Back to Journals » Drug Design, Development and Therapy » Volume 9
Targeting brain cells with glutathione-modulated nanoliposomes: in vitro and in vivo study
Authors Salem HF, Ahmed S, Hassaballah AS, Omar MM ![]()
Received 23 March 2015
Accepted for publication 15 May 2015
Published 20 July 2015 Volume 2015:9 Pages 3705—3727
DOI https://doi.org/10.2147/DDDT.S85302
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Shu-Feng Zhou
Heba F Salem,1 Sayed M Ahmed,2 Ashraf E Hassaballah,3 Mahmoud M Omar1,4
1Department of Pharmaceutics and Industrial Pharmacy, Beni-suef University, 2Department of Industrial Pharmacy, Assiut University, 3Department of Clinical Pathology, Faculty of Medicine, Assiut University, Assuit, 4Department of Pharmaceutics and Industrial Pharmacy, Deraya University, Egypt
Background: The blood–brain barrier prevents many drug moieties from reaching the central nervous system. Therefore, glutathione-modulated nanoliposomes have been engineered to enhance the targeting of flucytosine to the brain.
Methods: Glutathione-modulated nanoliposomes were prepared by thin-film hydration technique and evaluated in the primary brain cells of rats. Lecithin, cholesterol, and span 65 were mixed at 1:1:1 molar ratio. The molar percentage of PEGylated glutathione varied from 0 mol% to 0.75 mol%. The cellular binding and the uptake of the targeted liposomes were both monitored by epifluorescent microscope and flow cytometry techniques. A biodistribution and a pharmacokinetic study of flucytosine and flucytosine-loaded glutathione–modulated liposomes was carried out to evaluate the in vivo brain-targeting efficiency.
Results: The size of glutathione-modulated nanoliposomes was <100 nm and the zeta potential was more than -65 mV. The cumulative release reached 70% for certain formulations. The cellular uptake increased as molar percent of glutathione increased to reach the maximum at 0.75 mol%. The uptake of the targeted liposomes by brain cells of the rats was three times greater than that of the nontargeted liposomes. An in vivo study showed that the relative efficiency was 2.632±0.089 and the concentration efficiency was 1.590±0.049, and also, the drug-targeting index was 3.670±0.824.
Conclusion: Overall, these results revealed that glutathione-PEGylated nanoliposomes enhance the effective delivery of flucytosine to brain and could become a promising new therapeutic option for the treatment of the brain infections.
Keywords: nanoliposomes, glutathione-modulated nanoliposomes, brain-targeting
Introduction
Infection of central nervous system with fungi can cause abscesses, parenchymal brain lesions, acute and chronic meningitis, encephalitis, or myelopathy.1,2 Cryptococcal meningitis is the most common fungal meningitis. The antifungal flucytosine is recommended for central nervous system infections caused especially by Candida and Cryptococcus neoformans.3,4
Nonetheless, the drug delivery to brain is very challenging. These challenges are classified into chemical and physical barriers. The blood–brain barrier (BBB) is an example of both physical and chemical barriers, where tight junction among endothelial cells of BBB presents physical barrier and also endothelial cells of the BBB produce significant amount of adenosine triphosphate-driven drug efflux transporters, which present chemical barrier. The blood–brain barrier protects the brain by regulating transport in and out of the brain.5 Many receptors are overexpressed on the brain cells such as the cloned μ opioid receptor,6 the low-density lipoprotein receptor,7 N-methyl-D-aspartate receptor, insulin receptor,8 kinin-B2 receptors (BK-B2R),9 angiotensin II AT1 and AT2 receptors,10 and glutathione.11–13
Recently, many targeting moieties are being used to deliver drug into the brain efficiently. This was recognized with scopine,14 monoclonal antibody 2C5,15 the OX2616 or R17217 antibodies,16 glutathione,17 and anti-transferrin.16,18 These moieties were found to enhance the uptake of the pharmaceutical moieties up to 14.25-fold.
Glutathione receptors in the brain were largely localized in the white matter, more specifically in the neuroglial cells.19 Mechanism of glutathione transportation across the brain cells was proven to be performed through a specific mechanism.20 Adsorptive-mediated endocytosis is one of the mechanisms used for drug targeting the brain.21 Endocytic pathways are activated within the cells using a directing moiety or a ligand, which is used as a vector.22 It has recently been shown that glutathione is coupled with micelles and nanoparticles and showed enormous potential in both targeting and enhancing the drug moieties into brain.23
In literatures, liposomes were used extensively as drug carriers.24,25 Therefore, liposomes possess excellent criteria as drug carriers, because they are closed vesicles composed of an internal aqueous core and an external lipophilic bilayer of phospholipids molecules. External surface of liposomes can be modified by polyethyleneglycol (PEG) to produce stealth liposomes.26 Stealth liposomes are characterized by long circulation time in vivo. They are easily coupled to different targeting moieties to improve the targeting of administered drugs27 to their site of action.28–30
Drug targeting is usually used to achieve ultra-high concentrations of prepared targeted liposomes in an infected target organ as compared with an equivalent dose given either in nontargeted liposome or free drug. It is desirable to deliver drugs selectively to the brain tissue. To this end, glutathione receptors can be used as a target due to the high expression of this receptor on the surface of neurons.19,31–34
In this study, glutathione–maleimide–polyethyleneglycol–distearoyl phosphatidyl ethanolamine was attached to nanoliposomes to deliver flucytosine. Moreover, in this study the ability of glutathione-modulated liposomes to target neuron cells of rat brain primary cell culture was evaluated.
Materials and methods
Phosphatidylcholine from soya bean (PC), cholesterol (Ch), calcein green AM, and span 65 (SA) were purchased from Sigma Chemical Co. (St Louis, USA). Glutathione–maleimide–polyethylene glycol2000–distearoyl phosphatidyl ethanolamine (glutathione–maleimide–PEG2000–DSPE) was purchased from Dutch Biotechnology Company (Leiden, the Netherlands). Methyl alcohol was obtained from BDH Ltd (Poole, UK). Chloroform, diethyl ether, sodium hydrogen phosphate, disodium hydrogen phosphate, and sodium chloride were purchased from Adwic, El-Nasr Pharmaceutical Co. (Cairo, Egypt) and were prepared according to the methods of Prolabo (Paris, France). Primary rat brain cells were extracted from rats that were purchased from the animal house of Assiut University. Dulbecco’s Modified Eagle’s Medium and fetal bovine serum were purchased from Sigma Chemical Co. All reagents were of analytical grade and 99% pure. All chemicals were used as received.
Liposome preparation
Liposomes were prepared using thin-film hydrating technique.35–37 In this method, four different liposomal formulae were prepared. They were composed of soya bean phosphatidylethanolamine, cholesterol, span 65, and glutathione–maleimide–polyethyleneglycol–distearoyl phosphatidyl ethanolamine (Table 1). All the components were dissolved in an appropriate amount of chloroform and methanol mixture (3:2 v/v). The mixtures were dried up to thin lipid films under vacuum using a rotary evaporator (Buchi, Switzerland). These films were kept under high vacuum for at least 2 hours to remove traces of chloroform. The dry films were hydrated with aqueous solution of flucytosine (15 mL, 1.9 mg/mL) in PBS (pH 7.4) by rotation at 90 rpm for 1 hour. The resulting vesicles were kept for 4 hours at 25°C to form large multilamellar vesicles. The liposomal suspensions were agitated for 30 minutes using an ultrasonic bath (model 275 T manufactured by Crest Ultrasonics, Trenton, NJ, USA). The prepared suspensions were kept at 4°C overnight. The encapsulated flucytosine was separated by ultracentrifugation at 20,000 rpm at 4°C. Free flucytosine was separated from the liposomes by centrifugation (20,000 rpm, 1 hour) at 4°C using a high-speed centrifuge model T-70BL (Laby Instrument Industry, Haryana, India). The supernatant was aspirated and the concentration was calculated at λ276 (USP 2012). All steps of the liposome preparation were carried out under aseptic conditions using a laminar air flow hood (horizontal laminar flow hood, BZ Series, model BZ-3SSRX; Germfree, Ormond Beach, FL, USA).
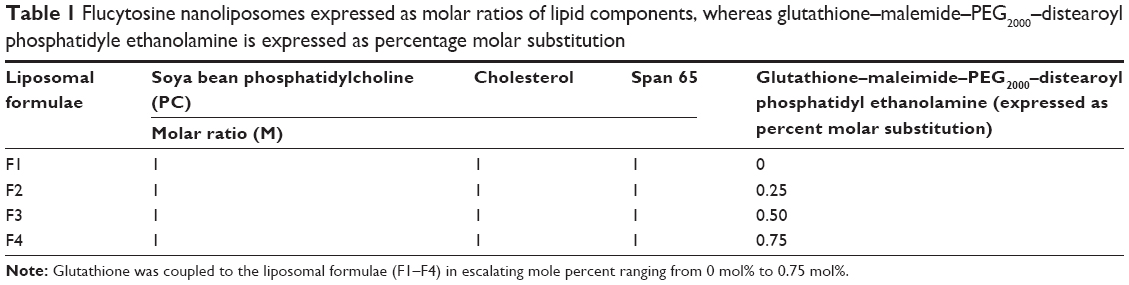 | Table 1 Flucytosine nanoliposomes expressed as molar ratios of lipid components, whereas glutathione–malemide–PEG2000–distearoyl phosphatidyle ethanolamine is expressed as percentage molar substitution |
Determination of entrapment efficiency of the liposomes
The liposomes were separated from the supernatant, and then 1 mL of the supernatant was diluted 10 times with methanol.38 The concentration of drug was determined spectrophotometrically at λ=285. The entrapped efficiency of flucytosine was expressed as weight percent. Loading efficiency is usually defined as the percent fraction of the total input drug encapsulated both in lipid bilayers and in aqueous compartments within the liposomes at a particular phospholipid concentration and expressed as weight percentage and was calculated by the following equation:
|
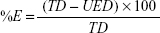
where %E is the percent encapsulation or loading efficiency, TD is the total drug added, and UED is the amount of unencapsulated drug.
Determination of the particle size and zeta potential of liposomes
The liposome dispersion aliquots (100 μL) were diluted with deionized water (900 μL) and were used to characterize both the particle size and the zeta potential of the liposomes.39–41 They were measured by the method of dynamic light scattering by Malvern Zetasizer, Malvern Instruments Corp (Nano ZS ZEN 3600, Worcestershire, UK).
Investigation of the morphology of the liposomes
The morphology of prepared liposomes was recognized by transmission electron microscopy, where the prepared liposomes were diluted with double-distilled water.42,43 One drop of prepared suspension was added onto hydrophobic carbon grids to adsorb liposome particles from the suspension. The samples were air dried for 1 minute at room temperature. The specimens were stained by 2.5% uranyl acetate for 4 minutes. The excess of uranyl acetate was blotted by filter paper. The specimens were observed under a TEM-1010 Transmission Electron microscope (JEOL, Japan) operated at 80 kV.
In vitro release study from the liposomes
In vitro release of the liposomes was carried out via a dialysis method according to the method described by Hao et al.44 The equivalent of 5 mg of the drug of flucytosine-loaded liposomal suspension was introduced into dialysis bags with a molecular weight cut-off 12,000 kDa. The dialysis bags were fully immersed under the surface 250 mL of isotonic phosphate buffer (pH 7.4) at speed of rotation 100 rpm and placed within the dissolution flask of the USP dissolution apparatus with a constant temperature of 37°C±0.5°C. The samples were measured spectrophotometrically at λ276. The concentration of flucytosine at time (t) was calculated using the following equation:
|
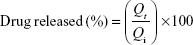
To elucidate the equation, Qi and Qt are the initial amounts of drug encapsulated in the liposomes and the amounts of drug released at time t, respectively.
Stability study of the flucytosine-loaded liposomes
For evaluating the aggregation of the prepared liposomes and leakage of flucytosine from the same during storage, a physical stability study of the prepared liposomes was carried out. A protocol developed by Du Plessis et al was adopted with some modifications.45 The prepared liposomal formulations were stored in tightly sealed 20 mL glass vials at 4°C±1°C, 25°C±1°C (room temperature, RT) and 37°C±1°C (physiologic temperature) for 3 months. The particle size and entrapment efficiency of the prepared liposomes were evaluated over the period of the physical stability study. Samples of liposomal dispersions were regularly examined for evaluating their particle size and encapsulation efficiency. Signs of sedimentation, creaming (if any), or any changes in color of the examined dispersions were evaluated.
Preparation of rat brain primary cell culture
Protocol of the performed experiments was approved by the animal ethical committee of the Faculty of Medicine, Assiut University. The rats were humanely executed by decapitation. The preparation of brain cells was carried out using the following procedures, as reported by Watson et al with some modifications.46 The dorsal side of rat head is pushed toward a dissecting board and ethanol 70% v/v is sprayed to decrease a possible contamination. All steps of the experiment were carried under laminar flow cabinet. The skin from the head was removed by using sharp curved scissors. A circular cut was made to remove the top of the skull, revealing the brain and the two olfactory bulbs at the tip of the nose.47–51 The olfactory bulbs were gently released from the brain, and they were placed in a Petri dish containing 5–10 mL ethanol 70% v/v for 1 minute, followed by inoculation with 5–10 mL of fresh rat brain tissue washing medium for 5 minutes. The olfactory bulbs were then chopped into small pieces of size 3–4 mm. Brain tissue pieces were washed with an isotonic phosphate salt solution free from calcium and magnesium three times. Dissociation of cells from primary tissue was carried out mechanically by forcing tissue suspension using syringe. Cell suspension was centrifuged for 5 minutes at 3,000 rpm. The primary cell culture media was prepared by the addition of fetal bovine serum (10% fetal bovine serum, 5 mL) to Dulbecco’s Modified Eagle’s Medium (45 mL), which contains glutamine (2 mL). A mixture of penicillin (1 mL, 100 IU) and streptomycin (0.1 mg/mL) was added to the prepared cell culture medium. Cells 1×106 were incubated at 37°C±0.5°C in a CO2 incubator.
Qualitative binding and uptake of nanoliposomes
Half milliliter of glutathione-modulated liposomes and glutathione-free liposomes were added to each well at a concentration of 75 m mole per 1×106 seeded cells and incubated with the cells for 24 hours at 37°C in cell culture medium. Unbounded liposomes were then removed by washing with phosphate-buffered saline (PBS). The cells were fixed in 4% paraformaldehyde in PBS for 10 minutes, stained with excess 4′,6-diamidino-2-phenylindole stain, and mounted with coverslips. 4′,6-diamidino-2-phenylindole was used to stain nucleic acid of cell’s nucleus. The cells were visualized under epifluorescence microscope (excitation wavelength, 495 nm; emission wavelength, 516 nm) and images were obtained with IX81 (Olympus, Tokyo, Japan) and photographed using Nikon D7000 camera (Tokyo, Japan).52
The samples were taken for microscopic fluorescence evaluation at 0 and 6-hour time intervals.
