Back to Journals » Journal of Blood Medicine » Volume 11
Understanding Sideroblastic Anemia: An Overview of Genetics, Epidemiology, Pathophysiology and Current Therapeutic Options
Authors Abu-Zeinah G, DeSancho MT ![]()
Received 18 June 2020
Accepted for publication 18 August 2020
Published 25 September 2020 Volume 2020:11 Pages 305—318
DOI https://doi.org/10.2147/JBM.S232644
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Martin H Bluth
Ghaith Abu-Zeinah, Maria T DeSancho
Division of Hematology and Oncology, Department of Medicine, Weill Cornell Medicine, New York Presbyterian Hospital, New York, NY, USA
Correspondence: Ghaith Abu-Zeinah
Weill Cornell Medicine, New York Presbyterian Hospital, 1300 York Ave. C610-D, New York, NY 10065, USA
Tel +1 646 962 2700
Fax +1 646 962 0115
Email [email protected]
Abstract: Sideroblastic anemia (SA) consists of a group of inherited and acquired anemias of ineffective erythropoiesis characterized by the accumulation of ring sideroblasts in the bone marrow due to disrupted heme biosynthesis. Congenital sideroblastic anemia (CSA) is rare and has three modes of inheritance: X-linked (XLSA), autosomal recessive (ARCSA), and maternal. Acquired SA is more common and can be a result of myelodysplastic syndromes (MDS) or other, generally reversible causes. The diagnostic approach to SA includes a work-up for reversible causes and genetic testing for CSA based on clinical suspicion, family history and genetic pedigree. The treatment of SA depends on the underlying etiology but remains primarily supportive with vitamin B6 supplementation for select cases of XLSA, thiamine for thiamine-responsive megaloblastic anemia subtype, red blood cell transfusions for symptomatic patients and iron chelation therapy for iron overload. The management of anemia in MDS subtypes with ring sideroblasts remains unique and includes the recently approved erythroid maturation agent, Luspatercept. Although there is currently no curative therapy for CSA, anecdotal reports of hematopoietic stem cell transplant demonstrate remissions in selective, non-syndromic cases. This review summarizes the genetics, pathophysiology, diagnosis and treatment of SA for general practitioners and clinical hematologists.
Keywords: ring sideroblasts, heme biosynthesis, congenital sideroblastic anemia, acquired sideroblastic anemia, vitamin B6, iron chelation
Introduction
Sideroblastic anemia (SA) includes a group of inherited and acquired anemias of ineffective erythropoiesis characterized by an accumulation of ring sideroblasts (RS) in the bone marrow and decreased production of mature red blood cells.1 Ring sideroblasts are nucleated erythroblasts with a pathologic accumulation of iron granules in the mitochondrial matrix. They are detected, by Prussian blue staining, as blue perinuclear aggregates in bone marrow aspirate erythroblasts of all SA patients (Figure 1A–C).
 |
Figure 1 Bone marrow and peripheral blood histomorphology in sideroblastic anemia from a 65-year-old female with XLSA (A and B) Iron-stained bone marrow aspirate showing ring sideroblasts by the presence of iron granules (blue) in a perinuclear ring formation in erythroblasts (black arrows indicate the ringed sideroblasts). (C) H&E stained bone marrow aspirate showing several erythroblasts and myeloid precursors. (D) Wright-Giemsa stained peripheral blood smear showing basophilic stippling of a hypochromic red blood cell (black arrow indicates the presence of a red cell with basophilic stippling). The red cell morphology is defined by microcytosis, anisocytosis and poikilocytosis. Echinocytes are additionally present. |
Mechanisms in the pathogenesis of SA include impaired synthesis of heme, impaired iron-Sulfur (Fe-S) cluster assembly and transport, or impaired synthesis of mitochondrial and cytosolic proteins essential for heme synthesis. These mechanisms culminate in the build-up of iron granules rather than the normal incorporation of iron into protoporphyrin IX (PPIX) in the mitochondrion. The ensuing deficiency in cellular heme available to form hemoglobin results in red cell maturation arrest and accumulation of sideroblasts. Mature red blood cells, however, adapt to the deficiency in hemoglobin by maintaining a low mean corpuscular volume (MCV) to preserve the mean corpuscular hemoglobin concentration (MCHC) by homeostatic mechanisms that optimize their function as oxygen transporters. These are similar mechanisms that drive microcytosis in patients with iron deficiency. However, they are morphologically distinguished from iron deficient red cells on a Wright-Giemsa stain of a blood smear by the presence of basophilic stippling (Figure 1D), which represents aggregates of ribosomal RNA with degenerating mitochondria and siderosomes. Macrocytic anemia, or normocytic anemia with a high red cell distribution width, may also be observed due to the accelerated release of reticulocytes from the marrow due to the erythropoietin drive of anemic hypoxia in SA patients.2 The macrocytic red cells and circulating reticulocytes are typically the cells with sufficient hemoglobin.
SA can be inherited or acquired. Congenital sideroblastic anemia (CSA) is rare and characterized by three modes of inheritance: X-linked (XLSA), autosomal recessive (ARCSA), or maternal. Acquired SA is more common and can be observed in the setting of dysplastic hematopoiesis of mutated stem and progenitor cells in MDS. Acquired SA is a heterogenous category that also includes heavy metal poisoning (Lead, Arsenic), overdose (Zinc), or deficiency (Copper); Vitamin B6 deficiency and alcohol abuse; and treatment with drugs such as Isoniazid (INH), chloramphenicol, or linezolid that interfere with heme biosynthesis. These acquired causes of SA are often reversible, whereas SA due to MDS and CSA have a genetic basis and are potentially treatable but not curable. This review will summarize the causes of congenital and acquired sideroblastic anemia with a detailed overview of genetics, pathophysiology, diagnostic and treatment approaches.
