Back to Journals » Clinical Ophthalmology » Volume 10
Vogt–Koyanagi–Harada syndrome – current perspectives
Authors Baltmr A, Lightman S, Tomkins-Netzer O
Received 5 June 2016
Accepted for publication 22 September 2016
Published 24 November 2016 Volume 2016:10 Pages 2345—2361
DOI https://doi.org/10.2147/OPTH.S94866
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Dr Scott Fraser
Abeir Baltmr,1 Sue Lightman,1,2 Oren Tomkins-Netzer1–3
1Uveitis Service, Moorfields Eye Hospital, London, UK; 2Department of Clinical Ophthalmology, UCL Institute of Ophthalmology, London, UK; 3Faculty of Medicine, Technion, Israel Institute of Technology, Haifa, Israel
Abstract: Vogt–Koyanagi–Harada syndrome is a cause of noninfectious panuveitis, leading to significant vision loss in many patients. It is an autoimmune disease occurring in genetically susceptible individuals and clinically presents as bilateral panuveitis with serous retinal detachments and hyperemic, swollen optic discs, which are associated with neurological and auditory manifestations. Early diagnosis and prompt and adequate treatment with immunosuppressive agents (corticosteroids and other immunosuppressive drugs) may halt disease progression and prevent recurrences and vision loss. This review summarizes the current knowledge on the variable clinical aspects of this disease, highlighting diagnostic and treatment strategies.
Keywords: multifocal choroiditis, serous retinal detachment, panuveitis, sunset glow fundus, starry sky, corticosteroid
Introduction
Vogt–Koyanagi–Harada disease (VKHD) is a sight-threatening disease and a common cause of noninfectious panuveitis.1,2 It is described as a multisystemic, granulomatous inflammation related to T-cell-mediated autoimmune dysregulation, targeting melanocytic self-antigens. The tyrosinase family proteins and gp100 are common antigenic targets for VKHD.1,3–5 The disease has a predilection for affecting melanocyte-containing tissues in the eye, the central nervous system (CNS), the inner ear and the skin, appears in genetically susceptible individuals and is related to HLA-DRB1*0405.6–8 Patients usually present with bilateral panuveitis preceded by a mild prodromal illness, associated with neurological and auditory features. However, it is common for patients to present with isolated ocular involvement during the early phases of the disease, with the choroid being the main site of ocular inflammation together with potential involvement of the iris and ciliary body. During the early acute phase of the disease, the inflammation is characterized by non-necrotizing diffuse granulomas, which persist following recurrences (chronic recurrent phase). As the disease progresses, it undergoes a chronic convalescent phase, which is characterized by a non-granulomatous inflammation, with later involvement of the choriocapillaris in the chronic recurrent phase.9,10 The disease has a variable clinical presentation, and clinical diagnosis is aided by the help of ophthalmic imaging such as fundus fluorescein angiography (FFA), indocyanine green angiography (ICG), and optical coherence tomography (OCT). Novel diagnostic techniques, which can demonstrate choroidal structure and precise retinal function, are now employed to assist in early diagnosis, leading to prompt treatment and monitoring of ongoing ocular inflammation.
Historical aspects and epidemiology
Ocular inflammation with poliosis was described in the first century AD by Ali-ibn-Isa.11 In 1906, Vogt12 reported a case of a 16-year-old patient with iridocyclitis and poliosis. In 1929, Yoshizo Koyanagi described 16 cases with similar features, including six new cases.13 In addition to the anterior uveitis and extraocular manifestations described by Vogt and Koyanagi, Harada14 described in 1926 a primarily posterior uveitis with exudative retinal detachments and pleocytosis of cerebrospinal fluid (CSF): an abnormal increase in the amount of lymphocytes and monocytes with normal glucose, as well as the prodromal phase of the disease and the integumentary signs. These findings were later noted by Babel, Bruno, and McPherson to represent a single, progressive syndrome, and the name was combined into VKHD syndrome.15
VKHD has variable incidence; it appears to be more prevalent in people with Asian, Hispanic, Indian, Native American, or Mediterranean origin, accounting for 7%–22.4% of uveitis referrals.16–21 It is uncommon in Caucasians and people of Turkish descent, where it accounts for <3% of uveitis referrals.22–25 VKHD commonly affects young females26–28 between 20 and 50 years of age,1 at a ratio of 2:1 in most large series.29 However, pediatric cases,30–32 elderly onset33 and equal sex predilection have been reported in some studies.34
Classification and clinical features
According to the revised diagnostic criteria proposed by the American Uveitis Society,35 in its complete form, VKHD is defined as a non-traumatic bilateral panuveitis and is associated with integumentary, neurological/auditory signs. Probable (ocular) VKHD is characterized as matching ocular manifestations but in the absence of extraocular manifestations. Patients with the incomplete form of the disease usually present with bilateral ocular involvement with either integumentary features or neurologic/auditory manifestations.35 In all forms of the disease, there should be no history of ocular surgery and no clinical or laboratory evidence suggestive of other ocular diseases.35 It is important to recognize that although classically VKHD is a multisystemic disease, patients may fail to develop the complete or incomplete forms, and isolated ocular involvement has been reported in 45% of patients with acute VKHD and up to 58% of those presenting with chronic disease.36
VKHD is a multiphasic disease that classically has four clinical phases or stages with variable clinical features in different ethnic groups: 1) prodromal; 2) acute; 3) chronic convalescent; and 4) chronic recurrent stage.
The prodromal phase, which may mimic a viral infection, generally precedes the ocular inflammation by a few days and lasts for 1–2 weeks. Patients typically complain of CNS and auditory symptoms including headaches, neck stiffness, photophobia, fever, orbital pain, scalp or skin sensitivity, and focal neurological deficits such as cranial nerve palsies.1,28 They may also have hearing loss, particularly for higher frequencies, dysacousia, vertigo, and tinnitus,1,37,38 with less frequent auditory features observed in Hispanic patients.39 CSF samples from lumbar punctures show pleocytosis in >80% of cases,40,41 and brain imaging studies can occasionally demonstrate focal parenchymal lesions.42
In the eye, the acute phase of VKHD is characterized by a panuveitis or posterior uveitis with multifocal serous retinal detachments (SRDs). Vitreous inflammation is commonly noted with a low-grade non-granulomatous anterior uveitis seen in some patients.10,34,41 The diffuse choroidal thickening seen on OCT scans during the acute phase of VKHD correlates with diffuse mononuclear inflammatory cellular infiltration of the uvea with sparing of the choriocapillaris9 and the appearance of Dalen-Fuchs nodules on histopathological studies. The Dalen-Fuchs nodules are aggregates of lymphocytes and pigment-laden macrophages that appear mainly in the posterior pole between Bruch membrane and the retinal pigment epithelium (RPE).9,10 The inflammation lasts several weeks, and early treatment may prevent the later development of the chronic phases of the disease in up to a third of cases.26,43,44 However, progression to the chronic convalescence phase occurs in 28%–62% of patients who present with acute VKHD, possibly due to more severe and rapid disease onset.26,45
The chronic convalescence phase usually develops 3–4 months after the onset of the disease and lasts from several weeks to years, during which time patients usually present with non-granulomatous panuveitis, poliosis of the eyebrows and eyelashes, vitiligo, and alopecia, as well as depigmentation of the choroid resulting in a bright-orange hue. The optic nerve remains pale, leading to the appearance of sunset glow fundus.1,2 This depigmentation of the integumentary system usually follows ocular and CNS involvement in VKHD and is more commonly seen in Japanese than in Hispanic patients.1,39 Despite early high-dose corticosteroid treatment, progression to the chronic recurrent phase may occur in up to 79% of the total cases who presented with acute VKHD.44
During the chronic recurrent phase, patients present with recurrent granulomatous anterior uveitis (Figure 1), and choroidal thickening develops.46–48 Approximately 25% of VKHD patients develop posterior and anterior segment recurrences, mainly in cases of more aggressive disease with poor response to immunosuppressive treatment.27 Anterior segment recurrences were found to be associated with choroidal inflammation and insufficient choroidal circulation demonstrated on ICG and OCT scans.48,49
 | Figure 1 Color photograph of a patient in the chronic recurrent phase of the VKHD showing acute anterior uveitis and mild poliosis (white arrow). |
Ocular findings in VKHD
In the eye, orbital pain, photosensitivity and tearing have been reported during the prodromal phase of VKHD.1 As the disease progresses to the acute phase, panuveitis manifests and patients commonly present with transient attacks of panuveitis or posterior uveitis with multifocal choroiditis lesions (Figure 2A), which progress to multifocal serous detachments and can coalesce into diffuse SRD (Figure 2B),41,50 resulting in reduced vision. SRD might initially be misdiagnosed as central serous chorioretinopathy (CSCR).51 However, in the absence of ocular inflammation and other systemic features of VKHD, the presence of optic disc hyperemia and disc leakage on FFA support the diagnosis of VKHD rather than CSCR.51
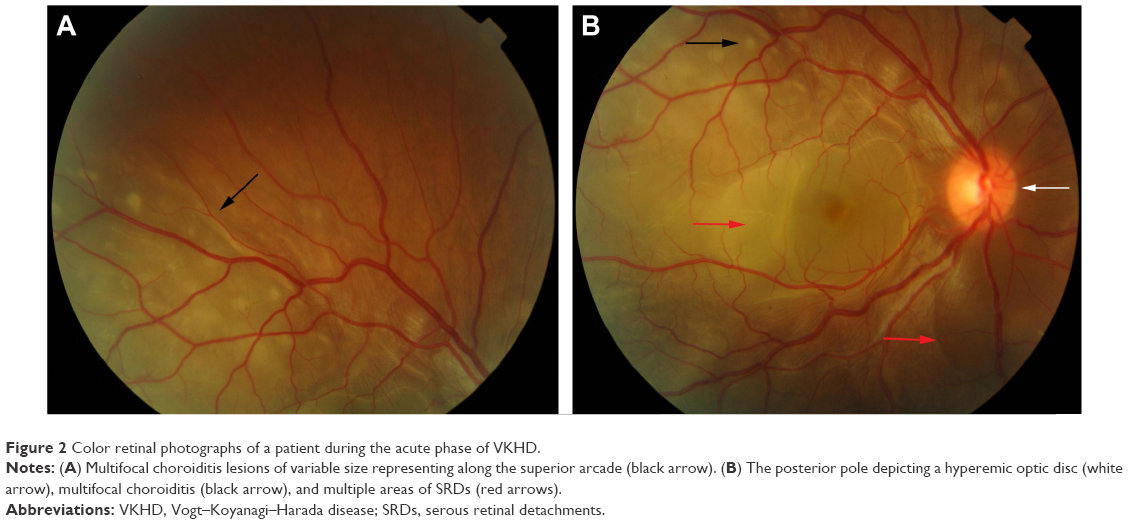 | Figure 2 Color retinal photographs of a patient during the acute phase of VKHD. |
Although bilateral simultaneous involvement has been observed in the majority of patients, involvement of the second eye can take up to 2 weeks, with delayed involvement reported to occur up to 6 years after the first eye.52 Sporadic cases of uniocular disease have also been reported with a follow-up time of 16 years;32,53,54 therefore, uniocular presentation should not exclude the diagnosis or delay starting treatment.