Binding and uptake measurements of liposomes using flow cytometry
Both the cellular binding and uptake of liposomes were calculated using the number of cells bound to the labeled liposomes. Calcein-AM was used as labeling material for the liposomes. Flow cytometry was used to monitor the interactions between targeted liposomes and the rat brain primary cells.53,54 Rat primary cells 1×106 per mL were trypsinized (trypsin 0.25% v/v with EDTA 0.1% v/v) and resuspended in ice-cold flow buffer PBS.
One million rat brain primary cells per mL were incubated with both conventional liposomes (500 μL, F1) and with glutathione-modulated liposomes (500 μL, F2, F3, and F4). Free calcein-AM (200 μL) was considered as control I. The liposomes were incubated for different time interval with the cells. Aliquots of rat brain primary cells (100 μL) were withdrawn from the tubes at 1 hour, 2.5 hours, 5 hours, 6 hours, and 24 hours. The cells were taken and centrifuged at 3,000 rpm for 5 minutes. The precipitated cells were washed three times with ice-cold PBS. Flow cytometric analysis was conducted, followed by immediate binding and the uptake of the liposomes was measured. Ten thousand events were acquired. The gates exclude all debris and dead cells. All expressions were assessed by percentage of positive cells (green fluorescence).
Flow cytometric analysis for all the above experiments was performed at the flow cytometry laboratory (BD FACSCalibur, BD Cell Quest Pro software on Mac® OS 9, version 5.1; Biosciences, San Jose, CA, USA). In the competition assays, excess of free glutathione (0.1 mol) was incubated with 1×106 of rat brain primary cells 2 hours before adding the targeted liposomes. This was followed by washing the cells twice with PBS. Then the glutathione-modulated liposome (500 μL, F4), which is functionalized with 0.75 molar percent (mol%) glutathione, was added to the cells. The cell population was described using the signals produced from the overlap of the forward scatter and the side scatters, as a measurement for both the cell size and granularity of the cells, respectively.
In vivo targeting study
Animal
Male Sprague-Dawley rats (200–250 g) were provided by the animal house of the Faculty of Medicine, Assiut University. They were kept at standard condition (25°C±1°C, 40% RH). All steps of animal study were approved by Assiut University Ethical Experimentation Committee.
Chromatographic conditions
An HPLC assay was adopted to determine the concentration of flucytosine in plasma, brain, and other tissue biosamples. HPLC (Schimadzu Instruments, Japan) system consisted of LC-10AD Schimadzu liquid chromatograph pumps (pump A and pump B), DGU-12A Schimadzu degasser, SIL-10AD Schimadzu autoinjector, SCL-10A Schimadzu system controller, CTO-10A Schimadzu column oven fixed with reverse-phase C18 column (MICRA Scientific column, 4.6 mm ×250 mm), and CTO-10A Schimadzu UV-Vis detector with response time 0.5 second, using Schimadzu Class-VP V6.12SP5 computer software, for instrument control and data analysis. A reverse-phase precolumn (50×4.6 mm) was added to the HPLC system to prevent contamination of the column with plasma traces remaining after plasma precipitation procedures. Analyses were performed using methanol 0.25 M KH2PO4, pH 2.5 (5:95), as mobile phase.
Biodistribution and pharmacokinetic studies of flucytosine and its loaded glutathione-modulated liposome
Efficiency of glutathione-modulated liposomes in targeting the brain was evaluated in vivo.55 A biodistribution and pharmacokinetic study was performed using 63 Sprague-Dawley rats (200–250 g), provided by the animal house of the Faculty of Medicine, Assiut University. They were randomly divided into five groups. In the first group, 3 rats were treated with flucytosine solution (10 mg/kg); in the second group, 27 rats were treated with flucytosine-loaded liposome (F1; 10 mg/kg); in the third group, 3 rats were treated with glutathione-modulated liposome (F2; 10 mg/kg); in the fourth group, 3 rats were treated with glutathione-modulated liposome (F3; 10 mg/kg); in the fifth group rats, 27 rats were treated with glutathione-modulated liposome (F4; 10 mg/kg). A sterile flucytosine solution and its loaded glutathione-modulated liposomal formulae (F1, F2, F3, or F4) were injected via the caudal vein. For the biodistribution study, the rats were humanely killed at 30 minutes after dosing (n=3). Tissue samples of brain were removed and flushed with saline solution (0.9% w/v). Homogenization of the tissue samples was carried out using saline (0.9% w/v) at 1:2 ratio. The tissue homogenate samples were stored at −20°C until HPLC assay was performed. For the pharmacokinetic study, only the second and the fifth group were included. As soon as blood samples were collected from the ocular artery at 30 minutes, 45 minutes, 60 minutes, 120 minutes, 240 minutes, 360 minutes, 480 minutes, 600 minutes, and 720 minutes postdose, they were centrifuged at 3,000 rpm for 10 minutes. The plasma samples were stored at −20°C till HPLC analysis was conducted.
Processing of biosamples
A 200 μL volume of plasma or 500 μL volume of tissue homogenate was mixed with 200 μL volume of 10 μg/mL 5-chlorouracil solution. The mixture was transferred into tubes containing 20 μL of 4 M trichloroacetic acid; they were centrifuged for 10 minutes at 0°C to separate the precipitated protein. The supernatant was diluted (1:4) with distilled water and 20 μL was assayed using HPLC system.
Data analysis
A two-compartment open model was applied for calculating the pharmacokinetic parameters of flucytosine and the prepared liposomal formulae (F1–F4) using winLIN 3.2 software package (Colo-Parmer Co. Ltd.). The maximal concentration (Cmax), area under the curve (AUC), and the mean residence time were calculated for either plasma or tissue homogenate samples separately.
For evaluating the brain-targeting efficiency of glutathione-modulated liposomes, the relative uptake efficiency (RE), concentration efficiency (CE), and drug-targeting index (DTI) were calculated using the following equations:
|
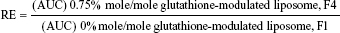
|
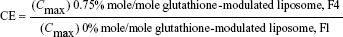
|

Statistical analysis
All experiments were carried out in triplicate. The differences were evaluated for statistical significance using Student’s t-test.
Results
Entrapment efficiency
Flucytosine entrapment efficiency in liposomes was studied, when different mole percentages of glutathione–maleimide–polyethyleneglycol–distearoyl phosphatidyl ethanolamine were used. Incorporation of glutathione–maleimide–PEG2000–DSPE at different mole% ranging from 0 mole% to 0.75 mole% did not show any significant difference in the entrapment efficiency as shown in Table 2. These results may suggest that the glutathione was attached onto the surface of the liposomes rather than interfering with the liposome bilayers. Optimization study for selection of the optimum ratio of the different liposomal ingredients was summarized in Supplementary material.
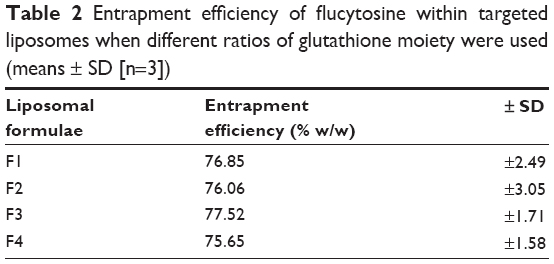 | Table 2 Entrapment efficiency of flucytosine within targeted liposomes when different ratios of glutathione moiety were used (means ± SD [n=3]) |
Determination of zeta potential measurements for the prepared liposomes
The entire charge of the targeted liposome could be affected by the charge of the glutathione. Incorporation of glutathione–maleimide–PEG2000–DSPE at different mole% ratios (0.25 mole%, 0.50 mole%, and 0.75 mole%) led to increased zeta potential values of liposomal surfaces as shown in Table 3. The zeta potential of formula 1 (F1), which did not have glutathione–maleimide–PEG2000–DSPE among its components, showed the lowest value of zeta potential when compared to the other formulae (F2, F3, and F4). This effect suggested that increasing the content of glutathione–maleimide–PEG2000–DSPE in liposomal preparations led to increasing the zeta potential values of liposomal formulae (F2–F4). The effect of glutathione–maleimide–PEG2000–DSPE on zeta potential could be explained taking into account the effect of both polyethylene glycol and distearoyl phosphatidylethanolamine. The zeta potential value for PEG–DSPE-containing liposomes (F2–F4) was more negative than that of the conventional liposome (F1) as shown in Table 3. A carbamate linkage, which is typically a conjugation between PEG and DSPE, is responsible for a net negative charge on the phosphate moiety at physiological pH.56 Increasing negative values of zeta potential of prepared liposomes (F1–F4) may be due to the presence of thiole groups of glutathione.57,58 The presence of escalated negative charges could enhance the stability of the liposomes according to what was reported in literature.59 To this end, the obtained results indicated that all liposomal preparations are colloidally stable and are concurrent with the articles reported in the literature.60
 | Table 3 Zeta potential values of glutathione-targeted nanoliposomes (measurements ± SD [n=3]) |
Particle size and ultrastructure study of the prepared liposomes
Particle size measurements showed that incorporation of glutathione–maleimide–PEG2000–DSPE into liposomal preparation led to a significant decrease (P>0.05) in the particle size. The particle size of F2, F3, and F4 was measured and seems to be smaller than that of formula (F1). The only difference between F1 and the rest of the formulae is that F1 is deficient in glutathione–maleimide–PEG2000–DSPE. Particle size of glutathione–maleimide–PEG2000–DSPE liposomes decreased when glutathione–maleimide–PEG2000–DSPE content increased. These results may be attributed to incorporation of glutathione–maleimide–PEG2000–DSPE. The incorporation of glutathione moiety might lead to enhance the packaging of the phospholipids within the liposome bilayer. The polydispersity index (PDI) is dimensionless. The maximum value is arbitrarily limited to 1.0. PDI value of 1.0 indicates that the sample has a very broad size distribution and may contain large particles or aggregates.61 Incorporation of glutathione–maleimide–PEG2000–DSPE into liposomal formulation might lead to lower PDI, and thus, it enhanced the uniformity of the system. Low particle size and low PDI are very important parameters in enhancing the binding and the uptake of the liposomes within the brain cells and their transportation across the BBB. The findings of the current study are concurrent with those of Sriwongsitanont Supaporn and Ueno Masaharu (2002), who have found that the particle size of DMPC liposomes containing DSPE–PEG2000 decreases significantly when DSPE–PEG2000 content increases.62
Figure 1 represents transmission electron micrographs of free and modulated glutathione nanoliposomes. Both conventional (Figure 1A) and glutathione–maleimide–PEG2000–DSPE liposomes (Figure 1B–D) are closed bilayer structure. They are spherical and hollow structures ranging in size from 80 nm to 150 nm. These findings were consistent with results obtained from the particle size measurement shown in Table 4. The particles seem to be colloidally stable because formation of aggregates is not detected within the investigated fields. These outcomes are concurrent with the results of the zeta potential.
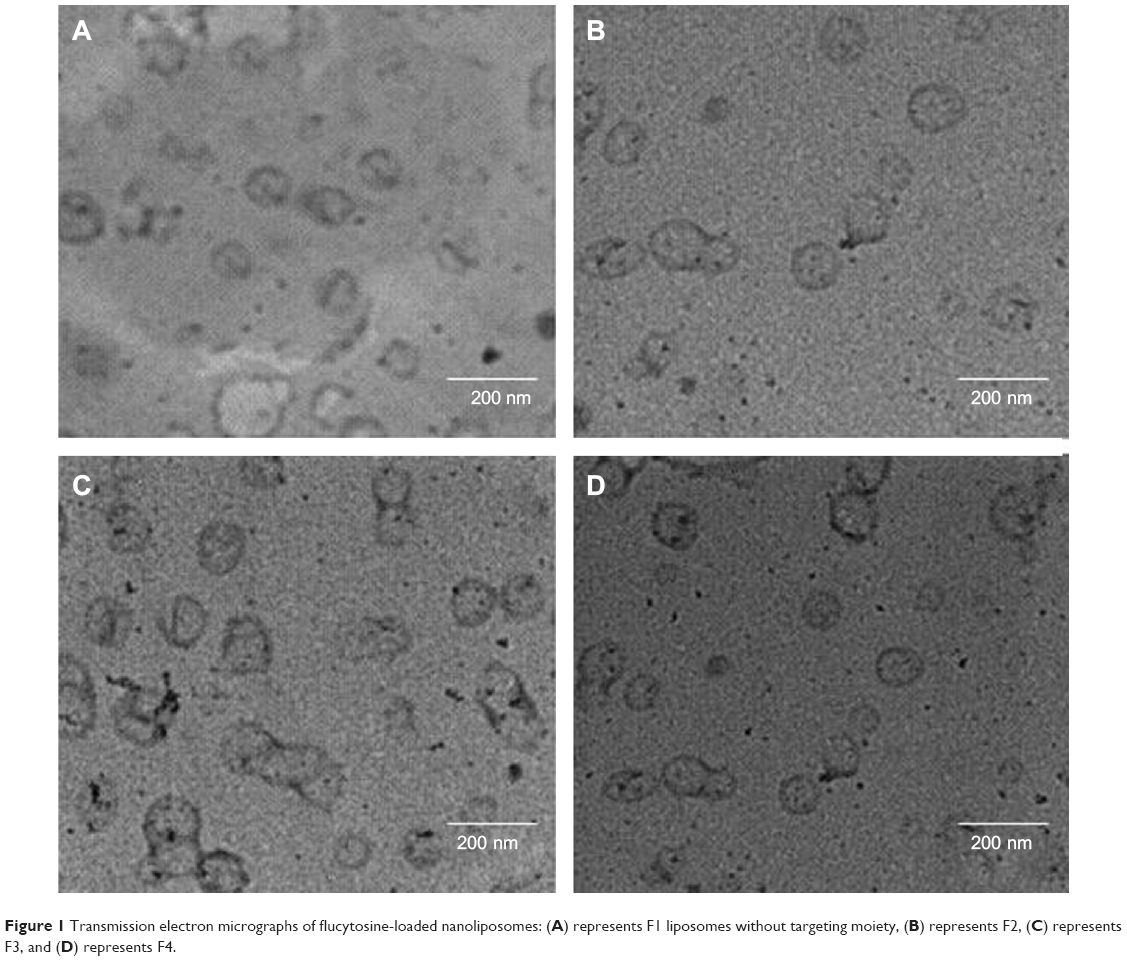 | Figure 1 Transmission electron micrographs of flucytosine-loaded nanoliposomes: (A) represents F1 liposomes without targeting moiety, (B) represents F2, (C) represents F3, and (D) represents F4. |
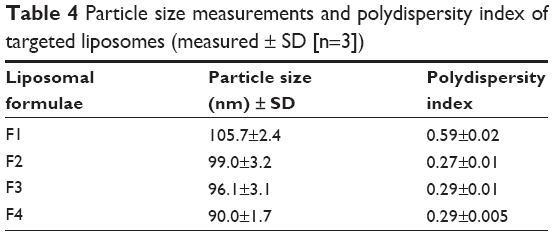 | Table 4 Particle size measurements and polydispersity index of targeted liposomes (measured ± SD [n=3]) |
Kinetic studies of the drugs released: in vitro study
The in vitro release kinetics of flucytosine out of both glutathione-modulated liposome and conventional liposome is shown in Figure 2. The data were represented as cumulative percent of drug release versus time over a period of 12 hours. The measurements were taken at 0 hour, 1 hours, 2 hours, 3 hours, 4 hours, 6 hours, 8 hours, 10 hours, and 12 hours. Dialysis method was used for evaluation of the in vitro drug released from the encapsulated liposomes.63
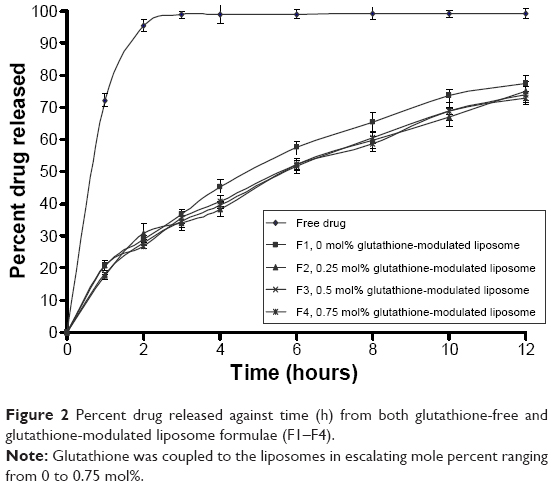 | Figure 2 Percent drug released against time (h) from both glutathione-free and glutathione-modulated liposome formulae (F1–F4). |
The release of flucytosine from glutathione-modulated liposomes with a final molar% of 0.25, 0.50, and 0.75 was lower than that of nonmodulated liposomes (F1; Figure 2). Glutathione–maleimide–PEG2000–DSPE moiety may play a role in reducing the release value. This may be due to the effect of the alkyl chain of the DSPE polymer in improving the packaging within the liposomal bilayers. The earlier results showed that DSPE–PEG2000 decreased the diffusion of hydrophilic drugs.64 The hydrophobic long alkyl chains of the polymer increased the entrapment of the drug in the polymers.65
Stability study
The particle size and entrapment efficiency measurements obtained from different liposomal formulations during the stability study are shown in Tables 5 and 6. There were insignificant changes (P>0.05) in the entrapment efficiency and vesicle size of the liposomal formulations stored at 4°C±1°C in refrigerated conditions.
