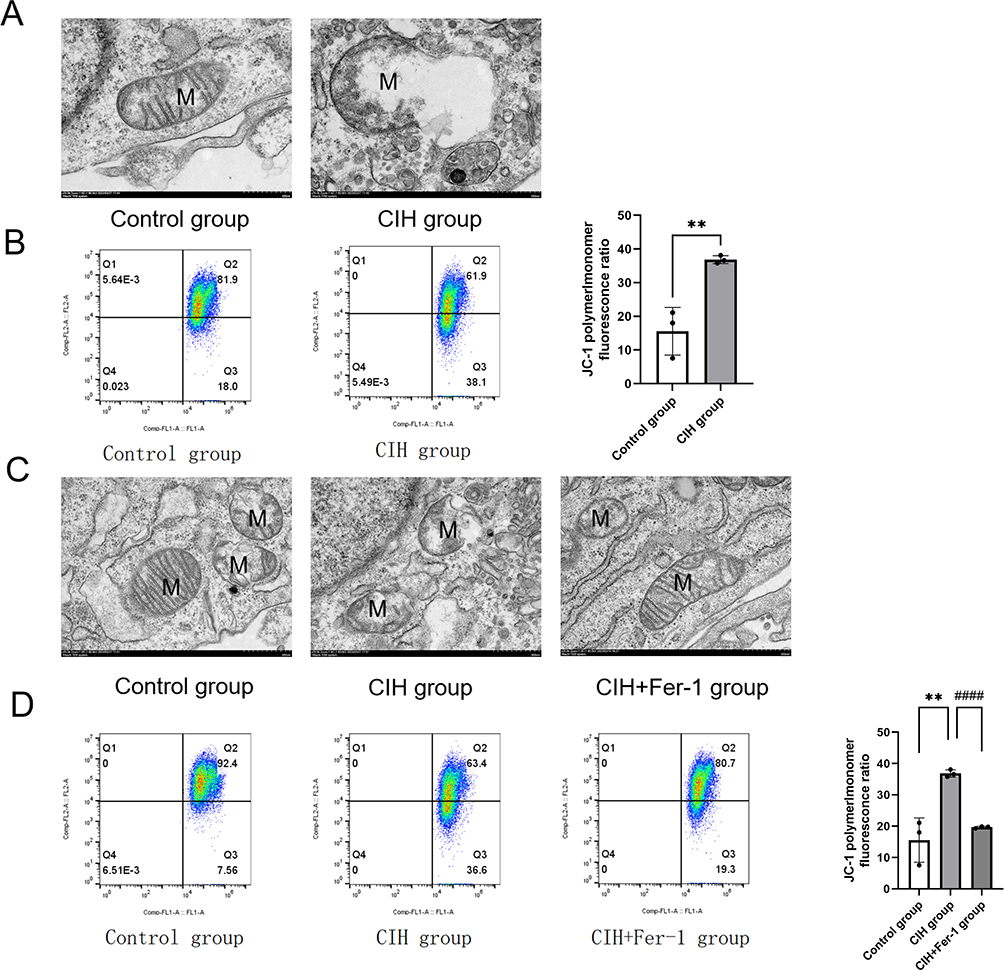 |
Figure 3 Fer-1 alleviated mitochondrial injury in CIH treated ROAEC. (A), Transmission electron microscopy (TEM) photo of the control group and CIH group; (B), Levels of mitochondrial membrane potential (MMP) in the control group and CIH group. (C), TEM photo of the control group, CIH group, and CIH+Fer-1 group. (D), Levels of MMP in the control group, CIH group, and CIH+Fer-1 group. Data are shown as the mean ± SD. Compared with the control group, **p < 0. 01; Compared with the CIH group, ####p < 0. 0001. |
Fer-1 Alleviated CIH-Induced Metabolism Disorder in Mitochondria
Mitochondria are the important sites for energy metabolism in cells. To further explore the role of mitochondria energy metabolism in CIH-related ferroptosis, a high-throughput target metabolomics- central carbon metabolism was performed. Hierarchical clustering was performed to reveal the different expression levels of the carbon metabolism products in the CIH group and control group, as shown in Figure 4A. Metabolites of central carbon metabolism include the major metabolites of TCA cycle, glycolysis, and pentose phosphate pathways (PPP). The study found that after Fer-1 treatment, the metabolites of the TCA cycle decreased significantly; however, metabolites of glycolysis and PPP were generally elevated, as shown in Figure 4C.