Genetics, Epidemiology and Pathophysiology
Sideroblastic anemia (SA) describes a heterogenous group of inherited and acquired anemias with ring sideroblasts that include a few common subtypes and several uncommon subtypes. The prevalence of SA has not been defined but it is considered a rare disease. A rare disease is defined as a condition that affects fewer than 200,000 people in the United States.3 The National Organization for Rare Disorders acknowledges SA as a group of rare blood disorders4 while the Genetic and Rare Diseases Information Center (GARD) program of the United States National Institute of Health includes sideroblastic anemia because of its genetic basis but not because it is a rare disease.5
Sideroblastic anemia can be congenital or acquired. The CSA is further classified as syndromic or non-syndromic. The most common form of CSA is the non-syndromic form of X-linked SA (XLSA) first described by Cooley in 1945.6 The prevalence is undetermined but approximately 200 cases with less than 100 unrelated probands are described in the literature.7 Approximately two-thirds of cases present in males during early childhood or adolescence and a third in young to middle-aged females. XLSA is most often due to mutations in the δ-aminolevulinic acid synthase 2 (ALAS2) gene on Xp11.21 that encodes 5-aminolevulinate synthase,8–10 the first enzyme in the heme biosynthesis pathway in erythroid precursors. Deficiency of ALAS2 enzyme constitutes approximately 40% of all CSA cases.9 More than 80 mutations in ALAS2 have been reported in patients with XLSA to this date.11 The majority encode missense mutations, but several nonsense and frameshift mutations have been identified among clinically affected female carriers.8,10,12 The mutation most often occurs in the catalytic domain or the pyridoxal phosphate (active vitamin B6 co-factor) binding domain, but may also affect the ALAS2 promoter region, enhancer region, or mitochondrial targeting sequence.13,14 Due to an X-linked recessive pattern of inheritance, XLSA typically affects younger males and the inherited ALAS2 mutation is almost uniformly a partial loss-of-function missense mutation.8,13 Female carriers of ALAS2 mutations may have a late presentation of CSA as a result of familial-skewed inactivation of the normal X-chromosome, also referred to as “unfortunate skewing”.2,15 The type and location of the ALAS2 gene mutation may play a role in disease severity in females with unfortunate skewing later in life as the mutant hematopoietic clone with markedly defective ALAS2 enzyme predominates. Unlike males, females may have null mutations that prematurely truncate the ALAS2 enzyme leading to a complete loss-of-function and more severe disease than partial loss-of-function mutations observed in males or less symptomatic females.16 In one study of a XLSA family, 62–82% of the active X-chromosomes of affected females contained the mutant ALAS2 allele as a result of unfortunate skewing.17,18 However, the clone expressing mutant ALAS2 was at a significant survival disadvantage during erythropoiesis both in vivo and in vitro. This observation was supported by the finding that only wild-type allele transcripts were isolated from peripheral blood reticulocyte mRNA and erythroid maturation cultures. Macrocytic anemia is subsequently observed because of the accelerated release of this minority of normal reticulocytes under anemic hypoxia.2 Several other cases of XLSA in young adults and older females have been described,19 and we have emphasized the importance of maintaining diagnostic acumen for XLSA in young or older females with anemia and iron overload.12
Less frequently, XLSA is attributed to inherited mutations in the ATP-binding cassette subfamily B7 (ABCB7) and NADH:ubiquinone Oxidoreductase Subunit B1 (NDUFB11), both of which are associated with syndromic forms of XLSA; “XLSA with ataxia” (XLSA/A) and “Mitochondrial Myopathy, Lactic Acidosis and SA” (MLASA) respectively. ABCB7 is a mitochondrial ATP-cassette binding membrane transporter involved in the transfer of iron-sulfur (Fe-S) clusters into the cytoplasm, an essential step in the process of assembling Fe-S cluster-containing proteins such as hemoglobin. Therefore, loss of normal ABCB7 function can directly and indirectly impair heme biosynthesis and lead to ineffective erythropoiesis.20–22 There are at least 4 identified missense mutations in ABCB7 that cause XLSA/A, a syndrome of SA with spinocerebellar ataxia.23 On the other hand, NDUFB11 is one of several genes, but the only X-linked gene, known to cause MLASA when mutated. It is a nuclear-encoded mitochondrial complex I protein essential for oxidative phosphorylation. Partial loss of function of the protein has potentially severe multiorgan manifestations that include but are not limited to myopathy, lactic acidosis and SA.8,24
