Back to Journals » International Journal of Nanomedicine » Volume 12
Synthetic high-density lipoprotein nanodisks for targeted withalongolide delivery to adrenocortical carcinoma
Authors Kuai R, Subramanian C, White PT, Timmermann BN, Moon JJ, Cohen MS, Schwendeman A ![]()
Received 27 April 2017
Accepted for publication 3 August 2017
Published 6 September 2017 Volume 2017:12 Pages 6581—6594
DOI https://doi.org/10.2147/IJN.S140591
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Thomas Webster
Rui Kuai,1,2,* Chitra Subramanian,3,* Peter T White,3,* Barbara N Timmermann,4 James J Moon,1,2,5 Mark S Cohen,3,6 Anna Schwendeman1,2
1Department of Pharmaceutical Sciences, College of Pharmacy, 2Biointerfaces Institute, University of Michigan, 3Department of Surgery, University of Michigan, Ann Arbor, MI, 4Department of Medicinal Chemistry, University of Kansas, Lawrence, KS, 5Department of Biomedical Engineering, 6Department of Pharmacology, University of Michigan, Ann Arbor, MI, USA
*These authors contributed equally to this work
Abstract: Adrenocortical carcinoma (ACC) is a rare endocrine malignancy and has a 5-year survival rate of <35%. ACC cells require cholesterol for steroid hormone production, and this requirement is met via expression on the cell surface of a high level of SRB1, responsible for the uptake of high-density lipoproteins (HDLs), which carry and transport cholesterol in vivo. Here, we describe how this natural lipid carrier function of SRB1 can be utilized to improve the tumor-targeted delivery of a novel natural product derivative – withalongolide A 4,19,27-triacetate (WGA-TA) – which has shown potent antitumor efficacy, but poor aqueous solubility. Our strategy was to use synthetic HDL (sHDL) nanodisks, which are effective in tumor-targeted delivery due to their smallness, long circulation half-life, documented safety, and ability to bind to SRB1. In this study, we prepared sHDL nanodisks using an optimized phospholipid composition combined with ApoA1 mimetic peptide (22A), which has previously been tested in clinical trials, to load WGA-TA. Following optimization, WGA-TA nanodisks showed drug encapsulation efficiency of 78%, a narrow particle size distribution (9.81±0.41 nm), discoid shape, and sustained drug release in phosphate buffered saline. WGA-TA-sHDL nanodisks exhibited higher cytotoxicity in the ACC cell line H295R half maximal inhibitory concentration ([IC50] 0.26±0.045 µM) than free WGA-TA (IC50 0.492±0.115 µM, P<0.05). Fluorescent dye-loaded sHDL nanodisks efficiently accumulated in H295R adrenal carcinoma xenografts 24 hours following dosing. Moreover, daily intraperitoneal administration of 7 mg/kg WGA-TA-loaded sHDL nanodisks significantly inhibited tumor growth during 21-day administration to H295R xenograft-bearing mice compared to placebo (P<0.01). Collectively, these results suggest that WGA-TA-loaded nanodisks may represent a novel and beneficial therapeutic strategy for the treatment of ACC.
Keywords: synthetic high-density lipoproteins, scavenger receptor class B1, targeted delivery, nanodisks, withalongolides, adrenocortical carcinomas
Introduction
Adrenocortical carcinoma (ACC) is a rare endocrine malignancy with a poor prognosis.1 The majority of ACC patients have metastasis at the time of diagnosis, resulting in a 5-year survival rate of <35%.2 Complete surgical resection remains the standard of care today. Other commonly used pharmacological interventions include the adrenotoxic drug mitotane (o,p′-DDD), which is administered either alone or in combination with other cytotoxic chemotherapy, such as etoposide, doxorubicin, and platinum agents.3 Because this method of treatment has a relatively low response rate and carries significant systemic toxicity, better treatment methods are critically needed for more effective targeting and inhibition of ACC.
Recently, we showed that the novel semisynthetic withalongolide A 4,19,27-triacetate (WGA-TA), from the plant Physalis longifolia, targets several oncogenic pathway proteins implicated in ACC.4 This toxicity occurs through a combined mechanism of induction of an oxidative stress response in cancer cells and functional inhibition of the molecular chaperone HSP90 through its interaction with the cochaperone CDC37.4 Although WGA-TA has shown great efficacy both in vitro and in vivo, one significant limitation of its translational development has been its low aqueous solubility.5
ACC cells require cholesterol for steroidogenesis and are known to express SRB1 to take up cholesterol from circulating high-density lipoprotein (HDL).6 Many patients with advanced ACC develop steroid oversecretion,7 and specific patient-derived ACC cell lines have been classified as “high cortisol secretors” (H295R) or “steroid nonsecretors” (SW13).8,9 In addition to ACC cells, lymphoma, prostate, breast, and ovarian cancers have also been found to overexpress SRB1 receptors.10–12 In fact, in recent years, high SRB1 expression has emerged as an indicator of aggression in both prostate and breast cancers and a predictor of poor survival. Therefore, nanoparticles that recognize SRB1 receptors can potentially improve the targeted delivery of drugs to a wide range of different cancers.
HDL is an endogenous nanoparticle of 8–14 nm diameter and mainly composed of lipids and apolipoproteins, with apolipoprotein A1 (ApoA1) acting as the main protein component.13 The primary function of endogenous HDL involves the efflux of excess cholesterol from cells in peripheral tissues and delivering it to the liver for metabolism. In addition to cholesterol, hormones, vitamins, signaling lipids, microRNAs, enzymes, and even hydrophobic drugs have been found to associate with HDL, by partitioning into the hydrophobic core of nanoparticles, insertion in the lipid layer, or binding to the negatively charged particle surface.14,15 Therefore, with its smallness and large surface area, HDL has emerged as an important vascular carrier of lipophilic products.
Several groups have utilized biomimetic HDL nanodisks for drug delivery purposes.16–19 Most biomimetic HDL is prepared with full-length ApoA1 protein, either purified from plasma or produced by recombinant means, which is then combined with lipids and drugs in the presence of surfactant to form HDL nanodisks. In most cases, the resulting nanoparticles have heterogeneous size distribution with average particles larger than endogenous HDL, and thus require purification prior to use.20 The resulting ApoA1 expression/purification and HDL assembly processes are technically complex and expensive; moreover, the process does not assure the removal of clinically unacceptable impurities, such as endotoxins, surfactants, and particulates.
To circumvent some of these technical issues, our laboratory utilizes a fully synthetic ApoA1-mimetic peptide, 22A, to prepare highly homogeneous synthetic HDL (sHDL) with an average diameter of 10–12 nm. The surfactant-free assembly process involves colyophilization of ApoA1 peptide, phospholipids, and anticancer or imaging agents from organic solvent solution, followed by hydration with buffer to form sHDL.21–23 This specific ApoA1 peptide and the same process for nanoparticle assembly were used to prepare ETC642, an HDL product for treatment of cardiovascular diseases.24,25 In clinical studies, ETC642 was found to have long plasma residence (~8 hours) and to be safe when administered at 30 mg/kg peptide (90 mg/kg total components) by weekly intravenous (IV) administration.15,26 Prior clinical safety data with ETC642 can facilitate clinical translation of sHDL-mediated drug delivery for treatment of adrenal diseases.
In the present study, we first characterized expression levels of SRB1 in various ACC cell lines. Next, we explored cellular uptake of fluorescently labeled HDL nanoparticles in both high (H295R) and low (SW13) SRB1-expressing ACC cell lines and examined the extent to which SRB1 mediated cellular uptake. We then confirmed accumulation of sHDL in ACC tumor xenografts in vivo. For this, we optimized a phospholipid composition of biomimetic sHDL to encapsulate the novel natural product derivative WGA-TA and characterized nanoparticle size, morphology, drug-loading, and drug-release kinetics. The cytotoxicity of WGA-TA-sHDL was confirmed in vitro and then compared in vivo with free drug, blank HDL, and a model standard-of-care chemotherapy treatment using H295R tumor xenografts.
Materials and methods
Materials
Phospholipids, including 1,2-dipalmitoyl-sn-glycero-3-phosphocholine (DPPC), egg sphingomyelin (SM), 1,2-dimyristoyl-sn-glycero-3-phosphocholine (DMPC), 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC), 1,2-dioleoyl-sn-glycero-3-phosphocholine (DOPC), and “egg-yolk phosphocholine” (EPC) were purchased from NOF America (White Plains, NY, USA). ApoA1-mimetic peptide was synthesized by GenScript (Piscataway, NJ, USA). WGA-TA was provided by Dr Barbara N Timmermann (University of Kansas). The fluorescent dyes DiO and DiR were purchased from Thermo Fisher Scientific (Waltham, MA, USA). All other chemical reagents were obtained commercially and were of analytical grade.