 |
Figure 4 Fer-1 ameliorated CIH-induced metabolism abnormalities in the TCA cycle. Hierarchical clustering of significantly regulated metabolites of central carbon metabolism among the control group and CIH group (n = 6, biological replicates).(A) color scale indicates relative metabolite levels: red for up-regulated, blue for down-regulated. (B), Schematic of altered metabolites in TCA cycle.(C), Hierarchical clustering of significantly regulated metabolites of central carbon metabolism among the control group, CIH group, and CIH+Fer-1 group (n = 6, biological replicates). A color scale indicates relative metabolite levels: red for up-regulated, blue for down-regulated. (D), CIH increased the levels of metabolites in the TCA cycle. Fer-1 treatment alleviated the increase of metabolites induced by CIH. Data are shown as the mean ± SD. Compared with the control group, **p < 0. 01, ***p < 0. 001, ****p < 0. 0001; Compared with the CIH group, ### p < 0. 001, ####p < 0. 0001. |
The tricarboxylic acid (TCA) cycle is an enzyme pathway located in the mitochondrial matrix. Differentially expressed key metabolites in the TCA cycle were screened, as shown in Figure 4B. The expression levels of alpha-ketoglutaric acid (αKG), succinic acid, fumaric acid, malic acid, 2-Oxobutanoic acid, and cis-Aconitic acid were compared among each group. CIH exposure increased the levels of metabolite in the TCA cycle. The metabolite levels in the TCA cycle decreased in the Fer-1+CIH group, compared to the CIH group, as shown in Figure 4D (all p < 0.05).
Discussion
The current investigation demonstrated that CIH exposure induced ferroptosis in ROAEC. The injury can be effectively inhibited by ferroptosis inhibitor Fer-1. Furthermore, mitochondrial morphology, function, and the metabolites in the TCA cycle were changed in the CIH treated ROAEC, which could be reversed by Fer-1.
Ferroptosis, a new type of cell-programmed death, featured Fe2+ overload, with excess ROS, lipid peroxidation, and Fenton response.23 Previous studies confirmed ferroptosis involved in CIH-induced liver injury and lung injury, but few research referred to CIH-related arterial endothelial cell ferroptosis. In this study, we established a CIH cell model to simulate OSA. Changes in apoptosis rate and cell viability confirmed the occurrence of CIH-related ROAEC injury. Compared to the control group, we found that the mRNA and protein levels of GPX4 and SLC7A11 changed in the CIH group. GPX4 is considered a central inhibitor of ferroptosis dominated by selenium and GSH,24 and its activity depends on glutathione produced by SLC7A11 activation.25 SLC7A11, a cystine/glutamate antitransporter component, is involved in regulating the exchange of equal amounts of intracellular glutamate and extracellular cystine and in regulating ferroptosis induced by reactive oxygen species (ROS).26,27 In this study, down-regulated GPX4 and SLC7A11 genes, elevated levels of ROS, lipid ROS, MDA, and Fe2+, confirmed the occurrence of ferroptosis after CIH treatment. The morphological changes of mitochondria are important features of ferroptosis. In this study, the mitochondrial membrane destruction, matrix efflux, and MMP changes all suggested the mitochondrial destruction caused by CIH, which further confirmed the occurrence of ferroptosis.
Mitochondria are the “energy factories” of cells, supplying them with ATP through oxidative phosphorylation. Mitochondria-dependent vascular endothelial cell injury was an important early pathogenesis of CIH-associated atherosclerosis.28 Increasing evidences suggested that mitochondrial damage caused by the mitochondrial metabolic disorder and mitochondria-mediated ROS production were necessary for lipid peroxidation and ferroptosis.14,24,29–31 ROS affected metabolism through TCA cycling and electron transport complex by interfering with the activity of iron-containing enzymes in mitochondria.32,33 Liu et al found that mitochondrial function promoted erastin-induced ferroptosis cells in non-small cell lung cancer cells.34 In our study, we found that mitochondrial structure and function were injured, and the expression of several metabolites αKG, succinic acid, fumaric acid, and malic acid in the TCA cycle increased. There are a few studies that focus on the relationship between the TCA metabolites and CIH-related ferroptosis. This study has a certain novelty. The metabolite analysis in our study found that the key metabolites in the TCA cycle were up-regulated. αKG, as the TCA cycle metabolite immediately downstream of glutaminolysis, could replace glutamine for cysteine deprivation-induced lipid ROS accumulation and ferroptosis. Succinic acid, fumaric acid, and malic acid were the TCA metabolites downstream of αKG.29 They all could replace the role of glutamine in lipid ROS accumulation. The canonical metabolic function of mitochondria was lipid ROS generation, which was the prerequisite of ferroptosis; and inhibition of the TCA cycle relieved ferroptosis.29 In addition, increased lipid ROS and decreased NAD+/NADH in the CIH group further confirmed that the increased activity of important metabolites in the TCA cycle led to ROS accumulation. Then the large amounts of ROS accumulation led to MMP decrease, mitochondrial structure destruction,35 and ferroptosis.