Autosomal recessive CSA or ARCSA is the second most common form of inherited SA (~35–40%).9 Several autosomal genes have been implicated. Homozygous or compound heterozygous inheritance of SLC19A2 mutations constitute approximately 26% of all CSA and are associated with thiamine-responsive megaloblastic anemia (TRMA), or Rogers syndrome. TRMA is characterized by megaloblastic anemia, progressive sensorineural hearing loss, and diabetes mellitus. Additional visual, neurologic, cardiac and multiorgan impairment have been described.25 Manifestations of TRMA are a result of any of non-sense, missense, deletion, insertion or indel mutations in SLC19A2,11 located on chromosome 1 (1q23.3), that lead to a decrease in the cell membrane transporter of thiamine, Slc19A2 or thiamine carrier 1 (TC1). The decrease in cellular uptake of thiamine is associated with megaloblastosis in erythroid, myeloid and megakaryocytic lineages and with multi-organ manifestations. Thiamine is an important cofactor for enzymes in the pentose and tricarboxylic acid (TCA) cycles crucial for cellular metabolism and DNA, RNA and heme biosynthesis.26,27 Thiamine’s role in the production of succinyl-coenzyme A, the substrate of ALAS2, is the hypothesized mechanistic link to the development of ring sideroblasts in TRMA.28
The second most common ARCSA is the non-syndromic SA with inherited mutations in SLC25A38 located on chromosome 3 (3p 22.1) that constitutes <10% of all CSA cases.9 SLC25A38 encodes Mitochondrial glycine transporter that is highly and preferentially expressed in CD71 positive erythroid cells and serves an important role in the biosynthesis of heme in eukaryotes.29 Several other autosomal genes have been implicated in the pathogenesis of ARCSA including TRNT1, PUS1, LARS2, YARS2, FECH, GLRX5, HSPA9, and HSCB, but constitute a small proportion of all CSA cases. The inheritance of such genes can lead to syndromic CSA or non-syndromic CSA through various mechanisms that ultimately lead to impaired synthesis of heme, mitochondrial proteins, and iron-sulfur (Fe-S) clusters (Table 1). Of the rare syndromic forms of CSA, the most common is probably SA with B-cell immunodeficiency, periodic fevers and developmental delay (SIFD) associated with biallelic mutations in TRNT1. The gene encodes the CCA-adding enzyme essential for assembly of nuclear and mitochondrial tRNA. Mutations in TRNT1 cause a partial loss of function that impair metabolic activities in both the cytosol and mitochondria including essential pathways for heme biosynthesis in the erythroid lineage.30 Another syndromic ARCSA to recognize is MLASA, which can be caused by mutations in PUS1, LARS2, YARS2. The SA is hypothesized to result from impaired post-transcriptional modification of mitochondrial and cytosolic tRNA and translation of mitochondrial respiratory complex proteins. Finally, SA may manifest in patients with erythropoietic protoporphyria (EPP) due to biallelic mutations in the ferrochelatase gene (FECH), that encodes the final enzyme in the heme synthetic pathway.31 The rare, non-syndromic forms of ARCSA can result from mutations in GLXR5, HSPA9 and HSCB which encode proteins required for formation of Fe-S clusters.32–34 Fe-S clusters are essential components of various proteins such as hemoglobin, FECH, and iron regulatory protein 1 (IRP1), a protein involved in iron metabolism and the translation of ALAS2.35
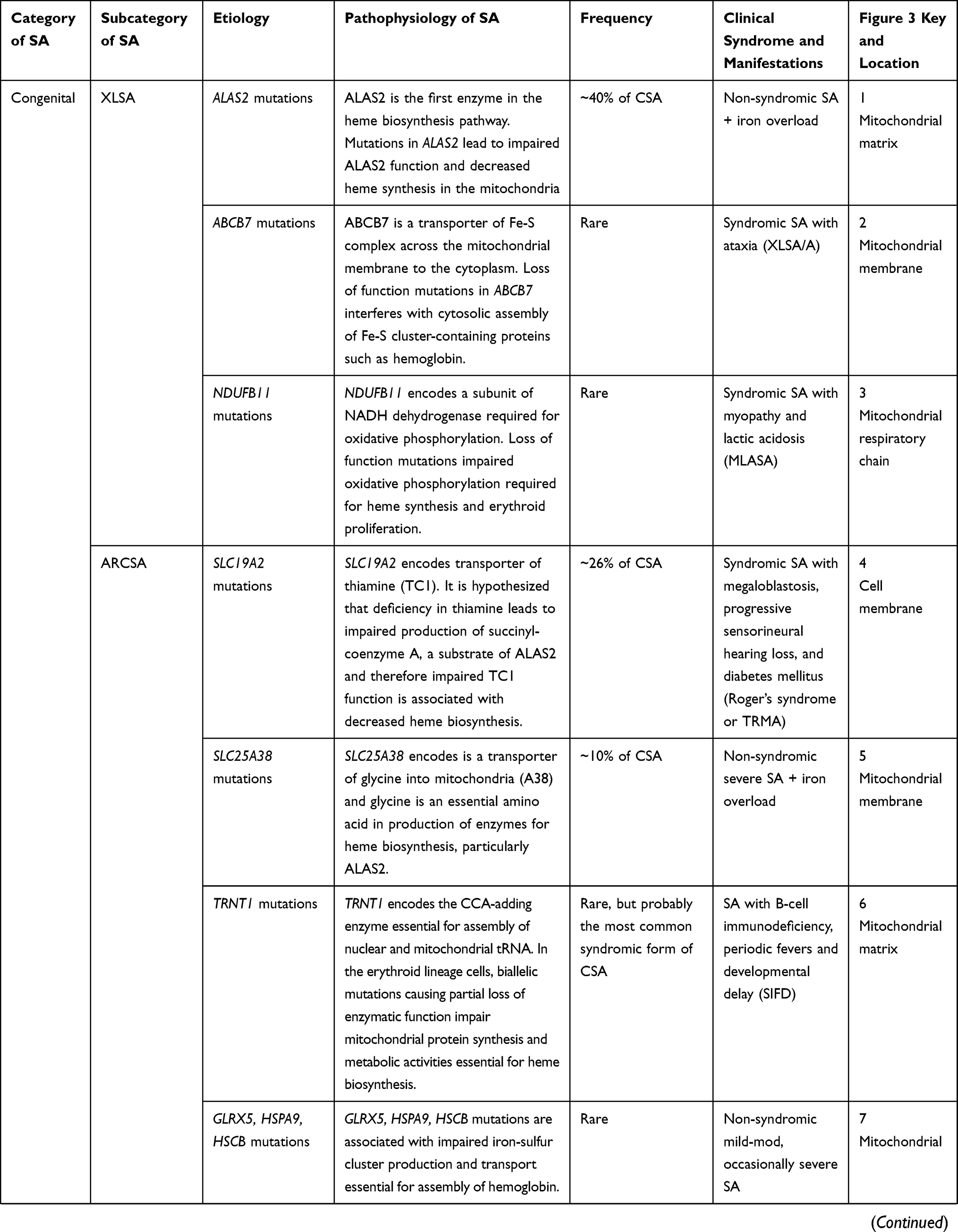 | 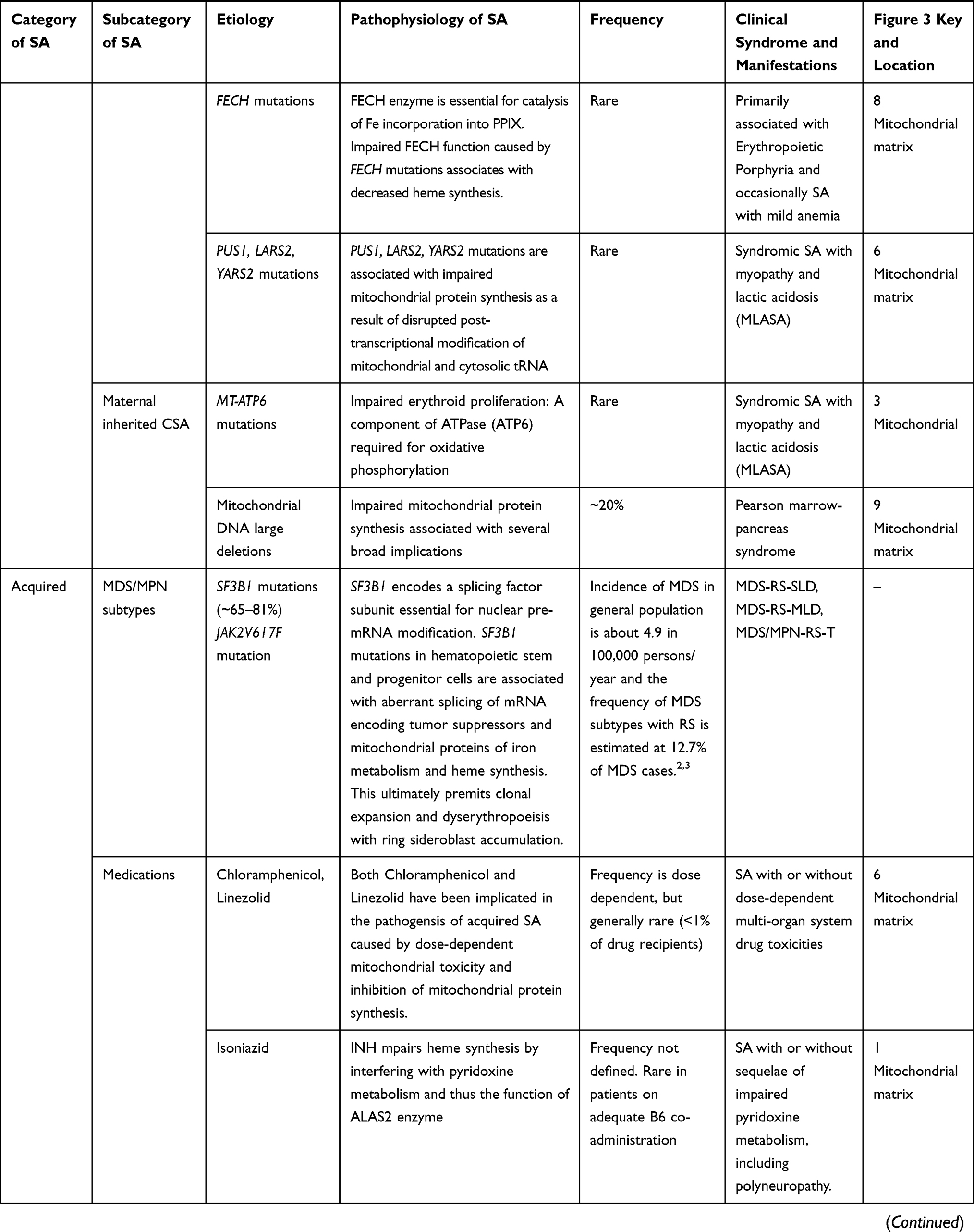 |  |
Table 1 Sideroblastic Anemia Congenital and Acquired Subtypes, Pathophysiology, Epidemiology and Clinical Manifestations |
The third mode of CSA inheritance is maternal, from mitochondrial DNA point mutations (MT-ATP6) or large deletions (Pearson marrow-pancreas syndrome), and constitutes approximately 20% of all CSA cases.9 MT-ATP6 is a mitochondrial gene that encodes ATPase 6, a component of the mitochondrial respiratory chain complex V. Mutations in MT-ATP6 are associated with MLASA phenotype.36 Conversely, a heteroplasmy of deletions, duplications or rearrangements in mitochondrial DNA encoding proteins and enzymes of the respiratory complex, iron metabolism and heme biosynthesis pathways may have more profound effects. Pearson marrow-pancreas syndrome is characterized by early-onset severe SA with cytopenias, pancreatic insufficiency, lactic acidosis and a failure to thrive.37,38
Acquired SA can be categorized into SA with clonal hematopoiesis, specifically MDS subtypes, and reversible SA from environmental factors. MDS and Myeloproliferative Neoplasms (MPN) are hematologic malignancies driven by clonal expansion of hematopoietic stem, progenitor and mature cells with an acquired somatic driver mutation. The driver mutations involve several mRNA splicing, epigenetic regulators, cytokine signaling and other genes that cause dysplastic and/or proliferative hematopoiesis. However, RS are seen in only specific subtypes of MDS and MPN that are often associated with mutations in the mRNA splicing gene SF3B1 (65–81% of cases according to some studies39,40) and less commonly the JAK2V617F mutation. Namely, the subtypes of MDS/MPN with RS are defined by the World Health Organization as MDS with RS and single lineage dysplasia (MDS-RS-SLD); MDS with RS and multilineage dysplasia (MDS-RS-MLD); and MDS/MPN with RS and thrombocytosis (MDS/MPN-RS-T).41