Unilateral and bilateral optic disc hyperemia (Figure 3) without visual field loss is a frequent presentation in acute VKHD55–57 and is the most common finding in acute VKHD patients with no SRD at presentation.58
 | Figure 3 Color retinal photographs of right (A) and left eye (B) showing swollen and hyperemic optic discs (white arrows), with choroidal folds disc (black arrows), in the acute phase of VKHD. |
Optic disc involvement in acute VKHD is found to be more common in females and patients with a mean age of 55 years, who also have a greater likelihood of vision loss or progression to the chronic phases.50 In addition to older age at presentation, optic disc swelling is found to correlate significantly with the morphology of the optic nerve (a smaller cup-to-disc ratio and a higher disc–macula distance to disc diameter).57 Swollen optic discs may be the only presenting sign and should be differentiated from other conditions,56 such as optic neuritis,59 diabetic papillopathy,58 central retinal vein occlusion, or anterior ischemic optic neuropathy (AION). Extensive optic disc swelling can itself lead to further optic nerve pathology, and patients can develop AION following the resolution of the optic disc swelling and SRD. This is thought to be due to an impaired posterior ciliary artery supply to the optic nerve, secondary to severe choroidal inflammation.60–62 Involvement of the optic nerve leading to fixed dilated pupils has also been observed in a 28-year-old patient with acute VKHD.63
Choroidal folds are ripples in the RPE, Bruch membrane and the inner aspect of the choroid and have been described to accompany or precede the development of SRD in up to 71% of eyes with acute VKHD.64 These folds (Figure 3) can be detected on fundus examination and are more easily identifiable on FFA, ICG, and OCT scans.65–67 Their reappearance on OCT is predictive of recurrence of SRD.68 In studies examining VKHD patients, choroidal folds were noted in between 12% and 57% of cases. Their occurrence was related with poor visual acuity at presentation, as well as poor visual outcome,65,66,69 disc swelling, and the development of choroidal thinning and sunset glow fundus later in the course of the disease.69
Scleral involvement in patients in the acute phase of VKHD has been observed with case reports of patients presented with unilateral or bilateral posterior scleritis and choroidal thickening.70 Extraocular features of VKHD with bilateral posterior scleritis, bilateral SRD, and optic disc swelling were also observed.71
Rare presentations of acute VKHD include angle closure glaucoma, moderately elevated intraocular pressure (IOP) with variable degrees of intraocular inflammation72–74 or RPE tears that can be seen bilaterally without choroidal neovascularization (CNV) and are thought to be due to acute choroidal inflammation.75 Vitreomacular traction (VMT) that rapidly progresses to full-thickness macular hole (MH) formation has also been noted to develop during acute VKHD,76 possibly due to active inflammation.
In eyes that progress to the chronic convalescence stage, resolution of SRDs with the development of chorioretinal depigmentation results in an orange-red discoloration of the fundus giving the distinct sunset glow fundal appearance (Figure 4) and perilimbal vitiligo (Sugiura’s sign), which suggest ongoing uveal inflammation. Sunset glow fundus has been reported to develop as soon as 4 months after the initial presentation, despite treatment with high-dose immunosuppressive agents.10,45 Histological studies suggest that this is secondary to choroidal melanocyte loss from diffuse granulomatous choroiditis.10,77 The development of sunset glow fundus was found to be related to increased numbers of inflammatory cells in CSF samples and severe CNS involvement at presentation.78
 | Figure 4 Color retinal photographs of right (A) and left eye (B) during the convalescent phase of VKHD, showing sunset glow fundus with pale optic discs (white arrows) and bright-orange choroids (black arrows). Note the peripapillary atrophy (gray arrow) and macular scaring (blue arrow) in (A). |
The convalescent phase is further characterized by the appearance of numerous small choroidal depigmented atrophic and hyperpigmented lesions in the retinal periphery (Figure 5)79 and peripapillary subretinal fibrosis lesions (Figure 4A), as well as the Dalen-Fuchs nodules that can be seen in all phases of the disease.9,10 Choroidal atrophy due to subfoveal loss of the choriocapillaris has been confirmed on spectral domain OCT (SD-OCT) scans and correlated with duration of the disease, lower visual acuity, and atrophy of overlying retina and RPE.80
 | Figure 5 Color retinal photograph displaying sunset glow fundus with a numerous small choroidal depigmented atrophic lesions (black arrow) and hyperpigmented lesions (white arrow) in the retinal periphery during the convalescent phase of VKHD. |
Other uncommon findings in this phase of the disease include progressive scleral changes and posterior scleritis possibly due to ongoing chronic choroidal inflammation,81–83 as well as VMT and epiretinal membranes (ERMs), possibly due to RPE migration later in the course of the disease.84
The chronic recurrent phase is characterized by episodes of granulomatous anterior uveitis with mutton fat keratic precipitates, iris nodules, and posterior synechiae.46 A prospective study that examined 70 eyes with acute VKHD and 92 eyes with recurrent disease, before and after treatment with immunosuppressive agents, showed a significantly higher anterior segment cell count and flare level in the eyes during the chronic recurrent phase, possibly reflecting a refractory inflammation with lasting breakdown of the blood–aqueous barrier.85 Peripheral iris stromal and pigment epithelium depigmentation may develop due to ciliary body inflammation, associated with the severe lasting anterior uveitis.86,87 RPE clumping and disruption seen as multiple foci of hypopigmentation, in addition to sunset glow fundus and chorioretinal atrophy, are prominent findings on fundus examination. Though they do not appear to be active chorioretinal inflammation, OCT and ICG studies have demonstrated increased choroidal thickness during this stage, reflecting subclinical inflammation that may require additional immunosuppression.47,48
Imaging in VKHD
Imaging plays a major role in the diagnosis of VKHD, monitoring of the intraocular inflammation and response to treatment, as well as the development of complications.