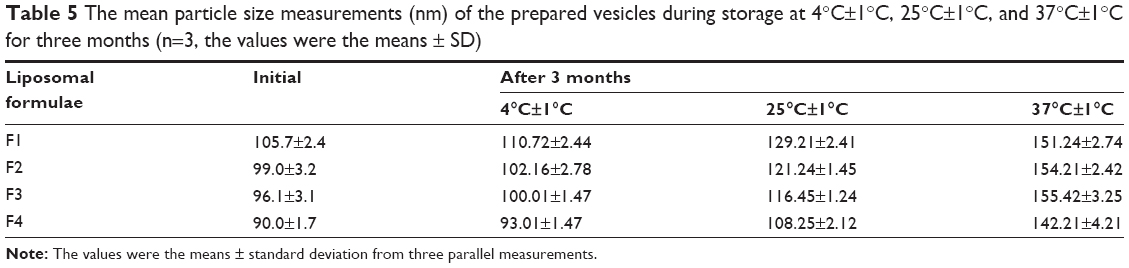 | Table 5 The mean particle size measurements (nm) of the prepared vesicles during storage at 4°C±1°C, 25°C±1°C, and 37°C±1°C for three months (n=3, the values were the means ± SD) |
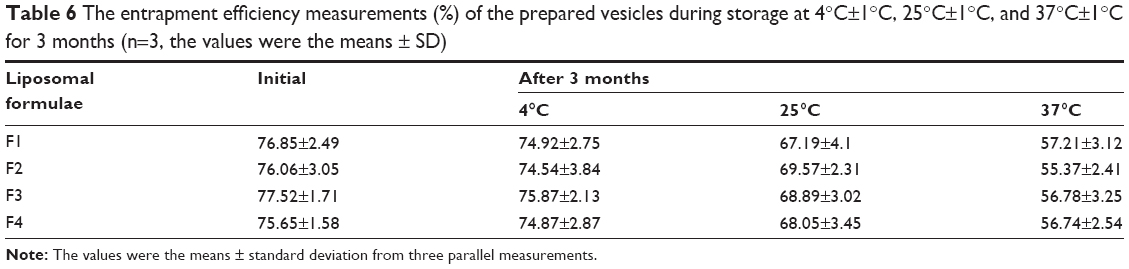 | Table 6 The entrapment efficiency measurements (%) of the prepared vesicles during storage at 4°C±1°C, 25°C±1°C, and 37°C±1°C for 3 months (n=3, the values were the means ± SD) |
On the other hand, evaluation of the liposomal formulations stored at higher temperature conditions showed significant changes (P<0.05) in the measuring parameters. A dramatic increase in the vesicle size and drug leakage was observed at both room temperature and physiologic temperature. Lower percentage of flucytosine was leaked from the liposomal formulations stored at 4°C compared to the others stored at higher temperatures. By the end of the study, all liposomal formulations stored at refrigerated temperature (4°C±1°C) were physically stable. No sedimentation or change in color was observed. The liposomal formulations stored at 25°C±1°C showed slight sedimentation, which can be redispersed easily on shaking. The color of the liposomal formulations stored at 37°C±1°C was changed to greenish yellow which cannot be redispersed on shaking. Formation of big clumps was observed.
Qualitative binding and uptake of glutathione-modulated nanoliposomes using epifluorescent microscope
Specific glutathione binding sites in the brain are widely localized in the white matter, indicating the presence of glutathione receptors on neuroglial cells.66,67 To explore the effect of increasing the mole percentage of glutathione moiety on the efficiency of cellular uptake, binding experiments were conducted.
The cellular binding and the uptake of different liposomal formulations were investigated by fluorescence microscopy.68
The conventional liposome (F1) was used as zero percent glutathione moiety-modulated liposome. When F1 was incubated with the cells, the green fluorescence of calcein-loaded liposomes showed a relatively low signal. This phenomenon could point to the low concentration of liposomes that are delivered within the cells in the case of the nontargeted liposomes.
In contrast, glutathione-modulated liposomes (F2–F4) were accumulated to significant extents in the cytoplasm of the brain cells. All the glutathione-modulated liposomal formulae were found to be efficient in delivering drug. However, this was in an escalating manner reaching maximum when glutathione moiety 0.75 mole%-modulated liposome was used (F4) as shown in Figure 3.
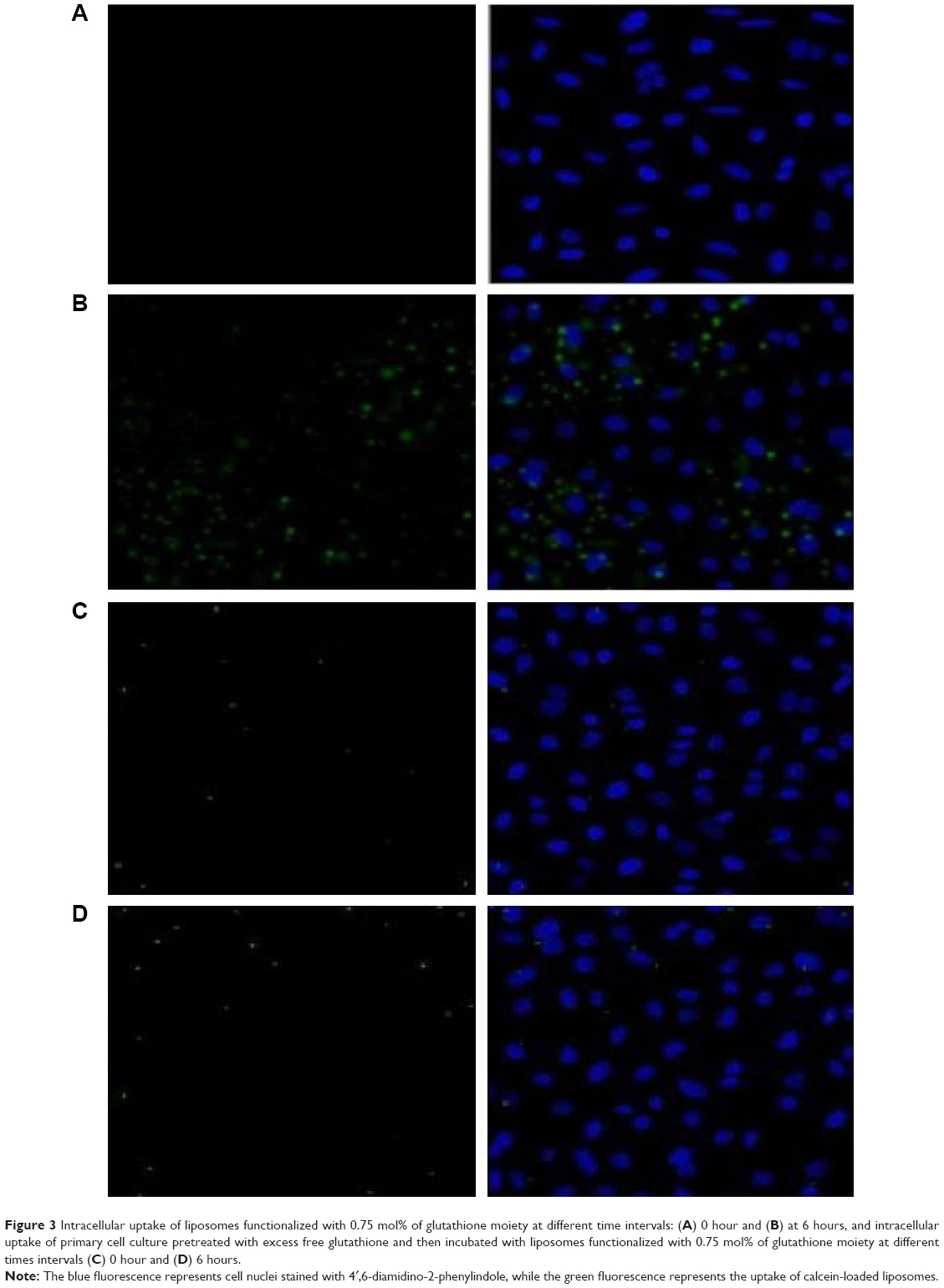 | Figure 3 Intracellular uptake of liposomes functionalized with 0.75 mol% of glutathione moiety at different time intervals: (A) 0 hour and (B) at 6 hours, and intracellular uptake of primary cell culture pretreated with excess free glutathione and then incubated with liposomes functionalized with 0.75 mol% of glutathione moiety at different times intervals (C) 0 hour and (D) 6 hours. |
The binding and the uptake results revealed that the increase in molar percentage of glutathione moiety caused the increase in the cellular uptake of liposomes. This conclusion suggested that glutathione moiety enhanced the uptake of liposomes especially when escalating molar percentage of glutathione was used.
Competition study was conducted to explore the mechanism of binding. This experiment was conducted to show whether the binding sites were specific or nonspecific. As shown in Figure 3, intracellular uptake of primary cell culture incubated with liposomes functionalized with 0.75 mol% of glutathione moiety became very weak as a result of pretreating with excess free glutathione.
The quenching of fluorescent signal occurred due to saturation of the receptor with the excess free glutathione, which suggested that the binding of the glutathione-modulated nanoliposomes to glutathione receptors was specific. In contrast to that, free calcein-AM solution showed a very weak fluorescent signal within the cytoplasm, which boosts the suggestion that the binding and the uptake of glutathione-modulated liposomes was due to the presence of the targeting moiety and not due to the hydrophobic nature of the labeling agent or the nature of the liposomes (Figure 3).
The quantitative uptake of glutathione-targeted liposomes using flow cytometry
The main objective of current study was to quantitatively measure the difference in binding and the uptake of both naked and glutathione-modulated liposomes at different mole percentages ranging from 0 to 0.75. Flow cytometric analysis was used to identify the cell populations that bound and absorbed glutathione-modulated nanoliposomes. Therefore, the cells were incubated with the escalating concentration of glutathione-modulated nanoliposomes.
Increasing the mole percentage of glutathione–maleimide–PEG2000–DSPE within liposomal bilayers enhanced the cellular uptake of liposomes. The highest cellular uptake of liposome was observed when 0.75 mol% of glutathione–maleimide–PEG2000–DSPE was incorporated within the bilayer liposomal formula (F4). The cellular uptake of liposome conjugated with 0.75 mol% glutathione–maleimide–PEG2000–DSPE was threefold higher than that of conventional liposomes (F1), as shown in Figures 4 and 5. This finding suggested that increasing the concentration of glutathione moieties on the surface of liposomes led to the increase in the cellular uptake of liposome.