Preparation of WGA-TA-loaded sHDL
Lipid mixtures (DPPC, SM, DMPC, POPC, DOPC, and EPC), 22A peptide, and the anticancer drug WGA-TA were dissolved in glacial acetic acid at predetermined ratios (Table 1). Acetic acid was removed by freeze drying, and the powder was hydrated at 15 mg/mL 22A peptide concentration with phosphate buffered saline (PBS) (pH 7.4). The suspension was cycled three times between 50°C (3 minutes) and 20°C (3 minutes) with gentle shaking to obtain drug-loaded sHDL nanoparticles. The fluorophore-loaded sHDL was prepared using the same protocol, but the anticancer drug was instead replaced with DiO or DiR dye at a 2:1:0.02 weight ratio of phospholipids to 22A peptide to dye.
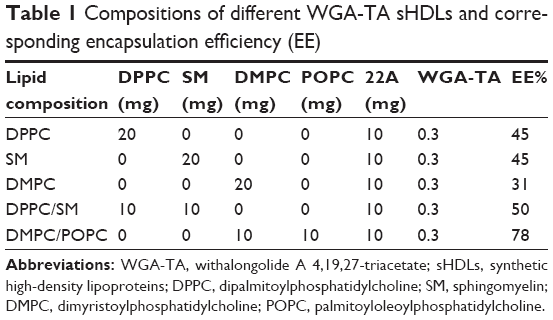 | Table 1 Compositions of different WGA-TA sHDLs and corresponding encapsulation efficiency (EE) |
Characterization of WGA-TA-loaded sHDL
Drug encapsulation efficiency was determined using desalting column centrifugation. Briefly, WGA-TA-sHDL was passed through the desalting column (cutoff 7,000 Da) to remove any unencapsulated drug. Ethanol was added to break sHDL and dissolve the drug before high-performance liquid chromatography (HPLC) analysis.27 WGA-TA concentration was determined by HPLC using isocratic elution with 35% A (water with 0.1% acetic acid) and 65% B (methanol with 0.1% acetic acid) at a flow rate of 0.7 mL/min. A C18 column (250×4.6 mm) was used for separation, and the detector wavelength was set at 230 nm. Dynamic light scattering (DLS) and gel permeation chromatography (GPC) were employed for analysis of average size, size distribution, and purity of WGA-TA-loaded sHDL nanoparticles. For DLS, 10 μL of the sHDL was diluted to 1 mL with PBS before measurement on a Malvern Zetasizer Nano. For GPC, 40 μL of the sHDL formulation (0.5 mg/mL 22A peptide) was injected into the HPLC equipped with a TSK 2000 GPC column. The flow rate was set at 0.7 mL/min, and the detector wavelength was 220 nm. Morphology of sHDL particles was further characterized by transmission electron microscopy (TEM). All images were acquired on a JEM-1200EX electron microscope (JEOL, Tokyo, Japan) equipped with an AMT XR-60 digital camera (Advanced Microscopy Techniques, Woburn, MA, USA).28
To evaluate the in vitro release profile of drugs, 1 mL of 0.15 mg/mL free drug (dissolved at 0.3 mg/mL in dimethyl sulfoxide and diluted to 1 mL with PBS) or 1 mL of 0.15 mg/mL WGA-TA-loaded sHDL was added to a dialysis bag (6–8 kDa), which was sealed and placed in 200 mL PBS (pH 7.4) containing 0.1% Tween 80.29,30 The contents were shaken gently at 100 rpm in a 37°C air bath shaker. An equal amount of free drug was directly added to the release medium to mimic complete drug release. At predetermined time points, 2 mL of the release medium was sampled, and an equal volume of fresh release medium was added back. The amount of drug in the media was quantified by reverse-phase HPLC. Cumulative release was calculated:
| (1) |
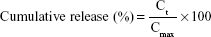
where Ct is the concentration of WGA-TA in the release media and Cmax is the concentration of WGA-TA when the drug was completely released.
Cell culture
Human ACC cell lines H295R (high cortisol secretor) and SW13 (nonsecretor) were obtained from ATCC. RL251 (nonsecretor) was derived using a clinical protocol approved by the Institutional Review Board at the University of Michigan Hospital and validated by the provider.31 H295R cells were grown in 1:1 DDMEM:F12 nutrient mixture (Thermo Fisher Scientific) supplemented with 5% FBS, 100 U/mL penicillin, 100 μg/mL streptomycin, and ITS (final concentrations 0.001 mg/mL bovine insulin, 0.0055 mg/mL human transferrin, and 6.7 ng/mL sodium selenite; Thermo Fisher Scientific). ACC cell lines SW13 and RL251 were grown in DMEM supplemented with 10% FBS. All cell lines were cultured at 37°C in a humidified incubator with 5% CO2.
Quantification of SRB1 expression
mRNA and protein expression levels of SRB1 in the ACC cell lines were evaluated by reverse-transcription polymerase chain reaction (RT-PCR) and Western blot, respectively. For RT-PCR, RNA was isolated from cells grown in culture using RNEasy kit as per the manufacturer’s protocol (Qiagen, Venlo, the Netherlands). mRNA was reverse transcribed, and expression of SRB1 and control β-actin examined with specific primer sets by the standard curve method in a Viia7 real-time PCR system (Thermo Fisher Scientific). For Western blot, cells were lysed using radioimmunoprecipitation assay buffer (50 mM Tris–HCI, pH 7.4, 150 mM NaCI, 1% [v:v] NP-40, 0.5% [w:v] sodium deoxycholate, 10 mM sodium fluoride, 1 mM sodium orthovanadate, 1 mM PMSF, 10 mM sodium pyrophosphate 0.1% [w:v]), and sodium dodecyl sulfate supplemented with protease inhibitor solution (EMD Millipore, Billerica, MA, USA). Lysates were centrifuged at 14,000 rpm for 20 minutes and proteins quantified using protein assay reagent (Thermo Fisher Scientific). Equal amounts of total proteins were separated using sodium dodecyl sulfate polyacrylamide gel electrophoresis and then transferred onto a Hybond nitrocellulose membrane (GE Healthcare, Little Chalfont, UK). Membranes were blocked using 5% milk and probed overnight with appropriate dilutions of the primary antibodies (SRB1 or actin) for proteins. Blots were then washed three times with PBST and incubated with horseradish peroxidase (HRP)-conjugated secondary antibodies (1:5,000 dilution) from Santa Cruz Biotechnology (Dallas, TX, USA). To ensure equal loading of proteins, actin was used as a control. Bands were visualized using enhanced chemiluminescence reagent (Thermo Fisher Scientific). Images were captured on Kodak X-ray film. ImageJ software was used for quantification of Western blot.
Cellular uptake of fluorophore-loaded sHDL
The ability of ACC H295R and SW13 cells to recognize sHDL nanodisks and internalize sHDL cargo through the SRB1 receptor was examined after a 2-hour incubation of cells with DiO-labeled sHDL. For confocal microscopy, cells were fixed with 4% paraformaldehyde, permeabilized with 0.1% Triton X-100, and stained with DAPI. Cellular uptake was then visualized using an A1 spectral confocal microscopy system (Nikon, Tokyo, Japan). Quantification of cellular fluorescent signals was determined using flow cytometry (Cyan5) at an excitation wavelength of 488 nm. To investigate whether sHDL uptake was mediated by SRB1, H295R cells were either pretreated with anti-SRB1 antibody (NB400-113; Novus Biological, Littleton, CO, USA) at 1:100 dilution or preincubated with 100-fold excess of unlabeled sHDL for 1 hour at 37°C.
Cytotoxicity of drug-loaded sHDL on adrenal cancer cells
For in vitro testing of drug efficacy, H295R was used and viability determined by CellTiter 96 aqueous nonradioactive cell proliferation assay. Approximately 8,000 H295R cells/well were seeded in 96-well microtiter plates in 90 μL of growth media and cells allowed to attach overnight. On the second day, 10 μL serial dilutions of sHDL nanoparticles, WGA-TA, and sHDL-WGA-TA were added to the plates. The cells were then incubated for 72 hours at 37°C in a CO2 humidified chamber. In some experiments, H295R cells were pretreated with SRB1 antibody (1:100 dilution) or PBS for 1 hour before incubation with WGA-TA-sHDL for 24 hours. The number of viable cells was determined by measuring absorbance at 490 nm wavelength of the dissolved formazan reagent on a Synergy Neo plate reader (BioTek, Winooski, VT, USA) after the addition of 20 μL of MTS reagent for 2 hours, as per the manufacturer’s protocol (Promega, Fitchburg, WI, USA). All experiments were carried out in triplicate, and the viability of the cells is expressed as the ratio of the number of viable cells with treatment compared to untreated cells. Half maximal inhibitory concentration (IC50) values were calculated from MTS assay curves using GraphPad Prism 5 software.