Fundus fluorescein angiography
FFA is used to aid in the diagnosis, during follow up,88,89 and as a prognostic tool in VKHD.90 (Figure 6A; color retinal photograph showing deep yellow lesions of variable size and multiple SRD). During the early venous phase of FFA, active inflammation in the acute phase leads to delayed choroidal perfusion in the posterior pole and retinal periphery, displayed as areas of hypofluorescent (Figure 6B).91 Numerous hyperfluorescent pinpoint foci of leakage, which coincide with areas of choroiditis, are manifested at the level of RPE (Figure 6C).91 This is accompanied by multilobular dye pooling in the subretinal space, each lobule surrounded by a dark rim (Figure 6D) that is thought to be related to the formation of subretinal septa as demonstrated on previously published OCT scans.88,92,93 Homogeneous staining on FFA is usually seen in areas of retinal detachment and optic disc leakage.88,90 A hot disc can be observed in up to 94.4% of patients with acute VKHD and 66.6% of those in the chronic convalescent phase (Figure 7).88 Numerous hyperfluorescent pinpoint foci of leakage at the level of RPE result in a classic “starry sky” appearance on the FFA that is highly suggestive of VKHD (Figure 8). Hypofluorescent lines corresponding to choroidal folds are also seen.65,91 In a series of 95 patients with VKHD, 11 (12%) showed choroidal folds manifested as hypofluorescent bands radiating from the optic disc.65 During the chronic phases of VKHD, FFA usually shows RPE window defects, spotted hyper- and hypofluorescence and blockage of choroidal fluorescence due to RPE migration.1,88 Complications such as CNV may also be seen on FFA (Figure 9).91
 | Figure 6 Color retinal and FFA photographs of a patient in the acute phase of VKHD showing (A) hyperemic optic disc with deep yellow lesions of variable size and multiple SRD, (B) early FFA showing delayed choroidal filling, (C) mid phase of FFA showing pinpoint hyperfluorescent leakage at the RPE level (D) with pooling of the dye in the subretinal space in the late phase of the angiogram. |
 | Figure 7 Color retinal photograph of the right eye showing swollen and hyperemic optic discs (A) and FFA showing hot disc (B) (white arrows), in the acute phase of VKHD. |
 | Figure 8 FFA photograph of a patient in the acute phase of VKHD showing optic disc leakage and numerous hyperfluorescent pinpoint foci of leakage at the level of RPE leading to the classic “starry sky” appearance. |
 | Figure 9 Color retinal (A) and FFA (B) photographs of a VKHD patient with macular CNV (white arrows). |
FFA is also found to have a prognostic value in acute VKHD, where the absence of early pinpoint peripapillary hyperfluorescence on pretreatment FFA is a poor prognostic factor for developing chronic VKHD, and its presence is observed in up to 85% of eyes that resolved.90
Indocyanine green angiography
ICG is well established as a sensitive method to delineate the choroidal circulation and visualize the choroid.94 In VKHD, it is used to confirm the presence of choroidal inflammation, monitor choroidal activity and response to therapy.95,96 However, it is an invasive and time-consuming imaging method, in which it is difficult to distinguish stromal scarring rather than active choroiditis,97 and pretreatment ICG findings have been shown to have limited prognostic value for the development of chronic VKHD.90
The main ICG features in acute VKHD are: 1) a patchy hypofluorescence pattern at the posterior pole and retinal periphery during the early angiographic phase due to choroidal inflammatory vasculopathy and delayed choroidal perfusion (Figure 10A); 2) large choroidal stromal vessel hyperfluorescence in the early phase (Figure 10B); 3) diffusely leaking fuzzy choroidal vessels possibly due to vasculitis (Figure 10B); 4) hypofluorescent dark dots during the intermediate phase of angiography (Figure 10C), representing partial or full-thickness choroidal scaring and granulomas observed in all eyes in a series of 36 patients with acute VKHD.98 These dark spots are noted to regress with treatment at 2 months follow-up96 and even to completely resolve in some patients;95 5) diffuse choroidal hyperfluorescence in the late phase (22–28 minutes) (Figure 10D).96 Unlike in healthy eyes, where the optic disc remains hypofluorescent, VKHD eyes exhibit optic disc hyperfluorescence indicating severe papillitis and corresponding to the “hot disc” appearance on FFA, which resolves following treatment.95,98 Hypofluorescent patches in areas of exudative retinal detachments have been described during the early phases of the angiogram.98 Choroidal folds are also seen in the late phase of the ICG as long hyperfluorescent lines radiating from the optic disc.91 Patients with chronic VKHD usually display similar but less severe ICG findings to those seen in the acute phase, which are fuzzy vessels, late diffuse choroidal hyperfluorescence, and hypofluorescent lesions corresponding to areas of chorioretinal atrophy.99 Evidence of active ongoing choroidal inflammation can be detected even in the absence of manifest clinical inflammation. Signs of choroidal inflammation were found in ICG scans of 72.5% (31 of 51 eyes) of patients with chronic VKHD, regardless of their clinical appearance and duration of treatment, as clinically active disease was observed in only 41.2% (21 of 51 eyes) of those eyes.99
 | Figure 10 ICG photographs of the right eye of a patient in the acute phase of VKHD showing (A) patchy hypofluorescence during the early angiographic phase, (B) large choroidal stromal vessel hyperfluorescence with fuzzy choroidal vessels in the early phase, (C) hypofluorescent dark dots during the intermediate phase of angiography, and (D) diffuse choroidal hyperfluorescence in the late phase. |
B-scan ultrasonography
Ultrasound (US) is a non-contact diagnostic imaging technique that uses high-frequency acoustic (sound) waves to provide a dynamic view of ocular structures. It has been found to be useful in diagnosing different stages of VKHD, particularly in cases with severe media opacities and a poor fundal view. Diffuse low-to-medium reflective choroidal thickening and SRD are evident during acute VKHD, especially when the SRD is too extensive.89 Swollen ciliary body with shallow anterior chamber, vitreous condensations, and scleral or episcleral thickening have also been seen on US in the acute stage.100–102 Subretinal fibrosis in chronic VKHD was described on US in 64.8% of the eyes with chronic VKHD as dome shaped, heterogeneous lesion; however, no other clinical signs of the chronic disease were detected on the US.103 Therefore, it is relatively unreliable in detecting subclinical recurrences and minor changes in choroidal thickness due to its limited resolution in comparison to OCT.104
Fundus autofluorescence (FAF)
FAF is a noninvasive efficient imaging technique that provides valuable information on the RPE and outer retinal layers.105
Metabolic and functional activity of the RPE can be detected in different stages of VKHD using the short wavelength light (blue) FAF that has been used to detect lipofuscin abnormalities and the near-infrared light FAF that has been used for melanin and melanin compound abnormalities.106 Different patterns of FAF have been demonstrated in different phases of VKHD, which supports the role of the RPE in the disease and allows repeated close monitoring of disease progression. In the acute phase, FAF demonstrates mild and uniform hyperautofluorescence in the macula and areas of hypoautofluorescence, corresponding to the areas of SRD.106,107 Resolution of the SRD with treatment after 1 month is associated with the appearance of placoid areas of hyperautofluorescence in the macula and peripapillary regions. These FAF changes are more evident on the near-infrared FAF and typically resolve after 6 months of treatment.106 In the chronic phase of VKHD, areas of hypoautofluorescence (Figure 11A), hyperautofluorescence (Figure 11B), and lattice-like pattern in the peripheral fundus are observed.108 Areas of hypoautofluorescence on FAF corresponded with peripapillary atrophy and RPE loss, while increased autofluorescence matches areas of RPE proliferation on SD-OCT in chronic VKHD.109
 | Figure 11 Short wavelength light (blue) FAF photographs of eyes of a patient with chronic VKHD. |
Optical coherence tomography
Recent advances in OCT imaging allows visualization of the choroid and choriocapillaris by providing cross-sectional images of the choroid.110 It is a noninvasive method, which is used to measure choroidal thickness for frequent, quantitative assessment of disease activity and response to treatment in different phases of VKHD. The presence of SRDs is seen on OCT scans as areas of subretinal fluid.92,93 This is usually associated with formation of fibrinous subretinal septa dividing the subretinal space into several compartments.92 Intraretinal edema, RPE undulation, choroidal folds, and choroidal hyperreflective dots can also be seen on the OCT scan in acute VKHD.67,111,112 Following resolution of the inflammation, with corticosteroid treatment, these signs resolve (Figure 12).113 Damage to the ellipsoid zone, persistent choroidal folds, and RPE undulation have been correlated with poor visual prognosis.69,93,114 OCT can be used to differentiate VKHD from other similar conditions, such as CSCR. In a retrospective study that compared the SD-OCT features of acute VKHD and acute CSCR, SRD was found to be present in both diseases, while RPE folds, fluctuations (rising and falling) of the internal limiting membrane (ILM) and fibrinous subretinal septa were only present in VKHD (Figure 12). The CSCR cases were found to have significantly more pigment epithelial detachments and bulges of the RPE.115 High-penetration OCT (HP-OCT) using a light source with a long wavelength (1,060 nm) may be superior to SD-OCT in visualizing structures beneath the RPE.116 HP-OCT is used to study 42 eyes of 21 patients with acute VKHD, of whom 24 eyes of 13 patients (57.1%) developed choroidal folds at presentation.69 The presence of these folds significantly correlated with older age, presence of disc swelling, increased choroidal thickness, and the development of sunset glow fundus at 6 months follow-up.69
 | Figure 12 OCT scan of the macula of a patient in the acute phase of VKHD before and after starting corticosteroid treatment. |
Enhanced depth imaging (EDI)-OCT demonstrates that patients with the acute phase of VKHD have a marked increase in the average subfoveal choroidal thickness, possibly due to inflammatory infiltration and exudation, which reverses following systemic corticosteroid therapy and resolution of the inflammation.104,117 Choroidal thickness continues to be abnormally increased during the chronic recurrent phase.118 An increase in choroidal thickness of >100 μm during corticosteroid tapering, even in the presence of apparently good clinical control, has been defined as rebound choroidal thickening suggesting recurrent subclinical inflammation.117 While this can be accompanied by other ocular signs, such as anterior chamber cells and/or optic disc hyperfluorescence on FFA and/or dark dots on ICG,119 it can also precede them.47 Rebound choroidal thickening on EDI-OCT was the only sign of ocular inflammation in a 71-year-old patient who presented 6 months after his initial diagnosis with headache, tinnitus, and bilateral sensorineural hearing loss.120 Conversely, in chronic VKHD, sunglow fundus correlates with choroidal thinning on EDI-OCT scans.121 The mean subfoveal choroidal thickness is significantly reduced in patients with severe depigmentation of the fundus, in comparison to patients with no-or-mild fundus depigmentation or healthy controls.122 Subfoveal choroidal thinning and focal loss of the choriocapillaris, with preservation of medium (Sattler’s layer) and large choroidal vessels (Haller’s layer), have also been observed on SD-OCT scans of 26 eyes of 13 consecutive patients during the convalescent stage of the disease.80
Ancillary tests
Electroretinography
Full-field electroretinography (ERG), which reflects the total electrophysiological activity of the entire retina, and the multifocal ERG, which represents the simultaneous electrical response of photoreceptors and the inner retina from different regions, are used to evaluate visual function in patients with VKHD.123,124 Extensive chorioretinal atrophy in chronic VKHD is associated with abnormal ERG recordings, possibly due to inflammatory retinal changes. A marked diffuse reduction in the amplitude of both scotopic and photopic phases of full-field ERG is correlated with severe fundus depigmentation and fibrosis in those patients.123 Reduction in multifocal ERG is correlated with reduced vision before treatment in chronic VKHD and shows good recovery with immunosuppressive therapy, reflecting improvement in the visual function.124 Improvement in the N1 and P1 waves of multifocal ERG is observed after treatment in a prospective study that involved 22 eyes with VKHD, which suggests that ERG may be a useful tool to monitor peripheral retinal disease activity and response to treatment.125
Microperimetry
Microperimetry, which determines differential light sensitivity at a certain retinal area, is used to assess the effect of VKHD on the retina and determine response to treatment. It provides a quantitative measurement of the visual function of a specific part of the retina and correlates that to OCT structural changes in that area. In patients with chronic VKHD, macular sensitivity on microperimetry and functional changes (best corrected visual acuity) match RPE thickening and other structural retinal changes on OCT scans. This could help in monitoring disease activity by assessing the visual function in those patients.126 Chronic recurrent disease with sunset glow fundus is associated with worse retinal sensitivity and poor visual outcome.127
Differential diagnosis of VKHD
The differential diagnosis of VKHD includes many other causes of panuveitis and posterior scleritis. Sympathetic ophthalmia (SO) has a similar clinical and histopathological appearance to VKHD, with swollen hyperemic optic discs and SRDs, and is in differential diagnosis of VKHD. However, bilateral granulomatous panuveitis in eyes with SO follow previous penetrating trauma or surgery to one eye.128,129
Posterior scleritis may present with SRD, optic disc swelling and choroidal or retinal folds and may be a presenting feature of VKHD. However, in cases of posterior scleritis, pain is a common feature classically radiating to the forehead or face and waking the patient up during the night. In addition, posterior scleritis is usually unilateral presenting often with reduced vision and ocular pain. Scleral and choroidal thickening, fluid in the Tenon’s capsule, and optic nerve head with the resulting “T sign” appearance are characteristic features on US.70,71,83
CSCR is an idiopathic condition that commonly affects healthy middle-aged men and is characterized by the development of SRDs, and patients may present with headaches that might be mistaken for the prodromal stage of VKHD. However, in CSCR, visual acuity remains good, patients do not complain of ocular pain and there is no evidence of granulomatous ocular inflammation or a hot optic disc on FFA.51,130 Infectious choroiditis caused by syphilis,131 tuberculosis,132 and Bartonella henselae infection133,134 should also been considered in the differential diagnosis of VKHD. Cat scratch disease caused by B. henselae can present with headache, tinnitus, panuveitis, optic disc swelling, and diffuse choroidal thickening mimicking VKHD. However, in cat scratch disease, the CSF is usually normal with raised antibody titers against B. henselae in the serum.134
Other less frequent causes of panuveitis include intraocular lymphoma that can present with neurological features and/or panuveitis masquerade appearance with thickened choroid and multifocal yellowish subretinal lesions.135 The rare occurrence of idiopathic uveal effusion syndrome is another differential that presents with scleral thickening, subacute onset of exudative detachments of choroid and retina but with no intraocular inflammation.136 Finally, ocular sarcoidosis can present as a granulomatous anterior uveitis or panuveitis with choroidal and optic disc granulomas, segmental phlebitis, and SRDs.137 Pulmonary findings, as well as elevated serum angiotensin-converting enzyme levels, will help differentiate the two conditions.