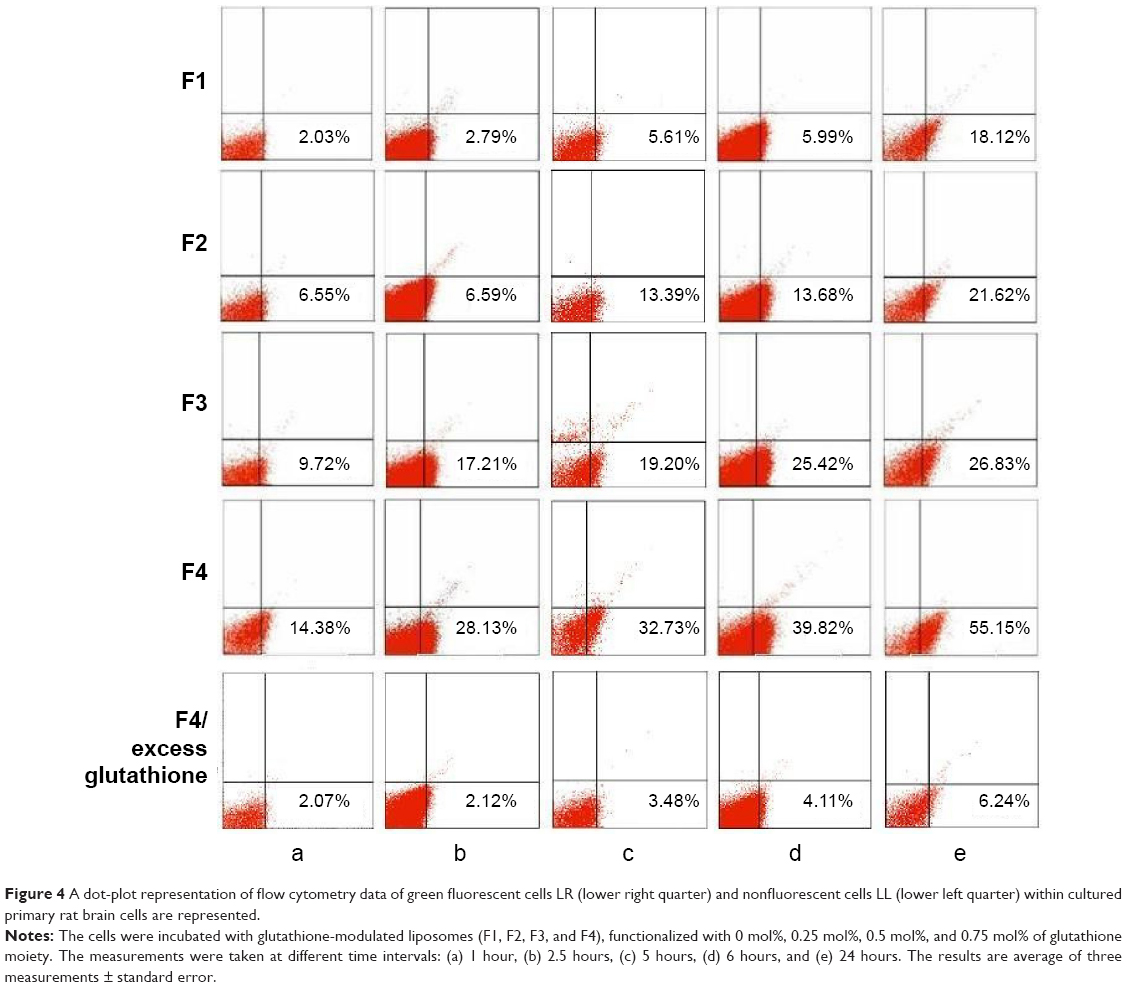 | Figure 4 A dot-plot representation of flow cytometry data of green fluorescent cells LR (lower right quarter) and nonfluorescent cells LL (lower left quarter) within cultured primary rat brain cells are represented. |
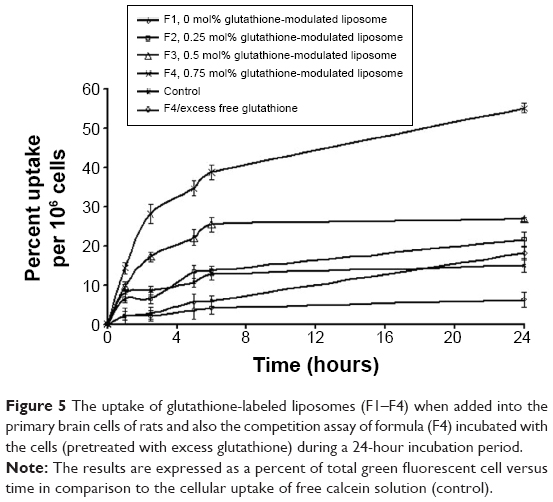 | Figure 5 The uptake of glutathione-labeled liposomes (F1–F4) when added into the primary brain cells of rats and also the competition assay of formula (F4) incubated with the cells (pretreated with excess glutathione) during a 24-hour incubation period. |
In the competition assay, excess free glutathione was added in the medium in order to examine the specifity of the targeting moiety to the glutathione receptor. Excess free glutathione was used in this study to grant saturation of the glutathione receptors. After saturation of the receptors, the cells were incubated with glutathione-modulated liposome (F4) over a period of 24 hours. The cellular uptake of glutathione-modulated liposomes (F4) in the absence of free glutathione was compared to their uptake in the presence of excess free glutathione. The result showed a steep decline in the percent cells that absorbed the liposomes in the presence of free glutathione. This may indicate saturation of the receptors with free glutathione, which competed for the glutathione receptor when glutathione-modulated liposomes were added. The uptake of the modulated nanoliposomes in the presence of free glutathione was 11.31% of that obtained in the absence of free glutathione (Figure 6). The uptake of the conventional liposome (F1) was achieved by 18.12% of the cell population when it was incubated for 24 hours with the cells. This observation suggests that the uptake of the liposomes could be achieved either specifically through targeting of the liposomes with glutathione moiety or nonspecifically through the hydrophobic nature of the liposomes.
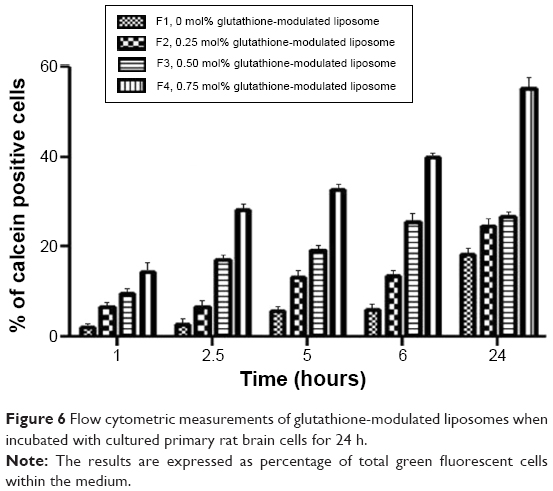 | Figure 6 Flow cytometric measurements of glutathione-modulated liposomes when incubated with cultured primary rat brain cells for 24 h. |
In vivo targeting efficiency study
A biodistribution and pharmacokinetic study of flucytosine and flucytosine-loaded glutathione-modulated liposomes was carried out to evaluate in vivo the brain-targeting efficiency. The pharmacokinetic measurements in the present study only indirectly respond to the targeted accumulation of the drug loaded into brain. The concentration of flucytosine in the brain versus that in the plasma (Cbrain/Cplasma)flucytosine was used to evaluate the brain-targeting efficiency of the liposomal formulations. The concentration of flucytosine in the plasma and brain 30 minutes after intravenous injection of flucytosine solution or flucytosine-loaded glutathione-modulated liposomes (F1–F4; equivalent to 10 mg of flucytosine) was determined as shown in Figure 7. The liposomal formulations displayed increased drug concentrations in the brain (Figure 8, P<0.05). The calculated (Cbrain/Cplasma)flucytosine ratios 30 minutes postinjection of flucytosine or flucytosine-loaded glutathione–modulated liposomes (F1, F2, F3 and F4) were 0.147±0.018, 0.266±0.107, 0.382±0.082, 0.495±0.107, and 0.734±0.095, respectively, as shown in Figure 8 (n=3).
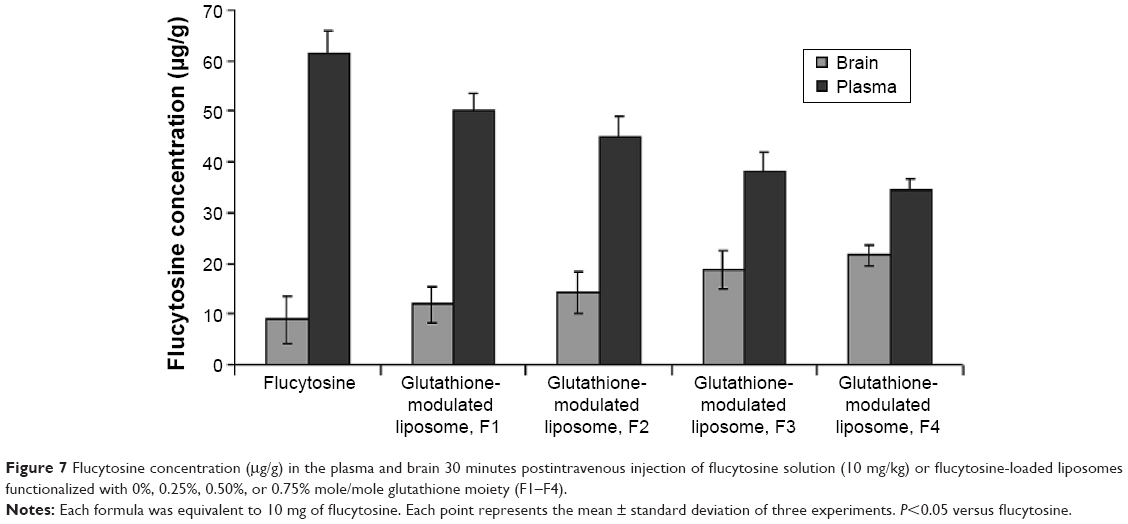 | Figure 7 Flucytosine concentration (μg/g) in the plasma and brain 30 minutes postintravenous injection of flucytosine solution (10 mg/kg) or flucytosine-loaded liposomes functionalized with 0%, 0.25%, 0.50%, or 0.75% mole/mole glutathione moiety (F1–F4). |
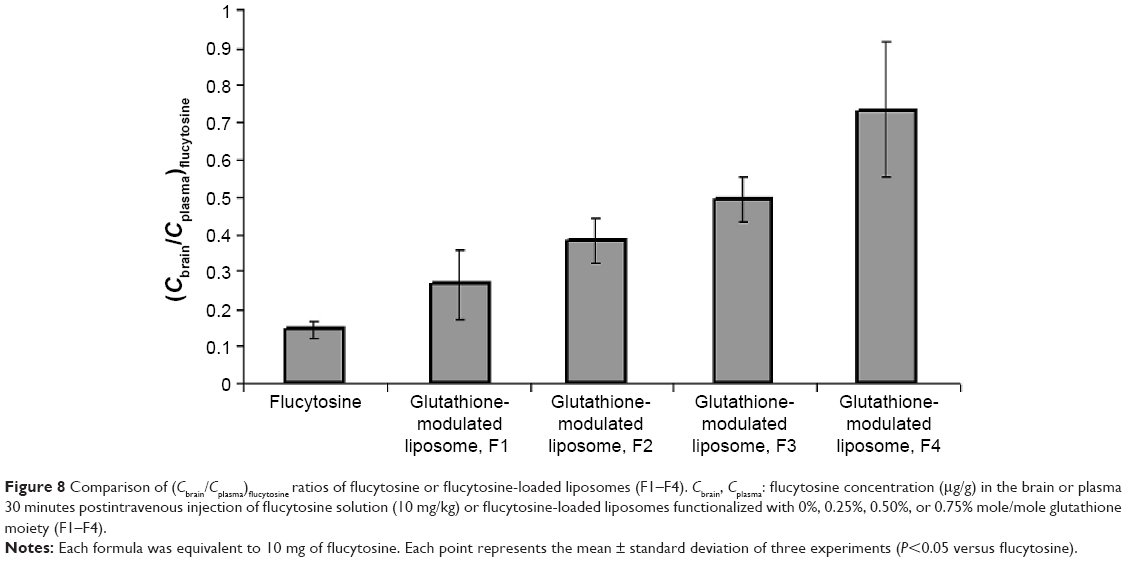 | Figure 8 Comparison of (Cbrain/Cplasma)flucytosine ratios of flucytosine or flucytosine-loaded liposomes (F1–F4). Cbrain, Cplasma: flucytosine concentration (μg/g) in the brain or plasma 30 minutes postintravenous injection of flucytosine solution (10 mg/kg) or flucytosine-loaded liposomes functionalized with 0%, 0.25%, 0.50%, or 0.75% mole/mole glutathione moiety (F1–F4). |
To evaluate the brain-targeting efficiency of flucytosine-loaded glutathione-modulated liposomes, ratios of the flucytosine concentration in the brain versus its concentration in plasma (Cbrain/Cplasma) were determined and plotted as shown in Figure 8. The (Cbrain/Cplasma) ratios were moderate (not >0.2666) postinjection of the conventional liposomal formula (F1) as shown in Figure 8. The AUCbrain/AUCplasma ratio of conventional liposomal formula (F1) was only 0.142. Those results indicated that although conventional liposomal formula (F1) could pass the BBB, the concentration of flucytosine in brain is still low. In comparison with the injection of conventional liposomal formula (F1), the brain concentration of flucytosine postinjection of flucytosine-loaded glutathione-modulated liposome (F4) was significantly increased (P<0.05).
In comparison with intravenous injection of conventional liposomal formula (F1), the flucytosine plasma concentration postinjection of flucytosine-loaded liposome (F4) was moderate as shown in Figure 9. The concentration of flucytosine in plasma was 29.47±2.49 μg/g 30 minutes postinjection of flucytosine-loaded liposome (F4), which was lower than that of conventional liposomal formula (F1) (51.118±6.75 μg/g, P<0.05). The (Cbrain/Cplasma)flucytosine ratio of loaded liposome (F4) was 0.734±0.095 at 30 minutes, much higher than that of the (Cbrain/Cplasma)flucytosine ratio of conventional liposomal formula (F1). While the (Cbrain/Cplasma)flucytosine ratio of conventional liposomal formula (F1) was only 0.266±0.091 at 30 minutes (Figure 10; P<0.05). These findings show a diminished effect of the BBB in response to glutathione-modulated liposome (F4), and also, the observed high concentration of flucytosine in brain was slowly decreased, with a mean residence time of 1.62±0.089 hour. The (Cbrain/Cplasma)flucytosine ratios of glutathione-modulated liposomes (F2–F4) were 1.43, 1.86, and 2.76-fold higher, respectively, than those of conventional liposomal formula (F1). So, the glutathione-modulated liposomes showed good brain-targeting efficiency (P<0.05). The relative brain-targeting efficiency could be arranged as follow (F4>F3>F2>F1). The plasma and the brain concentrations of the flucytosine versus time postintravenous injection (10 mg) were determined. The pharmacokinetic parameters of flucytosine and its loaded liposome (F4, liposome functionalized with 0.75% mole/mole glutathione moiety) were performed.