In vivo biodistribution of fluorophore-loaded sHDL in H295R xenografts
Animal experiments were performed in accordance with federal, state, and local guidelines. All work performed on animals was in accordance with and approved by the University Committee on Use and Care of Animals at the University of Michigan, Ann Arbor, Michigan. When H295R cells reached 60%–70% confluence, cells were collected using 0.25% trypsin–0.02% EDTA (Sigma-Aldrich, St Louis, MO, USA) and mechanical dissociation, then suspended at 6×107 cells/mL in PBS and placed on ice. Within 30 minutes, athymic nude mice (Foxn1nu; Harlan Laboratories, Indianapolis, IN, USA) were anesthetized with isoflurane, and 6×106 cells suspended in 100 μL PBS were injected subcutaneously into the right flank.
For immunohistochemical analysis of SRB1 receptors, tumors were collected when they were ~6 mm in diameter and fixed in 10% buffered formalin (Fisher Diagnostics). Immunohistochemical staining was performed on an IntelliPath FLX (Biocare Medical, Pacheco, CA, USA) at the Unit for Laboratory Animal Medicine, In Vivo Animal Core, University of Michigan. Briefly, slides were deparaffinized in xylene and rehydrated through graded alcohols to water; then heat-induced epitope retrieval was performed in a Decloaking Chamber (Biocare Medical) with Diva (Biocare Medical). Slides were incubated in Peroxidazed 1 (Biocare Medical) for 5 minutes, followed by 30 minutes of incubation in Rodent Block M (Biocare Medical). Rabbit antimouse SRB1 (Novus Biological) antibody was applied for 30 minutes; then SRB1 was detected with Rabbit-on-Rodent HRP polymer (Biocare Medical) for 30 minutes. HRP staining was visualized with DAB (Biocare Medical). Slides were counterstained in hematoxylin (Biocare Medical), rinsed in deionized water, dehydrated through graded alcohols, cleared in xylene, and mounted with Micromount (Leica Biosystems, Nussloch, Germany). Stained slides were imaged with microscopy (Mantra; PerkinElmer, Waltham, MA, USA).
To characterize the biodistribution and tumor-targeting efficacy of sHDL-WGA-TA nanoparticles in vivo, imaging studies were performed in the ACC (H295R) tumor-bearing mice when tumors were ~6 mm in diameter. DiR-labeled sHDL nanoparticles (20 μg/mL DiR) were injected into the tail vein or intraperitoneally at 0.2 mL/dose. After 24 hours, animals were killed, and organs (spleen, liver, heart, lung, kidney, brain) and tumors harvested and imaged to characterize biodistribution using a Xenogen IVIS spectrum imaging system (PerkinElmer).32,33
Inhibition of tumor growth by WGA-TA sHDL nanodisks in H295R xenografts
The ability of WGA-TA-sHDL nanodisks to inhibit tumor growth was examined in H295R xenografts in athymic mice. Tumors were implanted as described earlier, and treatment was started when tumors had reached ~100 mm3. Mice were randomized into five groups with eight animals per group: WGA-TA-sHDL (6 mg/kg/day drug, 150 mg/kg/day 22A peptide), WGA-TA (6 mg/kg/day), blank sHDL (150 mg/kg/day 22A peptide), Italian standard-of-care protocol (etoposide 7.5 mg/kg/day, doxorubicin 1.5 mg/kg/day, cisplatin 2.5 mg/kg/day, and mitotane 300 mg/kg/day), and no-treatment control. Mice were treated with daily intraperitoneal (IP) injections for 21 days. Tumor length and width measurements were recorded by digital caliper every 3 days for the duration of treatment. Some mice became moribund during treatment and were killed. Average tumor volume was calculated:
| (2) |
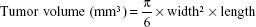
Following 21 days of treatment, serum was collected for analysis of AST and ALT levels, which were performed at the In Vivo Animal Core Animal Diagnostic Laboratory at the University of Michigan. The normal range of AST and ALT was 39.55–386.05 U/L and 24.3–115.25 U/L, respectively. Histologic analysis was performed following 21 days of treatment to evaluate the liver toxicity of different formulations. Signs of chronic inflammation (defined by the presence of lymphocytes and plasma cells), cellular necrosis, and steatohepatitis were assessed in the liver. “Normal” was assigned to specimens with essentially no pathology, except for a small degree of inflammation comparable to what is seen physiologically. “Mild/moderate/severe” was assigned to specimens with <10%, ≥10%–50%, and >50% increase in inflammatory cells compared to physiologic state, respectively.
Statistical analysis
Data were analyzed statistically using one-way ANOVA Kruskal–Wallis nonparametric tests and two-way ANOVA for comparisons of three or more variables with Dunn or Holm–Sidak multiple comparison tests and two-tailed Student’s t-tests for comparisons of two values. P<0.05 was considered statistically significant.
Results
Preparation and characterization of sHDL
A schematic of WGA-TA- and dye-loaded sHDL nanoparticle preparation is shown in Figure 1. Phospholipids, 22A peptide, and drug/dye were first dissolved in organic solvent and lyophilized. The resulting powder was hydrated above phospholipid melting temperature (Tm) to facilitate nanoparticle assembly and drug incorporation. A 1:2 weight ratio of 22A to phospholipids was used, because it is known to result in the formation of homogeneous HDLs 10–12 nm in diameter.34 The theoretical loading of WGA-TA was set at 1% (0.3 mg/30 mg peptide–lipid mixture). Following assembly, sHDL-drug nanoparticles were passed through the desalting column (7,000 Da) to remove unincorporated drug and determine WGA-TA encapsulation efficiency (Table 1).
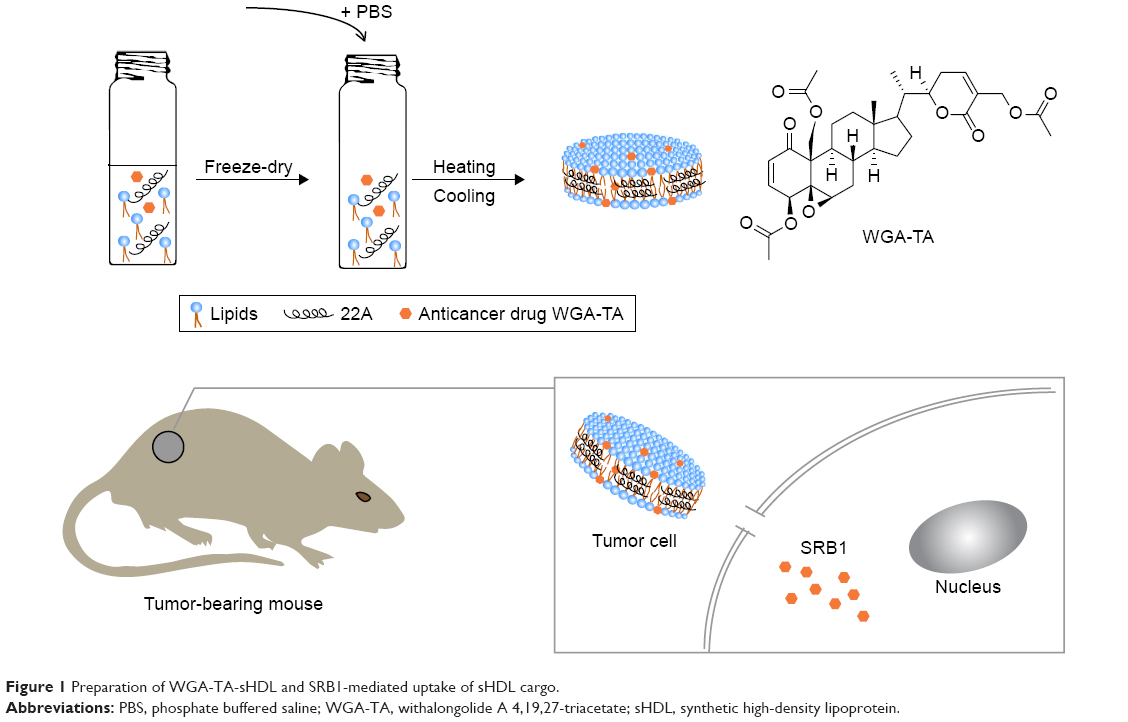 | Figure 1 Preparation of WGA-TA-sHDL and SRB1-mediated uptake of sHDL cargo. |
Due to the limited space available for incorporating drug inside sHDL, molecular interactions between phospholipids and WGA-TA are rather important. Fatty-acid chain length and saturation determine Tm and the resulting fluidity of the phospholipid bilayer, which is important for incorporation and retention of WGA-TA in sHDL. We used phospholipids of different Tm to prepare WGA-TA-sHDL nanoparticles: DPPC (Tm 41°C), SM (Tm 38°C), DMPC (Tm 23°C), POPC (Tm −3°C), and their mixtures.21 The visual appearance of the resulting suspension correlated with both the size and the quality of the resulting nanoparticles, with clear solutions correlating with the formation of 10–12 nm sHDL particles and cloudy solutions indicating HDL aggregation and drug precipitation. When WGA-TA-sHDL was prepared with DPPC and/or SM, nanoparticle solutions were clear at 50°C, but cloudy at both 37°C and 20°C, indicating drug precipitation at temperatures below Tm. When DMPC (Tm 23°C) was used to prepare nanoparticles, drug-loaded sHDL was clear at 50°C and 37°C but cloudy at 20°C. Samples with cloudy solutions showed a loss of drug upon passage through the desalting column and low encapsulation efficiency of 31%–50% (Table 1).