Fer-1, a synthetic antioxidant, acts via a reductive mechanism to prevent the injury to membrane lipids and thereby inhibits cell death. In this study, the biomarkers of ferroptosis and cell viability were reversed after co-culturing with Fer-1, which further confirmed that ferroptosis modulated CIH-associated vascular endothelial cell injury in ROAEC. We further analyzed the potential mechanism of Fer-1 treatment. Previous studies reported that the inhibition of the mitochondrial TCA cycle or electron transport chain (ETC) reduced MMP hyperpolarization, lipid peroxidation accumulation, and ferroptosis.29 Similar to the previous study, the Fer-1 treatment reversed the levels of metabolites in the TCA cycle, the levels of ROS, and the levels of lipid ROS in the manuscript. The central carbon metabolites in this study included the major metabolites of TCA cycle, glycolysis, and pentose phosphate pathways (PPP). The study found that after Fer-1 treatment, the metabolites of the TCA cycle decreased significantly. However, metabolites of glycolysis and PPP were generally elevated. These results suggested that Fer-1 did not reverse glycolysis and PPP. The level of lipid ROS is closely related to the intensity of the Fenton reaction. The reduction of lipid ROS thus reduced the MMP and alleviated mitochondrial structure injury. The reprogramming of mitochondrial function finally reversed CIH-associated ferroptosis. The possible mechanisms are shown in Figure 5. Fer-1 may be a potentially promising compound for CIH-associated cardiovascular injury.
 |
Figure 5 Possible molecular mechanisms of Fer-1 reversing CIH-induced ferroptosis in aortic endothelial cells. In ROAEC, after CIH treatment, the activity of important metabolites in the TCA cycle increased, which led to the accumulation of lipid ROS, the changes in mitochondrial membrane potential, and disruption of mitochondrial structure, eventually led to ferroptosis. Co-treatment with Fer-1 reversed ferroptosis via reprogramming the metabolites of the TCA cycle and mitochondrial function. |
There are some limitations in this study. The study was limited to cell experiments. There may be some differences in the results of in vitro and in vivo studies. So, we will confirm the role of Fer-1 in CIH-associated cardiovascular injury through animal experiments in the future.
Conclusion
CIH-induced ROAEC ferroptosis. Fer-1 reversed the levels of TCA metabolites, then decreased ROS accumulation and mitochondrial structure breakage, and in the end reversed the occurrence of CIH-induced ferroptosis in ROAEC.
Abbreviations
OSA, obstructive sleep apnea; CIH, chronic intermittent hypoxia; Fer-1, ferrostatin-1; ROAEC, Rat arterial endothelial cells; SLC7A11, cystine/glutamate antiporter solute carrier family 7 member 11; GPX4, glutathione peroxidase 4; CCK-8, cell counting kit-8; ROS, reactive oxygen species; COPD, chronic obstructive pulmonary disease; DMEM, Dulbecco’s Modified Eagle’s Medium; DCFH-DA, 2’,7’-Dichlorodihydrofluorescein diacetate; TEM, transmission electron microscope; RIPA, Radio Immunoprecipitation Assay; SDS-PAGE, sodium dodecyl sulfate-polyacrylamide gel electrophoresis; TBST, Tris Buffer Solution Tween; PBS, phosphate-buffered saline; TCA, tricarboxylic acid cycle; MMP, mitochondrial membrane potential; αKG, alpha-Ketoglutaric acid; NAD+, nicotinamide adenine dinucleotide; MDA, Malondialdehyde; C11-BODIPY 581/591, 4,4-difluoro-5-(4-phenyl-1,3-butadienyl)-4-bora-3a,4a-diazas-indacene-3-undecanoic acid; PPP, pentose phosphate pathways.
Data Sharing Statement
The data used in this study can be obtained through the corresponding author.