Acquired SA from reversible causes is similarly linked to mechanisms of impaired heme biosynthesis and accumulation of siderosomes. Alcoholics, for instance, may develop anemia through several mechanisms but approximately 23%42 will have RS as a consequence of alcohol’s suppressive effects on vitamin B6 metabolism to pyridoxal phosphate,43 an essential cofactor for ALAS2. Nevertheless, it is important to note that anemia in alcoholics is almost always multifactorial and that the sideroblastic component, when evident, is almost never the sole abnormality. Because of the important role of pyridoxal phosphate (the active form of vitamin B6) for ALAS2 enzymatic activity, a severe deficiency in vitamin B6 due to alcohol, malnutrition or malabsorption is therefore likely to cause SA. Similarly, INH, which has been extensively used to treat tuberculosis for extended courses, interferes with pyridoxine metabolism and thereby causes neuropathy and sideroblastic anemia in cases where vitamin B6 co-administration is inadequate.44 Unlike INH, Chloramphenicol and Linezolid are hypothesized to cause sideroblastic anemia through their dose-dependent mitochondrial toxicity and inhibition of mitochondrial protein synthesis that impairs heme biosynthesis.45,46
Heavy metal toxicity, specifically from lead poisoning or zinc overdose is associated with SA. Under such circumstances, there is an increased likelihood of zinc incorporation into PPIX at the expense of iron. This reduces functional hemoglobin and the displaced iron accumulates in siderosomes. While this may be true for zinc toxicity, the presence of authentic sideroblasts in lead poisoning anemia has been questioned by several.48 The anemia in lead poisoning has been mechanistically linked to inhibition of various enzymes involved in heme synthesis including FECH, δ-aminolevulinic acid ALA dehydratase (ALAD) and coproporphyrinogen oxidase which would theoretically cause SA.49 Zinc is additionally necessary for the full activity of various enzymes including ALAD and is therefore an essential dietary mineral. Excess zinc ingestion, however, not only competes with iron incorporation into protoporphyrin but also induces intestinal metal-binding protein metallothionein which prevents intestinal absorption of copper.50,51 Copper is a well-established cause of acquired SA because copper is an essential cofactor for mitochondrial redox enzyme superoxide dismutase and reduced activity of this enzyme can lead to mitochondrial iron accumulation.51–53
Diagnosis and Treatment
Despite the genetic heterogeneity and pathophysiologic diversity among SA subtypes, the diagnostic approach for SA follows the same basic principles. Reversible causes of SA and MDS must be excluded first and then a clinical and genetic assessment for congenital SA should be performed (Figure 2). The principles of SA treatment include management of reversible causes or MDS; administration of vitamin B6 for responsive subtypes or thiamine for TRMA; iron chelation to prevent organ damage from iron overload; and red cell transfusions for symptomatic anemia.
 |
Figure 2 Diagnostic algorithm for sideroblastic anemia. |
The diagnosis of all forms of SA by definition requires the detection of RS in the bone marrow aspirate by iron staining (Figure 1). Next-generation sequencing for acquired mutations in MDS or inherited mutations in CSA is necessary once reversible causes have been ruled out, but detecting a mutation does not preclude a bone marrow evaluation. Clues to the diagnosis of SA that would prompt a bone marrow evaluation and confirmation of SA include: unexplained anemia that is often but not exclusively microcytic with distinct features on a blood smear such as basophilic stippling; anemia in a male infant, male child, or female adolescent or young adult with iron overload; anemia in older patients with clonal hematopoiesis or predisposition to MDS; and anemia in patients with a family history of SA or those with reversible factors associated with SA. Reversible factors that should be carefully reviewed when eliciting a history include excess alcohol consumption; heavy metal toxicity; treatment with INH, chloramphenicol or linezolid; and malnutrition with ensuing copper, vitamin B6 or thiamine deficiency as seen after bariatric surgery. When reversible causes of SA are suspected, one must consider unusual links that patients may not think to report. For instance, regular use of zinc-containing denture creams or a prolonged ingestion of high amounts of zinc from fad diets may not be obvious causes of zinc poisoning and sideroblastic anemia. This justifies inclusion of copper, ceruloplasmin, zinc and lead levels in the diagnostic evaluation even in cases without an obvious link.