Lipids with a low transition temperature, such as POPC and DOPC, failed to form sHDL, so they were not used for the preparation of drug-loaded sHDL. However, the mixture of DMPC and POPC resulted in successful loading of WGA-TA into sHDL and clear sHDL solutions at all temperatures tested. For this formulation, drug encapsulation efficiency of 78% was obtained, which corresponded to 0.78% of WGA-TA weight loading. This formulation was selected for additional analytical characterization and was used in cell culture and animal experiments.
Characterization of WGA-TA-sHDL nanoparticles
To examine the purity and size of the WGA-TA-sHDL nanoparticles, blank and drug-loaded sHDL nanoparticles were analyzed by GPC, DLS, and TEM. GPC confirmed the formation of homogeneous WGA-TA-sHDL distribution without the presence of large nanoparticle aggregates or unbound 22A peptide (Figure 2A). DLS measurement showed narrow size distribution for sHDL and WGA-TA-sHDL of 9.7±0.61 nm and 9.81±0.41 nm, respectively. Consistent with DLS and GPC data, TEM results also showed that the formulation had homogeneous discoid WGA-TA-sHDL nanoparticles with an average size of ~10 nm (Figure 2B and C). This size and shape closely resembled the characteristics of endogenous HDL nanoparticles.15
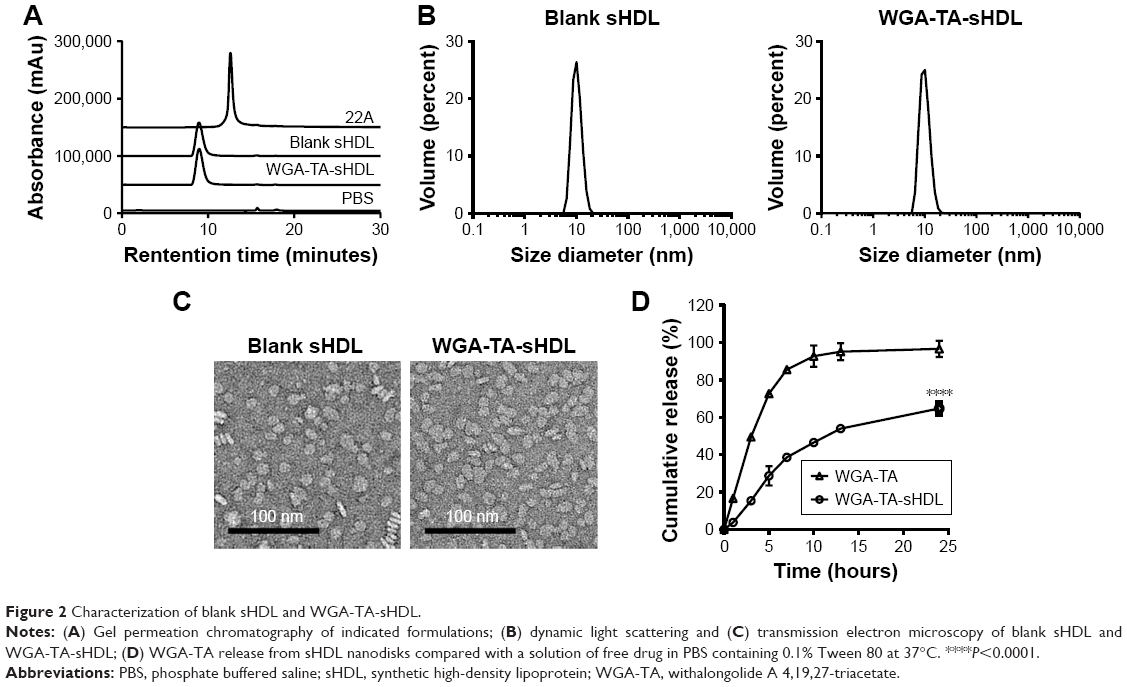 | Figure 2 Characterization of blank sHDL and WGA-TA-sHDL. |
To examine drug-release kinetics, WGA-TA-sHDL nanoparticles were placed in a dialysis bag and incubated in sink conditions in 200 mL of PBS containing 0.1% Tween 80 for 24 hours. To compare nanoparticle release with free drug, the same amount of WGA-TA-sHDL was dissolved in 50% dimethyl sulfoxide and placed in a dialysis bag. The free drug was rapidly released to the media, with almost 100% release within 10 hours (Figure 2D). In contrast, the release of WGA-TA from sHDL was relatively slow, with ~46% released to the media within 10 hours and 65% released within 24 hours. These results indicated that the encapsulation of WGA-TA into sHDL significantly altered drug-release behavior.
Quantification of SRB1 on ACC cells
To select ACC cell lines for evaluation of sHDL cellular uptake and WGA-TA-sHDL cytotoxicity, we examined SRB1 expression in various ACC cell lines by measuring mRNA levels using RT-PCR and protein levels by Western blot analysis (Figure 3). Actin was used as internal control for RT-PCR, and actin immunoblot confirmed equal loading of protein per well. Steroid hormone-synthesizing H295R cells were found to have much higher mRNA and protein expression levels of SRB1 compared to the other two human ACC cell lines, SW13 and RL251 (P<0.0001), which do not synthesize steroid hormones. Therefore, H295R was selected to assess cellular uptake and cytotoxicity of WGA-TA-sHDL.
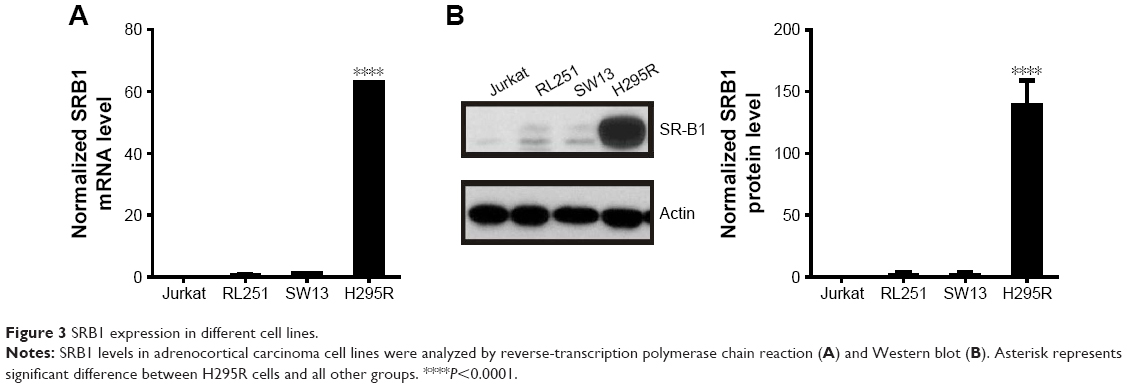 | Figure 3 SRB1 expression in different cell lines. |
Cellular uptake of sHDL
To determine if our sHDL was recognized by SRB1 and capable of delivering its cargo to the target cells efficiently, DiO-loaded sHDLs were incubated with high (H295R) and low (SW13) SRB1-expressing ACC cell lines for 2 hours (Figure 4). After incubation, cells were imaged by confocal microscopy, and DiO fluorescence in cells was quantified by flow cytometry. It is clear from the image that SRB1-positive H295R cells effectively internalized DiO-labeled sHDL, in contrast to SW13 cells, which showed limited dye uptake. The addition of anti-SRB1 antibody or excess of blank sHDL nanoparticles reduced dye uptake in H295R cells, confirming the involvement of SRB1 (Figure 4A). Fluorescence quantification by flow cytometry confirmed visual observations (Figure 4B). More than 80% of H295R cells showed a positive DiO signal, while <10% of SW13 cells showed DiO uptake. Preincubation of H295R cells with anti-SRB1 antibody or blank sHDL reduced dye uptake by 2.5-fold and 20%, respectively, confirming the interaction between SRB1 and sHDL nanodisks. Based on these results, H295R cells were selected to confirm cytotoxicity and in vivo efficacy of WGA-TA-sHDL nanodisks in ACC.