Treatment
The goal of treatment in VKHD is to suppress active ocular inflammation, prevent disease relapse and avoid sight-threatening complications. As such, early diagnosis and rapid commencement of treatment are important in preserving vision in these young patients. Multiple therapeutic regimens are used combining both systemic immunosuppression agents together with locally administered corticosteroids and anti-vascular endothelial growth factor drugs (anti-VEGF).138–142 Because VKHD can involve multiple organs, the mainstay of treatment is based on high-dose systemic corticosteroids,43 administered either orally or intravenously.143 Oral prednisone at a dose of 1–2 mg/kg/day started early in the course of the disease followed by slow tapering to avoid recurrences is the generally accepted regimen,144,145 while pulse intravenous corticosteroid therapy of 1 g/day of methylprednisolone for 3–5 days followed by oral prednisolone is usually reserved for cases with severe inflammation.43 Slow tapering of the corticosteroid dose, with frequent follow-up examinations, is warranted in order to avoid recurrence of posterior segment inflammation.144 Aiming to reach a dose of <7.5 mg/day over several months and keeping patients on long-term treatment may reduce the risk of relapses, but nonetheless, these occur even when inflammatory control is achieved at high doses.27,44 Treatment at these low, safe doses with no evident ocular inflammation should be maintained for several years before cessation of therapy is considered. Topical corticosteroids and cycloplegic agents are effective in controlling the anterior segment inflammation.34 The addition of second-line immunosuppressive agents including mycophenolate mofetil, methotrexate, cyclosporine, or azathioprine are usually considered in patients who are not controlled with corticosteroids alone or cannot be tapered to a safe low dose for the extended period of time they require to control their inflammation.146–148 Use of these agents has been found to shorten the duration of the acute stage and to reduce the risk of progression to the chronic stages and development of sight-threatening complications.138,149,150 Their addition early in the disease has been found to be associated with a better visual outcome in comparison to corticosteroids alone or if their addition is delayed.138,151
While intravitreal and posterior subtenon triamcinolone acetonide injections have been found to be effective in short-term control of inflammation during the acute phase of VKHD,139,140 intravenous infusion of anti-tumor necrosis factor α agents (adalimumab and infliximab) and rituximab, which is an anti-CD20 monoclonal antibody, has been used in refractory cases and found to be effective in long-term disease control.152–154
Intravitreal injections of anti-VEGF have been used successfully for the treatment of CNVs,155 and combined treatment of triamcinolone acetate and anti-VEGF has also been tried to treat recurrent CNVs.156 Persistent SRDs involving the fovea in four patients with bilateral VKHD were successfully resolved using intravitreal injections of anti-VEGF.142
Complications associated with VKHD
Although the inflammation can be controlled by corticosteroids and other immunosuppressive agents, some patients may still have a poor visual outcome due to development of other sight-threatening complications such as glaucoma, cataract (which can be treated), CNV, and subretinal fibrosis (Table 1). These complications tend to occur during the chronic stage of the disease and are associated with a poor visual outcome,1,2 especially if there is delayed presentation.86
 | Table 1 Complications associated with VKHD |
Glaucoma is the most common complication of VKHD with an incidence of up to 45%.157–159 Secondary glaucoma has been attributed to a combination of ciliary body inflammation, pupillary block, formation of anterior synechiae leading to trabecular meshwork obstruction and uveal effusion.159,160 In one study, glaucoma occurred in up to 38% of VKHD patients with the majority requiring surgical intervention to control their IOP.159 A lower incidence of 15.8% has been reported by another group, in whom IOP was controlled mainly by medical treatment. The authors propose that aggressive use of steroid-sparing immunosuppressive agents to control the inflammatory process in their cohort, as well as the effectiveness of the currently available topical anti-glaucoma medication, prevented the need for glaucoma surgery.160
Cataract is the second most common complication of VKHD,157,158,161 with an incidence of up to 42%.2,34 Prolonged steroid treatment and recurrent ocular inflammation are the main risk factors for its development.1,30 Cataract surgery is usually recommended only when the inflammation is quiescent for at least 3 months and should be performed with preoperative steroid cover for up to 2 weeks.162,163 Other less common complications that might be responsible for visual loss in patients with VKHD include peripapillary and macular CNVs,164 which develop in up to 14.7%,165 and subretinal fibrosis.2,30,34 Subretinal fibrosis can occur in up to 40% of VKHD patients165 and may lead to substantial visual impairment when the fovea is involved.166 Unlike other ocular inflammatory conditions, CME appears to be a relatively rare complication of VKHD.160,167 Other uncommon complications (Table 1) includes posterior synechiae,158 rhegmatogenous retinal detachment and ERM160 and MH formation,84 band keratopathy,168 and pseudotumoral RPE proliferation.169,170 The high number of complications together with delayed treatment and poor visual acuity at presentation especially in older patients is found to be associated with poor prognosis.2,171
Conclusion
VKHD is a multisystemic autoimmune disease affecting mainly middle-aged patients; it has four distinct phases that may not present in sequential order and detection of subclinical inflammation is still a challenge. Therefore, in order to avoid delayed treatment and development of sight-threatening complications that tend to occur mostly in patients with long-standing ocular inflammation, it is important to recognize the variable clinical presentations of the disease. Utilizing ocular imaging for diagnosis and repeat use of noninvasive modalities, such as OCT to monitor disease activity, can promote early treatment and prevent the development of ocular complications. Early aggressive treatment with systemic corticosteroids during the acute stage is highly recommended. The use of immunosuppressive agents has reduced the risk of developing ocular complications; however, a considerable number of patients still progress to the chronic stages, suffer recurrences and progress to visual loss, probably due to undetectable and undertreated subclinical inflammation. Further prospective studies using sensitive imaging methods are needed to help clarify the role of ongoing choroidal inflammation and RPE activity on the clinical course of the disease and its effect on retinal function and visual outcome.
Disclosure
The authors report no conflicts of interest in this work.