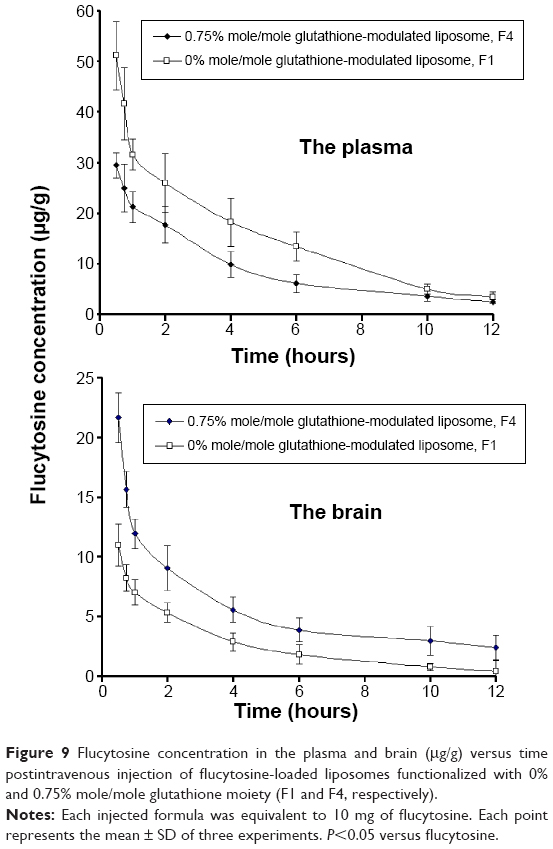 | Figure 9 Flucytosine concentration in the plasma and brain (μg/g) versus time postintravenous injection of flucytosine-loaded liposomes functionalized with 0% and 0.75% mole/mole glutathione moiety (F1 and F4, respectively). |
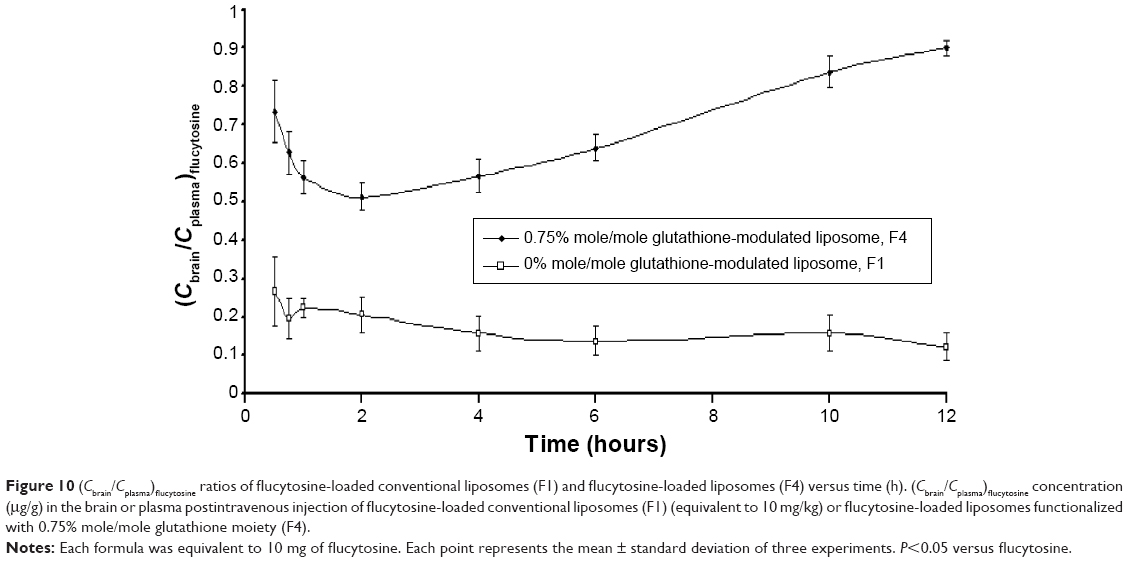 | Figure 10 (Cbrain/Cplasma)flucytosine ratios of flucytosine-loaded conventional liposomes (F1) and flucytosine-loaded liposomes (F4) versus time (h). (Cbrain/Cplasma)flucytosine concentration (μg/g) in the brain or plasma postintravenous injection of flucytosine-loaded conventional liposomes (F1) (equivalent to 10 mg/kg) or flucytosine-loaded liposomes functionalized with 0.75% mole/mole glutathione moiety (F4). |
The AUC0–12 values in the brain postintravenous injection of conventional liposomal formula (F1) and flucytosine-loaded liposome (F4) were 25.011±5.74 μg/g h and 65.82±12.24 μg/g h, respectively. The (AUC0–12brain/AUC0–12plasma) ratio of flucytosine postinjection conventional liposomal formula (F1) was 0.14199. The observed finding was attributed to role of the BBB in decreasing the entry of conventional liposomal formula (F1) into the brain. In the brain, the values of Cmax postintravenous injection of conventional liposomal formula (F1) and flucytosine-loaded liposome (F4) were 13.61±2.75 μg/g and 21.64±4.81 μg/g, respectively, as shown in Table 7. As listed in Table 8, in the brain, the RE (RE = AUCliposomal formulation F4/AUCliposomal formulation F1) was 2.632±0.089 and the CE (CE = (Cmax)liposomal formulation F4/(Cmax)liposomal formulation F1) was 1.590±0.049. And also, the DTI was calculated as 3.670±0.824 from:
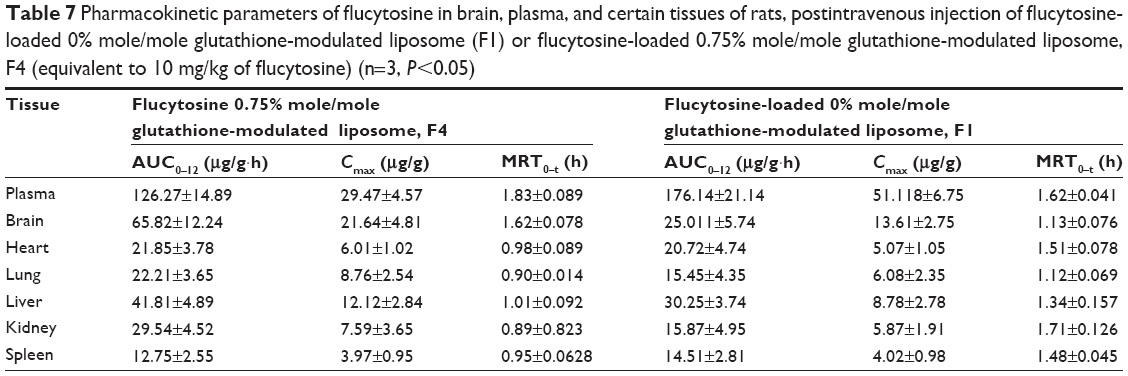 | Table 7 Pharmacokinetic parameters of flucytosine in brain, plasma, and certain tissues of rats, postintravenous injection of flucytosine-loaded 0% mole/mole glutathione-modulated liposome (F1) or flucytosine-loaded 0.75% mole/mole glutathione-modulated liposome, F4 (equivalent to 10 mg/kg of flucytosine) (n=3, P<0.05) |
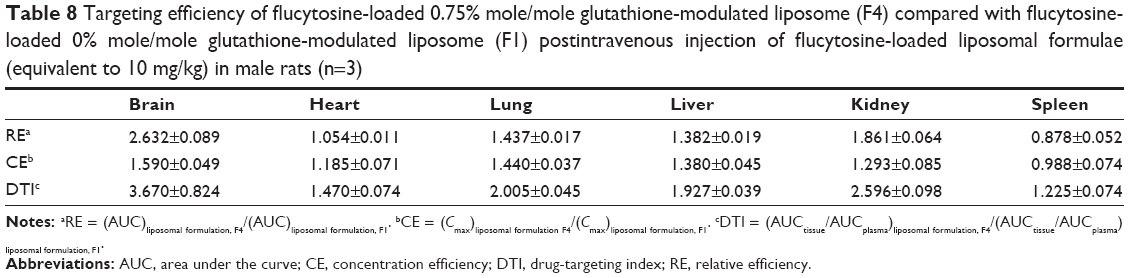 | Table 8 Targeting efficiency of flucytosine-loaded 0.75% mole/mole glutathione-modulated liposome (F4) compared with flucytosine-loaded 0% mole/mole glutathione-modulated liposome (F1) postintravenous injection of flucytosine-loaded liposomal formulae (equivalent to 10 mg/kg) in male rats (n=3) |
|

Discussion
Recently, several delivery systems have been developed to improve drug targeting to the brain.11,13 In an attempt to prepare the most suitable liposomal formula, the optimization study using phospholipids, cholesterol, and span 65 was conducted. The selected liposomal formula to be modified with glutathione moiety should be characterized with a high encapsulation efficiency, small particle size, and slow release rate. According to the recorded findings of the particle size and encapsulation efficiency, this result could be due to the surfactant chemical structure. All span types have the same head group and different alkyl chain. Increasing the alkyl chain number leads to increase entrapment efficiency.44 It is known that span 65 has three alkyl chains (stearate alkyl chain). Consequently, it may play a role in decreasing the permeability of the membrane and increases the encapsulation efficiency. In addition, span 65 has higher transition temperature (Tc). The span surfactant having the highest Tc provides the highest entrapment efficiency for the drug.69 Also the ordered gel state and higher phase transition temperature exhibited by span 65 plays a role in the observed outcomes.23,70 Incorporation of cholesterol led to increase the membrane rigidity and reduce its permeability in the liquid–crystalline state.71 The encapsulation efficiency results may suggest that glutathione is attached onto the surface of the liposomes rather than interfering with the liposomes bilayers. Being amphipathic, cholesterol can insert itself into the bilayer membrane with its hydrophilic head oriented toward the aqueous surface and aliphatic chain line up parallel to the hydrocarbon chains in the center of the bilayer. It is known that cholesterol increases the chain order of the liquid-state bilayer and strengthens the nonpolar tail of the nonionic surfactant.72,73 At low cholesterol concentration, cholesterol leads to a close packing of surfactant monomers with increasing curvature and reducing the size. Cholesterol also increases the membrane thickness and the liposome’s overall size.19,74,75 Polydispersity index value of 1 indicates that the sample has a very broad size distribution and may contain large particles or aggregates.61 The incorporation of glutathione moiety might lead to enhance the packaging of the phospholipids within the liposome bilayer and lower the PDI so it enhanced the uniformity of the system. Low particle size and low PDI are very important parameters in enhancing the binding and the uptake of the liposomes within the brain cells. These findings of current study are concurrent with those of Supaporn and Masaharu, who have found that the particle size of DMPC liposomes containing DSPE–PEG2000 decreases significantly when the DSPE–PEG2000 content increases.62 The particles seem to be colloidally stable because no formation of aggregate is detected within the investigated fields. These outcomes are concurrent with the results of the zeta potential.
According to zeta potential measurements, increasing the content of glutathione–maleimide–PEG2000–DSPE in liposomal preparations led to increasing the zeta potential values of liposomal formulae. The effect of glutathione–maleimide–PEG2000–DSPE on zeta potential could be explained taking into account the effect of both polyethylene glycol and distearoyl phosphatidylethanolamine. The zeta potential value for PEG–DSPE-containing liposomes was more negative than that of conventional liposomes. A carbamate linkage, which is typically a conjugation between PEG and DSPE is responsible for a net negative charge on the phosphate moiety at physiological pH.56 Increasing the negative values of zeta potential of the prepared liposomes (F2–F4) may be due to the presence of thiole groups of glutathione.57,58 The presence of escalated negative charges could enhance the stability of the liposomes according to what was reported in literature.59 To this end, the obtained results indicated that all liposomal preparations are colloidally stable and are concurrent with the articles reported in the literature.60,76
On the basis of hydrophilic–lipophilic balance of the surfactant which is used in the liposomal preparations, it was found that an increase of hydrophilicity of the surfactant leads to an increase of zeta potential values. Consequently, the more stable liposome was produced. These results are in agreement with what was reported in the literature.77
Glutathione–maleimide–PEG2000–DSPE moiety may play a role in reducing the release value. This may be due to the effect of the alkyl chain of the DSPE polymer in improving the packaging within the liposomal bilayers. The earlier results showed the fact that DSPE–PEG2000 decreases the hydrophilic drug diffusion.64 The hydrophobic long alkyl chains of the polymer increases the entrapment of the drug in the polymers.65
The stability study of liposomal formulations is an important factor to be considered in the development of an efficient drug delivery system. In the present study, the stability of different liposomal vesicles at different temperatures was carried out to mimic physiological conditions. The obtained findings have revealed that the prepared liposomal vesicles were physically and chemically stable at refrigerated temperature (4°C±1°C) for 3 months. At refrigerated condition, the liposomal formulations were found to be reasonably stable in terms of aggregation and fusion. No significant changes in the physical appearance, particle size, and entrapment efficiency were determined for different liposomal formulations during the stability study at refrigerated condition (P>0.05).
However, the liposomal formulations stored at 25°C±1°C and 37°C±1°C showed an increase in particle size, which may be attributed to the aggregation or swelling of vesicles. The decrease in the entrapment efficiency was reported, which shows a significant leakage of flucytosine from different liposomal formulations over time of the stability study. The decrease in encapsulation efficiency at 37°C±1°C may be explained on the basis of the fluidity of vesicular membrane. The fluidity of vesicular membrane is increased at higher temperature, resulting in a significant leakage of the loaded drug.78 Possible chemical degradation of the phospholipids at 37°C±1°C may be another reason. It leads to defects in the vesicular membrane packing.79 Those findings are in accordance with other scientists, who found that clofazimine-loaded liposomes were stable at refrigeration temperature (4°C±1°C).80 Above absolute zero temperature, colloidal particles in the bilayer of liposomes are in a dynamic state. As the temperature increases, the motion and vibration of colloidal particles increases.78
The binding and the uptake results revealed that the increase in molar percentage of glutathione moiety caused an increase in the cellular uptake of liposomes. This conclusion suggested that glutathione moiety enhanced the uptake of liposomes, especially when escalating molar percentage of glutathione was used.
The quenching of fluorescent signal that occurred due to saturation of the receptor with the excess free glutathione suggested that the binding of the glutathione-modulated nanoliposomes to glutathione receptors was specific. In contrast to that, free calcein-AM solution showed a very weak fluorescent signal within the cytoplasm, which boosts the suggestion that the binding and the uptake of glutathione-modulated liposomes was due to the presence of the targeting moiety, and not due to the hydrophobic nature of the labeling agent or the nature of the liposomes (Figure 3).