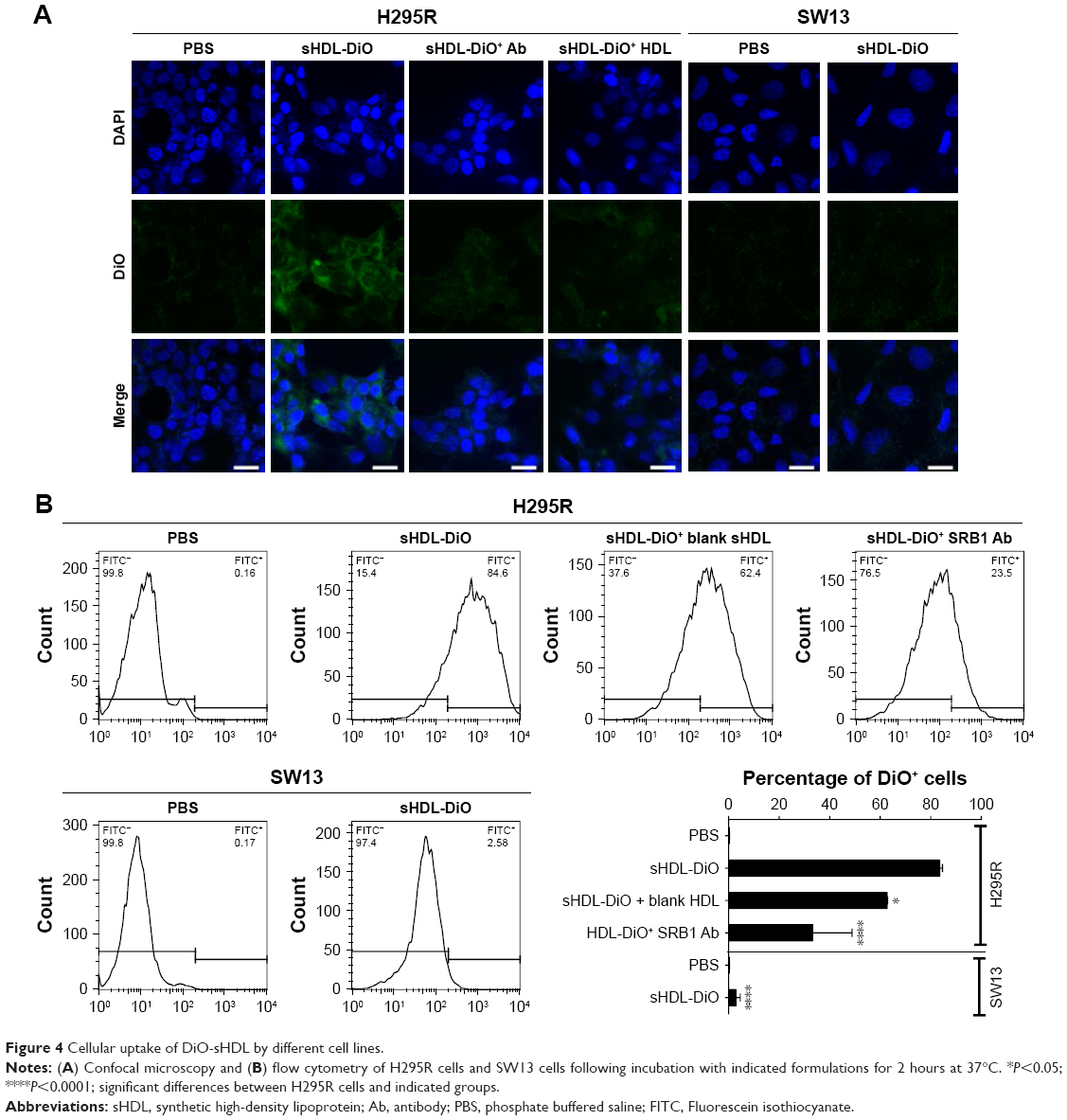 | Figure 4 Cellular uptake of DiO-sHDL by different cell lines. |
Cytotoxicity of WGA-TA-loaded sHDL
The cytotoxicity of WGA-TA-loaded sHDL nanodisks was evaluated by MTS-proliferation assay in the H295R cell line. Cells were treated with varying concentrations of sHDL, free WGA-TA, or WGA-TA-loaded sHDL nanodisks for 72 hours, and the viability of the cells was then analyzed (Figure 5). The results indicated that compared to either sHDL or WGA-TA alone, WGA-TA-loaded sHDL nanoparticles had improved cytotoxicity (reduction in cell viability with lower IC50 levels by quantitative curve fitting using GraphPad 5.0). IC50 calculated by quantitative curve fitting using GraphPad 5.0 was 0.26±0.045 μM for WGA-TA-sHDL and 0.492±0.115 μM for WGA-TA (P<0.05). Blank-sHDL treatment showed some cytotoxicity, likely due to the ability of sHDL for cholesterol efflux and inhibition of corticosteroid synthesis, as previously reported,35 but sHDL cytotoxicity was observed at a higher concentration relative to WGA-TA-sHDL (IC50 corresponding to 8.63±1.52 μM WGA-TA [P<0.05] and equivalent to 0.13±0.02 mg/mL of 22A peptide). The presence of SRB1 antibody significantly decreased the cytotoxicity of WGA-TA-sHDL in H295 cells, with a 2-fold decrease and 4.8-fold decrease in viability for 2.5 μM and 5 μM WGA-TA-sHDL, respectively, indicating the uptake of WGA-TA-sHDL was at least partially mediated by SRB1 receptors.
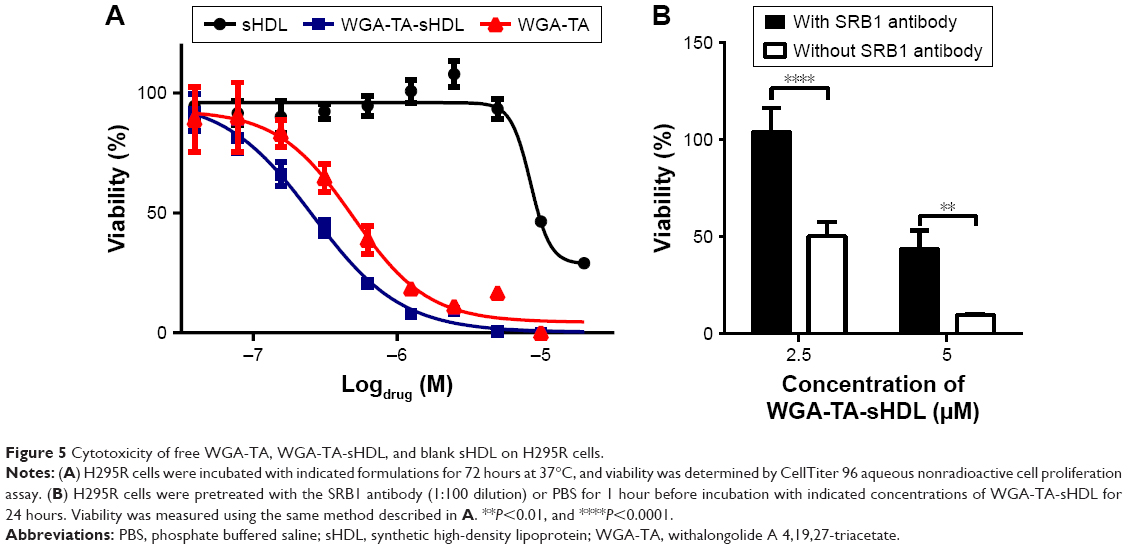 | Figure 5 Cytotoxicity of free WGA-TA, WGA-TA-sHDL, and blank sHDL on H295R cells. |
In vivo biodistribution of fluorophore-loaded sHDL
Efficient accumulation throughout individual tumors is important for the targeted delivery of anticancer drugs. In order to test the ability of our sHDL to accumulate in tumor regions efficiently, DiR-loaded sHDL nanodisks were prepared and administered by IV or IP routes into athymic mice bearing H295R tumors, which express high levels of SRB1 receptors (Figure S1). At 24 hours following the injection, biodistribution of DiR-sHDL was examined by imaging individual organs (Figure 6). The imaging clearly demonstrated that tumors harvested 24 hours after IV or IP injection had efficient accumulation of sHDL-cargo DiR, providing direct evidence of in vivo ACC targeting with sHDL. As expected, strong fluorescence signals from the liver were observed, given that the liver is the major organ for elimination of nanoparticles. Accumulation of sHDL nanodisks in other organs was significantly less compared to that in the liver or tumors. Since IV and IP injection showed similar distribution profiles of sHDL-DiR (P>0.05), for subsequent efficacy study in H295R xenografts, WGA-TA-sHDL was administered by the IP route.