Funding
This study was funded by the Natural Science Foundation of Fujian Province (No. 2023J01554), Joint Funds for the Fujian Provincial Finance Project (No: BPB-LNF2021), National Natural Science Foundation of China (No. 82170101).
Disclosure
The authors report no conflicts of interest in this work.
References
1. Gottlieb DJ, Punjabi NM. Diagnosis and management of obstructive sleep apnea: a review. JAMA. 2020;323(14):1389–1400. doi:10.1001/jama.2020.3514
2. Drager LF, Togeiro SM, Polotsky VY, et al. Obstructive sleep apnea: a cardiometabolic risk in obesity and the metabolic syndrome. J Am Coll Cardiol. 2013;62(7):569–576. doi:10.1016/j.jacc.2013.05.045
3. Chen L, Zou S, Wang J. Association of obstructive sleep apnea syndrome (OSA/OSAHS) with coronary atherosclerosis risk: systematic review and meta-analysis. Comput Math Methods Med. 2022;2022:8905736. doi:10.1155/2022/8905736
4. Badran M, Ayas N, Laher I. Cardiovascular complications of sleep apnea: role of oxidative stress. Oxid Med Cell Longev. 2014;2014:985258. doi:10.1155/2014/985258
5. Badran M, Gozal D. PAI-1: a major player in the vascular dysfunction in obstructive sleep apnea?. Int J Mol Sci. 2022;23(10):5516. doi:10.3390/ijms23105516
6. Dikalov SI, Nazarewicz RR. Angiotensin II-induced production of mitochondrial reactive oxygen species: potential mechanisms and relevance for cardiovascular disease. Antioxid Redox Signal. 2013;19(10):1085–1094. doi:10.1089/ars.2012.4604
7. Puddu P, Puddu GM, Galletti L, et al. Mitochondrial dysfunction as an initiating event in atherogenesis: a plausible hypothesis. Cardiology. 2005;103(3):137–141. doi:10.1159/000083440
8. Zhao L, Liu T, Dou ZJ, et al. CB1 receptor antagonist rimonabant protects against chronic intermittent hypoxia-induced renal injury in rats. BMC Nephrol. 2021;22(1):153. doi:10.1186/s12882-021-02362-6
9. Song JQ, Jiang LY, Fu CP, et al. Heterozygous SOD2 deletion deteriorated chronic intermittent hypoxia-induced lung inflammation and vascular remodeling through mtROS-NLRP3 signaling pathway. Acta Pharmacol Sin. 2020;41(9):1197–1207. doi:10.1038/s41401-019-0349-y
10. Pai PY, Lin YY, Yu SH, et al. Angiotensin II receptor blocker irbesartan attenuates sleep apnea-induced cardiac apoptosis and enhances cardiac survival and Sirtuin 1 upregulation. Sleep Breath. 2022;26(3):1161–1172. doi:10.1007/s11325-021-02499-6
11. Shi C, Guo H, Liu X. Platelet mitochondria transplantation rescues hypoxia/reoxygenation-induced mitochondrial dysfunction and neuronal cell death involving the FUNDC2/PIP3/Akt/FOXO3a axis. Cell Transplant. 2021;30:9636897211024210. doi:10.1177/09636897211024210
12. Chen J, Zhu H, Chen Q, et al. The role of ferroptosis in chronic intermittent hypoxia-induced lung injury. BMC Pulm Med. 2022;221:488. BMC Pulmonary Medicine. 10.1186/s12890-022-02262-x
13. Arnaud C, Billoir E, De Melo Junior AF, et al. Chronic intermittent hypoxia-induced cardiovascular and renal dysfunction: from adaptation to maladaptation. J Physiol. 2023;601(24):5553–5577. doi:10.1113/JP284166
14. Dixon SJ, Lemberg KM, Lamprecht MR, et al. Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell. 2012;149(5):1060–1072. doi:10.1016/j.cell.2012.03.042
15. Li J, Cao F, Yin HL, et al. Ferroptosis: past, present and future. Cell Death Dis. 2020;11(2):88. doi:10.1038/s41419-020-2298-2
16. Zhao WK, Zhou Y, Xu TT, et al. Ferroptosis: opportunities and challenges in myocardial ischemia-reperfusion injury. Oxid Med Cell Longev. 2021;2021:9929687. doi:10.1155/2021/9929687