For the majority of reversible causes, a bone marrow biopsy to confirm the presence of RS in the anemic patient may not be required and the diagnosis of SA may never be established. This is true in cases where withdrawal of the causative agent or treatment of the reversible cause is therapeutic by itself, with or without vitamin B6 supplementation. In the rare cases of INH induced SA, for instance, stopping treatment or ensuring adequate vitamin B6 co-intake, often in doses as large as 200mg per day, restores normal erythropoiesis.54 A bone marrow biopsy in such cases is rarely indicated. However, when acquired SA from MDS is suspected, a bone marrow biopsy is always necessary after reversible causes such as copper deficiency have been excluded. Currently, with the widespread availability of next-generation sequencing assays for SF3B1 and other gene mutations in clonal hematopoiesis and MDS, sequencing is often performed routinely in the earlier stages of diagnostic work-up of MDS. Nevertheless, confirming or ruling out SF3B1 mutations by molecular analysis of peripheral blood leukocytes does not preclude histomorphology assessment of the bone marrow. Bone marrow histology is an essential diagnostic criterion for the 3 subtypes of MDS with RS: MDS-RS-SLD, MDS-RS-MLD, and MDS/MPN-RS-T.41 In MDS-RS, either erythroid lineage dysplasia alone (MDS-RS-SLD) or multilineage dysplasia (MDS-RS-MLD) is observed in the bone marrow, along with ≥15% ring sideroblasts or ≥5% ring sideroblasts in the presence of an SF3B1 mutation and <5% blasts.41 In MDS/MPN-RS-T, anemia with erythroid lineage dysplasia, with or without multilineage dysplasia, ≥15% ring sideroblasts, and <5% in the bone marrow is a major criterion along with thrombocytosis (platelets ≥ 450,000/uL) and ideally in the presence of a mutation in SF3B1, JAK2, CALR or MPL.41
Treatment of MDS with RS includes the use of erythropoiesis-stimulating agents (ESA), transfusion support, and iron chelation for red cell transfusion-dependent patients on a case-by-case basis. Characteristics that predict response to ESA in MDS patients include transfusion of less than 2 units of pack red blood cells per month and a baseline erythropoietin level of less than 500 IU/mL. On the other hand, the use of iron chelation therapy in MDS-RS requires prospective evaluation for routine use. Lenalidomide, an immunomodulatory agent that is effective in treating anemia of MDS with deletion 5-q and lower-risk MDS with normal karyotype, is effective in treating anemia of MDS-RS-SLD with 35% of patients in one study achieving transfusion independence.55–57 While lenalidomide treatment may be effective for eligible MDS patients, we encourage clinical trial participation and treatment with novel agents that have disease-modifying potential. Luspatercept is a novel agent recently FDA approved for treating patients with very low- to intermediate-risk MDS-RS or MDS/MPN-RS-T. Luspatercept is a recombinant fusion protein that binds transforming growth factor β (TGF-β) superfamily ligands to reduce SMAD2 and SMAD3 dependent signaling implicated in the pathogenesis of anemia. It was FDA approved after the MEDALIST trial, a randomized, multi-center, placebo-controlled, Phase III clinical trial demonstrated higher rates of transfusion independence in very low- to intermediate-risk MDS-RS or MDS/MPN-RS-T treated with luspatercept compared to those treated with placebo (38% vs 13%, respectively).58 Other novel agents for treatment of anemia in MDS are currently being investigated in preclinical studies and clinical trials.
In SA patients without an identifiable reversible cause, a thorough medical history, family history, and physical exam are important to establish the diagnosis of CSA. The congenital form is suspected in patients with early onset of anemia in the presence of a family history of anemia. Further history and physical exam findings provide clues to whether CSA is syndromic or non-syndromic. At this point, genetic testing for CSA is an essential component of the diagnostic work-up. In our published case report of a woman in her mid-sixties with macrocytic anemia, iron overload with hepatic deposition, and bone marrow RS, genetic testing revealed a novel heterozygous frame shift mutation c.633delC, p.A211fs of the ALAS2 gene.48 This case highlights that congenital causes cannot be excluded based solely on the age of presentation.
In patients with a known family history of SA, testing can be limited to the inherited gene. When this is unknown, a family pedigree can help narrow the mode of inheritance to X-linked, autosomal recessive or maternal and testing can be tailored accordingly. Additional clinical manifestations in children with SA can provide clues to inform genetic testing for syndromic CSA (Table 1). This includes sequencing ABCB7 in children with spinocerebellar ataxia to establish the diagnosis of XLSA/A or sequencing PUS1, LARS2, YARS2, NDUFB11 and/or mitochondrial MT-ATP6 in patients with myopathy, lactic acidosis, and developmental delay to establish the diagnosis of MLASA. In the aforementioned syndromic forms of CSA in children, the clinical course is largely dominated by the nonhematologic manifestations. There are anecdotal reports of the efficacy of coenzyme Q10 in the treatment of MLASA with improvement in both the myopathy and the transfusion-dependent anemia.59,60 Beyond anecdotes and supportive care, there is no standard treatment for the anemia in syndromic cases. Treatment with corticosteroids or ESA, for instance, have failed to demonstrate efficacy in some of the severe syndromic forms of CSA such as Pearson marrow-pancreas syndrome.61 Nevertheless, particular and prompt attention should be given to those suspected of having TRMA (or Roger’s syndrome), based on clinical manifestations such as diabetes and sensorineural hearing loss, and sequencing of the implicated gene SLC19A2 should be performed. When TRMA is identified early in childhood, supraphysiologic doses of thiamine given orally are effective at treating the anemia and may also treat the nonhematologic manifestations.
In the absence of family history, genetic pedigree, or clinical clues of syndromic CSA, a genetic test should include the more commonly mutated genes in non-syndromic CSA such as ALAS2. Simultaneous or subsequent testing for SLC25A38, HSPA9, HSCB, GLRX5 can be considered. In approximately two-thirds of XLSA due to ALAS2 mutations, supplementation with vitamin B6 is sufficient to treat the anemia at variable degrees with an infrequent need for red cell transfusions.62 Therefore, and in accordance to the published literature, we recommend that patients with genetically defined or suspected XLSA receive a 3-month trial of oral vitamin B6 therapy at a dose of 50–100 mg daily prior to determining responsiveness to vitamin B6. Responders should be treated with maintenance therapy to preserve an adequate pool of pyridoxal phosphate.
Patients with SA are prone to iron overload, regardless of red blood cell transfusions. This is partly attributed to reduced hepcidin levels because of ineffective erythropoiesis which promotes increased intestinal absorption of iron.63 It has been reported that iron overload may suppress pyridoxine responsiveness and that iron depletion by phlebotomy results in higher hemoglobin concentrations64 or improved pyridoxine response.65 In GLRX5 deficiency, for instance, there is a strong correlation between iron depletion and improved hemoglobin levels.66 Iron depletion has also proven useful in increasing hemoglobin levels in case reports of MDS.67 Thus, in cases of sideroblastic anemia with iron overload, iron chelation should be strongly considered.