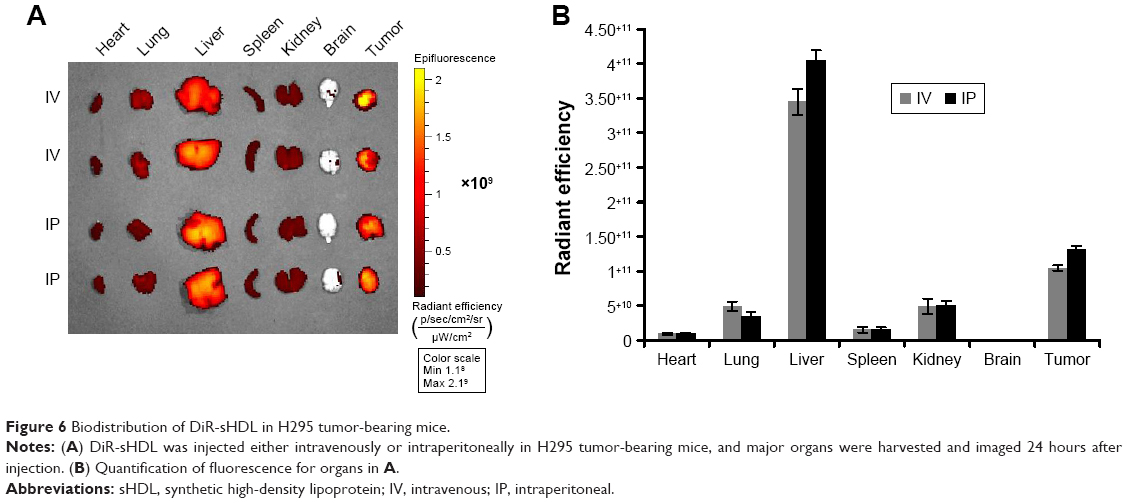 | Figure 6 Biodistribution of DiR-sHDL in H295 tumor-bearing mice. |
Tumor growth inhibition in vivo
The ability of WGA-TA-sHDL nanodisks to inhibit ACC growth in vivo was tested in H295R xenografts, which exhibited high levels of SRB1 expression in tumors (Figure S1). Once tumor volume had reached ~100 mm3, the tumor-bearing mice were randomly assigned to one of the five treatment groups. The control group received no treatment, while the other groups received blank sHDL, free WGA-TA, WGA-TA-sHDL, or Italian standard-of-care protocol (etoposide, doxorubicin, cisplatin, and mitotane) by IP administration daily for 21 days. Following the start of treatment, tumor volumes were assessed every 3 days (Figure 7). H295R is a very aggressive and rapidly growing tumor with high animal–animal variability and rapid growth kinetics, making this tumor very difficult to cure. The results from the study, however, showed significant differences in tumor volume between the no-treatment control and the WGA-TA-sHDL group at the end of treatment (P<0.01). No statistical differences between other groups were observed. Importantly, we did not see any significant change in serum ALT and AST levels, as both were in the normal range for all treated groups following 21-day treatment (Figure 7B). Moreover, livers from all experimental groups were normal (Figure 7C). Although mice treated with both free WGA-TA and WGA-TA-HDL had lower body weights than other groups, it should be noted that part of the body weight decrease for WGA-TA-HDL can be ascribed to tumor volume decrease following treatment. Compared to the free drug-treated animals that had much larger tumors and lower body weights, WGA-TA-sHDL treatment decreased side effects of WGA-TA in vivo (Figure S2).
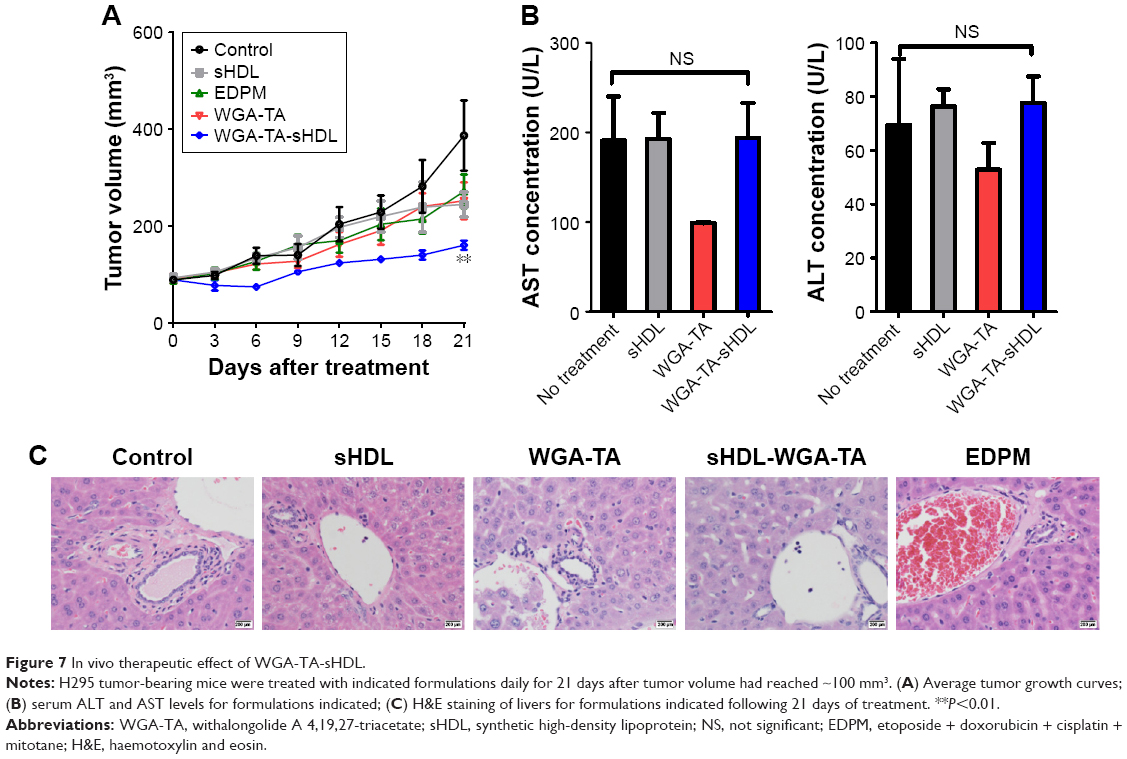 | Figure 7 In vivo therapeutic effect of WGA-TA-sHDL. |
Discussion
Here, we encapsulated WGA-TA, a novel natural product, in sHDL nanodisks and showed its utility in the translational treatment of ACC. The composition of sHDL was optimized based on the encapsulation efficiency of WGA-TA in sHDL. WGA-TA-sHDL nanodisks were prepared using the same methodology and ApoA1 peptide (22A):phospholipid ratio that was previously optimized for the manufacturing of clinical-grade sHDL cardiovascular treatments. By utilizing clinically tested methodologies and sHDL compositions, we obtained sHDL nanodisks with a high level of purity and homogeneous size distribution, while avoiding the laborious steps of purification. We also identified an ACC cell line that shows a high level of SRB1 expression and confirmed the ability of these tumor cells to take up sHDL cargo both in vitro and in vivo. These features make sHDL a promising platform for the targeted delivery of therapeutic drugs to ACC.