References
Moorthy RS, Inomata H, Rao NA. Vogt–Koyanagi–Harada syndrome. Surv Ophthalmol. 1995;39(4):265–292. | ||
Read RW, Rechodouni A, Butani N, et al. Complications and prognostic factors in Vogt–Koyanagi–Harada disease. Am J Ophthalmol. 2001;131(5):599–606. | ||
Norose K, Yano A. Melanoma specific Th1 cytotoxic T lymphocyte lines in Vogt–Koyanagi–Harada disease. Br J Ophthalmol. 1996;80(11):1002–1008. | ||
Yamaki K, Gocho K, Hayakawa K, Kondo I, Sakuragi S. Tyrosinase family proteins are antigens specific to Vogt–Koyanagi–Harada disease. J Immunol. 2000;165(12):7323–7329. | ||
Sugita S, Takase H, Taguchi C, et al. Ocular infiltrating CD4+ T cells from patients with Vogt–Koyanagi–Harada disease recognize human melanocyte antigens. Invest Ophthalmol Vis Sci. 2006;47(6):2547–2554. | ||
Shindo Y, Inoko H, Yamamoto T, Ohno S. HLA-DRB1 typing of Vogt–Koyanagi–Harada’s disease by PCR-RFLP and the strong association with DRB1*0405 and DRB1*0410. Br J Ophthalmol. 1994;78(3):223–226. | ||
Shi T, Lv W, Zhang L, Chen J, Chen H. Association of HLA-DR4/HLA-DRB1*04 with Vogt–Koyanagi–Harada disease: a systematic review and meta-analysis. Sci Rep. 2014;4:6887. | ||
Damico FM, Cunha-Neto E, Goldberg AC, et al. T-cell recognition and cytokine profile induced by melanocyte epitopes in patients with HLA-DRB1*0405-positive and -negative Vogt–Koyanagi–Harada uveitis. Invest Ophthalmol Vis Sci. 2005;46(7):2465–2471. | ||
Rao NA. Pathology of Vogt–Koyanagi–Harada disease. Int Ophthalmol. 2007;27(2–3):81–85. | ||
Oellers P, Jaffe GJ, Proia AD. Clinical-pathological correlation of Vogt–Koyanagi–Harada disease. JAMA Ophthalmol. 2016;7:1–3. | ||
Pattison EM. Uveomeningoencephalitic syndrome (Vogt–Koyanagi–Harada). Arch Neurol. 1965;12:197–205. | ||
Vogt A. Frühzeitiges Ergrauen der Zilien und Bemerkungen über den sogenannten plötzlichen Eintritt dieser Veränderung [Early graying of cilia and remarks about the so-called sudden occurrence of these change]. Klin Monatsbl Augenheilkd. 1906;44:228–242. German. | ||
Koyanagi YD. Alopecia und Poliosis bei schwerer Uveitis nicht traumatischen Ursprungs [Dysacusis, alopecia and poliosis in severe uveitis without traumatic origin]. Klin Monatsbl Augenheilkd. 1929;82:194–211. German. | ||
Harada E. Clinical study of nonsuppurative choroiditis: a report of acute diffuse choroiditis. Acta Soc Opthalmol Jpn. 1926;30:356–378. | ||
Herbort CP, Mochizuki M. Vogt–Koyanagi–Harada disease: inquiry into the genesis of a disease name in the historical context of Switzerland and Japan. Int Ophthalmol. 2007;27(2–3):67–79. | ||
Yang P, Zhang Z, Zhou H, et al. Clinical patterns and characteristics of uveitis in a tertiary center for uveitis in China. Curr Eye Res. 2005;30(11):943–948. | ||
Ohguro N, Sonoda KH, Takeuchi M, Matsumura M, Mochizuki M. The 2009 prospective multi-center epidemiologic survey of uveitis in Japan. Jpn J Ophthalmol. 2012;56(5):432–435. | ||
Silpa-Archa S, Noonpradej S, Amphornphruet A. Pattern of uveitis in a referral ophthalmology center in the central district of Thailand. Ocul Immunol Inflamm. 2014:1–9. Epub 2014 Aug 11. | ||
Al Dhahri H, Al Rubaie K, Hemachandran S, et al. Patterns of uveitis in a University-Based Tertiary Referral Center in Riyadh, Saudi Arabia. Ocul Immunol Inflamm. 2014:1–9. Epub 2014 Jul 24. | ||
Liberman P, Gauro F, Berger O, Urzua CA. Causes of uveitis in a tertiary center in Chile: a cross-sectional retrospective review. Ocul Immunol Inflamm. 2014:1–7. Epub 2014 Dec 1. | ||
Nussenblatt RB. Clinical studies of Vogt–Koyanagi–Harada’s disease at the National Eye Institute, NIH, USA. Jpn J Ophthalmol. 1988;32(3):330–333. | ||
Perkins ES, Folk J. Uveitis in London and Iowa. Ophthalmologica. 1984;189(1–2):36–40. | ||
Smit RL, Baarsma GS, de Vries J. Classification of 750 consecutive uveitis patients in the Rotterdam Eye Hospital. Int Ophthalmol. 1993;17(2):71–76. | ||
Cimino L, Aldigeri R, Salvarani C, et al. The causes of uveitis in a referral centre of Northern Italy. Int Ophthalmol. 2010;30(5):521–529. | ||
Tugal-Tutkun I, Ozyazgan Y, Akova YA, et al. The spectrum of Vogt–Koyanagi–Harada disease in Turkey: VKH in Turkey. Int Ophthalmol. 2007;27(2–3):117–123. | ||
Chee SP, Jap A, Bacsal K. Spectrum of Vogt–Koyanagi–Harada disease in Singapore. Int Ophthalmol. 2007;27(2–3):137–142. | ||
Iwahashi C, Okuno K, Hashida N, Nakai K, Ohguro N, Nishida K. Incidence and clinical features of recurrent Vogt–Koyanagi–Harada disease in Japanese individuals. Jpn J Ophthalmol. 2015;59(3):157–163. | ||
Rao NA, Gupta A, Dustin L, et al. Frequency of distinguishing clinical features in Vogt–Koyanagi–Harada disease. Ophthalmology. 2010;117(3):591–599,599.e1. | ||
Wang Y, Chan CC. Gender differences in Vogt–Koyanagi–Harada disease and sympathetic ophthalmia. J Ophthalmol. 2014;2014:157803. | ||
Abu El-Asrar AM, Al-Kharashi AS, Aldibhi H, Al-Fraykh H, Kangave D. Vogt–Koyanagi–Harada disease in children. Eye. 2008;22(9):1124–1131. | ||
Martin TD, Rathinam SR, Cunningham ET Jr. Prevalence, clinical characteristics, and causes of vision loss in children with Vogt–Koyanagi–Harada disease in South India. Retina. 2010;30(7):1113–1121. | ||
Forster DJ, Green RL, Rao NA. Unilateral manifestation of the Vogt–Koyanagi–Harada syndrome in a 7-year-old child. Am J Ophthalmol. 1991;111(3):380–382. | ||
Yamamoto Y, Fukushima A, Nishino K, Koura Y, Komatsu T, Ueno H. Vogt–Koyanagi–Harada disease with onset in elderly patients aged 68 to 89 years. Jpn J Ophthalmol. 2007;51(1):60–63. | ||
Yang P, Ren Y, Li B, Fang W, Meng Q, Kijlstra A. Clinical characteristics of Vogt–Koyanagi–Harada syndrome in Chinese patients. Ophthalmology. 2007;114(3):606–614. | ||
Read RW, Holland GN, Rao NA, et al. Revised diagnostic criteria for Vogt–Koyanagi–Harada disease: report of an international committee on nomenclature. Am J Ophthalmol. 2001;131(5):647–652. | ||
Rao NA, Sukavatcharin S, Tsai JH. Vogt–Koyanagi–Harada disease diagnostic criteria. Int Ophthalmol. 2007;27(2–3):195–199. | ||
Morita S, Nakamaru Y, Obara N, Masuya M, Fukuda S. Characteristics and prognosis of hearing loss associated with Vogt–Koyanagi–Harada disease. Audiol Neurootol. 2014;19(1):49–56. | ||
Gaudreau P, Moy J, Lindsay F. An unusual cause of vertigo, tinnitus, and hyperacusis: Vogt–Koyanagi–Harada syndrome. Ear Nose Throat J. 2012;91(12):E7–E9. | ||
Sukavatcharin S, Tsai JH, Rao NA. Vogt–Koyanagi–Harada disease in Hispanic patients. Int Ophthalmol. 2007;27(2–3):143–148. | ||
Tsai JH, Sukavatcharin S, Rao NA. Utility of lumbar puncture in diagnosis of Vogt–Koyanagi–Harada disease. Int Ophthalmol. 2007;27(2–3):189–194. | ||
Beniz J, Forster DJ, Lean JS, Smith RE, Rao NA. Variations in clinical features of the Vogt–Koyanagi–Harada syndrome. Retina. 1991;11(3):275–280. | ||
Han HJ, Kim HY, Park JH, Lee EJ, Kim do G, Shin DI. Magnetic resonance imaging of pachymeningeal enhancement in Vogt–Koyanagi–Harada disease. Neurol Sci. 2010;31(6):785–788. | ||
Rao NA. Treatment of Vogt–Koyanagi–Harada disease by corticosteroids and immunosuppressive agents. Ocul Immunol Inflamm. 2006;14(2):71–72. | ||
Sakata VM, da Silva FT, Hirata CE, et al. High rate of clinical recurrence in patients with Vogt–Koyanagi–Harada disease treated with early high-dose corticosteroids. Graefes Arch Clin Exp Ophthalmol. 2015;253(5):785–790. | ||
Keino H, Goto H, Usui M. Sunset glow fundus in Vogt–Koyanagi–Harada disease with or without chronic ocular inflammation. Graefes Arch Clin Exp Ophthalmol. 2002;240(10):878–882. | ||
Taylor S, Lightman S. Recurrent anterior uveitis in patients with Vogt–Koyanagi–Harada syndrome. Arch Ophthalmol. 2004;122(6):922–923. | ||
Tagawa Y, Namba K, Mizuuchi K, et al. Choroidal thickening prior to anterior recurrence in patients with Vogt–Koyanagi–Harada disease. Br J Ophthalmol. 2016;100(4):473–477. | ||