The therapeutic options beyond vitamin B6 supplementation are limited. Supportive care with red cell transfusions can be considered for those with severe and symptomatic anemia but the risk of worsening iron overload on the long term is high. Significant iron overload causing micronodular cirrhosis and hepatic fibrosis has been demonstrated in asymptomatic individuals with XLSA later in adulthood.12 Therefore, monitoring for significant iron overload should be done routinely by hepatic and cardiac magnetic resonance imaging. We recommend this be done in accordance to guidelines for hemochromatosis patients. Red cell transfusions should therefore be limited to patients with symptomatic anemia or hemoglobin levels less than 7 g/dL. Iron chelation should be initiated in patients requiring transfusions after at least 10 transfusions or if serum ferritin is over 1000 mcg/L.68 Available chelators in the United States are parenteral deferoxamine (Desferal), or oral agents deferasirox (Exjade, Jadenu) and deferiprone (Ferriprox). Combinations of these drugs can be used in severe iron overload. Iron depletion by phlebotomy is considered when the anemia is mild or moderate (hemoglobin ≥ 9 g/dL), or when a patient with XLSA has responded to pyridoxine. Maintenance phlebotomies are ultimately continued for life to control iron re-accumulation.69
The management of SA remains primarily supportive rather than definitive. There is currently no cure for inherited sideroblastic anemias. However, anecdotal reports of hematopoietic stem cell transplant (HSCT) in non-syndromic CSA describe an effective remission.70 Developing definitive treatments for SA is clearly an area of need. Preclinical studies and clinical trials are essential to determine whether novel agents such as luspatercept or approaches involving gene therapy for CSA would be beneficial in the treatment of SA.
Summary and Conclusion
Sideroblastic anemia (SA) is due to ineffective erythropoiesis with formation of ring sideroblasts. It occurs as a consequence of decreased biosynthesis of heme by various mechanisms. These are attributed to the inheritance of genetic defects (CSA), acquisition of mutations in hematopoietic stem and progenitor cells (MDS subtypes with RS), and reversible factors such as drug, alcohol and heavy metal toxicities. Reversible factors should be identified or eliminated first because treatment is definitive. Congenital SA, on the other hand, includes a few common and several rare inherited disorders. Inheritance patterns include X-linked, autosomal recessive and maternal. Some forms are syndromic and present in childhood while others are non-syndromic and may present in older females. X-linked SA due to mutations in ALAS2 gene is the most common CSA and is important to recognize as it may respond favorably to vitamin B6 treatment. Additional management approaches include red blood cell transfusion, iron chelation or phlebotomy, erythropoietin stimulating agents, and consideration of novel agents and investigational treatments in clinical trials.
Acknowledgments
We acknowledge Dr. Julia T Geyer from the Division of Hematopathology at WCM for providing the high-resolution images of peripheral blood and bone marrow specimens. We also acknowledge Biorender.com for providing us the platform to design Figure 3.
 |
Figure 3 Cell and mitochondrial mechanisms in heme biosynthesis and localization of pathway defects in sideroblastic anemia subtypes. Labels 1–10 encode the localization of mechanistic defects in heme biosynthesis in various subtypes of sideroblastic anemia detailed in Table 1. |
Disclosure
Dr Maria T DeSancho participated in advisory boards for Apellis Pharamceuticals, Bio Product Laboratory, and Sanofi-Genzyme, outside the submitted work. The authors report no other conflicts of interest related to this work.
References
1. Cartwright GE, Deiss A. Sideroblasts, siderocytes, and sideroblastic anemia. N Engl J Med. 1975;292(4):185–193. doi:10.1056/NEJM197501232920405
2. Aivado M, Gattermann N, Rong A, et al. X-linked sideroblastic anemia associated with a novel ALAS2 mutation and unfortunate skewed X-chromosome inactivation patterns. Blood Cells Mol Dis. 2006;37:40–45. doi:10.1016/j.bcmd.2006.04.003
3. Congress 97th. Public Law 97–414 - Jan.4, 1983; 1983. Available from: https://www.fda.gov/media/99546/download.
4. Anemias, Sideroblastic. National Organization for Rare Disorders (NORD); 2007. Available from: https://rarediseases.org/rare-diseases/anemias-sideroblastic/.
5. Sideroblastic anemia Genetic and Rare Diseases Information Center (GARD) – an NCATS Program. Available from: https://rarediseases.info.nih.gov/diseases/667/sideroblastic-anemia.
6. Cooley. T. A severe type of hereditary anemia with elliptocytosis. Interesting sequence of splenectomy. Am J Med Sci. 1945;209:561–568. doi:10.1097/00000441-194505000-00001
7. Orphanet: X linked sideroblastic anemia. Available from: https://www.orpha.net/consor/cgi-bin/Disease_Search.php?lng=EN&data_id=11087&Disease_Disease_Search_diseaseGroup=x-linked-sideroblastic-anemia&Disease_Disease_Search_diseaseType=Pat&Disease(s)/group of diseases=X-linked-sideroblastic-anemia&title=X-linked sideroblastic anemia&search=Disease_Search_Simple.
8. Fleming MD. Congenital sideroblastic anemias: iron and heme lost in mitochondrial translation. Hematol Am Soc Hematol Educ Program. 2011;2011(1):525–531. doi:10.1182/asheducation-2011.1.525
9. Bergmann AK, Campagna DR, McLoughlin EM, et al. Systematic molecular genetic analysis of congenital sideroblastic anemia: evidence for genetic heterogeneity and identification of novel mutations. Pediatr Blood Cancer. 2010;54(2):273–278. doi:10.1002/pbc.22244
10. Ducamp S, Kannengiesser C, Touati M, et al. Sideroblastic anemia: molecular analysis of the ALAS2 gene in a series of 29 probands and functional studies of 10 missense mutations. Hum Mutat. 2011;32(6):590–597. doi:10.1002/humu.21455
11. The Human Gene Mutation Database; HGMD® gene result. Available from: http://www.hgmd.cf.ac.uk/ac/gene.php?gene=ALAS2.
12. Abu-Zeinah G, DeSancho MT, Al-Kawaaz M, Geyer J. Delayed diagnosis of congenital sideroblastic anemia. Semin Hematol. 2018;55(4):177–178. doi:10.1053/j.seminhematol.2017.09.001
13. Campagna DR, de Bie CI, Schmitz-Abe K, et al. X-linked sideroblastic anemia due to ALAS2 intron 1 enhancer element GATA-binding site mutations. Am J Hematol. 2014;89(3):315–319. doi:10.1002/ajh.23616
14. Kaneko K, Furuyama K, Fujiwara T, et al. Identification of a novel erythroid-specific enhancer for the ALAS2 gene and its loss-of-function mutation which is associated with congenital sideroblastic anemia. Haematologica. 2014;99(2):252–261. doi:10.3324/haematol.2013.085449
15. Cazzola M, May A, Bergamaschi G, Cerani P, Rosti VBD. Familial-skewed X-chromosome inactivation as a predisposing factor for late-onset X-linked sideroblastic anemia in carrier females. Blood. 2000;96(13):4363–4365. doi:10.1182/blood.V96.13.4363
16. Aivado M, Gattermann N, Bottomley S, Cazzola M, Bergamaschi G. X chromosome inactivation ratios in female carriers of X-linked sideroblastic anemia [3] (multiple letters). Blood. 2001;97(12):4000–4002. doi:10.1182/blood.V97.12.4000
17. Sankaran VG, Ulirsch JC, Tchaikovskii V, et al. X-linked macrocytic dyserythropoietic anemia in females with an ALAS2 mutation. J Clin Invest. 2015;125(4):1665–1669. doi:10.1172/JCI78619
18. Sankaran VG, Ulirsch JC, Tchaikovskii V, et al. Erratum: X-linked macrocytic dyserythropoietic anemia in females with an ALAS2 mutation (Journal of Clinical Investigation (2015) 125:4 (1665–1669) DOI: 10.1172/JCI78619). J Clin Invest. 2020;130(1):552. doi:10.1172/JCI132538
19. Cotter PD, May A, Fitzsimons EJ, et al. Late-onset X-linked sideroblastic anemia. Missense mutations in the erythroid delta-aminolevulinate synthase (ALAS2) gene in two pyridoxine-responsive patients initially diagnosed with acquired refractory anemia and ringed sideroblasts. J Clin Invest. 1995;96(4):2090–2096. doi:10.1172/JCI118258
20. Pondarre C, Campagna DR, Antiochos B, Sikorski L, Mulhern H, Fleming MD. Abcb7, the gene responsible for X-linked sideroblastic anemia with ataxia, is essential for hematopoiesis. Blood. 2007;109(8):3567–3569. doi:10.1182/blood-2006-04-015768
21. Allikmets R, Raskind WH, Hutchinson A, Schueck ND, Dean M, Koeller DM. Mutation of a putative mitochondrial iron transporter gene (ABC7) in X-linked sideroblastic anemia and ataxia (XLSA/A). Hum Mol Genet. 1999;8(5):743–749. doi:10.1093/hmg/8.5.743
22. Bekri S, Kispal G, Lange H, et al. Human ABC7 transporter: gene structure and mutation causing X-linked sideroblastic anemia with ataxia with disruption of cytosolic iron-sulfur protein maturation. Blood. 2000;96(9):3256–3264. doi:10.1182/blood.v96.9.3256.h8003256_3256_3264
23. The Human Gene Mutation Database; HGMD® gene result. Available from: http://www.hgmd.cf.ac.uk/ac/gene.php?gene=ABCB7.