ACC is usually a fatal disease, due to its late stage at the time of diagnosis and lack of effective treatment options available for preventing tumor progression.3 We have recently identified novel naturally derived withanolides as potential ACC therapeutics, due to their ability simultaneously to target multiple signaling pathways involved in ACC development and growth. Our results showed that our lead compound, WGA-TA, had potent antiproliferative activity against ACC in vitro, inducing cell cycle arrest and apoptosis, as well as modulating key pathway proteins in the Notch, MAPK, and PI3K–Akt–mTOR signaling pathways in ACC, along with downregulation of HSP90 function and expression.4 Although WGA-TA showed high potency and selectivity in ACC in vitro, these compounds have low aqueous solubility and thus could benefit from incorporation into the lipid core of sHDL nanodisks for improved and more targeted delivery to ACC tumor cells.25
The successful incorporation and retention of drugs in sHDL nanodisks depends on the interaction between the cargo and sHDL itself. For example, the lipid composition of sHDL can affect drug loading.21 In addition, hydrophobic molecules can be preferentially incorporated in sHDL, while hydrophilic molecules require covalent attachment to sHDL or chemical modification in order to be loaded in sHDL.15,36 In the current study, we optimized the sHDL lipid composition to maximize encapsulation efficiency.
Next, we examined the expression of SRB1 at both the mRNA and protein levels. Both RT-PCR and Western blot protein analysis confirmed that SRB1 receptors were overexpressed in ACC, particularly in the H295R cell line. When DiO-sHDL was incubated with cells expressing different levels of SRB1, only cells with elevated SRB1 expression exhibited significant uptake of DiO, while cells with low SRB1 expression exhibited limited uptake of DiO. Moreover, H295R DiO-sHDL uptake was reduced by preincubation with anti-SRB1 antibody or excess blank sHDL. We also confirmed the effective accumulation of DiR-sHDL in the tumor regions of H295R tumor-bearing mice. However, in all cases, the selective uptake of sHDL cargo both in vitro and in vivo by SRB1-expressing tumor cells was demonstrated only for DiO dye, rather than WGA-TA, due to analytical limitations in detection of the drug molecule. The selective delivery of WGA-TA to ACC cells and tumors in vivo, as well as any changes in WGA-TA plasma half-life, will be evaluated in future studies following the establishment of a mass spectrometry-based drug detection method.
WGA-TA-sHDL nanodisks showed superior cytotoxicity compared to free WGA-TA or blank sHDL in the H295R ACC cell line, possibly indicating an advantage in using sHDL for WGA-TA delivery. Surprisingly, blank sHDL nanodisks were still able to inhibit ACC cell viability at high-enough concentrations. This effect may be due to cholesterol efflux by sHDL from H295R cells through SRB1, which is known for being a bidirectional transporter, or via passive efflux from cell plasma membranes.37,38 The ability of sHDL for cholesterol efflux via SRB1 or by passive efflux has been confirmed in previous studies.39 In addition, sHDL has been shown to cause cholesterol efflux and inhibition of cortisol production in noncancerous HAC16 adrenocortical cells.35 The synergy between sHDL-induced cytotoxicity and traditional chemotherapeutic agents in H295R and SW13 cells has also been previously reported.28 Therefore, some of the improved cytotoxicity of WGA-TA-sHDL could be due to synergistic effects between sHDL and WGA-TA.
While several other chemotherapeutic agents have been incorporated in HDL nanoparticles for targeted delivery,29,30,40–42 the approach described here offers several distinct advantages with respect to the ease of clinical translation. Most biomimetic HDL nanoparticles are assembled using the full-length ApoA1 protein,15 which is technically difficult and costly to produce, making ApoA1-based HDL delivery not commercially feasible. In addition, given the amphiphilic nature of the protein, ApoA1 tends to aggregate and bind to endotoxin and host cell proteins, resulting in low production yields and difficulty in scaling up.43,44 Moreover, most drug-loaded sHDL is prepared by a cholate dialysis process, which requires purification postassembly in order to isolate nanoparticles of the desired size range and remove residual cholate that could be toxic to the liver in clinical settings.45 In contrast, we used an ApoA1 peptide, which was selected from nearly 200 peptide sequences and produced at kilogram scale by chemical synthesis. Moreover, this peptide has completed toxicology and Phase I clinical evaluations.26,46 The sHDL particles were assembled by a simple process that involves colyophilization of 22A peptide, phospholipids, and WGA-TA in organic solvent and self-assembly of sHDL nanodisks upon hydration in buffer. In addition, the same methodology has been used to prepare ETC642-blank sHDL, which has been examined in single- and multiple-dose clinical trials for treatment of cardiovascular diseases.15,24,25 Therefore, prior clinical experience could facilitate the translation of 22A peptide-based sHDL drug delivery nanodisks.
In summary, we optimized the composition of sHDL for the delivery of a novel lipophilic natural product – WGA-TA. When WGA-TA was loaded into sHDL, this formulation was shown to kill SRB1-expressing H295R cancer cells more efficiently than free drug alone in vitro. In addition, sHDL cargo molecules accumulated in tumor regions of ACC in vivo, and the WGA-TA-sHDL formulation was more effective in halting the growth of ACC tumors than the Italian standard-of-care protocol. Taken together, sHDL delivery could represent a more useful drug delivery platform for hydrophobic chemotherapeutics in such tumors as ACC that overexpress SRB1 and should be further evaluated for potential clinical application. Additionally, this unique drug delivery strategy could be applied in the delivery of hormones, steroids, and other therapeutic agents in treating other adrenal diseases.
Acknowledgments
The authors acknowledge the following funding sources for support of this work: University of Michigan, Michigan Translational Research and Commercialization (MTRAC) grant (MSC and AS), University of Michigan Comprehensive Cancer Center support grant F030221 (MSC), and University of Michigan Department of Surgery, Research Advisory Committee (RAC) grant (CS and MSC). This work was partly funded by NIH T32 CA009672, R01 CA173292, R01-AI127070, R01-EB022563, R01-CA210273, U01-CA-120458, and R21 NS091555. Rui Kuai was supported in part by American Heart Association (AHA) predoctoral fellowship 15PRE25090050. The authors also acknowledge Grace Wang (University of Michigan) for help with histological analyses.
Disclosure
The authors report no conflicts of interest in this work.