Bacsal K, Wen DS, Chee SP. Concomitant choroidal inflammation during anterior segment recurrence in Vogt–Koyanagi–Harada disease. Am J Ophthalmol. 2008;145(3):480–486. | ||
Takemoto Y, Namba K, Mizuuchi K, et al. Choroidal circulation impairment during the anterior recurrence of Vogt–Koyanagi–Harada disease confirmed with indocyanine green angiography and laser speckle flowgraphy. Acta Ophthalmol. 2016. Epub 2016 Apr 15. | ||
Okunuki Y, Tsubota K, Kezuka T, Goto H. Differences in the clinical features of two types of Vogt–Koyanagi–Harada disease: serous retinal detachment and optic disc swelling. Jpn J Ophthalmol. 2015;59(2):103–108. | ||
Shin WB, Kim MK, Lee CS, Lee SC, Kim H. Comparison of the Clinical Manifestations between Acute Vogt–Koyanagi–Harada Disease and Acute Bilateral Central Serous Chorioretinopathy. Korean J Ophthalmol. 2015;29(6):389–395. | ||
Roe RH, Rathinam SR, Wong RW, McDonald HR, Jumper JM, Cunningham ET Jr. Delayed fellow eye involvement in patients with Vogt–Koyanagi–Harada disease. Br J Ophthalmol. 2009;93(5):701–702. | ||
Neves A, Cardoso A, Almeida M, Campos J, Campos A, Castro Sousa JP. Unilateral Vogt–Koyanagi–Harada Disease: a Clinical Case Report. Case Rep Ophthalmol. 2015;6(3):361–365. | ||
Usui Y, Goto H, Sakai J, Takeuchi M, Usui M, Rao NA. Presumed Vogt–Koyanagi–Harada disease with unilateral ocular involvement: report of three cases. Graefes Arch Clin Exp Ophthalmol. 2009;247(8):1127–1132. | ||
Ohno S, Minakawa R, Matsuda H. Clinical studies of Vogt–Koyanagi–Harada’s disease. Jpn J Ophthalmol. 1988;32(3):334–343. | ||
Yang HK, Park KH, Kim JS, Hwang JM. Bilateral disc edema in a patient with Vogt–Koyanagi–Harada disease. Can J Ophthalmol. 2014;49(2):e54–e56. | ||
Nakao K, Abematsu N, Mizushima Y, Sakamoto T. Optic disc swelling in Vogt–Koyanagi–Harada disease. Invest Ophthalmol Vis Sci. 2012;53(4):1917–1922. | ||
Attia S, Khochtali S, Kahloun R, et al. Clinical and multimodal imaging characteristics of acute Vogt–Koyanagi–Harada disease unassociated with clinically evident exudative retinal detachment. Int Ophthalmol. 2016;36(1):37–44. | ||
Rajendram R, Evans M, Khurana RN, Tsai JH, Rao NA. Vogt–Koyanagi–Harada disease presenting as optic neuritis. Int Ophthalmol. 2007;27(2–3):217–220. | ||
Nakao K, Mizushima Y, Abematsu N, Goh N, Sakamoto T. Anterior ischemic optic neuropathy associated with Vogt–Koyanagi–Harada disease. Graefes Arch Clin Exp Ophthalmol. 2009;247(10):1417–1425. | ||
Abematsu N, Shimonagano Y, Nakao K, Sakamoto T, Shimizu K, Hirashima S. [A case of anterior ischemic optic neuropathy associated with Vogt–Koyanagi–Harada disease]. Nippon Ganka Gakkai Zasshi. 2006;110(8):601–606. Japanese. | ||
Yokoyama A, Ohta K, Kojima H, Yoshimura N. Vogt–Koyanagi–Harada disease masquerading anterior ischaemic optic neuropathy. Br J Ophthalmol. 1999;83(1):123. | ||
Narang S, Sood S, Malik A. Probable Vogt–Koyanagi–Harada’s syndrome associated with tonic pupils. Nepal J Ophthalmol. 2010;2(2):154–156. | ||
Kato Y, Yamamoto Y, Tabuchi H, Fukushima A. Retinal pigment epithelium folds as a diagnostic finding of Vogt–Koyanagi–Harada disease. Jpn J Ophthalmol. 2013;57(1):90–94. | ||
Wu W, Wen F, Huang S, Luo G, Wu D. Choroidal folds in Vogt–Koyanagi–Harada disease. Am J Ophthalmol. 2007;143(5):900–901. | ||
Zhao C, Zhang M, Wen X, Dong F, Han B, Du H. Choroidal folds in acute Vogt–Koyanagi–Harada disease. Ocul Immunol Inflamm. 2009;17(4):282–288. | ||
Gupta V, Gupta A, Gupta P, Sharma A. Spectral-domain cirrus optical coherence tomography of choroidal striations seen in the acute stage of Vogt–Koyanagi–Harada disease. Am J Ophthalmol. 2009;147(1):148–153.e142. | ||
Tanigawa M, Ochiai H, Tsukahara Y, Ochiai Y, Yamanaka H. Choroidal folds in acute-stage Vogt–Koyanagi–Harada disease patients with relatively short axial length. Case Rep Ophthalmol. 2012;3(1):38–45. | ||
Tsuboi K, Nakai K, Iwahashi C, Gomi F, Ikuno Y, Nishida K. Analysis of choroidal folds in acute Vogt–Koyanagi–Harada disease using high-penetration optical coherence tomography. Graefes Arch Clin Exp Ophthalmol. 2015;253(6):959–964. | ||
Kouda N, Sasaki H, Harada S, Yamada Y, Takahashi N, Sasaki K. Early manifestation of Vogt–Koyanagi–Harada disease as unilateral posterior scleritis. Jpn J Ophthalmol. 2002;46(5):590–593. | ||
Watanabe K, Kato T, Hayasaka S. Concurrent bilateral posterior scleritis and Vogt–Koyanagi–Harada disease in a patient with positive rheumatoid factor. Ophthalmologica. 1997;211(5):316–319. | ||
Rathinam SR, Namperumalsamy P, Nozik RA, Cunningham ET Jr. Angle closure glaucoma as a presenting sign of Vogt–Koyanagi–Harada syndrome. Br J Ophthalmol. 1997;81(7):608–609. | ||
Yao J, Chen Y, Shao T, Ling Z, Wang W, Qian S. Bilateral acute angle closure glaucoma as a presentation of Vogt–Koyanagi–Harada syndrome in four Chinese patients: a small case series. Ocul Immunol Inflamm. 2013;21(4):286–291. | ||
Yang P, Liu X, Zhou H, Guo W, Zhou C, Kijlstra A. Vogt–Koyanagi–Harada disease presenting as acute angle closure glaucoma at onset. Clin Experiment Ophthalmol. 2011;39(7):639–647. | ||
Mili-Boussen I, Letaief I, Dridi H, Ouertani A. Bilateral retinal pigment epithelium tears in acute Vogt–Koyanagi–Harada disease. Retin Cases Brief Rep. 2013;7(4):350–354. | ||
Mizuno M, Fujinami K, Watanabe K, Akiyama K. Macular hole associated with Vogt–Koyanagi–Harada Disease at the Acute Uveitic Stage. Case Rep Ophthalmol. 2015;6(3):328–332. | ||
Inomata H, Sakamoto T. Immunohistochemical studies of Vogt–Koyanagi–Harada disease with sunset sky fundus. Curr Eye Res. 1990;9(suppl):35–40. | ||
Keino H, Goto H, Mori H, Iwasaki T, Usui M. Association between severity of inflammation in CNS and development of sunset glow fundus in Vogt–Koyanagi–Harada disease. Am J Ophthalmol. 2006;141(6):1140–1142. | ||
Inomata H, Rao NA. Depigmented atrophic lesions in sunset glow fundi of Vogt–Koyanagi–Harada disease. Am J Ophthalmol. 2001;131(5):607–614. | ||
Nazari H, Hariri A, Hu Z, Ouyang Y, Sadda S, Rao NA. Choroidal atrophy and loss of choriocapillaris in convalescent stage of Vogt–Koyanagi–Harada disease: in vivo documentation. J Ophthalmic Inflamm Infect. 2014;4:9. | ||
Harada Y, Bhat P, Munk MR, Goldstein DA. Changes in Scleral Architecture in Chronic Vogt–Koyanagi–Harada disease. Ocul Immunol Inflamm. 2015:1–8. Epub 2015 Dec 8. | ||
Moon SY, Yoon WT, Park SP. Recurrent unilateral Vogt–Koyanagi–Harada disease with posterior scleritis. Korean J Ophthalmol. 2015;29(5):352–354. | ||
Sambhav K, Majumder PD, Biswas J. Necrotizing scleritis in a case of Vogt–Koyanagi–Harada disease. Oman J Ophthalmol. 2015;8(3):216. | ||
Kobayashi I, Inoue M, Okada AA, Keino H, Wakabayashi T, Hirakata A. Vitreous surgery for macular hole in patients with Vogt–Koyanagi–Harada disease. Clin Experiment Ophthalmol. 2008;36(9):861–864. | ||
Fang W, Zhou H, Yang P, Huang X, Wang L, Kijlstra A. Longitudinal quantification of aqueous flare and cells in Vogt–Koyanagi–Harada disease. Br J Ophthalmol. 2008;92(2):182–185. | ||
Attia S, Zaouali S, Bettaieb A, Yahia SB, Khairallah M. Peripheral iris depigmentation and ocular hypotony: result of the natural course of non-treated Vogt–Koyanagi–Harada (VKH) disease. Int Ophthalmol. 2007;27(2–3):221–222. | ||
Suhler EB, Buggage RR, Nussenblatt RB, Neumann R. Symmetric peripheral iris depigmentation in Vogt–Koyanagi–Harada syndrome. Arch Ophthalmol. 2002;120(8):1104–1105. | ||
Arellanes-Garcia L, Hernandez-Barrios M, Fromow-Guerra J, Cervantes-Fanning P. Fluorescein fundus angiographic findings in Vogt–Koyanagi–Harada syndrome. Int Ophthalmol. 2007;27(2–3):155–161. | ||
Jap A, Chee SP. Imaging in the diagnosis and management of Vogt–Koyanagi–Harada disease. Int Ophthalmol Clin. 2012;52(4):163–172. | ||