24. Lichtenstein DA, Crispin AW, Sendamarai AK, et al. A recurring mutation in the respiratory complex 1 protein NDUFB11 is responsible for a novel form of X-linked sideroblastic anemia. Blood. 2016;128(15):913–917. doi:10.1182/blood-2016-05-719062
25. Bergmann AK, Sahai I, Falcone JF, et al. Thiamine-responsive megaloblastic anemia: identification of novel compound heterozygotes and mutation update. J Pediatr. 2009;155(6):888. doi:10.1016/j.jpeds.2009.06.017
26. Oishi K, Hofmann S, Diaz GA, et al. Targeted disruption of Slc19a2, the gene encoding the high-affinity thiamin transporter Thtr-1, causes diabetes mellitus, sensorineural deafness and megaloblastosis in mice. Hum Mol Genet. 2002;11(23):2951–2960. doi:10.1093/hmg/11.23.2951
27. Boros LG, Steinkamp MP, Fleming JC, Lee WNP, Cascante M, Neufeld EJ. Defective RNA ribose synthesis in fibroblasts from patients with thiamine-responsive megaloblastic anemia (TRMA). Blood. 2003;102(10):3556–3561. doi:10.1182/blood-2003-05-1537
28. Abboud MR, Alexander D, Najjar SS. Diabetes mellitus, thiamine-dependentmegaloblastic anemia, and sensorineural deafness associated with deficient α-ketoglutarate dehydrogenase activity. J Pediatr. 1985;107(4):537–541. doi:10.1016/S0022-3476(85)80011-1
29. Guernsey DL, Jiang H, Campagna DR, et al. Mutations in mitochondrial carrier family gene SLC25A38 cause nonsyndromic autosomal recessive congenital sideroblastic anemia. Nat Genet. 2009;41(6):651–653. doi:10.1038/ng.359
30. Chakraborty PK, Schmitz-Abe K, Kennedy EK, et al. Mutations in TRNT1 cause congenital sideroblastic anemia with immunodeficiency, fevers, and developmental delay (SIFD). Blood. 2014;124(18):2867–2871. doi:10.1182/blood-2014-08-591370
31. Rademakers LHPM, Koningsberger JC, Sorber CWJ, La Faille HBD, Van Hattum J, Marx JJM. Accumulation of iron in erythroblasts of patients with erythropoietic protoporphyria. Eur J Clin Invest. 1993;23(2):130–138. doi:10.1111/j.1365-2362.1993.tb00752.x
32. Ye H, Jeong SY, Ghosh MC, et al. Glutaredoxin 5 deficiency causes sideroblastic anemia by specifically impairing heme biosynthesis and depleting cytosolic iron in human erythroblasts. J Clin Invest. 2010;120(5):1749–1761. doi:10.1172/JCI40372
33. Crispin A, Schmidt P, Campagna D, et al. Hscb, a mitochondrial iron-sulfur cluster assembly co-chaperone, is a novel candidate gene for congenital sideroblastic anemia. Blood. 2017;130(Supplement 1):79. doi:10.1182/BLOOD.V130.SUPPL_1.79.79
34. Schmitz-Abe K, Ciesielski SJ, Schmidt PJ, et al. Congenital sideroblastic anemia due to mutations in the mitochondrial HSP70 homologue HSPA9. Blood. 2015;126(25):2734–2738. doi:10.1182/blood-2015-09-659854
35. Wingert RA, Galloway JL, Barut B, et al. Deficiency of glutaredoxin 5 reveals Fe-S clusters are required for vertebrate haem synthesis. Nature. 2005;436(7053):1035–1039. doi:10.1038/nature03887
36. Burrage LC, Tang S, Wang J, et al. Mitochondrial myopathy, lactic acidosis, and sideroblastic anemia (MLASA) plus associated with a novel de novo mutation (m.8969G>A) in the mitochondrial encoded ATP6 gene. Mol Genet Metab. 2014;113(3):207–212. doi:10.1016/j.ymgme.2014.06.004
37. Pearson HA, Lobel JS, Kocoshis SA, et al. A new syndrome of refractory sideroblastic anemia with vacuolization of marrow precursors and exocrine pancreatic dysfunction. J Pediatr. 1979;95(6):976–984. doi:10.1016/S0022-3476(79)80286-3
38. Rötig A, Bourgeron T, Chretien D, Rustin P, Munnich A. Spectrum of mitochondrial DNA rearrangements in the pearson marrow-pancreas syndrome. Hum Mol Genet. 1995;4(8):1327–1330. doi:10.1093/hmg/4.8.1327
39. Malcovati L, Karimi M, Papaemmanuil E, et al. SF3B1 mutation identifies a distinct subset of myelodysplastic syndrome with ring sideroblasts. Blood. 2015;126(2):233–241. doi:10.1182/blood-2015-03-633537
40. Papaemmanuil E, Cazzola M, Boultwood J, et al. Somatic SF3B1 Mutation in Myelodysplasia with Ring Sideroblasts. N Engl J Med. 2011;365(15):1384–1395. doi:10.1056/NEJMoa1103283
41. Arber DA, Orazi A, Hasserjian R, et al. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood. 2016;127(20):2391–2405. doi:10.1182/blood-2016-03-643544
42. Savage D, Lindenbaum J. Anemia in alcoholics. Med (United States). 1986;65(5):322–338. doi:10.1097/00005792-198609000-00005
43. Hines JD, Cowan DH. Studies on the pathogenesis of alcohol-induced sideroblastic bone-marrow abnormalities. N Engl J Med. 1970;283(9):441–446. doi:10.1056/NEJM197008272830901
44. Konopka L, Hoffbrand AV. Haem synthesis in sideroblastic anaemia. Br J Haematol. 1979;42(1):73–83. doi:10.1111/j.1365-2141.1979.tb03699.x
45. Willekens C, Dumezy F, Boyer T, et al. Linezolid induces ring sideroblasts. Haematologica. 2013;98(11):e138–e140. doi:10.3324/haematol.2013.092395
46. Beck EA, Ziegler G, Schmid R, Lüdin H. Reversible sideroblastic anemia caused by chloramphenicol. Acta Haematol. 1967;38(1):1–10. doi:10.1159/000208994
47. Hastka J, Lasserre JJ, Schwarzbeck AHR. Central role of zinc protoporphyrin in staging iron deficiency. Clin Chem. 1994;40(5):768–773. doi:10.1093/clinchem/40.5.768