According to quantitative uptake of glutathione-targeted liposomes using flow cytometry, the cellular uptake of liposome conjugated with 0.75 mol% glutathione–maleimide–PEG2000–DSPE was threefold higher than that of conventional liposomes (F1). This finding suggested that increasing the concentration of glutathione moieties on the surface of liposomes led to the increase in the cellular uptake of liposome. This observation suggests that the uptake of the liposomes could be achieved either specifically through targeting of the liposomes with glutathione moiety or nonspecifically through the hydrophobic nature of the liposomes.
In the competition assay, the results may indicate saturation of the receptors with free glutathione, which competed for the glutathione receptor when glutathione-modulated liposomes were added.
On the basis of the represented overall data, these results indicated significant brain-targeting efficiency of glutathione-modulated liposome and a decreased plasma biodistribution. Hence, the formulation of flucytosine in glutathione-modulated liposomes led to improved brain targeting of flucytosine and a decrease in peripheral side effects. Those results are concurrent with the studies of many scientists, who found that glutathione–PEGylated liposomes may be an efficient brain drug delivery system.81–83
PEG2000 in the glutathione–maleimide–PEG2000–DSPE has important properties, which may augment the targeting efficiency of the glutathione–PEGylated liposomes. PEG polymer gave the liposome surface kinetic rather than thermodynamic protection.84,85 de Gennes has reported that the polymer linked to a surface follows one of two regimens relying on the graft density of the polymer.86 The mushroom regimen can be followed when the polymer graft density is low, while brush regimen can be followed when the polymer graft density is high. The degree of surface coverage and the distance between graft sites were governed by both the molecular weight of the polymer as well as the graft density. Bedu-Addo et al have proposed that PEG chains interact via interchain hydrogen bonding and van der Waals forces interchain hydrogen bonding leading to chain entanglement generating “PEG-rich” and “PEG-poor” domains.87 Allen et al have reported that the circulation residence time of cholesterol-containing liposomes increased depending on the mole fraction of the incorporated PEG and also on the length or molecular weight of the PEG.88 In most of the studies, it has reported that the circulation residence time of liposomes was increased because the PEG chains inhibit the interaction of the PEGylated liposomes with proteins of plasma or cell surface.89–93 Allen et al have reported that insertion of the PEG chains into liposomes leads to a decrease in the mononuclear phagocytic system uptake of the PEGylated liposomes.88 However, several studies have reported that although an inclusion of the PEG chains leads to extend the circulation time of the PEGylated liposomes, it does not affect the cumulative cellular uptake of the mononuclear phagocytic system.94–96
Another formula component that may contribute in increasing the brain concentration of the formulated drug was the surfactant. In the present study, span 65 was used in the preparation of liposome. Troster et al, Lockman et al, and Koziara et al have reported that the inclusion of certain surfactants might enhance the brain-targeting efficiency.97–101 The transmembrane proteins located in the brain are involved in active transport across the BBB. Those proteins interact with the glutathione molecule. A grafting of the liposome with glutathione could potentiate the brain-targeting efficiency through interaction with transmembrane proteins, one of them being the P-glycoprotein (Pgp). The permeation of flucytosine-loaded glutathione-modulated liposomes across the endothelial cell membrane might be carried out via adenosine triphosphate-coupled reactions with Pgp and also a number of other proteins are implicated in a similar mechanism.102,103 An elevated retention of the glutathione-modulated liposomes in the blood capillaries of the brain is due to the adsorption of the glutathione-modulated liposomes to the capillary walls. Hence, a higher concentration gradient which is created enforces the permeation across the BBB, and as a result, the delivery to the brain is achieved. Incorporated surfactant span 65 might suppress the efflux system, especially Pgp, and also it may solubilize the lipids of the endothelial cell membrane, and as a result, the fluidization of endothelial cell membrane potentiates the transport across the BBB. The nanoliposomes might cause a gap in the tight junction between the endothelial cells; consequently, the permeation of the drug-loaded liposomes or the released drug through the endothelial cell is achieved. The endocytosis of glutathione-modulated liposomes by the endothelial cells might be one of the possible mechanisms in the delivery of drug-loaded glutathione-modulated liposomes.104–106 Finally, flucytosine-loaded glutathione-modulated liposomes could actively or passively transcytose through the layer of endothelial cells.107
Conclusion
Glutathione–maleimide–PEG2000–DSPE liposomes were evaluated in the present study to determine if they meet the criteria of in vitro and in vivo targeting ability to the brain cells. Both in vitro and in vivo, the brain-targeting evaluations unambiguously showed a significantly increased delivery of flucytosine to the brain with glutathione–maleimide–PEG2000–DSPE liposomes compared with conventional liposome or free flucytosine. Glutathione-modulated liposomes could be a valuable tool for delivery of flucytosine to the brain.
Acknowledgments
The authors thank Professor Doaa Mohammed Sayed for her assistance with flow cytometer at the flow cytometry laboratory in the South Egypt Cancer Institute of the University of Assiut.
Disclosure
The authors report no conflicts of interest in this work.
References
Jellinger KA, Setinek U, Drlicek M, Böhm G, Steurer A, Lintner F. Neuropathology and general autopsy findings in AIDS during the last 15 years. Acta Neuropathol. 2000;100(2):213–220. | ||
Baddley JW, Salzman D, Pappas PG. Fungal brain abscess in transplant recipients: epidemiologic, microbiologic, and clinical features. Clin Transplant. 2002;16(6):419–424. | ||
Dismukes WE, Cloud G, Gallis HA, et al. Treatment of cryptococcal meningitis with combination amphotericin B and flucytosine for four as compared with six weeks. N Engl J Med. 1987;317(6):334–341. | ||
Fennelly AM, Slenker AK, Murphy LC, Moussouttas M, DeSimone JA. Candida cerebral abscesses: a case report and review of the literature. Med Mycol. 2013;51(7):779–784. | ||
Ohtsuki S, Terasaki T. Contribution of carrier-mediated transport systems to the blood–brain barrier as a supporting and protecting interface for the brain; importance for CNS drug discovery and development. Pharm Res. 2007;24(9):1745–1758. | ||
Mansour A, Fox CA, Burke S, Akil H, Watson SJ. Immunohistochemical localization of the cloned μ opioid receptor in the rat CNS. J Chem Neuroanat. 1995;8(4):283–305. | ||
Shibata M, Yamada S, Kumar SR, et al. Clearance of Alzheimer’s amyloid-β1-40 peptide from brain by LDL receptor–related protein-1 at the blood–brain barrier. J Clin Invest. 2000;106(12): 1489–1499. | ||
Laron Z. Insulin and the brain. Arch Physiol Biochem. 2009;115(2):112–116. | ||
Côté J, Savard M, Bovenzi V, et al. Selective tumor blood–brain barrier opening with the kinin B2 receptor agonist [Phe(8)psi(CH(2)NH)Arg(9)]-BK in a F98 glioma rat model: an MRI study. Neuropeptides. 2010;44(2):177–185. | ||
Saavedra JM. Emerging features of brain angiotensin receptors. Regul Pept. 1999;85(1):31–45. | ||
Moscato EH, Peng X, Jain A, Parsons TD, Dalmau J, Balice-Gordon RJ. Acute mechanisms underlying antibody effects in anti–N-methyl-D-aspartate receptor encephalitis. Ann Neurol. 2014;76(1):108–119. | ||
Lanius RA, Shaw CA, Wagey R, Krieger C. Characterization, distribution, and protein kinase C-mediated regulation of [35S] glutathione binding sites in mouse and human spinal cord. J Neurochem. 1994;63(1):155–160. | ||
Geldenhuys W, Wehrung D, Groshev A, Hirani A, Sutariya V. Brain-targeted delivery of doxorubicin using glutathione-coated nanoparticles for brain cancers. Pharm Dev Technol. 2015;20(4):497–506. | ||
Wang X, Li J, Xu C, et al. Scopine as a novel brain-targeting moiety enhances the brain uptake of chlorambucil. Bioconjug Chem. 2014; 25(11):2046–2054. | ||
Gupta B, Levchenko TS, Mongayt DA, Torchilin VP. Monoclonal antibody 2C5-mediated binding of liposomes to brain tumor cells in vitro and in subcutaneous tumor model in vivo. J Drug Target. 2005;13(6):337–343. | ||
Ulbrich K, Hekmatara T, Herbert E, Kreuter J. Transferrin-and transferrin-receptor-antibody-modified nanoparticles enable drug delivery across the blood–brain barrier (BBB). Eur J Pharm Biopharm. 2009;71(2):251–256. | ||
Lee DH, Rötger C, Appeldoorn CC, et al. Glutathione PEGylated liposomal methylprednisolone (2B3-201) attenuates CNS inflammation and degeneration in murine myelin oligodendrocyte glycoprotein induced experimental autoimmune encephalomyelitis. J Neuroimmunol. 2014;274(1–2):96–101. | ||
Chang J, Jallouli Y, Kroubi M, et al. Characterization of endocytosis of transferrin-coated PLGA nanoparticles by the blood–brain barrier. Int J Pharm. 2009;379(2):285–292. | ||
Guo N, McIntosh C, Shaw C. Glutathione: new candidate neuropeptide in the central nervous system. Neuroscience. 1992;51(4):835–842. | ||
Kannan R, Kuhlenkamp JF, Jeandidier E, Trinh H, Ookhtens M, Kaplowitz N. Evidence for carrier-mediated transport of glutathione across the blood–brain barrier in the rat. J Clin Invest. 1990;85(6): 2009–2013. | ||
Béduneau A, Saulnier P, Benoit JP. Active targeting of brain tumors using nanocarriers. Biomaterials. 2007;28(33):4947–4967. | ||
Kang YS, Pardridge WM. Brain delivery of biotin bound to a conjugate of neutral avidin and cationized human albumin. Pharm Res. 1994; 11(9):1257–1264. | ||
Cheng R, Feng F, Meng F, Deng C, Feijen J, Zhong Z. Glutathione-responsive nano-vehicles as a promising platform for targeted intracellular drug and gene delivery. J Control Release. 2011;152(1): 2–12. | ||
Schaeffer HE, Krohn DL. Liposomes in topical drug delivery. Invest Ophthalmol Vis Sci. 1982;22(2):220–227. | ||
Torchilin VP. Liposomes as targetable drug carriers. Crit Rev Ther Drug Carrier Syst. 1984;2(1):65–115. | ||
Immordino ML, Dosio F, Cattel L. Stealth liposomes: review of the basic science, rationale, and clinical applications, existing and potential. Int J Nanomedicine. 2006;1(3):297. | ||
Turk MJ, Waters DJ, Low PS. Folate-conjugated liposomes preferentially target macrophages associated with ovarian carcinoma. Cancer Lett. 2004;213(2):165–172. | ||
Maruyama K. Intracellular targeting delivery of liposomal drugs to solid tumors based on EPR effects. Adv Drug Deliv Rev. 2011;63(3):161–169. | ||
Pornpattananangkul D, Zhang L, Olson S, et al. Bacterial toxin-triggered drug release from gold nanoparticle-stabilized liposomes for the treatment of bacterial infection. J Am Chem Soc. 2011;133(11):4132–4139. | ||
Pandit J, Garg M, Jain NK. Miconazole nitrate bearing ultraflexible liposomes for the treatment of fungal infection. J Liposome Res. 2014;24(2):163–172. | ||
Hirakawa Y, Kijima H. Behavioral analysis of glutathione receptor of Hydra. J Comp Physiol. 1980;137(1):73–81. | ||
Oja SS, Janáky R, Varga V, Saransaari P. Modulation of glutamate receptor functions by glutathione. Neurochem Int. 2000;37(2–3): 299–306. | ||
Ogita K, Yoneda Y. Possible presence of [3H] glutathione (GSH) binding sites in synaptic membranes from rat brain. Neurosci Res. 1987;4(6): 486–496. | ||
Ogita K, Yoneda Y. Temperature-dependent and -independent apparent binding activities of [3H] glutathione in brain synaptic membranes. Brain Res. 1988;463(1):37–46. | ||
Reeves JP, Dowben RM. Formation and properties of thin-walled phospholipid vesicles . J Cell Physiol. 1969;73(1):49–60. | ||
Szoka F Jr, Papahadjopoulos D. Comparative properties and methods of preparation of lipid vesicles (liposomes). Annu Rev Biophys Bioeng. 1980;9(1):467–508. | ||
Elhissi AM, O’Neill MA, Roberts SA, Taylor KM. A calorimetric study of dimyristoylphosphatidylcholine phase transitions and steroid–liposome interactions for liposomes prepared by thin film and proliposome methods. Int J Pharm. 2006;320(1):124–130. | ||
Guo J, Ping Q, Jiang G, Huang L, Tong Y. Chitosan-coated liposomes: characterization and interaction with leuprolide. Int J Pharm. 2003; 260(2):167–173. | ||
Egelhaaf SU, Wehrli E, Adrian M, Schurtenberger P. Determination of the size distribution of lecithin liposomes: a comparative study using freeze fracture, cryoelectron microscopy and dynamic light scattering. J Microsc. 1996;184(3):214–228. | ||
Matsuzaki K, Murase O, Sugishita K, et al. Optical characterization of liposomes by right angle light scattering and turbidity measurement. Biochim Biophys Acta. 2000;1467(1):219–226. | ||
Matos C, de Castro B, Gameiro P, Lima JL, Reis S. Zeta-potential measurements as a tool to quantify the effect of charged drugs on the surface potential of egg phosphatidylcholine liposomes. Langmuir. 2004;20(2):369–377. | ||
Anabousi S, Laue M, Lehr CM, Bakowsky U, Ehrhardt C. Assessing transferrin modification of liposomes by atomic force microscopy and transmission electron microscopy. Eur J Pharm Biopharm. 2005; 60(2):295–303. | ||
Kodama T, Tomita N, Horie S, et al. Morphological study of acoustic liposomes using transmission electron microscopy. J Electron Microsc (Tokyo). 2010;59(3):187–196. | ||
Hao Y, Zhao F, Li N, Yang Y, Li K. Studies on a high encapsulation of colchicine by a niosome system. Int J Pharm. 2002;244(1):73–80. | ||