Chee SP, Jap A, Cheung CM. The prognostic value of angiography in Vogt–Koyanagi–Harada disease. Am J Ophthalmol. 2010;150(6):888–893. | ||
Fardeau C, Tran TH, Gharbi B, Cassoux N, Bodaghi B, LeHoang P. Retinal fluorescein and indocyanine green angiography and optical coherence tomography in successive stages of Vogt–Koyanagi–Harada disease. Int Ophthalmol. 2007;27(2–3):163–172. | ||
Yamaguchi Y, Otani T, Kishi S. Tomographic features of serous retinal detachment with multilobular dye pooling in acute Vogt–Koyanagi–Harada disease. Am J Ophthalmol. 2007;144(2):260–265. | ||
Ishihara K, Hangai M, Kita M, Yoshimura N. Acute Vogt–Koyanagi–Harada disease in enhanced spectral-domain optical coherence tomography. Ophthalmology. 2009;116(9):1799–1807. | ||
Yannuzzi LA, Slakter JS, Sorenson JA, Guyer DR, Orlock DA. Digital indocyanine green videoangiography and choroidal neovascularization. 1992. Retina. 2012;32(suppl 1):191. | ||
Herbort CP, Mantovani A, Bouchenaki N. Indocyanine green angiography in Vogt–Koyanagi–Harada disease: angiographic signs and utility in patient follow-up. Int Ophthalmol. 2007;27(2–3):173–182. | ||
Bouchenaki N, Herbort CP. The contribution of indocyanine green angiography to the appraisal and management of Vogt–Koyanagi–Harada disease. Ophthalmology. 2001;108(1):54–64. | ||
Knecht PB, Mantovani A, Herbort CP. Indocyanine green angiography-guided management of Vogt–Koyanagi–Harada disease: differentiation between choroidal scars and active lesions. Int Ophthalmol. 2013;33(5):571–577. | ||
Abouammoh MA, Gupta V, Hemachandran S, Herbort CP, Abu El-Asrar AM. Indocyanine green angiographic findings in initial-onset acute Vogt–Koyanagi–Harada disease. Acta Ophthalmol. 2016;94(6):573–578. | ||
da Silva FT, Hirata CE, Sakata VM, et al. Indocyanine green angiography findings in patients with long-standing Vogt–Koyanagi–Harada disease: a cross-sectional study. BMC Ophthalmol. 2012;12:40. | ||
Forster DJ, Cano MR, Green RL, Rao NA. Echographic features of the Vogt–Koyanagi–Harada syndrome. Arch Ophthalmol. 1990;108(10):1421–1426. | ||
Kishi A, Nao-i N, Sawada A. Ultrasound biomicroscopic findings of acute angle-closure glaucoma in Vogt–Koyanagi–Harada syndrome. Am J Ophthalmol. 1996;122(5):735–737. | ||
Wada S, Kohno T, Yanagihara N, et al. Ultrasound biomicroscopic study of ciliary body changes in the post-treatment phase of Vogt–Koyanagi–Harada disease. Br J Ophthalmol. 2002;86(12):1374–1379. | ||
Mayorquín-Ruiz M, Cheja-Kalb R, Concha-del Río L, Arellanes-García L, Moragrega-Adame E. Echographic findings in the late stages of Vogt–Koyanagi–Harada disease in Mexican population. Rev Bras Oftalmol. 2014;73(6):348–350. | ||
Maruko I, Iida T, Sugano Y, et al. Subfoveal choroidal thickness after treatment of Vogt–Koyanagi–Harada disease. Retina. 2011;31(3):510–517. | ||
Schmitz-Valckenberg S, Holz FG, Bird AC, Spaide RF. Fundus autofluorescence imaging: review and perspectives. Retina. 2008;28(3):385–409. | ||
Koizumi H, Maruyama K, Kinoshita S. Blue light and near-infrared fundus autofluorescence in acute Vogt–Koyanagi–Harada disease. Br J Ophthalmol. 2010;94(11):1499–1505. | ||
Ayata A, Dogru S, Senol MG, Unal M, Ersanli D, Bilge AH. Autofluorescence findings in Vogt–Koyanagi–Harada disease. Eur J Ophthalmol. 2009;19(6):1094–1097. | ||
Heussen FM, Vasconcelos-Santos DV, Pappuru RR, Walsh AC, Rao NA, Sadda SR. Ultra-wide-field green-light (532-nm) autofluorescence imaging in chronic Vogt–Koyanagi–Harada disease. Ophthalmic Surg Lasers Imaging. 2011;42(4):272–277. | ||
Vasconcelos-Santos DV, Sohn EH, Sadda S, Rao NA. Retinal pigment epithelial changes in chronic Vogt–Koyanagi–Harada disease: fundus autofluorescence and spectral domain-optical coherence tomography findings. Retina. 2010;30(1):33–41. | ||
Mrejen S, Spaide RF. Optical coherence tomography: imaging of the choroid and beyond. Surv Ophthalmol. 2013;58(5):387–429. | ||
Fong AH, Li KK, Wong D. Choroidal evaluation using enhanced depth imaging spectral-domain optical coherence tomography in Vogt-Koyanagi-Harada disease. Retina. 2011;31(3):502–509. | ||
Zhao C, Zhang MF, Dong FT, et al. Spectral domain optical coherence tomography of Vogt–Koyanagi–Harada disease: novel findings and new insights into the pathogenesis. Chin Med Sci J. 2012;27(1):29–34. | ||
Yamanaka E, Ohguro N, Yamamoto S, Nakagawa Y, Imoto Y, Tano Y. Evaluation of pulse corticosteroid therapy for Vogt–Koyanagi–Harada disease assessed by optical coherence tomography. Am J Ophthalmol. 2002;134(3):454–456. | ||
Hosoda Y, Uji A, Hangai M, Morooka S, Nishijima K, Yoshimura N. Relationship between retinal lesions and inward choroidal bulging in Vogt–Koyanagi–Harada disease. Am J Ophthalmol. 2014;157(5):1056–1063. | ||
Lin D, Chen W, Zhang G, et al. Comparison of the optical coherence tomographic characters between acute Vogt–Koyanagi–Harada disease and acute central serous chorioretinopathy. BMC Ophthalmol. 2014;14:87. | ||
Nakai K, Gomi F, Ikuno Y, et al. Choroidal observations in Vogt–Koyanagi–Harada disease using high-penetration optical coherence tomography. Graefes Arch Clin Exp Ophthalmol. 2012;250(7):1089–1095. | ||
Nakayama M, Keino H, Okada AA, et al. Enhanced depth imaging optical coherence tomography of the choroid in Vogt–Koyanagi–Harada disease. Retina. 2012;32(10):2061–2069. | ||
Hashizume K, Imamura Y, Fujiwara T, Machida S, Ishida M, Kurosaka D. Choroidal thickness in eyes with posterior recurrence of Vogt–Koyanagi–Harada disease after high-dose steroid therapy. Acta Ophthalmol. 2014;92(6):e490–e491. | ||
Sakata VM, da Silva FT, Hirata CE, Takahashi WY, Costa RA, Yamamoto JH. Choroidal bulging in patients with Vogt–Koyanagi–Harada disease in the non-acute uveitic stage. J Ophthalmic Inflamm Infect. 2014;4(1):6. | ||
Ishibazawa A, Kinouchi R, Minami Y, Katada A, Yoshida A. Recurrent Vogt–Koyanagi–Harada disease with sensorineural hearing loss and choroidal thickening. Int Ophthalmol. 2014;34(3):679–684. | ||
da Silva FT, Sakata VM, Nakashima A, et al. Enhanced depth imaging optical coherence tomography in long-standing Vogt–Koyanagi–Harada disease. Br J Ophthalmol. 2013;97(1):70–74. | ||
Takahashi H, Takase H, Ishizuka A, et al. Choroidal thickness in convalescent Vogt–Koyanagi–Harada disease. Retina. 2014;34(4):775–780. | ||
da Silva FT, Hirata CE, Olivalves E, Oyamada MK, Yamamoto JH. Fundus-based and electroretinographic strategies for stratification of late-stage Vogt–Koyanagi–Harada disease patients. Am J Ophthalmol. 2009;148(6):939–945.e933. | ||
Chee SP, Luu CD, Cheng CL, Lim WK, Jap A. Visual function in Vogt–Koyanagi–Harada patients. Graefes Arch Clin Exp Ophthalmol. 2005;243(8):785–790. | ||
Yang P, Fang W, Wang L, Wen F, Wu W, Kijlstra A. Study of macular function by multifocal electroretinography in patients with Vogt–Koyanagi–Harada syndrome. Am J Ophthalmol. 2008;146(5):767–771. | ||
Zhou M, Jiang C, Gu R, Sun Z, Huynh N, Chang Q. Correlation between retinal changes and visual function in late-stage Vogt–Koyanagi–Harada disease: an Optical Coherence Tomography Study. J Ophthalmol. 2015;2015:916485. | ||
Abu El-Asrar AM, Mudhaiyan TA, Al Najashi AA, et al. Chronic recurrent Vogt–Koyanagi–Harada disease and development of ‘sunset glow fundus’ predict worse retinal sensitivity. Ocul Immunol Inflamm. 2016:1–11. Epub 2016 Mar 22. | ||
Goto H, Rao NA. Sympathetic ophthalmia and Vogt–Koyanagi–Harada syndrome. Int Ophthalmol Clin. 1990;30(4):279–285. | ||
Rao NA, Marak GE. Sympathetic ophthalmia simulating Vogt–Koyanagi–Harada’s disease: a clinico-pathologic study of four cases. Jpn J Ophthalmol. 1983;27(3):506–511. | ||
Komuku Y, Iwahashi C, Yano S, Tanaka C, Nakagawa T, Gomi F. En face optical coherence tomography imaging of the choroid in a case with central serous chorioretinopathy during the course of Vogt–Koyanagi–Harada Disease: a case report. Case Rep Ophthalmol. 2015;6(3):488–494. | ||