48. Bottomley SS. Pathophysiology of heme synthesis. Semin Hematol. 1988;25(4):282.
49. Moore MR, Goldberg A, Yeung-Laiwah AAC. Lead effects on the heme biosynthetic pathway relationship to toxicity. Ann N Y Acad Sci. 1987;514(1 Mechanisms of):191–203. doi:10.1111/j.1749-6632.1987.tb48774.x
50. Cousins RJ. Absorption, transport, and hepatic metabolism of copper and zinc: special reference to metallothionein and ceruloplasmin. Physiol Rev. 1985;65(2):238–309. doi:10.1152/physrev.1985.65.2.238
51. Fiske DN, McCoy HE, Kitchens CS. Zinc-induced sideroblastic anemia: report of a case, review of the literature, and description of the hematologic syndrome. Am J Hematol. 1994;46(2):147–150. doi:10.1002/ajh.2830460217
52. Simon SR, Branda RF, Tindle BH, Burns SL. Copper deficiency and sideroblastic anemia associated with zinc ingestion. Am J Hematol. 1988;28(3):181–183. doi:10.1002/ajh.2830280310
53. Williams DM, Loukopoulos D, Lee GR, Cartwright GE. Role of copper in mitochondrial iron metabolism. Blood. 1976;48(1):77–85. doi:10.1182/blood.V48.1.77.77
54. Piso RJ, Kriz K, Desax MC. Severe isoniazid related sideroblastic anemia. Hematol Rev. 2011;3(1):3–4. doi:10.4081/hr.2011.e2
55. Santini V, Almeida A, Giagounidis A, et al. Randomized Phase III Study of Lenalidomide Versus Placebo in RBC Transfusion-Dependent Patients With Lower-Risk Non-del(5q) Myelodysplastic Syndromes and Ineligible for or Refractory to Erythropoiesis-Stimulating Agents. J Clin Oncol. 2016;34(25):2988–2996. doi:10.1200/JCO.2015.66.0118
56. Raza A, Reeves JA, Feldman EJ, et al. Phase 2 study of lenalidomide in transfusion-dependent, low-risk, and intermediate-1-risk myelodysplastic syndromes with karyotypes other than deletion 5q. Blood. 2008;111(1):86–93. doi:10.1182/blood-2007-01-068833
57. Patnaik MM, Tefferi A. Refractory anemia with ring sideroblasts (RARS) and RARS with thrombocytosis: “2019 Update on Diagnosis, Risk-stratification, and Management.”. Am J Hematol. 2019;94(4):475–488. doi:10.1002/ajh.25397
58. Fenaux P, Platzbecker U, Mufti GJ, et al. Luspatercept in patients with lower-risk myelodysplastic syndromes. N Engl J Med. 2020;382(2):140–151. doi:10.1056/NEJMoa1908892
59. Bachmeyer C, Ferroir JP, Eymard B, Maïer-Redelsperger M, Lebre AS, Girot R. Coenzyme Q is effective on anemia in a patient with sideroblastic anemia and mitochondrial myopathy. Blood. 2010;116(18):3681–3682. doi:10.1182/blood-2010-07-299453
60. Kasapkara ÇS, Tümer L, Zanetti N, Ezgü F, Lamantea E, Zeviani M. A myopathy, lactic acidosis, sideroblastic anemia (MLASA) case due to a novel PUS1 mutation. Turk J Hematol. 2017;34(4):376–377. doi:10.4274/tjh.2017.0231
61. Gagne KE, Ghazvinian R, Yuan D, et al. Pearson marrow pancreas syndrome in patients suspected to have Diamond-Blackfan anemia. Blood. 2014;124(3):437–440. doi:10.1182/blood-2014-01-545830
62. Camaschella C. Recent advances in the understanding of inherited sideroblastic anaemia. Br J Haematol. 2008;143(1):27–38. doi:10.1111/j.1365-2141.2008.07290.x
63. Furuyama K, Kaneko K. Iron metabolism in erythroid cells and patients with congenital sideroblastic anemia. Int J Hematol. 2018;107(1):44–54. doi:10.1007/s12185-017-2368-0
64. Cotter PD, May A, Li L, et al. Four new mutations in the erythroid-specific 5-aminolevulinate synthase (ALAS2) gene causing X-linked sideroblastic anemia: increased pyridoxine responsiveness after removal of iron overload by phlebotomy and coinheritance of hereditary hemochromatosis. Blood. 1999;93(5):1757–1769. doi:10.1182/blood.V93.5.1757
65. Bottomley SS. Congenital sideroblastic anemias. Curr Hematol Rep. 2006;5(1):41–49.
66. Camaschella C, Campanella A, De Falco L, et al. The human counterpart of zebrafish shiraz shows sideroblastic-like microcytic anemia and iron overload. Blood. 2007;110(4):1353–1358. doi:10.1182/blood-2007-02-072520
67. Di Tucci AA, Murru R, Alberti D, Rabault B, Deplano S, Angelucci E. Correction of anemia in a transfusion-dependent patient with primary myelofibrosis receiving iron chelation therapy with deferasirox (Exjade, ICL670). Eur J Haematol. 2007;78(6):540–542. doi:10.1111/j.1600-0609.2007.00840.x
68. Angelucci E, Barosi G, Camaschella C, et al. Italian Society of Hematology practice guidelines for the management of iron overload in thalassemia major and related disorders. Haematologica. 2008;93(5):741–752. doi:10.3324/haematol.12413
69. Bottomley SS, Fleming MD. Sideroblastic anemia diagnosis and management. Hematol Oncol Clin North Am. 2014;28(4):653–670. doi:10.1016/j.hoc.2014.04.008
70. Kim MH, Shah S, Bottomley SS, Shah NC. Reduced-toxicity allogeneic hematopoietic stem cell transplantation in congenital sideroblastic anemia. Clin Case Rep. 2018;6(9):1841–1844. doi:10.1002/ccr3.1667
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.