References
Libé R. Adrenocortical carcinoma (ACC): diagnosis, prognosis, and treatment. Front Cell Dev Biol. 2015;3:45. | ||
Dy BM, Wise KB, Richards ML, et al. Operative intervention for recurrent adrenocortical cancer. Surgery. 2013;154(6):1292–1299. | ||
Else T, Kim AC, Sabolch A, et al. Adrenocortical carcinoma. Endocr Rev. 2014;35(2):282–326. | ||
Subramanian C, Zhang H, Gallagher R, Hammer G, Timmermann B, Cohen M. Withanolides are potent novel targeted therapeutic agents against adrenocortical carcinomas. World J Surg. 2014;38(6):1343–1352. | ||
White PT, Subramanian C, Motiwala HF, Cohen MS. Natural withanolides in the treatment of chronic diseases. In: Gupta SC, Prasad S, Aggarwal BB, editors. Anti-inflammatory Nutraceuticals and Chronic Diseases. Heidelberg: Springer; 2016:329–373. | ||
Miller WL, Auchus RJ. The molecular biology, biochemistry, and physiology of human steroidogenesis and its disorders. Endocr Rev. 2011;32(1):81–151. | ||
Lerario AM, Moraitis A, Hammer GD. Genetics and epigenetics of adrenocortical tumors. Mol Cell Endocrinol. 2014;386(1–2):67–84. | ||
Rainey WE, Bird IM, Mason JI. The NCI-H295 cell line: a pluripotent model for human adrenocortical studies. Mol Cell Endocrinol. 1994;100(1–2):45–50. | ||
Leibovitz A, McCombs WM 3rd, Johnston D, McCoy CE, Stinson JC. New human cancer cell culture lines – I – SW-13, small-cell carcinoma of the adrenal cortex. J Natl Cancer Inst. 1973;51(2):691–697. | ||
Leon CG, Locke JA, Adomat HH, et al. Alterations in cholesterol regulation contribute to the production of intratumoral androgens during progression to castration-resistant prostate cancer in a mouse xenograft model. Prostate. 2010;70(4):390–400. | ||
Pussinen PJ, Karten B, Wintersperger A, et al. The human breast carcinoma cell line HBL-100 acquires exogenous cholesterol from high-density lipoprotein via CLA-1 (CD-36 and LIMPII analogous 1)-mediated selective cholesteryl ester uptake. Biochem J. 2000;349(Pt 2):559–566. | ||
Imachi H, Murao K, Sayo Y, et al. Evidence for a potential role for HDL as an important source of cholesterol in human adrenocortical tumors via the CLA-1 pathway. Endocr J. 1999;46(1):27–34. | ||
Nofer JR, Kehrel B, Fobker M, Levkau B, Assmann G, von Eckardstein A. HDL and arteriosclerosis: beyond reverse cholesterol transport. Atherosclerosis. 2002;161(1):1–16. | ||
Vickers KC, Palmisano BT, Shoucri BM, Shamburek RD, Remaley AT. MicroRNAs are transported in plasma and delivered to recipient cells by high-density lipoproteins. Nat Cell Biol. 2011;13(4):423–433. | ||
Kuai R, Li D, Chen YE, Moon JJ, Schwendeman A. High-density lipoproteins: nature’s multifunctional nanoparticles. ACS Nano. 2016;10(3):3015–3041. | ||
Duivenvoorden R, Tang J, Cormode DP, et al. A statin-loaded reconstituted high-density lipoprotein nanoparticle inhibits atherosclerotic plaque inflammation. Nat Commun. 2014;5:3065. | ||
Zhang XB, Chen BS. Recombinant high density lipoprotein reconstituted with apolipoprotein AI cysteine mutants as delivery vehicles for 10-hydroxycamptothecin. Cancer Lett. 2010;298(1):26–33. | ||
Mooberry LK, Nair M, Paranjape S, McConathy WJ, Lacko AG. Receptor mediated uptake of paclitaxel from a synthetic high density lipoprotein nanocarrier. J Drug Target. 2010;18(1):53–58. | ||
Bricarello DA, Smilowitz JT, Zivkovic AM, German JB, Parikh AN. Reconstituted lipoprotein: a versatile class of biologically-inspired nanostructures. ACS Nano. 2011;5(1):42–57. | ||
Cao W, Ng KK, Corbin I, et al. Synthesis and evaluation of a stable bacteriochlorophyll-analog and its incorporation into high-density lipoprotein nanoparticles for tumor imaging. Bioconjug Chem. 2009;20(11):2023–2031. | ||
Yuan Y, Wen J, Tang J, et al. Synthetic high-density lipoproteins for delivery of 10-hydroxycamptothecin. Int J Nanomedicine. 2016;11:6229–6238. | ||
Tang J, Kuai R, Yuan W, Drake L, Moon JJ, Schwendeman A. Effect of size and PEGylation of liposomes and peptide-based synthetic lipoproteins on tumor targeting. Nanomedicine. 2017;13(6):1869–1878. | ||
Subramanian C, Kuai R, Zhu Q, et al. Synthetic high-density lipoprotein nanoparticles: a novel therapeutic strategy for adrenocortical carcinomas. Surgery. 2016;159(1):284–295. | ||
Di Bartolo BA, Nicholls SJ, Bao S, et al. The apolipoprotein A-I mimetic peptide ETC-642 exhibits anti-inflammatory properties that are comparable to high density lipoproteins. Atherosclerosis. 2011;217(2):395–400. | ||
Miles J, Khan M, Painchaud C, Lalwani N, Drake S, Dasseux JL. Single-dose tolerability, pharmacokinetics, and cholesterol mobilization in HDL-C fraction following intravenous administration of ETC-642, a 22-mer ApoA-I analogue and phospholipids complex, in atherosclerosis patients. Arterioscler Thromb Vasc Biol. 2004;24:E19. | ||
Li D, Gordon S, Schwendeman A, Remaley AT. Apolipoprotein mimetic peptides for stimulating cholesterol efflux. In: Anantharamaiah GM, Goldberg D, editors. Apolipoprotein Mimetics in the Management of Human Disease. Heidelberg: Springer; 2015:29–42. | ||
Zhang WL, Gu X, Bai H, Yang RH, Dong CD, Liu JP. Nanostructured lipid carriers constituted from high-density lipoprotein components for delivery of a lipophilic cardiovascular drug. Int J Pharm. 2010;391(1–2):313–321. | ||
Subramanian C, Kuai R, Zhu Q, et al. Synthetic high-density lipoprotein nanoparticles: a novel therapeutic strategy for adrenocortical carcinomas. Surgery. 2016;159(1):284–294. | ||
Yang T, Choi MK, Cui FD, et al. Preparation and evaluation of paclitaxel-loaded PEGylated immunoliposome. J Control Release. 2007;120(3):169–177. | ||
Zhang W, He H, Liu J, et al. Pharmacokinetics and atherosclerotic lesions targeting effects of tanshinone IIA discoidal and spherical biomimetic high density lipoproteins. Biomaterials. 2013;34(1):306–319. | ||
Schteingart DE, Giordano TJ, Benitez RS, et al. Overexpression of CXC chemokines by an adrenocortical carcinoma: a novel clinical syndrome. J Clin Endocrinol Metab. 2001;86(8):3968–3974. | ||
Kuai R, Yuan W, Li W, et al. Targeted delivery of cargoes into a murine solid tumor by a cell-penetrating peptide and cleavable poly(ethylene glycol) comodified liposomal delivery system via systemic administration. Mol Pharm. 2011;8(6):2151–2161. | ||
Yuan W, Kuai R, Ran R, et al. Increased delivery of doxorubicin into tumor cells using extracellularly activated TAT functionalized liposomes: in vitro and in vivo study. J Biomed Nanotechnol. 2014;10(8):1563–1573. | ||
Tang J, Li D, Drake L, et al. Influence of route of administration and lipidation of apolipoprotein A-I peptide on pharmacokinetics and cholesterol mobilization. J Lipid Res. 2017;58(1):124–136. | ||
Taylor MJ, Sanjanwala AR, Morin EE, et al. Synthetic high-density lipoprotein (sHDL) inhibits steroid production in HAC15 adrenal cells. Endocrinology. 2016;157(8):3122–3129. | ||
Kuai R, Ochyl LJ, Bahjat KS, Schwendeman A, Moon JJ. Designer vaccine nanodiscs for personalized cancer immunotherapy. Nat Mater. 2017;16(4):489–496. | ||
Wang M, Briggs MR. HDL: the metabolism, function, and therapeutic importance. Chem Rev. 2004;104(1):119–137. | ||
Silver DL, Wang N, Xiao X, Tall AR. High density lipoprotein (HDL) particle uptake mediated by scavenger receptor class B type 1 results in selective sorting of HDL cholesterol from protein and polarized cholesterol secretion. J Biol Chem. 2001;276(27):25287–25293. | ||
Schwendeman A, Sviridov DO, Yuan W, et al. The effect of phospholipid composition of reconstituted HDL on its cholesterol efflux and anti-inflammatory properties. J Lipid Res. 2015;56(9):1727–1737. | ||
Cui L, Lin Q, Jin CS, et al. A PEGylation-free biomimetic porphyrin nanoplatform for personalized cancer theranostics. ACS Nano. 2015;9(4):4484–4495. | ||
Singh A, Evens AM, Anderson R, et al. All trans retinoic acid nanodisks enhance retinoic acid receptor-mediated apoptosis and cell cycle arrest in mantle cell lymphoma. Blood. 2009;114(22):1433–1434. | ||
Ghosh M, Singh AT, Xu WW, Sulchek T, Gordon LI, Ryan RO. Curcumin nanodisks: formulation and characterization. Nanomedicine. 2011;7(2):162–167. | ||
Lerch PG, Förtsch V, Hodler G, Bolli R. Production and characterization of a reconstituted high density lipoprotein for therapeutic applications. Vox Sang. 1996;71(3):155–164. | ||
Brinkman N, Bigler D, Bolli R, Foertsch V, inventors; CSL Behring, assignee. Methods for purification of alpha-1-antitrypsin and apolipoprotein A-1. United States patent US 8962802 B2. 2015 Feb 24. | ||
Tardif JC, Gregoire J, L’Allier PL, et al. Effects of reconstituted high-density lipoprotein infusions on coronary atherosclerosis: a randomized controlled trial. JAMA. 2007;297(15):1675–1682. | ||
Dasseux JL, Sekul R, Buttner K, Cornut I, Metz G, inventors. Apolipoprotein A-I agonists and their use to treat dyslipidemic disorders. World Intellectual Property Organization patent WO 1999016408 A2. 1999 Apr 8. |
Supplementary materials
 | Figure S1 Immunohistochemical staining of SRB1 in H295R tumors. |
 | Figure S2 Body weights of animals treated with indicated formulations. Data represent mean ± SEM (n=4–6). |
 © 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.