Kim LA, Khurana RN, Parikh JG, Rao NA. Melanin-laden macrophages in the CSF to diagnose Vogt–Koyanagi–Harada simulating ocular syphilis. Ocul Immunol Inflamm. 2008;16(1):59–61. | ||
Gupta V, Gupta A, Rao NA. Intraocular tuberculosis – an update. Surv Ophthalmol. 2007;52(6):561–587. | ||
Jaumouille S, Mortemousque B. Neurorétinite avec œdème maculaire stellaire et œdème papillaire : infection à Bartonella henselae (maladie des griffes du chat) [Neuroretinitis with stellate macular edema and papilledema: Bartonella henselae infection (cat-scratch disease)]. Journal francais d’ophtalmologie. 2016;39(1):127–128. French. | ||
Khurana RN, Albini T, Green RL, Rao NA, Lim JI. Bartonella henselae infection presenting as a unilateral panuveitis simulating Vogt–Koyanagi–Harada syndrome. Am J Ophthalmol. 2004;138(6):1063–1065. | ||
Sagoo MS, Mehta H, Swampillai AJ, et al. Primary intraocular lymphoma. Surv Ophthalmol. 2014;59(5):503–516. | ||
Elagouz M, Stanescu-Segall D, Jackson TL. Uveal effusion syndrome. Surv Ophthalmol. 2010;55(2):134–145. | ||
Herbort CP, Rao NA, Mochizuki M; Members of Scientific Committee of First International Workshop on Ocular S. International criteria for the diagnosis of ocular sarcoidosis: results of the first International Workshop on Ocular Sarcoidosis (IWOS). Ocul Immunol Inflamm. 2009;17(3):160–169. | ||
Paredes I, Ahmed M, Foster CS. Immunomodulatory therapy for Vogt–Koyanagi–Harada patients as first-line therapy. Ocul Immunol Inflamm. 2006;14(2):87–90. | ||
Andrade RE, Muccioli C, Farah ME, Nussenblatt RB, Belfort R Jr. Intravitreal triamcinolone in the treatment of serous retinal detachment in Vogt–Koyanagi–Harada syndrome. Am J Ophthalmol. 2004;137(3):572–574. | ||
Hosoda Y, Hayashi H, Kuriyama S. Posterior subtenon triamcinolone acetonide injection as a primary treatment in eyes with acute Vogt–Koyanagi–Harada disease. Br J Ophthalmol. 2015;99(9):1211–1214. | ||
Latronico ME, Rigante D, Caso F, et al. Bilateral dexamethasone intravitreal implant in a young patient with Vogt–Koyanagi–Harada disease and refractory uveitis. Clin Rheumatol. 2015;34(6):1145–1148. | ||
Reibaldi M, Russo A, Avitabile T, et al. Treatment of persistent serous retinal detachment in Vogt–Koyanagi–Harada syndrome with intravitreal bevacizumab during the systemic steroid treatment. Retina. 2014;34(3):490–496. | ||
Read RW, Yu F, Accorinti M, et al. Evaluation of the effect on outcomes of the route of administration of corticosteroids in acute Vogt–Koyanagi–Harada disease. Am J Ophthalmol. 2006;142(1):119–124. | ||
Lai TY, Chan RP, Chan CK, Lam DS. Effects of the duration of initial oral corticosteroid treatment on the recurrence of inflammation in Vogt–Koyanagi–Harada disease. Eye. 2009;23(3):543–548. | ||
Errera MH, Fardeau C, Cohen D, et al. Effect of the duration of immunomodulatory therapy on the clinical features of recurrent episodes in Vogt–Koyanagi–Harada disease. Acta Ophthalmol. 2011;89(4):e357–e366. | ||
Jabs DA, Nussenblatt RB, Rosenbaum JT; Standardization of Uveitis Nomenclature Working G. Standardization of uveitis nomenclature for reporting clinical data. Results of the First International Workshop. Am J Ophthalmol. 2005;140(3):509–516. | ||
Tomkins-Netzer O, Talat L, Bar A, et al. Long-term clinical outcome and causes of vision loss in patients with uveitis. Ophthalmology. 2014;121(12):2387–2392. | ||
Tomkins-Netzer O, Talat L, Ismetova F, Samy A, Lightman S. Immunomodulatory therapy in uveitis. Dev Ophthalmol. 2016;55:265–275. | ||
Abu El-Asrar AM, Al Tamimi M, Hemachandran S, Al-Mezaine HS, Al-Muammar A, Kangave D. Prognostic factors for clinical outcomes in patients with Vogt–Koyanagi–Harada disease treated with high-dose corticosteroids. Acta Ophthalmol. 2013;91(6):e486–e493. | ||
Abu El-Asrar AM, Hemachandran S, Al-Mezaine HS, Kangave D, Al-Muammar AM. The outcomes of mycophenolate mofetil therapy combined with systemic corticosteroids in acute uveitis associated with Vogt–Koyanagi–Harada disease. Acta Ophthalmol. 2012;90(8):e603–e608. | ||
Urzua CA, Velasquez V, Sabat P, et al. Earlier immunomodulatory treatment is associated with better visual outcomes in a subset of patients with Vogt–Koyanagi–Harada disease. Acta Ophthalmol. 2015;93(6):e475–e480. | ||
Jeroudi A, Angeles-Han ST, Yeh S. Efficacy of adalimumab for pediatric Vogt–Koyanagi–Harada syndrome. Ophthalmic Surg Lasers Imaging Retina. 2014;45(4):332–334. | ||
Zmuda M, Tiev KP, Knoeri J, Héron E. Successful use of infliximab therapy in sight-threatening corticosteroid-resistant Vogt–Koyanagi–Harada disease. Ocul Immunol Inflamm. 2013;21(4):310–316. | ||
Dolz-Marco R, Gallego-Pinazo R, Diaz-Llopis M. Rituximab in refractory Vogt–Koyanagi–Harada disease. J Ophthalmic Inflamm Infect. 2011;1(4):177–180. | ||
Wu L, Evans T, Saravia M, Schlaen A, Couto C. Intravitreal bevacizumab for choroidal neovascularization secondary to Vogt–Koyanagi–Harada syndrome. Jpn J Ophthalmol. 2009;53(1):57–60. | ||
Pai SA, Hebri SP, Lootah AM. Management of recurrent inflammatory choroidal neovascular membrane secondary to Vogt–Koyanagi–Harada syndrome, using combined intravitreal injection of bevacizumab and triamcinolone acetate. Indian J Ophthalmol. 2012;60(6):551–552. | ||
Rubsamen PE, Gass JD. Vogt–Koyanagi–Harada syndrome. Clinical course, therapy, and long-term visual outcome. Arch Ophthalmol. 1991;109(5):682–687. | ||
Arevalo JF, Lasave AF, Gupta V, et al. Clinical outcomes of patients with Vogt–Koyanagi–Harada disease over 12 years at a tertiary center. Ocul Immunol Inflamm. 2015;23:1–9. | ||
Forster DJ, Rao NA, Hill RA, Nguyen QH, Baerveldt G. Incidence and management of glaucoma in Vogt–Koyanagi–Harada syndrome. Ophthalmology. 1993;100(5):613–618. | ||
Pandey A, Balekudaru S, Venkatramani DV, George AE, Lingam V, Biswas J. Incidence and management of glaucoma in Vogt Koyanagi Harada disease. J Glaucoma. 2016;25(8):674–680. | ||
Bykhovskaya I, Thorne JE, Kempen JH, Dunn JP, Jabs DA. Vogt–Koyanagi–Harada disease: clinical outcomes. Am J Ophthalmol. 2005;140(4):674–678. | ||
Meacock WR, Spalton DJ, Bender L, et al. Steroid prophylaxis in eyes with uveitis undergoing phacoemulsification. Br J Ophthalmol. 2004;88(9):1122–1124. | ||
Quek DT, Jap A, Chee SP. Risk factors for poor visual outcome following cataract surgery in Vogt–Koyanagi–Harada disease. Br J Ophthalmol. 2011;95(11):1542–1546. | ||
Moorthy RS, Chong LP, Smith RE, Rao NA. Subretinal neovascular membranes in Vogt–Koyanagi–Harada syndrome. Am J Ophthalmol. 1993;116(2):164–170. | ||
Lertsumitkul S, Whitcup SM, Nussenblatt RB, Chan CC. Subretinal fibrosis and choroidal neovascularization in Vogt–Koyanagi–Harada syndrome. Graefes Arch Clin Exp Ophthalmol. 1999;237(12):1039–1045. | ||
Kuo IC, Rechdouni A, Rao NA, Johnston RH, Margolis TP, Cunningham ET Jr. Subretinal fibrosis in patients with Vogt–Koyanagi–Harada disease. Ophthalmology. 2000;107(9):1721–1728. | ||
Rutzen AR, Ortega-Larrocea G, Frambach DA, Rao NA. Macular edema in chronic Vogt–Koyanagi–Harada syndrome. Retina. 1995;15(6):475–479. | ||
Yang P, Sun M. Band-shaped keratopathy in Chinese patients with Vogt–Koyanagi–Harada syndrome. Cornea. 2011;30(12):1336–1340. | ||
Khairallah M, Rao NA, Ben Yahia S, Zaouali S, Attia S. Pseudotumoral retinal pigment epithelium proliferation in a patient with Vogt–Koyanagi–Harada disease. Arch Ophthalmol. 2006;124(9):1366–1367. | ||
Yepez JB, Murati F, Petitto M, Arevalo JF. Pseudotumoral and multiple retinal pigment epithelium proliferation in Vogt–Koyanagi–Harada disease. Case Rep Ophthalmol Med. 2015;2015:153831. | ||
Chee SP, Jap A, Bacsal K. Prognostic factors of Vogt–Koyanagi–Harada disease in Singapore. Am J Ophthalmol. 2009;147(1):154–161.e151. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.