17. Li X, Ma N, Xu J, et al. Targeting ferroptosis: pathological mechanism and treatment of ischemia-reperfusion injury. Oxid Med Cell Longev. 2021;2021:1587922. doi:10.1155/2021/1587922
18. Chen LD, Wu RH, huang YZ, et al. The role of ferroptosis in chronic intermittent hypoxia-induced liver injury in rats. Sleep Breath. 2020;24(4):1767–1773. doi:10.1007/s11325-020-02091-4
19. Dan Dunn J, Alvarez LA, Zhang X, et al. Reactive oxygen species and mitochondria: a nexus of cellular homeostasis. Redox Biol. 2015;6:472–485. doi:10.1016/j.redox.2015.09.005
20. Paul BT, Manz DH, Torti FM, et al. Mitochondria and Iron: current questions. Expert Rev Hematol. 2017;10(1):65–79. doi:10.1080/17474086.2016.1268047
21. Stockwell BR, Friedmann Angeli JP, Bayir H, et al. Ferroptosis: a regulated cell death nexus linking metabolism, redox biology and, disease. Cell. 2017;171(2):273–285. doi:10.1016/j.cell.2017.09.021
22. Gao M, Jiang X. To eat or not to eat-The metabolic flavor of ferroptosis. Curr Opin Cell Biol. 2018;51:58–64. doi:10.1016/j.ceb.2017.11.001
23. Jiang X, Stockwell BR, Conrad M. Ferroptosis: mechanisms, biology and role in disease. Nat Rev Mol Cell Biol. 2021;22(4):266–282. doi:10.1038/s41580-020-00324-8
24. Tang D, Chen X, Kang R, et al. Ferroptosis: molecular mechanisms and health implications. Cell Res. 2021;31(2):107–125. doi:10.1038/s41422-020-00441-1
25. Sun Y, Berleth N, Wu W, et al. Fin56-induced ferroptosis is supported by autophagy-mediated GPX4 degradation and functions synergistically with mTOR inhibition to kill bladder cancer cells. Cell Death Dis. 2021;12(11):1028. doi:10.1038/s41419-021-04306-2
26. Chen D, Tavana O, Chu B, et al. NRF2 is a major target of ARF in p53-independent tumor suppression. Mol Cell. 2017;68(1):224–32.e4. doi:10.1016/j.molcel.2017.09.009
27. Lo M, Wang YZ, Gout PW. The x(c)- cystine/glutamate antiporter: a potential target for therapy of cancer and other diseases. J Cell Physiol. 2008;215(3):593–602. doi:10.1002/jcp.21366
28. Javaheri S, Barbe F, Campos-Rodriguez F, et al. Sleep apnea: types, mechanisms, and clinical cardiovascular consequences. J Am Coll Cardiol. 2017;69(7):841–858. doi:10.1016/j.jacc.2016.11.069
29. Gao M, Yi J, Zhu J, et al. Role of mitochondria in ferroptosis. Mol Cell. 2019;73(2):354–63.e3. doi:10.1016/j.molcel.2018.10.042
30. Li C, Zhang Y, Liu J, et al. Mitochondrial DNA stress triggers autophagy-dependent ferroptotic death. Autophagy. 2021;17(4):948–960. doi:10.1080/15548627.2020.1739447
31. Lee H, Zandkarimi F, Zhang Y, et al. Energy-stress-mediated AMPK activation inhibits ferroptosis. Nat Cell Biol. 2020;22(2):225–234. doi:10.1038/s41556-020-0461-8
32. Na YR, Je S, Seok SH. Metabolic features of macrophages in inflammatory diseases and cancer. Cancer Lett. 2018;413:46–58. doi:10.1016/j.canlet.2017.10.044
33. Bailey JD, Diotallevi M, Nicol T, et al. Nitric oxide modulates metabolic remodeling in inflammatory macrophages through TCA cycle regulation and itaconate accumulation. Cell Rep. 2019;28(1):218–30.e7. doi:10.1016/j.celrep.2019.06.018
34. Liu P, Wu D, Duan J, et al. NRF2 regulates the sensitivity of human NSCLC cells to cystine deprivation-induced ferroptosis via FOCAD-FAK signaling pathway. Redox Biol. 2020;37:101702. doi:10.1016/j.redox.2020.101702
35. Luo Y, Chen P, Yang L, et al. Metabolomic analysis and pharmacological validation of the cerebral protective effect of 3,4‑dihydroxybenzaldehyde on cerebral ischemia‑reperfusion injury. Mol Med Rep. 2023;27(1):9.
 © 2024 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2024 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.