Du Plessis J, Ramachandran C, Weiner N, Müller DG. The influence of lipid composition and lamellarity of liposomes on the physical stability of liposomes upon storage. Int J Pharm. 1996;127(2):273–278. | ||
Watson PM, Paterson JC, Thom G, Ginman U, Lundquist S, Webster CI. Modelling the endothelial blood-CNS barriers: a method for the production of robust in vitro models of the rat blood–brain barrier and blood-spinal cord barrier. BMC Neurosci. 2013;14(1):59. | ||
Adrian JE, Poelstra K, Scherphof GL, et al. Interaction of targeted liposomes with primary cultured hepatic stellate cells: involvement of multiple receptor systems. J Hepatol. 2006;44(3):560–567. | ||
Müller K, Nahde T, Fahr A, Müller R, Brüsselbach S. Highly efficient transduction of endothelial cells by targeted artificial virus-like particles. Cancer Gene Ther. 2001;8(2):107–117. | ||
De Mello TR, Busfield S, Dunlop SA, Plant GW. Culture conditions affect proliferative responsiveness of olfactory ensheathing glia to neuregulins. Glia. 2007;55(7):734–745. | ||
Moreno-Flores MT, Lim F, Martín-Bermejo MJ, Díaz-Nido J, Avila J, Wandosell F. Immortalized olfactory ensheathing glia promote axonal regeneration of rat retinal ganglion neurons. J Neurochem. 2003;85(4):861–871. | ||
Ramón-Cueto A, Nieto-Sampedro M. Glial cells from adult rat olfactory bulb: immunocytochemical properties of pure cultures of ensheathing cells. Neuroscience. 1992;47(1):213–220. | ||
Perillo E, Allard-Vannier E, Falanga A, et al. Quantitative and qualitative effect of gH625 on the nanoliposome-mediated delivery of mitoxantrone anticancer drug to HeLa cells. Int J Pharm. 2015;488(1–2):59–66. | ||
Huth US, Schubert R, Peschka-Süss R. Investigating the uptake and intracellular fate of pH-sensitive liposomes by flow cytometry and spectral bio-imaging. J Control Release. 2006;110(3):490–504. | ||
Chen WC, Completo GC, Sigal DS, Crocker PR, Saven A, Paulson JC. In vivo targeting of B-cell lymphoma with glycan ligands of CD22. Blood. 2010;115(23):4778–4786. | ||
Zhang X, Liu X, Gong T, Sun X, Zhang ZR. In vitro and in vivo investigation of dexibuprofen derivatives for CNS delivery. Acta Pharmacol Sin. 2012;33(2):279–288. | ||
Garbuzenko O, Zalipsky S, Qazen M, Barenholz Y. Electrostatics of PEGylated micelles and liposomes containing charged and neutral lipopolymers. Langmuir. 2005;21(6):2560–2568. | ||
Webb MS, Saxon D, Wong FM, et al. Comparison of different hydrophobic anchors conjugated to poly(ethylene glycol): effects on the pharmacokinetics of liposomal vincristine. Biochim Biophys Acta. 1998;1372(2):272–282. | ||
Kommareddy S, Amiji M. Preparation and evaluation of thiol-modified gelatin nanoparticles for intracellular DNA delivery in response to glutathione. Bioconjug Chem. 2005;16(6):1423–1432. | ||
Harris JM, Martin NE, Modi M. Pegylation. Clin Pharmacokinet. 2001;40(7):539–551. | ||
Xu X, Yu Z, Zhu Y, Wang B. Effect of sodium oleate adsorption on the colloidal stability and zeta potential of detonation synthesized diamond particles in aqueous solutions. Diam Relat Mater. 2005;14(2):206–212. | ||
Roberts MA, Locascio-Brown L, MacCrehan WA, Durst RA. Liposome behavior in capillary electrophoresis. Anal Chem. 1996;68(19): 3434–3440. | ||
Sriwongsitanont S, Ueno M. Physicochemical properties of PEG-grafted liposomes. Chem Pharm Bull (Tokyo). 2002;50(9):1238–1244. | ||
Liu D, Huang L. Role of cholesterol in the stability of pH-sensitive, large unilamellar liposomes prepared by the detergent-dialysis method. Biochim Biophys Acta. 1989;981(2):254–260. | ||
Xiao RZ, Zeng ZW, Zhou GL, Wang JJ, Li FZ, Wang AM. Recent advances in PEG-PLA block copolymer nanoparticles. Int J Nanomedicine. 2010;5:1057–1065. | ||
Panwar P, Pandey B, Lakhera PC, Singh KP. Preparation, characterization, and in vitro release study of albendazole-encapsulated nanosize liposomes. Int J Nanomedicine. 2010;5:101–108. | ||
Costantino L, Boraschi D. Is there a clinical future for polymeric nanoparticles as brain-targeting drug delivery agents? Drug Discov Today. 2012;17(7):367–378. | ||
Li F, Wang J, Ma C, et al. Glutamate receptor-like channel 3. 3 is involved in mediating glutathione-triggered cytosolic calcium transients, transcriptional changes, and innate immunity responses in Arabidopsis. Plant Physiol. 2013;162(3):1497–1509. | ||
Torchilin VP. Fluorescence microscopy to follow the targeting of liposomes and micelles to cells and their intracellular fate. Adv Drug Deliv Rev. 2005;57(1):95–109. | ||
Schreier H, Bouwstra J. Liposomes and niosomes as topical drug carriers: dermal and transdermal drug delivery. J Control Release. 1994; 30(1):1–15. | ||
Attia IA, El-Gizawy SA, Fouda MA, Donia AM. Influence of a niosomal formulation on the oral bioavailability of acyclovir in rabbits. AAPS Pharm Sci Tech. 2007;8(4):206–212. | ||
New R.R.C. In: New R.R.C, editor. Liposomes a Practical Approach. New York: Oxford University Press; 1990:1–32. | ||
Moriyama E, Saito T, Tokuoka Y, Takeuchi S, Kawashima N. Evaluation of the hardness of lipid bilayer membranes of liposomes by the ultrasound attenuation method. J Oleo Sci. 2003;52(8):433–437. | ||
Lee S-C, Lee K-E, Kim J-J, Lim S-H. The effect of cholesterol in the liposome bilayer on the stabilization of incorporated retinol. J Liposome Res. 2005;15(3–4):157–166. | ||
Fang J-Y, Hong C-T, Chiu W-T, Wang Y-Y. Effect of liposomes and niosomes on skin permeation of enoxacin. Int J Pharm. 2001; 219(1):61–72. | ||
Uchegbu IF, Bouwstra JA, Florence AT. Large disk-shaped structures (discomes) in nonionic surfactant vesicle to micelle transitions. J Phys Chem. 1992;96(25):10548–10553. | ||
Kronberg B, Dahlman A, Carlfors J, Karlsson J and Artursson P. Preparation and evaluation of sterically stabilized liposomes: Colloidal stability, serum stability, macrophage uptake, and toxicity. J Pharm Sci. 2006;79(8):667–671. | ||
Kato A, Arakawa M, Kondo T. Preparation and stability of liposome-type artificial red blood cells stabilized with carboxymethylchitin. J Microencapsul. 1984;1(2):105–112. | ||
Park SH, Oh SG, Mun JY, Han SS. Loading of gold nanoparticles inside the DPPC bilayers of liposome and their effects on membrane fluidities. Colloids Surf B Biointerfaces. 2006;48(2):112–118. | ||
Hernández-Caselles T, Villalain J, Gómez-Fernández JC. Stability of liposomes on long term storage. J Pharm Pharmacol. 1990;42(6):397–400. | ||
Patel VB, Misra AN. Encapsulation and stability of clofazimine liposomes. J Microencapsul. 1999;16(3):357–367. | ||
Rotman M, Welling MM, Bunschoten A, et al. Enhanced glutathione PEGylated liposomal brain delivery of an anti-amyloid single domain antibody fragment in a mouse model for Alzheimer’s disease. J Control Release. 2015;203(1):40–50. | ||
Rip J, Chen L, Hartman R, et al. Glutathione PEGylated liposomes: pharmacokinetics and delivery of cargo across the blood–brain barrier in rats. J Drug Target. 2014;22(5):460–467. | ||
Birngruber T, Raml R, Gladdines W, et al. Enhanced doxorubicin delivery to the brain administered through glutathione PEGylated liposomal doxorubicin (2B3-101) as compared with generic Caelyx,(®)/Doxil(®) – a cerebral open flow microperfusion pilot study. J Pharm Sci. 2014;103:1945–1948. | ||
Carignano MA, Szleifer I. Prevention of protein adsorption by flexible and rigid chain molecules. Colloids Surf B Biointerfaces. 2000; 18(3):169–182. | ||
Satulovsky J, MA C, Szleifer I. Kinetic and thermodynamic control of protein adsorption. Proc Natl Acad Sci U S A. 2000;97(16):9037–9041. | ||
de Gennes PG. Conformations of polymers attached to an interface. Macromolecules. 1980;13(5):1069–1075. | ||
Bedu-Addo FK, Tang P, Xu Y, Huang L. Effects of polyethyleneglycol chain length and phospholipid acyl chain composition on the interaction of polyethyleneglycol-phospholipid conjugates with phospholipid: implications in liposomal drug delivery. Pharm Res. 1996;13(5):710–717. | ||
Allen TM, Hansen C, Martin F, Redemann C, Yau-Young A. Liposomes containing synthetic lipid derivatives of poly (ethylene glycol) show prolonged circulation half-lives in vivo. Biochim Biophys Acta. 1991;1066(1):29–36. | ||
Blume G, Cevc G. Molecular mechanism of the lipid vesicle longevity in vivo. Biochim Biophys Acta. 1993;1146(2):157–168. | ||
Bazile D, Prud’homme C, Bassoullet MT, Marlard M, Spenlehauer G, Veillard M. Stealth Me. PEG-PLA nanoparticles avoid uptake by the mononuclear phagocytes system. J Pharm Sci. 1995;84(4):493–498. | ||
Peracchia MT, Fattal E, Desmaële D, et al. Stealth PEGylated polycyanoacrylate nanoparticles for intravenous administration and splenic targeting. J Control Release. 1999;60(1):121–128. | ||
Calvo P, Gouritin B, Chacun H, et al. Long-circulating PEGylated polycyanoacrylate nanoparticles as new drug cariers for brain delivery. Pharm Res. 2001;18(8):1157–1166. | ||
Brigger I, Morizet J, Aubert G, et al. Poly(ethylene glycol)-coated hexadecylcyanoacrylate nanospheres display a combined effect for brain tumor targeting. J Pharmacol Exp Ther. 2002;303(3):928–936. | ||
Lasic DD, Martin F, Gabizon A, Huang SK, Papahadjopoulos D. Sterically stabilized liposomes: a hypothesis on the molecular origin of the extended circulation times. Biochim Biophys Acta. 1991;1070(1): 187–192. | ||
Patel HM. Serum opsonins and liposomes: their interaction and opsonophagocytosis. Crit Rev Ther Drug Carrier Syst. 1991;9(1): 39–90. | ||
Mori A, Klibanov AL, Torchilin VP, Huang L. Influence of the steric barrier activity of amphipathic poly(ethyleneglycol) and ganglioside GM 1 on the circulation time of liposomes and on the target binding of immunoliposomes in vivo. FEBS Lett. 1991;284(2):263–266. | ||
Tröster SD, Müller U, Kreuter J. Modification of the body distribution of poly(methyl methacrylate) nanoparticles in rats by coating with surfactants. Int J Pharm. 1990;61(1–2):85–100. | ||
Lockman PR, Koziara J, Roder KE, et al. In vivo and in vitro assessment of baseline blood–brain barrier parameters in the presence of novel nanoparticles. Pharm Res. 2003;20(5):705–713. | ||
Lockman PR, Oyewumi MO, Koziara JM, Roder KE, Mumper RJ, Allen DD. Brain uptake of thiamine-coated nanoparticles. J Control Release. 2003;93(3):271–282. | ||
Koziara JM, Lockman PR, Allen DD, Mumper RJ. In situ blood–brain barrier transport of nanoparticles. Pharm Res. 2003;20(11): 1772–1778. | ||
Koziara JM, Lockman PR, Allen DD, Mumper RJ. Paclitaxel nanoparticles for the potential treatment of brain tumors. J Control Release. 2004;99(2):259–269. | ||
Smeyne M, Smeyne RJ. Glutathione metabolism and Parkinson’s disease. Free Radic Biol Med. 2013;62(1):13–25. | ||
Bellarosa C, Bortolussi G, Tiribelli C. The role of ABC transporters in protecting cells from bilirubin toxicity. Curr Pharm Des. 2009; 15(25):2884–2892. | ||
Geldenhuys W, Mbimba T, Bui T, Harrison K, Sutariya V. Brain-targeted delivery of paclitaxel using glutathione-coated nanoparticles for brain cancers. J Drug Target. 2011;19(9):837–845. | ||
Kreuter J. Nanoparticulate systems for brain delivery of drugs. Adv Drug Deliv Rev. 2001;47(1):65–81. | ||
Kreuter J. Transport of drugs across the blood–brain barrier by nanoparticles. Cent Nerv Syst Agents Med Chem. 2002;2(3):241–249. | ||
Kreuter J, Shamenkov D, Petrov V, et al. Apolipoprotein-mediated transport of nanoparticle-bound drugs across the blood–brain barrier. J Drug Target. 2002;10(4):317–325. |
Supplementary materials
Preparation of liposome samples
In attempt to prepare the most suitable liposomal formula, the optimization study was conducted. The selected liposomal formula to be modified with glutathione moiety should be characterized with a high encapsulation efficiency, small particle size, and slow release rate.
Liposomal formulae (S1–S9) were prepared using a thin-film hydrating technique as explained in main text. In this method, different ratios of phosphatidylcholine were used in formulating the different liposomal formulae. Span 65 and span 80 were also used in the optimization study as shown in Table S1. All the steps were performed under aseptic conditions. All glassware was sterilized by heating in a hot air oven over 120°C for 2 hours. Boiled double-distilled water was passed through a 0.22 μm disposable syringe filter (bacterial filter), and the entire procedure was performed in a laminar air flow hood in the presence of flame. Flucytosine liposomes were prepared using the thin-film hydration method. The prepared liposomes were in vitro characterized to evaluate their entrapment efficiency, particle size, zeta potential charge, and in vitro release as mentioned in the main text.
 | Table S1 Molar concentration of lipids and surfactants in the prepared liposomal formulations |
Results and discussion
Entrapment efficiency and particle size
From Table S2, it can be concluded that flucytosine has been successfully incorporated into liposomes. Encapsulation efficiency was ranging from 32.41%±2.41% to 76.85%±3.05%.
 | Table S2 Entrapment efficiency values of flucytosine in flucytosine-loaded liposomes formulae (means ± SD [n=3]) |
The effect of the cholesterol concentration and surfactant concentration was studied. From the results listed in Table S2, it could be concluded that an increase of cholesterol molar ratio led to an increase in the encapsulation efficiency, as shown with formulae S1–S3. This might be due to the fact that an increase of cholesterol concentration leads to a decrease in the membrane permeability and promotes the encapsulation efficiency. These results are in agreement with those reported in literature.1 It was observed that an inclusion of nonionic surfactant led to an increase in the entrapment efficiency and decrease in particle size, as shown with formulae S4–S9. But the effect of span 65 on decreasing the particle size or increasing the entrapment efficiency is more obvious than that of span 80.
This result could be due to the surfactant chemical structure. All span types have the same head group and different alkyl chain. Increasing the alkyl chain number leads to increased entrapment efficiency.2,3 It is observed that span 65 has three alkyl chains (three stearate alkyl chains) while span 80 has one (oleate alkyl chain). Consequently, span 65 decreased the permeability of the membrane and increased the encapsulation efficiency. In addition, span 65 has higher transition temperature (Tc) than span 80. The span having the highest Tc provides the highest entrapment efficiency for the drug.4 Also the ordered gel state and higher phase transition temperature exhibited by span 65 plays a role in the observed outcomes.5,6
Particle size of the liposomal formulae with polydispersity index was reported in Table S2. It could be noticed that an increase of cholesterol concentration led to an increase in the particle size of liposomes as is obvious with the formulae S1–S3.
This finding could be attributed to fact that incorporation of cholesterol led to an increase in the membrane rigidity and reduced its permeability in the liquid–crystalline state.7
Being amphipathic, cholesterol can insert itself into the bilayer membrane with its hydrophilic head oriented toward the aqueous surface and aliphatic chain line up parallel to the hydrocarbon chains in the center of the bilayer. It is known that cholesterol increases the chain order of the liquid-state bilayer and strengthens the nonpolar tail of the nonionic surfactant.8,9
At low cholesterol concentration, cholesterol leads to a close packing of surfactant monomers with increasing curvature and reducing size. Cholesterol also increases the membrane thickness and the liposome’s overall size.10–12
Zeta potential measurements
Nanoparticles with zeta potential values greater than +25 mV or less than −25 mV typically have high degrees of stability.
From Table S3, it can be noticed that the zeta potential values of liposomal preparations ranged from −25.7±1.02 to −59.4±2.13 mV. These results indicate that the liposomal formulae are stable according to what was reported in literatures.13,14
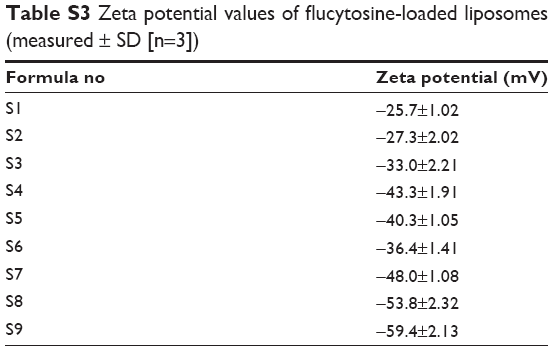 | Table S3 Zeta potential values of flucytosine-loaded liposomes (measured ± SD [n=3]) |
It could be noticed that an increase of cholesterol molar ratio led to an increase in the zeta potential of liposomal preparations, as shown with formulae S1–S3. The absolute zeta potential and the electrostatic repulsion between phosphatidylcholine liposomes increased as a result of an increase in the cholesterol molar ratio, thereby enhancing their stability.15–18 It could be noticed that an increase of surfactants concentration led to a different effect on liposomes zeta potential values. As increase of span 65 concentration led to a decrease in the negative charge of zeta potential values as shown with formulae S4–S6, whereas an increase of span 80 concentration led to an increase in negative charge of zeta potential values as shown with formulae S7–S9. This can be explained on the basis of hydrophilic–lipophilic balance (HLB) of the surfactant, which is used in liposomal preparations. It was found that an increase of hydrophilicity of the surfactant led to an increase of zeta potential values. Consequently, the effect of span 80 (HLB 4.3) on increasing the negative charge of zeta potential is an opposite to that of span 65 (HLB 2.1). These results are in agreement with what is reported in the literature.19
In vitro release study of flucytosine from flucytosine-loaded liposomes
It was found that, in vitro release of free flucytosine was the fastest as shown in Figure S1. As cholesterol and surfactant concentrations were increased, the release rate of drug decreased as is obvious with formulae S1–S9. Liposomal formula (S6) had the lowest drug release (77.49%±2.52%) while the fastest one was achieved by formula S1 (96.1%±2.45%) after 12 hours.
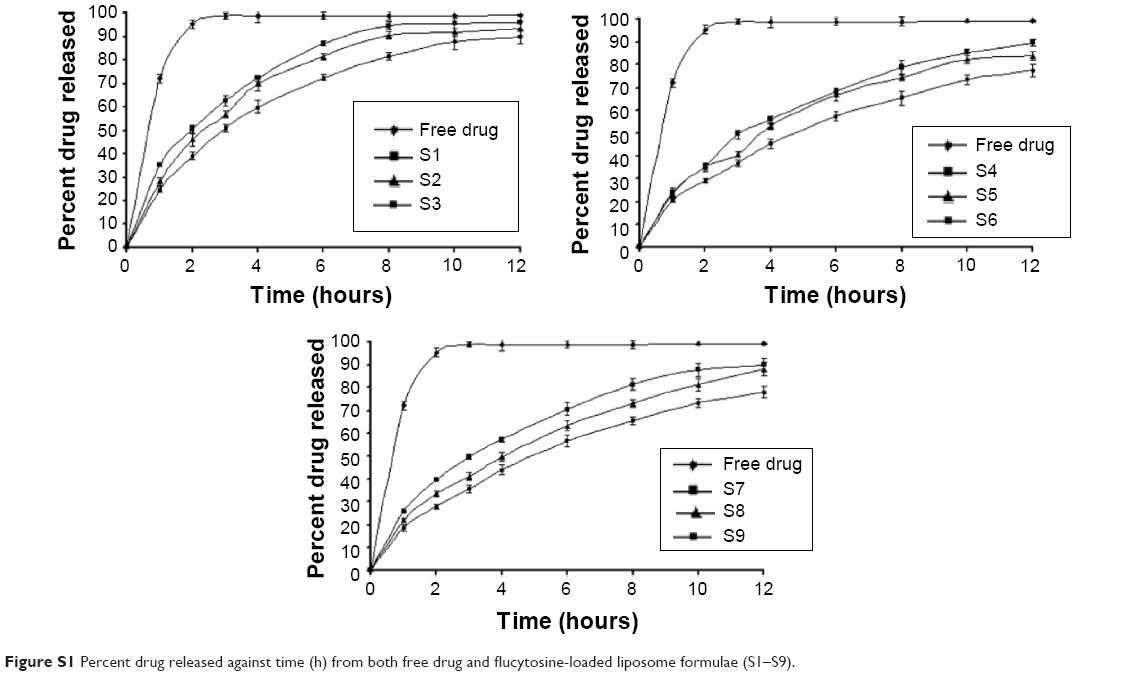 | Figure S1 Percent drug released against time (h) from both free drug and flucytosine-loaded liposome formulae (S1–S9). |
The current investigation showed that an increase of cholesterol concentration led to a decrease in the drug release from liposomal formulae. These findings could be attributed to an increase of lipophilicity and delayed release of the drug from liposome. These results are in agreement with what is reported in the literature.20 It was also noticed that surfactant concentrations and types have an effect on flucytosine release from flucytosine-loaded liposome. In general, addition of both span 65 and span 80 led to a decrease in the flucytosine release rate. This could be attributed to increased stability of vesicle membranes.21 The effect of increasing the concentration of span 65 was more obvious than that of span 80.
This could be explained based on the fact that they have strong hydrophilic moieties to form micelles in aqueous solutions. A mixture of cholesterol and span 65 or span 80 gave a high hydrophobic moiety as well as high hydrophilic moiety. This system was excellent to retard the release of flucytosine from flucytosine-loaded liposomes.22
Amount and type of surfactant
The type of surfactant is very important. The ester type surfactant is safer than the ether type. The safety of the ester type could be attributed to a degradation of these to fatty acids and triglycerides in vivo.23 An increase in the HLB of the surfactants such as span 65 (Sorbitan tristearate, HLB 2.1) compared to span 80 (Sorbitan monooleate, HLB 4.3) leads to an increase in the mean size of liposomes because an increase in surfactants’ hydrophobicity leads to a decrease in the surface free energy.24 The mean size of liposomes increases proportionally with an increase in the HLB of the surfactants such as span 65 (Sorbitan tristearate, HLB 2.1) compared to span 80 (Sorbitan monooleate, HLB 4.3) because the surface free energy decreases with an increase in hydrophobicity of surfactants.24 Depending on the temperature, the type of lipid or surfactant, and the presence of other components such as cholesterol, the vesicle bilayers are either in the so-called liquid or in the gel state. Consequently, alkyl chains of liposomes are well ordered in the gel state but they are more disordered in the liquid state. The gel–liquid phase transition temperature (TC) is considered an important physical characteristic for both the surfactants and lipids. The entrapment efficiency of the liposomes is affected by the gel–liquid phase transition temperature (TC) of the surfactant, ie, span 65 having higher TC provides better entrapment efficiency.
Cholesterol content
Inclusion of cholesterol in the liposomes increases its hydrodynamic diameter and entrapment efficiency.
The particle size and the entrapment efficiency of liposomes are increased as a result of inclusion of cholesterol. As cholesterol concentration is increased, the rigidity of the resulting bilayers of liposomes is increased. Consequently, the permeability of liposome membrane is decreased.24,25 A decrease in the release rate of encapsulated drug is determined. Hence, greater drug retention results.26
References
Frisken BJ, Asman C, Patty PJ. Studies of vesicle extrusion. Langmuir. 2000;16(3):928–933. | ||
Gillet A, Grammenos A, Compère P, Evrard B, Piel G. Development of a new topical system: drug-in-cyclodextrin-in-deformable liposome. Int J Pharm. 2009;380(1):174–180. | ||
Hao Y, Zhao F, Li N, Yang Y, Li K. Studies on a high encapsulation of colchicine by a niosome system. Int J Pharm. 2002;244(1):73–80. | ||
Schreier H, Bouwstra J. Liposomes and niosomes as topical drug carriers: dermal and transdermal drug delivery. J Control Release. 1994; 30(1):1–15. | ||
Attia IA, El-Gizawy SA, Fouda MA, Donia AM. Influence of a niosomal formulation on the oral bioavailability of acyclovir in rabbits. AAPS Pharm Sci Tech. 2007;8(4):206–212. | ||
Cheng R, Feng F, Meng F, Deng C, Feijen J, Zhong Z. Glutathione-responsive nano-vehicles as a promising platform for targeted intracellular drug and gene delivery. J Control Release. 2011;152(1):2–12. | ||
New R.R.C. In: New R.R.C, editors. Liposomes a Practical Approach. New York: Oxford University Press; 1990:1–32. | ||
Moriyama E, Saito T, Tokuoka Y, Takeuchi S, Kawashima N. Evaluation of the hardness of lipid bilayer membranes of liposomes by the ultrasound attenuation method. J Oleo Sci. 2003;52(8):433–437. | ||
Lee S-C, Lee K-E, Kim J-J, Lim S-H. The effect of cholesterol in the liposome bilayer on the stabilization of incorporated retinol. J Liposome Res. 2005;15(3–4):157–166. | ||
Fang J-Y, Hong C-T, Chiu W-T, Wang Y-Y. Effect of liposomes and niosomes on skin permeation of enoxacin. Int J Pharm. 2001; 219(1):61–72. | ||
Uchegbu IF, Bouwstra JA, Florence AT. Large disk-shaped structures (discomes) in nonionic surfactant vesicle to micelle transitions. J Phys Chem. 1992;96(25):10548–10553. | ||
Guo N, McIntosh C, Shaw C. Glutathione: new candidate neuropeptide in the central nervous system. Neuroscience. 1992;51(4):835–842. | ||
Xu X, Yu Z, Zhu Y, Wang B. Effect of sodium oleate adsorption on the colloidal stability and zeta potential of detonation synthesized diamond particles in aqueous solutions. Diam Relat Mater. 2005;14(2):206–212. | ||
Riddick TM. Control of colloid stability through zeta potential. Blood. 1968;10:1. | ||
Radko SP, Stastna M, Chrambach A. Capillary zone electrophoresis of sub-microm-sized particles in electrolyte solutions of various ionic strengths: size-dependent electrophoretic migration and separation efficiency. Electrophoresis. 2000;21(17):3583–3592. | ||
Liu D, Huang L. Role of cholesterol in the stability of pH-sensitive, large unilamellar liposomes prepared by the detergent-dialysis method. Biochim Biophys Acta. 1989;981(2):254–260. | ||
Vargha-Butler EI, Hurst EL. Study of liposomal drug delivery systems 1. Surface characterization of steroid loaded MLV liposomes. Colloids Surf B Biointerfaces. 1995;3(5):287–295. | ||
Ohvo-Rekilä H, Ramstedt B, Leppimäki P, Peter Slotte J. Cholesterol interactions with phospholipids in membranes. Prog Lipid Res. 2002;41(1):66–97. | ||
Kato A, Arakawa M, Kondo T. Preparation and stability of liposome-type artificial red blood cells stabilized with carboxymethylchitin. J Microencapsul. 1984;1(2):105–112. | ||
Jafari MR, Jones AB, Hikal AH, Williamson JS, Wyandt CM. Characterization of drug release from liposomal formulations in ocular fluid. Drug Deliv. 1998;5(4):227–238. | ||
Saesoo S, Sramala I, Soottitantawat A, Charinpanitkul T, Ruktanonchai UR. Enhanced stability and in vitro bioactivity of surfactant-loaded liposomes containing Asiatic pennywort extract. J Microencapsul. 2010;27(5):436–446. | ||
Carafa M, Marianecci C, Rinaldi F, Santucci E, Tampucci S, Monti D. Characterization of vesicles prepared with various non-ionic surfactants mixed with cholesterol. Colloids Surf B Biointerfaces. 2003;30(1):129–138. | ||
Hunter CA, Dolan TF, Coombs GH, Baillie AJ. Vesicular systems (niosomes and liposomes) for delivery of sodium stibogluconate in experimental murine visceral leishmaniasis. J Pharm Pharmacol. 1988;40(3):161–165. | ||
Yoshioka T, Sternberg B, Florence AT. Preparation and properties of vesicles (niosomes) of sorbitan monoesters (Span 20, 40, 60 and 80) and a sorbitan triester (Span 85). Int J Pharm. 1994;105(1):1–6. | ||
Du Plessis J, Ramachandran C, Weiner N, Müller DG. The influence of lipid composition and lamellarity of liposomes on the physical stability of liposomes upon storage. Int J Pharm. 1996;127(2):273–278. | ||
Perugini P, Pavanetto F. Liposomes containing boronophenylalanine for boron neutron capture therapy. J Microencapsul. 1998;15(4):473–483. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.